

Abstract
Triple-negative breast cancer (TNBC) patients exhibit the worst clinical outcome due to its aggressive clinical course, higher rate of recurrence, and a conspicuous lack of FDA approved targeted therapies. Here, we show that multilayered nanoparticles (NPs) carrying the metastasis suppressor microRNA miR-708 (miR708-NP) localize to orthotopic primary TNBC, and efficiently deliver the miR-708 cargo to reduce lung metastasis. Using a SOX2/OCT4 promoter reporter, we identified a population of miR-708low cancer cells with tumor-initiating properties, enhanced metastatic potential and marked sensitivity to miR-708 treatment. In vivo, miR708-NP directly targeted the SOX2/OCT4-mCherry+ miR-708low tumor cells to impair metastasis. Together, our preclinical findings provide a mechanism-based anti-metastatic therapeutic approach for TNBC, with a marked potential to generate miR-708 replacement therapy for high-risk TNBC patients in the clinic. To our knowledge, this gold nanoparticle-based delivery of microRNA-mimetic is the first oligonucleotide-based targeted therapy for TNBC.
Keywords: Triple negative breast cancer, Metastasis, microRNA, miR-708, gold nanoparticles
INTRODUCTION
Breast cancer is a chronic degenerative condition affecting more than 1.5 million individuals per year worldwide. In the US, of more than the 260,000 patients who are diagnosed with breast cancer each year, over 40,000 succumb to death (1). Breast cancer patients, expressing receptors for estrogen (ER), progesterone (PR), or human epidermal growth factor receptor 2 (Her2), have the availability of FDA-approved targeted therapies (Tamoxifen, Herceptin and Lapatinib) that have been developed against these receptors. However, triple-negative breast cancer (TNBC) patients lack these receptors (ER−, PR−, Her2−), and have the worst outcome due to high rates of recurrence and metastatic spread (2,3), further exacerbated by the lack of FDA approved targeted therapies. As such, a majority (~35%−40%) of TNBC patients relapse within 5 years of diagnosis (4,5). Standard chemotherapy and radiation regimens remain the only therapeutic options for women with TNBC (6,7). These treatments have adverse side effects and usually fail, resulting in an aggressive metastatic relapse and short overall survival (7). As a result, treatment of TNBC, which accounts for ~20% of all breast cancer cases remains a major challenge, and novel approaches are required to block widespread tumor dissemination and metastatic outgrowth for effective treatment of metastatic TNBC (7).
microRNAs (miRs) are endogenously encoded, highly conserved, non-coding RNAs (18-23 nts) that regulate gene expression by sequence-specific binding to messenger RNA (mRNA) and trigger translational repression or RNA degradation (8). The primary miRNA and precursor miRNA (60-100 nts) are cleaved by Drosha and Dicer, respectively, in a stepwise process, followed by incorporation of the single-stranded miRNA into RNA-induced silencing complex (RISC) to become mature miRNA (9,10). miRNAs regulate genes that are involved in a variety of cellular processes, including inflammation, cell-cycle regulation, stem cell fate, differentiation, apoptosis, and migration; and their dysregulation has been shown to play an essential role in the development and progression of cancer (11,12). In cancer, miRNAs may function either as oncogenes or as tumor suppressors (13).
Previously, we identified miR-708 as a “metastasis suppressor”, as it was significantly downregulated in metastatic lesions compared to matched primary tumors in both human and mouse TNBC (14). Mechanistically, miR-708 targeted endoplasmic reticulum membrane protein neuronatin (NNAT), to decrease intracellular calcium levels, resulting in reduced activation of ERK and FAK associated with decreased cell migration, and impaired metastases. Recent reports have also elucidated the tumor suppressor function of miR-708 in various cancers including prostate cancer (15), renal cancer (16), and chronic lymphocytic leukemia (17). These findings provided the rationale for developing miR-708 as a therapeutic agent for metastatic breast cancer.
Two main therapeutic strategies related to miRNAs include: 1) miRNAs that acquire a gain-of-function in the diseased tissue (e.g. oncogenic miRNAs) can be inhibited by using miRNA antagonists, such as anti-miRs and antagomiRs; and 2) miRNAs that show a loss-of-function can be delivered as miR-mimetics. However, nucleotide-based therapeutic approaches have been plagued with numerous challenges including stability, bioavailability, and therapeutic delivery (18–20). However, the recent approval of the antisense therapeutic nusinersen, for the treatment of spinal muscular atrophy (21), has reinvigorated enthusiasm in developing oligonucleotide technologies as next generation therapeutics.
Nanoparticle (NP)-based targeted delivery of miRNAs has emerged as an alternative approach in cancer therapy. NP offers several advantages as they can prevent degradation of oligonucleotides by endogenous nucleases, and provide sustained release overtime, which allows reduced frequency of therapeutic administration. Gold-based NPs provide many advantages, for instance tunable size, biocompatibility, and low cytotoxicity (22). Recently we have developed a layer-by-layer (LbL) fabrication method to deliver siRNA (23–26). The LbL fabrication method is a gentle and simple layering procedure based on the charge-charge interactions between the positively and the negatively charged polymers (27–29). Gold nanoparticles (AuNPs) were used as the core for their uniform size, electronic features, biocompatibility, and feasibility of surface modification (22,30). Onto an inert AuNPs, negatively charged siRNAs were layered between the positively charged poly-L-lysine (PLL) layers, which are sensitive to proteases such as cathepsin B which is upregulated in tumors (24). The degradation of PLL enabled the slow release of the siRNAs underneath. The assembled NP protected siRNAs from endogenous nuclease degradation, and prolonged their release and efficacy, which allowed reduced frequency of therapeutic administration. Using this innovative construct, miR-708 mimetic containing nanomedicines were able to restore tumor suppressive miR-708 in metastatic cells, and demonstrate the therapeutic efficacy for inhibiting TNBC metastasis in vivo.
MATERIALS AND METHODS
Chemicals and materials
All miR mimetics, siRNAs, and poly-L-lysine (PLL) (Mw = 30,000~70,000 g/mol) were obtained from Sigma-Aldrich (St. Louis, MO). Bare AuNPs (40 nm) particles were purchased from BB International (Cardiff, UK). D-Luciferin and CellTiter aqueous one solution was obtained from Promega (Madison, WI).
Preparation and characterization of multilayer AuNPs
Using our well-developed LbL nano-carrier fabrication method (23–26,31,32), multilayers of miR mimetic/siRNA and PLL were successfully deposited on Au surface. The sequences of miR mimetic against miR-708(14) are sense strand: 5’-AAGGAGCUUACAAUCUAGCUGGGdTdT-3’, antisense strand: 5’-CCCAGCUAGAUUGUAAGCUCCUUdTdT-3’, and of control nonsnese miR (scr) are sense strand: 5’-GCAAGCUCGCGUGGUAUAGAAUAdTdT-3’, antisense strand: 5’-UAUUCUAUACCACGCGAGCUUGCdTdT-3’. The sequence of siRNA against luciferase (33) are sense strand: 5’- UUAAUCAGAGACUUCAGGCGGUdTdT-3’, antisense strand: 5’-ACCGCCUGAAGUCUCUGAUUAAdTdT-3’. AuNP solution (3.15 × 109 particles in 0.7 ml) was added dropwise onto a PLL solution (0.5 ml of 5 mg/mL) in pure water. After incubating for 30 minutes in the dark with gentle shaking, the solution was centrifuged for 30 minutes at 16,100 g using a micro centrifuge (Eppendorf, Hauppauge, NY). The supernatant was removed, and the gel-like deep red pellet was re-suspended with pure water and centrifuged for 30 minutes at 16,100 g. PLL coated AuNPs were stored in pure water after an additional wash. Next polyelectrolyte layer was deposited by adding PLL coated AuNPs (in 0.5 ml pure water) to miR solution (4.0 nmole, 0.5 ml). The reaction solution was incubated in the dark for 30 minutes with gentle shaking, followed by three washes. The deposition procedures were repeated to yield a total of 5 layers of polyelectrolytes (1 layer of PLL, 2 layers of PLL-cy5.5, and 2 layers of miR mimetic or siRNA). Sizes and zeta potentials of AuNPs in water were measured by Zetasizer Nano-ZS (Malvern, Worcestershire, UK) according to the manufacturer’s instruction.
