

Summary
Objective
Brivaracetam (BRV) and levetiracetam (LEV) are antiepileptic drugs that bind synaptic vesicle glycoprotein 2A (SV2A). In vitro and in vivo animal studies suggest faster brain penetration and SV2A occupancy (SO) after dosing with BRV than LEV. We evaluated human brain penetration and SO time course of BRV and LEV at therapeutically relevant doses using the SV2A positron emission tomography (PET) tracer 11C‐UCB‐J (EP0074; NCT02602860).
Methods
Healthy volunteers were recruited into three cohorts. Cohort 1 (n = 4) was examined with PET at baseline and during displacement after intravenous BRV (100 mg) or LEV (1500 mg). Cohort 2 (n = 5) was studied during displacement and 4 hours postdose (BRV 50‐200 mg or LEV 1500 mg). Cohort 3 (n = 4) was examined at baseline and steady state after 4 days of twice‐daily oral dosing of BRV (50‐100 mg) and 4 hours postdose of LEV (250‐600 mg). Half‐time of 11C‐UCB‐J signal change was computed from displacement measurements. Half‐saturation concentrations (IC 50) were determined from calculated SO.
Results
Observed tracer displacement half‐times were 18 ± 6 minutes for BRV (100 mg, n = 4), 9.7 and 10.1 minutes for BRV (200 mg, n = 2), and 28 ± 6 minutes for LEV (1500 mg, n = 6). Estimated corrected half‐times were 8 minutes shorter. The SO was 66%‐70% for 100 mg intravenous BRV, 84%‐85% for 200 mg intravenous BRV, and 78%‐84% for intravenous 1500 mg LEV. The IC 50 of BRV (0.46 μg/mL) was 8.7‐fold lower than of LEV (4.02 μg/mL). BRV data fitted a single SO versus plasma concentration relationship. Steady state SO for 100 mg BRV was 86%‐87% (peak) and 76%‐82% (trough).
Significance
BRV achieves high SO more rapidly than LEV when intravenously administered at therapeutic doses. Thus, BRV may have utility in treating acute seizures; further clinical studies are needed for confirmation.
Keywords: 11C‐UCB‐J, brivaracetam, levetiracetam, positron emission tomography, synaptic vesicle glycoprotein 2A
1.
Key Points.
BRV enters the human brain faster than LEV
BRV has ninefold higher SV2A affinity than LEV in humans, consistent with in vitro binding studies and in vivo efficacy studies in animals
SV2A occupancy by BRV was consistent between acute and steady state conditions
2. INTRODUCTION
More than 60 million people are affected by epilepsy worldwide.1 The incidence of experiencing one epileptic seizure during a lifetime is 10%, and about one‐third of people who suffer one seizure will progress to recurrent seizures and a diagnosis of epilepsy.2 There are currently >20 antiepileptic drugs (AEDs) available for the treatment of different types of epilepsy; however, 20%‐40% of epilepsy patients develop drug‐resistant epilepsy3, 4, 5 and represent an unmet therapeutic need.
Brivaracetam (BRV) is a recently approved AED for monotherapy (in the United States) or adjunctive (in the European Union and the United States) treatment of focal seizures. Neither BRV nor levetiracetam (LEV) is indicated for status epilepticus (SE). BRV is structurally related to LEV, and both drugs bind to synaptic vesicle glycoprotein 2A (SV2A),6, 7, 8 an integral transmembrane glycoprotein specifically localized to secretory vesicles.9, 10 BRV has a 15–30‐fold increased affinity8 and greater selectivity toward high‐voltage–activated calcium channels and α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazoleproprionic acid receptors than LEV.11 BRV represents the first selective SV2A ligand for epilepsy treatment.11
In vitro studies have demonstrated that BRV has higher lipophilicity and passive diffusion permeability across Caco‐2 cell membranes than LEV.12 In audiogenic mice, BRV achieved maximal pharmacological activity 30 minutes postdose when plasma concentrations were maximum. In contrast, LEV peak activity was delayed by nearly 1 hour from peak plasma levels. Furthermore, brain entry of BRV was faster than for LEV in rats after single intravenous dosing, and the relatively slow brain penetration of LEV was confirmed in dogs. Together, these studies suggest that the rate of brain penetration and the onset of action were faster for BRV than for LEV.12
Positron emission tomography (PET) can be used noninvasively to determine drug target engagement and occupancy in living subjects. Several PET radioligands have recently been developed for imaging of SV2A,13, 14 with 11C‐UCB‐J being considered best in class thus far.15, 16 We previously demonstrated that 11C‐UCB‐J binding could be displaced by intravenous infusion of BRV (5 mg/kg) or LEV (30 mg/kg) in nonhuman primates. The doses were chosen in that study to provide plasma levels in rhesus monkeys similar to those achieved in humans after intravenous doses of 100 mg BRV and 1500 mg LEV. The mean estimated displacement half‐times of 11C‐UCB‐J binding were 10 minutes for BRV and 30 minutes for LEV,12 consistent with a faster brain penetration for BRV. The tracer displacement half‐times were expected to be an overestimation of the time taken for drug SV2A occupancy (SO), and an approximate correction yielded estimated drug entry half‐times of 3 and 23 minutes for BRV and LEV, respectively. In the current study, we determined brain entry rates for BRV and LEV using a displacement paradigm with 11C‐UCB‐J following single intravenous dosing in healthy volunteers. In addition, SO studies were performed to determine the half‐saturation concentrations (IC 50) of BRV and LEV.