Cellular uptake of miR708-NP
MDA-MB-231 cells were collected by trypsinization, counted and plated in a 96-well black clear bottom culture plate (Corning, Pittston, PA) at a density of 5 × 103 cells per well. After 1 day, miR708-NP (0.24 μM of miR-708) were added and incubated for 24 hours. Cells were then washed with PBS and imaged under EVOS FL Auto Cell Imaging System (Life Technologies, Carlsbad, CA).
Mice and cell lines
All animal work was conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee at Weill Cornell Medical College. CB-17 SCID mice and CB17.Cg-PrkdcscidHrhr/IcrCrl mice were obtained from Charles River (Wilmington, MA). BALB/cJ mice were obtained from Jackson Laboratory. 293T and MDA-MB-231 cells were obtained from ATCC and maintained in DMEM, 5% FBS, 1% Pennicilin/streptomycin and 1% L-glutamine. Lung metastatic derivative of human breast cancer cell line, MDA-MB-231-LM2 (LM2), was obtained from Dr. Joan Massague at MSKCC. The time between thawing and use of cell lines did not exceed one month. All animal experiments were performed within one week of thawing the cells. For bioluminescent tracking, these cells consists of a triple-fusion protein reporter construct encoding herpes simplex virus thymidine kinase 1, green fluorescent protein (GFP) and firefly luciferase (34). LM2 breast cell lines were maintained as described (14). SOX2/OCT4 response elements (SORE6), consisting of destabilized form of the mCherry fluorescent protein (dsmCherry), and assembled into pDest-663, a lentiviral destination vector based on the pFUGW lentiviral backbone with puromycin selection, were obtained from Wakefield lab (35). LM2 cells were infected with this lentivirus for all experiments. For in vitro experiments using NPs, cells were plated in 6-well plates 24 hours before the addition of NPs. After NP addition, cells were incubated for 48 hours before using them for further experiments.
Metastasis assay, bioluminescence imaging, and analysis
For initial in vivo NP efficacy analysis by testing luciferase gene silencing, 8-week old CB17.Cg-PrkdcscidHrhr/IcrCrl were injected in the fourth mammary fat pad with 106 LM2 cells in a total volume of 50 μl of PBS. Tumor growth was monitored by live animal bioluminescence imaging (BLI) with Xenogen (PerkinElmer, Waltham, MA) or Xtreme (Bruker, Billerica, MA) once per week. Briefly, mice were anesthetized and injected in the retroorbital plexus with 75 mg/kg of D-luciferin in 50 μl total volume. 19 days after orthotopic implantation, mice were injected with either PBS or AuNP coated with siRNA-Luciferase (siR-Luc-NP) and coated with cy5.5, on day 19 and day 22, via tail vein. BLIs and fluorescence was measured every alternate day for one week.
To monitor metastatic outgrowth from orthotopic tumors, 8-wk-old CB-17 SCID mice were injected in the fourth mammary fat pad with 1 × 106 LM2-SORE6 cells in a total volume of 50 μl of PBS. Tumor growth was monitored by live animal BLI (Xenogen) once per week. After 4 weeks of primary tumor growth, NPs (scr-NP or miR708-NP) were administered via tail vein twice per week for two weeks. Primary tumors were then resected at week 6, and animals were allowed to recover. Lung metastasis was measured by BLI at week 7, the endpoint at which all lungs were harvested for H&E staining.
Quantitative miR PCR
RNA, including miRNA, was extracted using mirVana™ miRNA isolation kit (Thermofisher), according to manufacturer’s protocol. miR-708 expression was measured by using Custom TaqMan microRNA Assays (Applied Biosystems, CA). RNU48 (Applied Biosystems) was used for normalization. 10 ng of RNA was used to make cDNA using TaqMan microRNA assays kit, and qPCR was performed as per the manufacturer’s protocol (Applied Biosystems, CA).
Quantitative RT-PCR analysis
Total RNA extracted, as described above, was converted to cDNA using Superscript II reverse transcriptase (Invitrogen). qPCR primers for NNAT (Forward: ACCGCATTCTGATCTGGACA; Reverse: ACCCTCCTTCCTCAACTGTG) and GAPDH (Forward: CTTCAACAGCGACACCCACTCCTC; Reverse: GTCCACCCTGTTGCTGTAG) were obtained from Integrated DNA Technologies (IDT), and qPCR was performed using iQ SYBR Green supermix (Bio-rad). Each sample was run in triplicates to minimize pipetting error. A standard protocol of initial denaturation at 95 °C for 3 minutes, 40 cycles of 95 °C for 20 sec, 60 °C for 30 sec, and 72 °C for 30 sec, followed by final extension at 72 °C for 5 minutes and melt curve analysis was applied on a Bio-rad CFX96 Real Time System (Bio-rad) coupled with Bio-Rad-CFX Manager software. The relative abundance of each transcript was compared with the control.
Cell Recovery from tumor xenografts
Freshly excised tumors were minced with scalpel blades, and digested using 1 mg/ml collagenase I (Sigma), and 1 mg/ml DNAse (Sigma) for 30 minutes at 37 °C. Cells were then passed through a 40 μm cell strainer (BD Bioscience), and suspended in FACS buffer. BD Influx cell sorter (BD Biosciences) was used to sort and collect GFP+ cy5.5+ tumor cells for mammosphere assay.
Wound healing assay
Cells (LM2, and FACS sorted SORE6− and SORE6+ cells) were plated in triplicates into 6-well plates (0.5 × 106 cells/well) and cultured in DMEM containing 10% FBS. A scratch wound was generated using sterile plastic 200 μl pipette tip, and floating debris was removed by washing with PBS, and supplemented with serum-free DMEM. Cell migration was observed at different time points (6, 24, 36, 48, and 72 hours). The area unoccupied was measured by phase-contrast microscopy. The area left unoccupied was measured using ImageJ program (NIH).
Mammosphere formation assay
To assess tumorsphere-forming ability, single-cell suspensions of tumor cells (collected from primary LM2 tumors or from in vitro NP treatment) were plated in ultra-low-attachment 24-well plates (Costar) at 1,000 cells/well in mammosphere media (in triplicates), which was prepared by combining 50 ml of phenol-free DMEM (Gibco) supplemented with 1% L-glutamine, 1% penicillin/streptomycin, 1X B27 supplement minus Vitamin A (Gibco), 20 ng/ml rhEGF (R&D Systems), and 20 ng/ml bFGF (Invitrogen). The plate was left undisturbed for two weeks in a 37 °C incubator. The mammospheres were then counted and measured using a computerized Zeiss fluorescent microscope (Axiovert 200M) at 10X magnification. For in vitro mammosphere assay using cultured cells, LM2, or FACS sorted SORE6− and SORE6+ cells were treated with either scr-NP or miR708-NP for 48 hours, and then plated for mammosphere assay as described above. The median mammosphere size (diameter) per experiment was used as the cut-off for a tumorsphere to be counted as one.
Immunostaining and microscopy
For immunofluorescence staining, cells were fixed in acetone, followed by staining of the nuclei with DAPI. Fluorescent images for GFP (tumor cells), mCherry (SORE6+ population), cy5.5, and DAPI were obtained using a computerized Zeiss fluorescent microscope (Axiovert 200M), fitted with an apotome and an HRM camera. Images were analyzed using Axiovision 4.6 software (Carl Zeiss Inc.).
Histologic and clinical pathologic analysis
Balb/cJ mice (8-week old) were injected with 1 × 105 4T1 cells in the fourth mammary fat pad. To examine in vivo cytotoxic effect of NP-delivery system, AuP (AuNP/PLL) or cy5.5 conjugated scr-NP were injected via tail vein in the mice (n = 3 per group). Three mice without any treatment were used as controls. For histologic analysis, the excised tumors and main organs of interest (heart, liver, kidneys, spleen, and bone marrow) in formaldehyde (10%) were dehydrated with ethanol, embedded in paraffin, and then slides containing 5 μm sections were prepared. The slides were subsequently stained with hematoxylin and eosin Y solution (H&E) to assess histological alterations via light microscopy, conducted by a single blinded pathologist. Blood samples were collected via retro-orbital sinus puncture into no anticoagulant or 3.2% citrate anticoagulant for the analysis of biochemical analytes (renal tests, liver, and muscle enzyme activity) and hemograms, respectively. The analysis was conducted in the Clinical Pathology Laboratory in the Animal Health Diagnostic Center at Cornell University, Ithaca, using automated equipment (Hitachi P modular chemistry analyzer, Roche Diagnostics, Indianapolis, IN, USA and ADVIA 2120 hematology analyzer, Siemens Healthcare Diagnostics Inc., Tarrytown, NJ) using manufacturer reagents, with the exception of GLDH (Randox Laboratories Ltd, Antrim, UK).
Measurement of plasma cardiac troponin I
Cardiac injury was assessed by measuring plasma levels of cardiac troponin I from control, AuP and scr-NP injected mice using a highly sensitive mouse cardiac troponin I ELISA kit (Life Diagnostics, West Chester, PA) according to the manufacturer’s instructions.
Cytotoxicity measurements
The cytotoxicity of prepared NPs was measured by MTT assay. Briefly, MDA-MB231 cells were collected by trypsinization, counted, and plated in a 96-well culture plate at a density of 5 × 103 cells per well. One day after seeding, various NPs (1.26 × 108 particles) were added, and the cells were further cultured for additional 24 or 72 hours. At day 2 or day 4, 20 μL of CellTiter solution (Promega, Madison, WI) was added to each well and incubated for 3 h. The absorbance of the solution was then measured at 490 nm using a plate reader (Tecan, Mannedorf, Switzerland).
Cell invasion
For transwell assays, 15,000 cells (bulk LM2 or FACS sorted SORE6− and SORE6+) were plated in triplicates onto the top chamber of 6.5 mm inserts in 24-well plates (Corning) with serum-free DMEM. The cells were either treated with scr-NP or miR708-NP. The bottom chamber contained complete DMEM media with 10% FBS. The cells migrated for 48 hours. After migration, the insert was washed twice with fresh PBS and fixed using Kwik-Diff™ Staining kit (ThermoFisher), according to the manufacturer’s protocol. After staining, images were obtained using a computerized Zeiss microscope (Axiovert 200M) and analyzed using Axiovision 4.6 software (Carl Zeiss Inc.).
Cell proliferation
For cell proliferation assays, 0.5×106 FACS sorted SORE6− and SORE6+ cells were cultured, either with miR-708 or scr treatment, separately in 6-well dishes for 2 days. EdU (10 nM) was administered to culture medium for 30 minutes to label proliferating cells. Cells were harvested, washed once with PBS, and fixed with 4% paraformadehyde for 15 minutes at RT. Fixed cells were permeabilized and stained for EdU incorporation and DNA content with the Click-iT® EdU Cell Proliferation Assay kit (Invitrogen Inc) according to the standard protocol. Cell cycle phases were analyzed using a dual parameter plot of Click-iT™ Plus EdU Alexa Fluor™ 647 and FxCycle™ Violet, according to the manufacturer’s protocol. Flow analysis was performed using BD Fortessa coupled with Diva software (BD Bioscience).
Apoptosis assay
For apoptosis detection, APC annexin V Apoptosis Detection Kit with 7-AAD (Biolegend) was utilized. 0.5×106 sorted SORE6− and SORE6+ populations from LM2 cells were either treated (in triplicates) with miR-708 or scr oligos using oligofectamine for 48 hours. These cells were then harvested, washed with FACS buffer, stained with annexinV and 7AAD for 15 minutes, before analyzing using BD Fortessa. Cells that were double positive for annexinV and 7AAD were counted as dead cells in the total population analyzed.
Mouse primary tumor flow cytometry staining
To study the immunogenicity of AuNPs, 4T1 breast cancer model was used. 8-week old Balbc/J mice were injected with 1 × 105 4T1 cells in the fourth mammary fat pad. The tumor sizes in all the mice were measured weekly using calipers. At week 2, when tumors reached 400-500mm3, treatment with AuNPs was started. To examine in vivo effect of NP-delivery system. AuP (AuNP/PLL) or cy5.5 conjugated scr-NP were injected via tail vein in a 4T1 breast tumor bearing mice (n = 3 per group). The mice were treated with two doses of either empty nanoparticles (AuP) or with scr-NPs. Three mice without any treatment were used as controls. After one week of treatment, the mice were euthanized and primary tumors were collected for determining the immunogenic effect of AuNPs. Primary tumors from all three groups were ground through a 140 μM wire mesh into RPMI-1640 media supplemented with 10%FBS, penicillin-streptomycin, and L-glutamine. Single-cell suspensions were obtained by subsequent filtration through 70 μM filters. Red blood cells were lysed using ACK lysing buffer. The cell suspension was then strained through a 40 μM filter. Cell pellets were washed with FACS buffer by centrifuging at 1500 RPM for 5 minutes. For surface stains, samples were blocked with anti-mouse CD16/32 for 15 minutes at room temperature, incubated with primary antibodies for 45 minutes in dark on ice, washed with FACS buffer, fixed with 1% formaldehyde for 30 minutes at room temperature in dark, washed with FACS buffer, resuspended in FACS buffer and stored at 4°C until analysis, which was performed within 24 hours of staining. For intracellular staining with stimulation, samples were stimulated with PMA (100ng/mL) and ionomycin (1μg/mL) for 4 hours in complete RPMI at 37°C in a humidified incubator. Brefeldin A (Biolegend) and Monensin (Biolegend) were also added to achieve golgi blocking. Samples were then blocked with anti-mouse CD16/32 for 15 minutes at room temperature, incubated with primary antibodies for 45 minutes on ice for surface staining, washed with FACS buffer, fixed with Fixation/Permeabilization Buffer (ebioscience) for 30 minutes in dark on ice, washed with permeabilization buffer and incubated with primary antibodies for cytokine staining for 30 minutes on ice. These were then washed 3 times with permeabilization buffer, resuspended in FACS buffer, and stored at 4°C for flow cytometric analysis within 24 hours. Acquisition was performed using Becton-Dickinson LSR II and analysis using FlowJo 10.
Myeloid population analysis
For distinguishing different myeloid-derived cells, single cell suspensions from primary tumors were stained with CD45 (clone 30-F11), CD11b (clone M1/70), CD11c (clone N418), F4/80 (clone BM8), Ly-6G (clone 1A8), Ly-6C (clone HK1.4), and MHC-II (clone M5/114.15.2). All antibodies were obtained from Biolegend and used at a dilution of 1:100. Granulocytes were defined as CD45+, CD11b+, Ly-6C(low), Ly-6G+, CD11c−, F4/80−. Dendritic cells were defined as: CD45+, CD11c+, F4/80−, Ly-6G−, MHC-II+. Finally, myeloid-derived suppressor cells were identified as follows: CD45+, Ly-6G+, CD11c−, F4/80−.
Immunogenic profile of AuNPs in primary tumors
For immunogenicity, single cell suspensions from primary tumors were stained with the following antibodies after stimulation: CD3 (clone 17A2), CD4 (clone RM4-5), CD8a (clone 53-6.7), FoxP3 (clone FJK-16s), PD-1 (clone 29F.1A12), IL-2 (clone JES6-5H4), TNFa (cloneMP6-XT22), IFNg (clone XMG1.2), and GzmB (clone GB11). Zombie dye (BV510) was used as a live-dead stain for fixed samples. All antibodies were obtained from Biolegend and used at a dilution of 1:100.
Statistical analysis
Results are expressed as mean ± SE, unless otherwise mentioned. Analyses of different treatment groups was performed by non-parametric Mann-Whitney test, using GraphPad Prism statistical program. When comparing 3 or more groups, we used a one-way ANOVA, together with Tukey post-test, to compare all treatment groups to each other. P values <0.05 will be considered statistically significant.
RESULTS
Synthetic miR-708, delivered using AuNPs, is functional
We fabricated AuNPs (40 nm) with miR-708 mimetic (miR708-NP) (14), or a scramble miRNA control (scr-NP) using a previously optimized protocol with minor modifications (24) (Fig. 1A). AuNP was sequentially coated with PLL, miRNA, PLL-cy5.5, miRNA and PLL-cy5.5 by electrostatic interactions. Cy5.5 fluorescent dye was added to PLL for tracking purposes. Hydrodynamic sizes of the formulated nano-vector were measured by dynamic light scattering (DLS), following each layer of coating (Fig. 1B). The size of the uncoated AuNPs was 40 nm, and the particle size increased steadily with the number of layers added; AuP (AuNP/PLL): 58 nm; miR1: 87 nm; miR1P: 110 nm; miR2: 123 nm; miR2P: 136 nm.