3. MATERIALS AND METHODS
The study was performed under a protocol approved by the Yale University Human Investigation Committee and the Yale New Haven Hospital Radiation Safety Committee, and in accordance with US federal policy for the protection of human research subjects contained in Title 45 Part 46 of the Code of Federal Regulations. Written informed consent was obtained from all participants after complete explanation of study procedures.
3.1. Study design and participants
Thirteen healthy subjects (seven males and six females) participated in the PET imaging studies using the SV2A‐selective tracer 11C‐UCB‐J (EP0074; NCT02602860). To be eligible, subjects had to be between 18 and 55 years of age and in good health as confirmed by comprehensive medical and psychiatric histories, physical examination, neurological and mental status examinations, routine laboratory studies, and electrocardiogram. Exclusions included any current or past clinically significant medical or neurological illness, a history of alcohol or substance abuse, current pregnancy (as documented by pregnancy testing at screening and on the day of the PET imaging study), breast‐feeding, and contraindications to magnetic resonance imaging.
In Cohort 1, four healthy subjects (two males and two females) participated in a baseline and displacement PET measurement. Each subject underwent two PET measurements (baseline and BRV [n = 2] or LEV [n = 2] displacement) on the same day. BRV (100 mg) or LEV (1500 mg) was intravenously infused over 5 minutes starting 60 minutes after the beginning of the 11C‐UCB‐J infusion17 (K bol = 150 minutes). One subject (Subject 3) underwent only a displacement PET measurement with LEV. The subject information and PET measurement data, including mass and radioactivity amounts, are presented in Table S1.
In Cohort 2, five healthy volunteers (three males and two females) participated in a displacement and postdose measurement. Each subject underwent a total of four PET measurements (BRV and LEV displacement and postdose). One subject (Subject 9) had only two PET measurements (BRV displacement and postdose). BRV (50 mg, n = 1; 100 mg, n = 2; 200 mg, n = 2) or LEV (1500 mg, n = 4) were intravenously administered at 60 minutes (80 minutes for BRV displacement, Subject 6) after 11C‐UCB‐J injection. The postdose PET measurement was performed approximately 4 hours postinfusion. The subject information and PET measurement data are presented in Table S2.
In Cohort 3, four healthy volunteers (two males and two females) participated in a baseline and three postdose measurements. Each subject had two PET measurements (baseline and 2 hours postdose for oral BRV administration) on Day 1. The subjects continued oral BRV dosing (50 mg twice daily [BID], n = 2; 100 mg BID, n = 2) for 4 days, that is, to reach steady state (SS). On Day 5, the subjects received the last oral BRV dosing, followed by two postdose PET measurements. One was at 2 hours postdose, at peak plasma concentration (SS peak) and the other was at 8 hours postdose (SS trough).
Three of the four volunteers in Cohort 3 (one male and two females) had an additional LEV postdose PET measurement at least 7 days after the last BRV postdose PET measurement. Each subject completed the fifth PET measurement 4 hours after intravenous administration of LEV (250 mg, n = 2; 600 mg, n = 1). The subject information and PET measurement data are presented in Table S3.
3.2. Magnetic resonance imaging
Magnetic resonance images were obtained on all subjects for PET image registration and definition of regions of interest. Magnetic resonance imaging was performed using a 3‐dimensional magnetization‐prepared rapid gradient‐echo pulse sequence with an echo time of 2.77 milliseconds, repetition time of 2530 milliseconds, inversion time of 1100 milliseconds, and flip angle of 7° on a 3‐T whole‐body scanner (TrioTrim; Siemens Medical Solutions) with a circularly polarized head coil.