Figure 1. Characterization and in vitro functional efficacy of miRAuNPs.
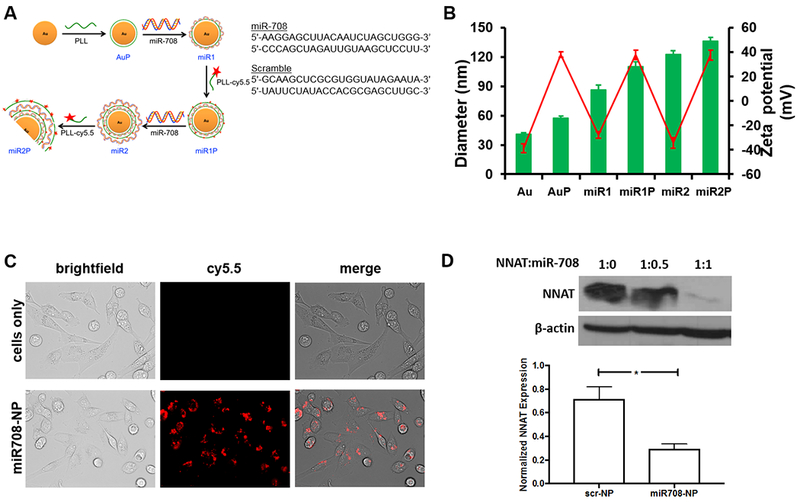
A) Schematic illustrating the process of preparing multilayered miRNA-coated AuNPs (miRAuNPs) by electrostatic interaction. B) The average size (green bar) and zeta potential (red line) of multilayered miRAuNPs. C) Cellular uptake of multilayered miR708-NP. Images of released miRNA from miRAuNPs were visualized using fluorescence microscopy with cy5.5 filters after 24 hours incubation with or without miR708-NP in MDA-MB-231 cells. D) NNAT expression after treatment with miR708-NP was significantly reduced (p = 0.02). 293T cells were transfected with NNAT. 48 hours after transfection, NPs (scrambled (scr-NP, scr: 0.24 μM) or miRNA-708 (miR708-NP, miR708: 0.24 μM)) were added to transfected 293T cells in triplicates. After 48 hours, protein and RNA content of NNAT was measured via western blotting (upper) and qPCR (lower). A 1:1 ratio of NNAT to miR-708 (by AuNPs) is defined at 0.24μM each. The experiment was performed in triplicates, at least three times. *p<0.05.
Since LbL fabrication is based on charge-charge interactions, the zeta-potential of the particle represents the surface polymer. The first layer of positively charged PLL converted the bare AuNPs from negatively (−39 mV) into positively charged particles (+38 mV). The subsequent coating of the negatively charged miRNA then converted the whole AuNP back to negatively charged particles (−28 mV). The overall charge of the final assembled NPs which have 3 layers of PLL and 2 layers of miR mimetics was approximately 37 mV. The zigzag pattern of the surface zeta-potential indicated the successful coating of each layer (Fig. 1 B).
Next, to determine if breast cancer cells can incorporate the assembled miR708-NP, we incubated MDA-MB-231 cells with miR708-NP (0.24 μM of miR-708); intracellular cy5.5 fluorescence signal suggested that the particles were effectively internalized by cells (Fig. 1C). To determine the functional efficacy of miR708-NP, 293T cells over-expressing NNAT, a downstream target of miR-708 were incubated either with miR708-NP or scr-NP for 24 hours. Notably, NNAT was significantly suppressed by miR708-NP, compared to scr-NP, as seen at the protein (Fig. 1D upper) and RNA (Fig. 1D lower) levels. These findings suggest that AuNPs can effectively deliver miR-708 cargo into cells.
Nanoparticle-mediated delivery of oligonucleotide in vivo
We first performed a pilot experiment to determine the efficacy of NP delivery in vivo. An orthotopic metastatic TNBC model, MDA-MB-231-LM2 (LM2), was used to examine the distribution of the particles and to assess if the oligonucleotide cargo was delivered to the primary tumors (Fig. 2A). LM2 cells consist of a triple-fusion protein reporter construct encoding herpes simplex virus thymidine kinase 1, green fluorescent protein (GFP) and firefly luciferase (34). Mice with LM2 -derived mammary tumors, expressing luciferase (luc) reporter gene, were administered AuNPs layered with cy5.5-PLL and siRNA targeting luciferase gene (siR-luc). Fluorescence imaging showed that the siR-luc-NP particles were localized in the primary tumors, as determined by cy5.5 fluorescence (Fig. 2C). Importantly, NP localization was associated with a concomitant suppression of luciferase gene expression (Fig. 2C–D), suggesting that the NPs homed to the primary tumor, and were able to deliver the siRNA cargo to the tumor site. As expected, there was no difference in tumor sizes between the controls and siR-luc-NP treated mice (Fig. 2B), suggesting that the luciferase suppression was specific to siRNA treated group, and not due to any cytotoxic effect of AuNPs. This is consistent with no significant toxicity observed with either miR708-NP or siR-luc-NP treated MDA-MB-231 cells (Fig. S1).
Figure 2. In vivo efficacy of NPs.
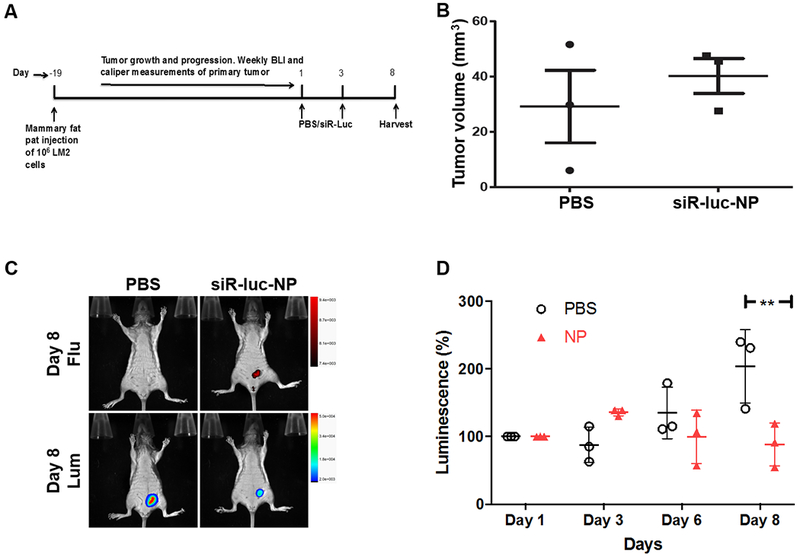
A) Primary tumor model. MDA-MB-231-LM2 cells were injected into the fourth mammary fat pad of female SCID mice. At 500 mm3 tumor volume, AuNPs were layered with siRNA against luciferase gene (sense strand: 5’-UUAAUCAGAGACUUCAGGCGGUdTdT-3′, antisense strand: 5’-ACCGCCUGAAGUCUCUGAUUAAdTdT-3’), labeled with cy5.5 (siR-luc-NP, siR-luc: 1.58 mg/kg) and injected via tail vein. The mice were imaged daily by Xtreme for luminescence and fluorescence. B) Tumor size of PBS versus siR-luc-NP treated groups. C) Accumulation of NPs in the tumor. 8 days following siR-luc-NP or PBS injection, siR-luc-NPs were specifically retained at the primary tumor site via enhanced permeability and retention (EPR) effect, evident in the localized fluorescent signal from mice injected with siR-luc-NP compared to the PBS cohort. D) In vivo gene-silencing effect of siR-luc-NP. siR-luc-NP or PBS was injected twice during 8 days, and the luminescence intensity were measured by Xtreme. The luminescence intensity of each mouse at day was set as 1. **p<0.01.
Together these findings suggest that miR-708 NPs may be used to determine the impact of therapeutic miR-708 delivery on breast cancer metastasis in vivo.