3.3. PET imaging with 11C‐UCB‐J
11C‐UCB‐J was radiolabeled as previously described.16 All subjects underwent PET examinations with 11C‐UCB‐J following an intravenous bolus plus constant infusion using a K bol value of 150 minutes to the end of the PET acquisition.15 PET examinations were conducted on the high‐resolution research tomograph (Siemens), which acquires 207 slices (1.2‐mm slice separation) with a reconstructed image resolution of ~3 mm at full width at half maximum. Before tracer injection, a 6‐minute transmission measurement was performed for attenuation correction. PET data were acquired in list‐mode for 120 minutes and reconstructed into 33 frames (6 × 0.5, 3 × 1, 2 × 2, and 22 × 5 minutes) with corrections for attenuation, normalization, scatter, randoms, and dead time using the MOLAR algorithm. Event‐by‐event motion correction was performed using the Polaris Vicra optical tracking system (NDI Systems) with reflectors mounted on a swim cap worn by the subject.18 PET images were corrected for any uncorrected motion using a mutual information algorithm (FSL‐FLIRT) by frame‐by‐frame registration to a summed image (0‐10 minutes postinjection).
3.4. PET image analysis
Magnetic resonance images were skull‐ and muscle‐stripped (FMRIB's Brain Extraction Tool, http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/BET) for coregistration with PET images and the template magnetic resonance image. An average PET image corresponding to the mean of the frames of 0‐10 minutes after 11C‐UCB‐J injection was aligned to each subject's magnetic resonance image via a rigid registration with mutual information. The individual magnetic resonance image was normalized to Montreal Neurological Institute (MNI) space using an affine linear plus nonlinear registration (BioImage Suite 2.5), to extract regions of interest (ROIs) from the automated anatomic labeling (AAL) template.19, 20 A number of ROIs were modified versions of the AAL template region (anterior insula, posterior insula, and ventromedial prefrontal cortex), defined on previous PET imaging data (substantia nigra and ventral striatum) or manually defined in the MNI atlas (centrum semiovale). The ROI for the centrum semiovale was eroded by two voxels on all borders to minimize partial volume effects. Regional time‐activity curves were obtained by applying the template ROIs to PET space using the transformations.
3.5. Blood analysis
All subjects underwent arterial cannulation, and blood was collected for measurement of the time course of 11C‐UCB‐J in plasma, including radiometabolite analysis. Samples were drawn every 10 seconds for the first 90 seconds and at 1.75, 2, 2.25, 2.5, 2.75, 3, 4, 5, 6, 8, 10, 15, 20, 25, 30, 45, 60, 75, 90, 105, and 120 minutes after 11C‐UCB‐J injection. For each sample, plasma was obtained by centrifugation at 4°C (2930 g for 5 minutes). Whole blood and plasma samples were counted in cross‐calibrated gamma‐counters (1480 Wizard; Perkin‐Elmer).
Radiometabolite analyses, performed for plasma samples at 3, 8, 15, 30, 60, and 90 minutes using an automatic column‐switching high‐performance liquid chromatography system, and blood processing were performed as described in our previous publication.21
Additional blood samples were collected for measurement of BRV or LEV plasma concentrations, immediately before, at the middle of, and at the end of the postdrug PET measurements.
3.6. Quantitative analysis
In Cohorts 1 and 2, five regional time‐activity curve (TAC) were generated to assess drug‐induced tracer displacement, including the centrum semiovale, cerebellum, frontal cortex, temporal cortex, and putamen.
The entry of BRV or LEV into brain and binding to SV2A will result in a reduction in the available binding sites for 11C‐UCB‐J. In the displacement studies, the rate of reduction in the 11C‐UCB‐J signal can be quantified and used as a measure of rate of entry and binding of BRV or LEV. Tracer displacement half‐times, the time it takes from drug administration to 50% maximal tracer displacement, were estimated using the change over time in the radioactivity concentration ratio between each brain region and metabolite‐corrected plasma; when brain and plasma concentrations are at equilibrium, this ratio represents the volume of distribution (V T, EQ). Regional time activity curves from end of infusion to end of PET measurement were fit by the following equation12:
| (1) |
where V ND accounts for nondisplaceable tracer uptake and p 2 (min−1) is the observed rate of change in specific binding. V ND was determined as the mean V T,EQ (t) value of the centrum semiovale region from 90 to 120 minutes after tracer injection. Tracer displacement half‐times (minutes) were estimated as ln(2)/p 2 in the four regions (cerebellum, frontal cortex, temporal cortex, and putamen) and averaged.