Nanoparticle-mediated delivery of miR-708 impairs metastasis
After having determined the delivery of AuNPs in the orthotopic tumor model, we explored the consequences of NP-mediated miR-708 delivery on primary tumor progression and metastasis. At week 4, mice were injected with AuNPs layered with either scrambled (scr-NP) or miR-708 mimetic (miR708-NP) twice weekly, for two weeks via tail vein (Fig. 3A). These NPs were also labeled with cy5.5 to track their localization, and mice were subsequently imaged for both luminescence and fluorescence. Both miR708-NP and scr-NP co-localized at the primary tumor site (Fig. 3B, Fig. S2), and did not show any reduction in primary tumor growth (Fig. 3C). To determine if miR708-NP efficiently delivered the miR-708 cargo in vivo, we analyzed resected primary tumors from the two cohorts (scr-NP and miR708-NP) for miR-708 levels. Using RNA extracted from bulk primary tumors, we found that the miR708-NP-treated group had higher levels of miR-708 expression (Fig. 3D) compared to the scr-NP cohort. Primary tumors were resected at 1 cm3. Metastases were analyzed via BLI (Xenogen). In the scr-NP group, mice developed significantly more lung metastases than the miR708-NP group (Fig. 4A–B). The number of metastatic nodules in the lungs were quantified by H&E, which, consistent with the BLI data, showed that miR708-NP delivery led to markedly fewer number of metastases as compared to scr-NP (Fig. 4C–D).
Figure 3. In vivo miR-708 delivery.
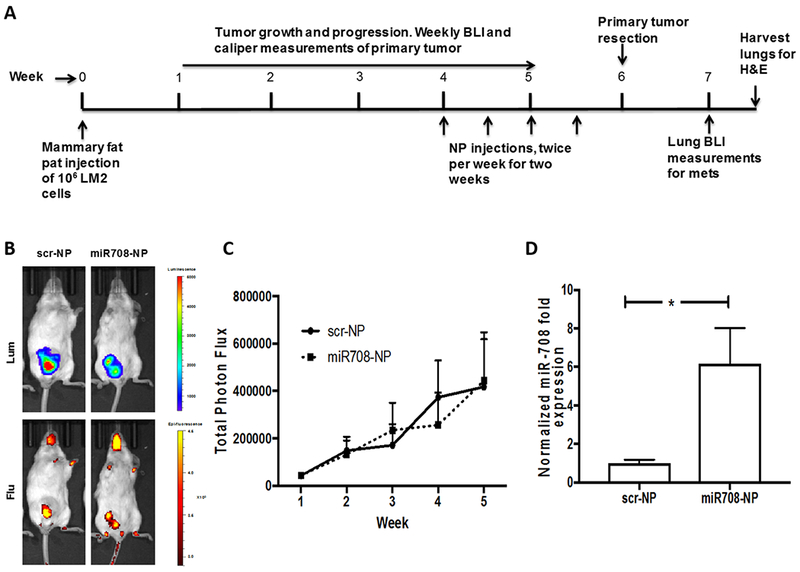
A) Schematic representation of the experiment design (n=10 per group). LM2 cells were injected into the fourth mammary fat pad of female SCID mice. At week 4, AuNPs layered with either with scr (scr-NP, scr: 1.58 mg/kg) or miR708 (miR708-NP, miR708: 1.58 mg/kg) oligos, and labeled with cy5.5 were injected via tail vein, twice weekly for two weeks. The mice were imaged for luminescence and fluorescence. B) Representative luminescence (Lum) and fluorescence (Flu) images of primary tumors. C) BLI quantification of primary tumors in the two groups. No change in primary tumor size in either group (p = 0.9) was observed. D) Bulk primary tumors (n = 5 per group) from the mice in each group (scr-NP or miR708-NP) were lysed, and analyzed for the expression of miR-708. Primary tumors from the mice treated with miR708-NP had higher miR-708 expression (p = 0.04). *p<0.05
Figure 4. Reduction in metastases by miR-708 delivery.

A-B) Primary tumors were resected at 1cm3. The mice (n = 10 per group) were monitored for lung metastasis by BLI, one week after resection. There was a significant reduction in total photon flux from lungs of mice treated with miR708-NP (P = 0.03). C-D) 1.5 weeks after resection, mice were sacrificed, and lungs were harvested for H&E analysis. Representative images of lung metastases (black arrows point to mets in lungs) and the quantification of the two cohorts, scr-NP (n=7 per group) and miR708-NP (n=8) (p = 0.003). *p<0.05, **p<0.01.
Together these findings suggest that NPs localize to the primary tumors, where they efficiently deliver the miR-708 mimetic resulting in reduced metastasis.
miR-708 targets a specific subset of invasive cancer cells in the primary tumor
To obtain insights by which NP-mediated delivery of miR-708 mimetic impairs metastasis without impacting primary tumor growth, we evaluated cell migration, a hallmark of the metastatic process using a wound-healing assay in vitro. As expected, treatment with miR-708 mimetic significantly reduced migration of LM2 cells as compared to scr-NP-treated cells (Fig. 5A). To determine if miR-708-mediated loss of cell migration impaired metastasis, we utilized a lentiviral-based stem cell reporter system that marks cells with higher metastatic potential (invasion, metastasis and mammosphere formation) in primary breast cancer cell lines (35). In this vector, six tandem repeats of composite core stem cell transcription factors SOX2 and OCT4 response element (SORE6) drives the expression of destabilized copepod mCherry reporter transgene (Fig 5B, upper), which allows accurate analysis of rapid/transient promoter activity (35). Following stable integration and selection, approximately 30% of GFP+ LM2 cells expressed mCherry consistent with that observed previously, compared to controls (a minimal CMV promoter lacking the SORE6 element) (35).
Figure 5. Specific targeting of a tumor subpopulation by miR-708 in vitro.
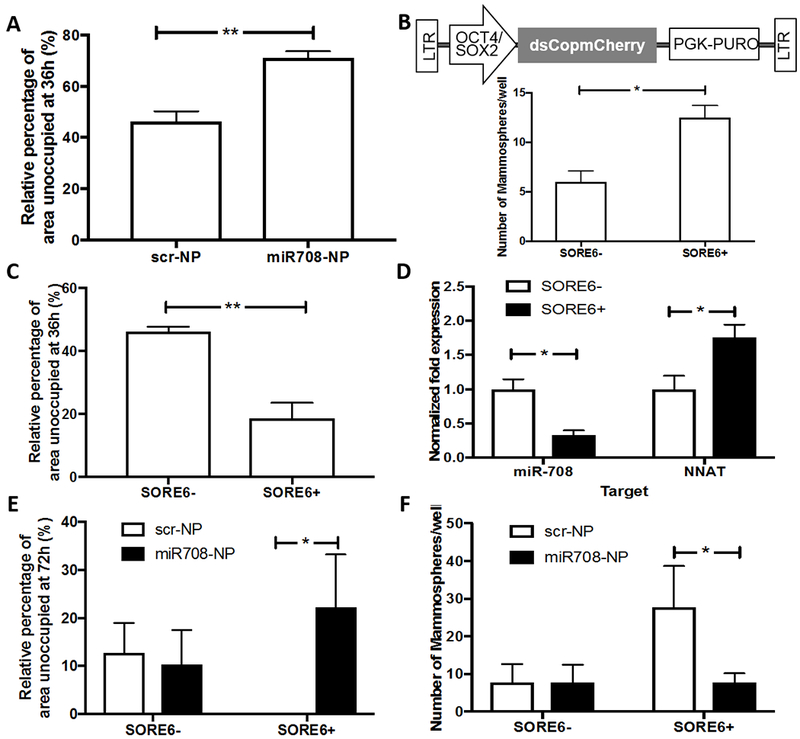
A) miR-708 overexpression in LM2 cells leads to a significant reduction in migration as compared to scr delivery in the same cells (p = 0.005). B) Reporter construct for SOX2/OCT4 (35) (upper panel); in vitro mammospheres formed by SORE6+ population is significantly higher than SORE6− cells (p = 0.03) (lower panel). C) A more migratory population (SORE6+) was detected using a SOX2/OCT4 reporter construct in SORE6+ cells (p = 0.005). D) SORE6+ and SORE6− cells were FACS sorted and analyzed for miR-708 (p = 0.03) and NNAT (p = 0.01) levels by qPCR. SORE6+ cells have inherently higher NNAT levels, with lower expression of miR-708 as compared to SORE6− cells. E) After treatment with either scr-NP or miR708-NP, migration in SORE6+ cells was reduced (p = 0.03), whereas SORE6− cells remained unaffected (p = 0.9). F) Mammosphere formation by sorted SORE6+ populations from in vitro cells in culture decreased (p = 0.011) after treatment with miR708-NP, whereas sorted SORE6− cells remained unaffected (p = 0.9). *p<0.05, **p<0.01.
FACS-sorted SORE6+ (GFP+ mCherry+), and SORE6− (GFP+ mCherry−) tumor cells were used for cell migration assay. As characterized, SORE6+ cells formed significantly higher numbers of mammospheres compared to SORE6− cells (Fig. 5B, lower), thus demonstrating their pluripotency. SORE6+ cells exhibited a higher migration potential as well, compared to SORE6− cells (Fig. 5C, Fig. S3A). We next analyzed SORE6+ and SORE6− cells for expression levels of miR-708 and its downstream target mRNA NNAT. Remarkably, miR-708 level was significantly lower in SORE6+ population than in SORE6− cells, which was inversely associated with NNAT expression (Fig. 5D). Following this observation, we determined the impact of miR-708 on the migration potential of SORE6− and SORE6+ cells. A higher migration rate, which was observed in SORE6+ cells, was significantly reduced following delivery of miR708-NP compared to scr-NP, in vitro (Fig. 5E, Fig. S3B). In transwell assays, although SORE6+ cells showed increased invasion potential (Fig. S4A), miR-708 treatment did not impact invasion (Fig. S4B).
SORE6+ cells displayed higher mammosphere forming potential compared to SORE6− cells, which is indicative of tumor initiating capacity (TIC)/pluripotency (Fig. 5B, lower). Importantly, these SORE6+-derived mammospheres were significantly reduced following delivery of miR708-NP compared to scr-NP (Fig. 5F).
Next, we generated primary breast tumors by administration of 1×106 GFP+ LM2 cells stably expressing the SORE6 reporter (mCherry+) to see the effect of our treatment on miR-708low and miR-708high cells in vivo. These mice were treated with NPs, as described above. Tumor cells (GFP+, cy5.5+: marking tumor cells that effectively received NP treatment), sorted from bulk primary tumors, were analyzed for mammosphere-forming capacity (Fig. 6A). The signal for cy5.5 shows that sorting of double positive (GFP+, cy5.5+) cells was effective (Fig. 6C). We observed that miR-708 treated tumor cells formed significantly less mammospheres, as compared to scr cohort (Fig. 6B). Analysis of primary tumors by immunofluorescence (IF) microscopy showed that amongst the GFP+ cancer cells a subset of cells expressed the mCherry reporter (Fig. 6E). Importantly, administration of miR708-NP markedly reduced the number of mCherry+ tumor cells compared to scr-NP treatment (Fig. 6D–E). As observed before miR-708 overexpression had no effect on proliferation of LM2 cells (Fig. S5). The reduction in mCherry+ cells was not due to enhanced apoptosis of sorted SORE6+ cells with either scr or miR-708 overexpression (Fig. S6) or decreased proliferation (Fig. S5).
Figure 6. In vivo targeting of SORE6+ cells by miR708-NPs.
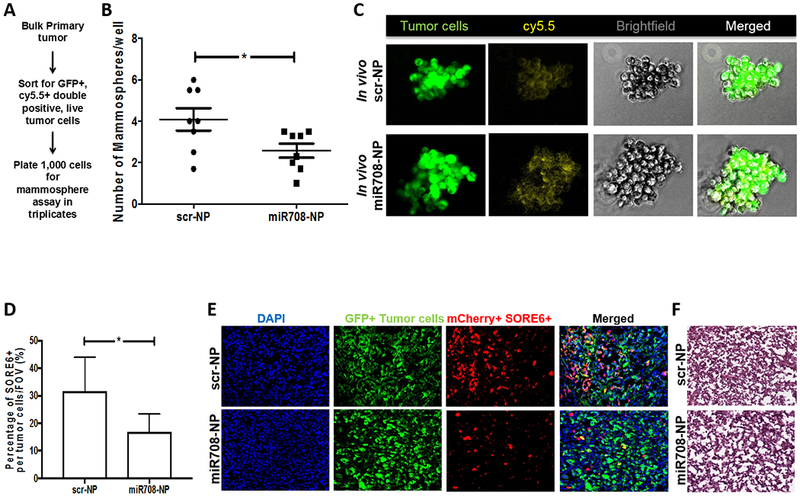
A) Schematic of cell sorting from bulk primary tumors. B) Mammosphere formation using tumor cells sorted (GFP+, cy5.5+) from bulk primary tumors (n = 8 per group) was also evaluated and was significantly reduced in the cohort treated with miR708-NP (p = 0.03). C) Images of representative mammospheres from scr-NP and miR-708 NP treated cohorts. D) Percentage of mCherry+ SORE6+ cells per GFP+ tumor cells from the IF images of primary tumors in scr-NP (n=5) and miR708-NP (n=5), quantified using ImageJ (p = 0.04). E) IF of primary tumors depict lesser mCherry+ SORE6+ cells in miR708-NP treated mice compared to scr-NP group. F) H&E staining of corresponding primary tumors from panel (C). *p<0.05
The results from our in vitro data suggest that the SOX2/OCT4-mCherry+ miR-708low tumor cells, which have an inherently higher mammosphere forming and migration properties (Fig. 5A–B), may be targeted by miR-708-NP treatment to reduce their motility (Fig. 5E) and tumor initiating capacity (Fig. 5F). Thus, miR-708 delivery may affect the more invasive SORE6+ population in the bulk tumors. When miR-708 mimetic is delivered to the bulk primary tumors, it may lead to an increased expression of miR-708 in these miR-708low mCherry+ SORE6 cells. This overexpression may subsequently decrease the pluripotency of these cells, as these cells then form significantly lesser mammospheres after treatment with miR-708. Upon cell division, a SORE6+ cell may divide into either a SORE6− or a SORE6+ cell. From our experimental observations, miR-708 delivery also affects the probability of SORE6+ cells dividing to form more of SORE6− daughter cells, leading to a dilution of mCherry signal in bulk tumors (Fig. 6D–E) and mammospheres (Fig. S7). Thus, dilution of this more metastatic population may be important for increasing therapeutic efficacy of other treatment regimens, for instance chemotherapeutic, which may be combined with miR-708 mimetics.
Biodistribution and toxicity profile of NP-mediated delivery system
To examine in vivo cytotoxic effect of NP-based delivery system, AuP or cy5.5 conjugated scr-NP were injected via tail vein in 4T1 breast tumor bearing Balb/cJ mice. Although biodistribution via optical imaging revealed the presence of some NPs in major organs of mice treated with scr-NP (Fig. 7A–B), no toxicity was observed after treatment of both AuP and scr-NP. Histopathologic studies showed that there was no morphological evidence of injury to the heart, liver, kidneys, spleen, or bone marrow (Fig. 7C). Next, to assess acute toxicity, we performed a blood biochemical analysis for the liver, renal, and muscle parameters in AuP and scr-NP injected mice. None of the treatments changed the number or morphology of blood cells (Fig. 7D). We also performed serum biochemistry to evaluate organ functions of the animals. No renal, hepatic, or muscle toxicity was observed in the animals treated with AuP and scr-NP, compared to the untreated cohort. The activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and glutamate dehydrogenase (GLDH) (for liver injury), creatine kinase (CK), lactate dehydrogenase (LDH) (for muscle injury), and urea (for renal function), as well as the concentration of Troponin I (for heart function), were comparable to the results observed in control mice (Fig. 7E–F). The number of myeloid-derived cells recruited to the primary tumor remained unaffected between treated (AuP or scr-NP) and untreated tumors in the immunocompetent mouse model (Fig. S8). The immunogenic profile after treatment with AuNP is not significantly affected when compared to untreated animals (Fig. S9–10). Overall, our results showed that our NP-mediated delivery system displays minimal host toxicity, thus conferring a great potential for inhibiting TNBC progression.
Figure 7. The biodistribution and toxicity profile of NP-mediated delivery system.