Because tracer displacement requires drug entry and tracer clearance, a corrected brain entry half‐time of the drugs was also estimated by subtracting the tracer clearance half‐time from the tracer displacement half‐time.12 The tracer clearance half‐time was calculated from the maximum tracer clearance rate as k 2 = K 1 / V ND, in which K 1 was obtained from a 1TC model fit to time‐activity curves of four regions (cerebellum, frontal cortex, temporal cortex, and putamen) and V ND was obtained as described above. The tracer clearance half‐time (minutes) was estimated as ln(2)/p 2 in the four regions and averaged across BRV and LEV displacement measurements. For a given drug, the observed rate of change in specific tracer binding is expected to depend on dose, as higher doses will reach a given occupancy faster.
In Cohorts 2 and 3, V T values were computed using the 1TC model to fit regional time‐activity curves.21, 22 The V T estimate was derived from the model fit parameters, K 1 and k 2. Regional V T values were computed for 23 regions, including amygdala, anterior cingulum, anterior insula, caudate nucleus, cerebellum, dorsolateral prefrontal cortex, frontal cortex, fusiform, globus pallidus, hippocampus, insula, occipital cortex, orbitofrontal cortex, parahippocampal gyrus, parietal cortex, posterior cingulum, posterior insula, putamen, substantia nigra, temporal cortex, thalamus, ventromedial prefrontal cortex, and ventral striatum.
In Cohort 2, a baseline V T was estimated from the first 60 minutes (before displacement) of the first PET measurement, and postdose V T was estimated from the postdose PET measurements. In Cohort 3, baseline V T was estimated from the baseline PET measurement, and postdose V T was estimated from each postdose PET measurement. These V T estimates were used to determine SO by BRV and LEV.
SO was determined using pairs of baseline and postdose PET measurements and the Lassen plot approach.23 This approach can be used to estimate brain occupancy when there is no region in the brain that is devoid of specific binding, in which the level of nonspecific binding can be estimated. It uses the principle that regions with higher specific binding (higher V T) at baseline will show a larger absolute reduction in binding during blockade. By regressing the change in V T against the baseline V T across regions, the SO and the nonspecific binding can be estimated.
| (2) |
Note that this equation is based on the assumption that SO and V ND are uniform across brain regions and PET measurements. Linear regression analysis is used to derive SO from the slope of the fitted line.
The pharmacokinetic‐brain target occupancy relationship between drug plasma concentration and SO was evaluated using the conventional E max model assuming that maximum occupancy is 100%. A two‐parameter (E max, IC 50) E max model (ie, maximal occupancy not constrained to 100%) was also evaluated. The extra‐sum‐of‐squares F test was used to determine which model more appropriately represented the data.
Drug concentration in plasma was calculated from an average of three blood samples acquired during the PET measurements (beginning, middle, and end). IC 50 was computed for each drug by grouping data from all subjects (“global”: Cohort 2, LEV, and BRV; Cohort 3, LEV, and BRV) and independently for each subject (“individual”: Cohort 3, BRV).
All image processing and kinetic modeling were performed with in‐house programs written within IDL 8.0 (ITT Visual Information Solutions). Tracer displacement half‐times were computed using GraphPad Prism.
4. RESULTS
4.1. Displacement of 11C‐UCB‐J
The radioactivity concentration ratio between brain region and metabolite‐corrected plasma (V T,EQ) reached an approximately constant value after 40‐50 minutes postinjection (Figure 1). The tracer displacement curves indicate a faster displacement by 100 mg BRV (n = 4, mean ± SD = 18.0 ± 6.3 minutes, range = 13.7‐27.3 minutes) and 200 mg BRV (9.7 and 10.1 minutes) than 1500 mg LEV (n = 6, mean ± SD = 28.5 ± 5.7 minutes, range = 21.7‐36.2 minutes) in all gray matter regions (Figure 1). Corrected half‐time values were lower, 10.0 ± 6.3 minutes for BRV (100 mg, n = 4), 1.7 and 2.1 minutes for BRV (200 mg, n = 2), and 20.5 ± 5.7 minutes for LEV (1500 mg, n = 6). The tracer displacement half‐times for all examined doses of BRV and LEV are listed in Table 1. The tracer displacement half‐times for BRV were dose‐dependent, as expected, and longer for lower BRV doses.
Figure 1.

Representative regional time‐activity curves of V T, EQ values in five brain regions (Cer, cerebellum; CS, centrum semiovale; FCx, frontal cortex; Put, putamen; TCx, temporal cortex) (Cohort 1). Brivaracetam (BRV) or levetiracetam (LEV) was administered intravenously (vertical dotted lines) and reduced the binding of 11C‐UCB‐J. Data were fitted to estimate tracer displacement half‐time (solid line, see Equation (1) in Materials and Methods section). Horizontal dotted lines indicate the V ND value estimated from the CS value from 90‐120 minutes postinjection
Table 1.