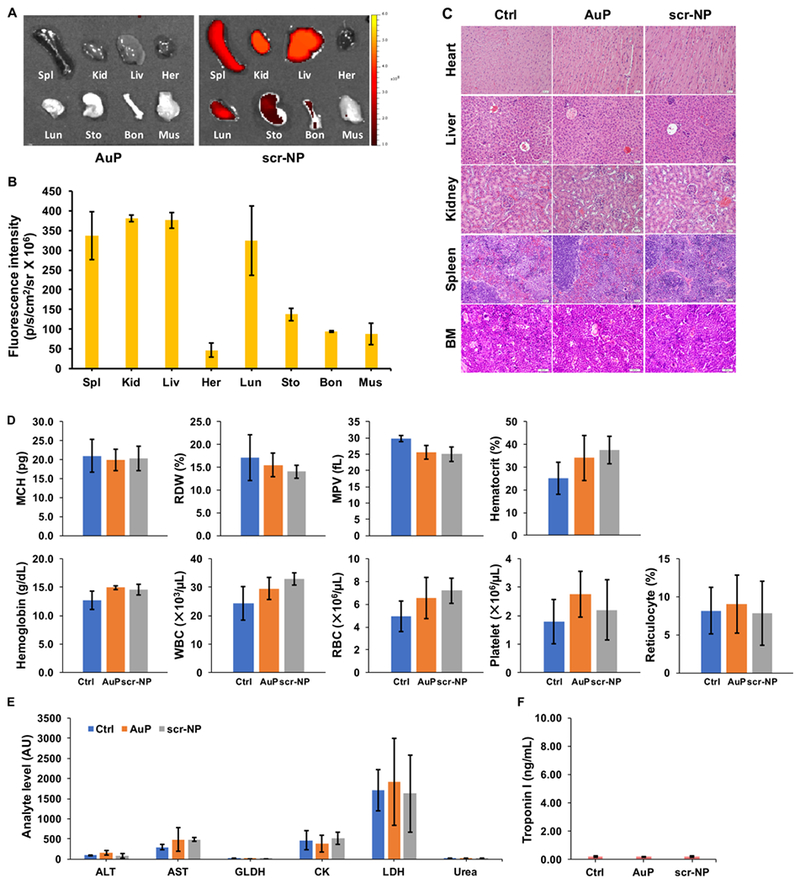
A-B) The biodistribution study. The fluorescence was measured by optical imaging (Xenogen) one week after treatment of AuP or cy5.5 conjugated scr-NP (n = 3 per group). (Spl = spleen, Kid = kidney, Liv = liver, Her = heart, Lun = lung, Sto = stomach, Bon = bone, Mus = muscle). C) Histopathologic evaluation of the major organs after any treatment. (BE = bone marrow). The scale bar represents 20 μm. D) Whole blood biochemistry indexes. (MCH = mean corpuscular hemoglobin, RDW = red cell distribution width, MPV = mean platelet volume, WBC = white blood cell, RBC = red blood cell). E) Serum analyte activity for liver (ALT, AST, GLDH) or muscle injury (CK, LDH). (ALT = alanine aminotransferase, AST = aspartate aminotransferase, GLDH = glutamate dehydrogenase, CK = creatine kinase, LDH = lactate dehydrogenase). F) The plasma levels of cardiac troponin I for cardiac injury.
DISCUSSION
Nucleotide-based therapeutic approaches (RNAi or miR-mimics) have been widely proposed as effective therapeutic strategies for over two decades (36). However, since their initial conception, one of the major challenges of miRNA-based cancer therapy is to achieve specific, efficient and safe systemic delivery of therapeutic miRNAs in vivo (37). Since miRNAs are naturally occurring molecules, and a single miRNA can regulate expression of multiple cancer-related genes, using miRNAs presents an attractive anti-cancer therapeutic approach. According to the functions of miRNAs, cancer types and stages, both miRNA-antagonists and mimics have the potential to be developed as therapeutic strategies to achieve tumor regression (38,39).
Our motivation for developing miR-708 as a therapeutic agent for TNBC was based on the premise that miR-708 is not only specifically downregulated in metastatic breast cancer cells lines, but also in breast cancer metastatic lesions (distant and lymph nodes) in both mouse models and TNBC patient samples (14). Mechanistically, miR-708 suppresses metastasis by targeting the expression of endoplasmic reticulum membrane protein NNAT to impact cell migration. Thus, its lower expression levels lead to an aggressive tumor phenotype.
The natural miRNAs, which are fully charged oligonucleotides, are not optimal drug candidates, as they exhibit sensitivity to nuclease, immunogenicity, and other major challenges (20). They need to be protected during circulation to facilitate later and effective release in designated locations; thus, various delivery vehicles have been proposed (40). The most described delivery vehicles, such as liposomes, micelles, and NPs are based on encapsulation, co-aggregation and surface conjugation, respectively. The loaded oligonucleotides usually are released at once when the particles encounter with the release triggers, such as acidic, redox, and/or proteolytic environments. Our LbL NP-mediated delivery system is prepared by sequential layering of well-defined biopolymers and nucleic acids. Its composition and release duration thus can be fine-tuned. Previously, we have used this strategy to load multiple therapeutic ingredients and prolong the gene silencing effect (23–26). LbL fabrication strategy that used in this development provides a flexible platform to prepare RNAi containing particles and also controls the intracellular release of RNAi.
In this study, we have optimized the delivery of a miR-708 mimetic, a “replacement therapy” for TNBC. The LbL NP was designed to overcome these biological hurdles. Intravenous administration of miR708-NP effectively accumulated at the tumor site via the enhanced permeability and retention (EPR) effect (41–43). Tumor-associated proteases, which were highly upregulated in the tumor, gradually hydrolyzed the PLL and release the layered miRNA. In the past, we have demonstrated that activated cathepsin B is effective in hydrolyzing PLL (44), and the release of underneath siRNA layers is PLL degradation dependent (24). Cells with higher level of protease activity release siRNA faster than cells with lower activity. Tumor-associated cathepsin B is highly upregulated in most tumors (45), therefore LbL NPs with PLL spacers can be potentially applied to many cancer types. The multilayered assembly made a sustained release possible, unlike other nanodelivery systems, such as liposomes, which only achieve a burst of release.
Our efforts to determine the cause of impaired metastasis in the absence of primary tumor regression led to the discovery that miR708-NP targets a unique OCT4/SOX2+ miR-708low subpopulation of cancer cells with increased mammosphere forming and migration potential. Given that these self-renewing, highly migratory cells have higher motility (Fig. 5C), miR-708 replacement therapy may help in the suppression of metastases formation, thus making the disease more manageable by standard regimens and less aggressive.
In malignant breast cancers, the polycomb repressive complex 2 (PRC2) binds to the miR-708 promoter to epigenetically suppress miR-708 by enhancing histone 3 lysine 27 trimethylation (H3K27me3) (14). In this context, it is important to note that miR-708 mimetics delivered through NP are refractory to PRC2-mediated suppression, and are therefore effective in achieving target suppression. While NPs protect miRNAs from degradation by nucleases in the circulation, there is potential to further enhance the efficacy of miR-708 in vivo, as chemical modifications (2’-OH, LNA modification, PNA) can enhance the stability of miRNA modulators and increase the resistance to degradation by nucleases at the tumor site.
Our study demonstrates a mechanism-based effectiveness of NP-mediated delivery of synthetic miR-708 in inhibiting breast cancer metastasis in preclinical TNBC mouse model, a paradigm shift in the treatment of this high-risk disease subset. miRNA-708 is a naturally occurring RNA molecule present in all normal human cells. Therefore, systemic treatment with miR-708 should elicit a strong and specific anti-metastatic effect on miR-708 deficient metastatic breast cancer cells, without non-specific side effects on normal cells. Together, these findings provide the rationale for developing the first miRNA replacement therapy for metastatic TNBC. Indeed, several miRNA-targeted therapeutics have reached clinical development (46), including the tumor suppressor miR-34a as a liposome encapsulated miR-34a replacement (MRX34) therapy for unresectable primary liver cancer (47,48). Although promising, this liposomal formulation of miR-34a was associated with several grade 1 and 2 toxicities causing difficulties in implementing the dosing regimen in all patients (49). Also, five patients experienced grade 4 adverse events that led to closing of this phase I clinical study. With the LbL fabrication technology, multiple layers of the oligonucleotide can be loaded on to a single AuNP core, which is essential for slow and gradual release in patients. Therefore, our NP-based delivery system has the potential to overcome the mild adverse events related to frequent dosing schedule. Moreover, due to their relative non-toxic properties (50), gold-based NP delivery systems have tremendous advantage over liposomes, with a greater scope for improvements in delivery (51). Many NP-based chemotherapeutics and diagnostic agents have been in clinic for quite sometimes (52), yet none specifically targeted the most aggressive breast cancer subtype, TNBC. Hence, with this study we aim to develop the first oligonucleotide-based therapeutic specifically for TNBC patients.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the US Department of Defense (BC123348, BC150875) and the Clinical and Translation Science Center at Weill Cornell Medical College (UL1TR000457) to V. Mittal.
Footnotes
The authors declare no potential conflicts of interest
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30 doi 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Dent R, Hanna WM, Trudeau M, Rawlinson E, Sun P, Narod SA. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res Treat 2009;115(2):423–8 doi 10.1007/s10549-008-0086-2. [DOI] [PubMed] [Google Scholar]
- 3.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA , et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research : an official journal of the American Association for Cancer Research 2007;13(15 Pt 1):4429–34 doi 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 4.Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer 2008;113(10):2638–45 doi 10.1002/cncr.23930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010;28(20):3271–7 doi 10.1200/JCO.2009.25.9820. [DOI] [PubMed] [Google Scholar]
- 6.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol 2012;23(9):2223–34 doi 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 7.Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist 2011;16 Suppl 1:1–11 doi 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. Metazoan MicroRNAs. Cell 2018;173(1):20–51 doi 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 2004;5(5):396–400 doi 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 10.Macfarlane LA, Murphy PR. MicroRNA: Biogenesis, Function and Role in Cancer. Curr Genomics 2010;11(7):537–61 doi 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet 2016;17(12):719–32 doi 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ. Biological functions of microRNAs: a review. J Physiol Biochem 2011;67(1):129–39 doi 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- 13.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol 2007;302(1):1–12 doi 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 14.Ryu S, McDonnell K, Choi H, Gao D, Hahn M, Joshi N, et al. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell 2013;23(1):63–76 doi 10.1016/j.ccr.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P, et al. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis 2015;4:e158 doi 10.1038/oncsis.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, et al. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res 2011;71(19):6208–19 doi 10.1158/0008-5472.CAN-11-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baer C, Oakes CC, Ruppert AS, Claus R, Kim-Wanner SZ, Mertens D, et al. Epigenetic silencing of miR-708 enhances NF-kappaB signaling in chronic lymphocytic leukemia. Int J Cancer 2015;137(6):1352–61 doi 10.1002/ijc.29491. [DOI] [PubMed] [Google Scholar]
- 18.Juliano RL. The delivery of therapeutic oligonucleotides. Nucleic Acids Res 2016;44(14):6518–48 doi 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 2017;35(3):238–48 doi 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol 2017;35(3):222–9 doi 10.1038/nbt.3802. [DOI] [PubMed] [Google Scholar]
- 21.Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy. N Engl J Med 2018;378(7):625–35 doi 10.1056/NEJMoa1710504. [DOI] [PubMed] [Google Scholar]
- 22.Ashraf S, Pelaz B, del Pino P, Carril M, Escudero A, Parak WJ, et al. Gold-Based Nanomaterials for Applications in Nanomedicine. Top Curr Chem 2016;370:169–202 doi 10.1007/978-3-319-22942-3_6. [DOI] [PubMed] [Google Scholar]
- 23.Lee SK, Han MS, Asokan S, Tung CH. Effective gene silencing by multilayered siRNA-coated gold nanoparticles. Small (Weinheim an der Bergstrasse, Germany) 2011;7(3):364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SK, Tung CH. A fabricated siRNA nanoparticle for ultra-long gene silencing in vivo. Adv Funct Mater 2013;23(28):3488–93 doi 10.1002/adfm.201202777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SK, Tung CH. siRNA Nanoparticles for Ultra-Long Gene Silencing In Vivo. Methods Mol Biol 2016;1372:113–20 doi 10.1007/978-1-4939-3148-4_9. [DOI] [PubMed] [Google Scholar]
- 26.Lee SK, Law B, Tung CH. Versatile Nanodelivery Platform to Maximize siRNA Combination Therapy. Macromol Biosci 2017;17(2) doi 10.1002/mabi.201600294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jewell CM, Lynn DM. Multilayered polyelectrolyte assemblies as platforms for the delivery of DNA and other nucleic acid-based therapeutics. Advanced drug delivery reviews 2008;60(9):979–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reum N, Fink-Straube C, Klein T, Hartmann RW, Lehr CM, Schneider M. Multilayer coating of gold nanoparticles with drug-polymer coadsorbates. Langmuir 2010;26(22):16901–8. [DOI] [PubMed] [Google Scholar]
- 29.Suma T, Miyata K, Anraku Y, Watanabe S, Christie RJ, Takemoto H, et al. Smart Multilayered Assembly for Biocompatible siRNA Delivery Featuring Dissolvable Silica, Endosome-Disrupting Polycation, and Detachable PEG. ACS nano 2012;6(8):6693–705. [DOI] [PubMed] [Google Scholar]
- 30.Daniel MC, Astruc D. Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chemical reviews 2004;104(1):293–346 doi 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 31.Lee SK, Han MS, Tung CH. Layered nanoprobe for long-lasting fluorescent cell label. Small (Weinheim an der Bergstrasse, Germany) 2012;8(21):3315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SK, Mortensen LJ, Lin CP, Tung CH. An authentic imaging probe to track cell fate from beginning to end. Nat Commun 2014;5:5216 doi 10.1038/ncomms6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang K, Elledge SJ, Hannon GJ. Lessons from Nature: microRNA-based shRNA libraries. Nature methods 2006;3(9):707–14. [DOI] [PubMed] [Google Scholar]
- 34.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436(7050):518–24 doi 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang B, Raviv A, Esposito D, Flanders KC, Daniel C, Nghiem BT, et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Reports 2015;4(1):155–69 doi 10.1016/j.stemcr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamore PD. RNA interference: big applause for silencing in Stockholm. Cell 2006;127(6):1083–6 doi 10.1016/j.cell.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Gao DY, Huang L. In vivo delivery of miRNAs for cancer therapy: challenges and strategies. Advanced drug delivery reviews 2015;81:128–41 doi 10.1016/j.addr.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elizabeth Labatut A, Mattheolabakis G. Non-Viral Based miR Delivery and Recent Developments. Eur J Pharm Biopharm 2018. doi 10.1016/j.ejpb.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrovic N, Ergun S. miRNAs as Potential Treatment Targets and Treatment Options in Cancer. Mol Diagn Ther 2018;22(2):157–68 doi 10.1007/s40291-017-0314-8. [DOI] [PubMed] [Google Scholar]
- 40.Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev 2016;104:61–77 doi 10.1016/j.addr.2016.06.011. [DOI] [PubMed] [Google Scholar]
- 41.Maeda H Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjugate chemistry 2010;21(5):797–802 doi 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- 42.Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced drug delivery reviews 2011;63(3):136–51 doi 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2013;4(1):81–9 doi 10.7150/thno.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res 2006;66(14):7225–9. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal N, Sloane BF. Cathepsin B: multiple roles in cancer. Proteomics Clin Appl 2014;8(5-6):427–37 doi 10.1002/prca.201300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacLeod AR, Crooke ST. RNA Therapeutics in Oncology: Advances, Challenges, and Future Directions. J Clin Pharmacol 2017;57 Suppl 10:S43–S59 doi 10.1002/jcph.957. [DOI] [PubMed] [Google Scholar]
- 47.Daige CL, Wiggins JF, Priddy L, Nelligan-Davis T, Zhao J, Brown D. Systemic delivery of a miR34a mimic as a potential therapeutic for liver cancer. Mol Cancer Ther 2014;13(10):2352–60 doi 10.1158/1535-7163.MCT-14-0209. [DOI] [PubMed] [Google Scholar]
- 48.Beg MS, Hong DS, Sachdev JC, Brenner AJ, Borad MJ, Lim HY, et al. First-in-human trial of microRNA cancer therapy with MRX34, a liposomal miR-34 mimic: Phase Ia expansion in patients with advanced solid tumors. Journal of Clinical Oncology 2016;34(15_suppl):TPS2597-TPS doi 10.1200/JCO.2016.34.15_suppl.TPS2597. [DOI] [Google Scholar]
- 49.Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs 2017;35(2):180–8 doi 10.1007/s10637-016-0407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khlebtsov N, Dykman L. Biodistribution and toxicity of engineered gold nanoparticles: a review of in vitro and in vivo studies. Chem Soc Rev 2011;40(3):1647–71 doi 10.1039/c0cs00018c. [DOI] [PubMed] [Google Scholar]
- 51.Cai W, Gao T, Hong H, Sun J. Applications of gold nanoparticles in cancer nanotechnology. Nanotechnol Sci Appl 2008;1:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med 2016;1(1):10–29 doi 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.