T1/2 and cT1/2, calculated as T1/2 minus mean tracer clearance half‐time (8 minutes), of 11C‐UCB‐J following intravenous infusion of BRV or LEV
| Cohort | ID | BRV | LEV | ||||
|---|---|---|---|---|---|---|---|
| Dose, mg | T1/2, min | cT1/2, min | Dose, mg | T1/2, min | cT1/2, min | ||
| 1 | 1 | 100 | 13.7 | 5.7 | |||
| 2 | 100 | 16.8 | 8.8 | ||||
| 3 | 1500 | 34.6 | 26.6 | ||||
| 4 | 1500 | 25.8 | 17.8 | ||||
| 2 | 5 | 100 | 27.3 | 19.3 | 1500 | 36.2 | 28.2 |
| 6 | 100 | 14.2 | 6.2 | 1500 | 27.4 | 19.4 | |
| 7 | 200 | 9.7 | 1.7 | 1500 | 21.7 | 13.7 | |
| 8 | 200 | 10.1 | 2.1 | 1500 | 25.0 | 17.0 | |
| 9 | 50 | 30.1 | 22.1 | ||||
BRV, brivaracetam; cT1/2, corrected half‐time; ID, subject identifier; LEV, levetiracetam; T1/2, tracer displacement half‐time.
It was noted that Subject 5 had a higher half‐time for BRV 100 mg compared to other subjects at the same dose. This same subject showed a relatively high half‐time for LEV 1500 mg also. We do not have a definite explanation for these observations in this one subject. The plasma concentrations and occupancies observed for this subject at these doses were similar to those of other subjects. The observation may be due to measurement variability, but the difference is considerable compared to the rest of the dataset. Because BRV and LEV are not P‐glycoprotein substrates, it cannot be explained on that basis.
4.2. SV2A occupancy
SO was estimated from the regional V T values using the Lassen plot approach. In Figure 2, two examples of Lassen plots are shown for each drug representing a higher dose (circles, BRV = 200 mg, LEV = 1500 mg) and a lower dose (squares, BRV = 50 mg, LEV = 250 mg). The plots show linear relationships for all doses, indicating good data fitting. V ND values, determined as the x‐intercepts from the Lassen plots in the postdose studies, were similar for BRV (mean ± SD = 2.80 ± 0.66, n = 9) and LEV (2.80 ± 0.59, n = 7). The SO values were dose‐dependent for both BRV and LEV and are shown in Table 2. The occupancy was 66% and 70% for intravenous 100 mg BRV, 84% and 85% for intravenous 200 mg BRV, and 78%‐84% for intravenous 1500 mg LEV (median = 78%, n = 4) approximately 4 hours postdose.
Figure 2.

Representative Lassen occupancy plots for brivaracetam (BRV; red) and levetiracetam (LEV; blue) for two different doses. The values on the x‐axis represent regional V T values during baseline and on the y‐axis represent the difference between V T values during baseline and V T values postdrug. The SV2A occupancy was derived from the slope of the linear fit, that is, 50% for BRV (50 mg), 85% for BRV (200 mg), 52% for LEV (250 mg), and 84% for LEV (1500 mg)
Table 2.
Plasma concentration, SV2A occupancy, V ND and IC 50 for BRV or LEV
| Cohort | ID | Drug | Study state | Dose, mg | Plasma conc., μg/mL | SV2A occupancy, % | V ND, mL/cm3 | IC 50, μg/mL, individual | IC 50, μg/mL, global |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 5 | BRV | Postdose | 100 | 1.04 | 66 | 2.28 | N/A | 0.51 |
| 6 | 100 | 0.98 | 70 | 3.06 | |||||
| 7 | 200 | 3.61 | 85 | 2.93 | |||||
| 8 | 200 | 2.94 | 84 | 2.36 | |||||
| 9 | 50 | 0.53 | 50 | 1.50 | |||||
| 5 | LEV | 1500 | 16.2 | 78 | 2.57 | N/A | 4.90 | ||
| 6 | 1500 | 16.5 | 78 | 2.79 | |||||
| 7 | 1500 | 23.1 | 84 | 2.86 | |||||
| 8 | 1500 | 23.6 | 78 | 2.76 | |||||
| 3 | 10 | BRV | Postdose | 100 | 1.63 | 74 | 3.25 | 0.54 | 0.45 |
| SS peak | 2.91 | 86 | 3.05 | ||||||
| SS trough | 1.7 | 76 | 3.80 | ||||||
| 11 | Postdose | 50 | 0.98 | 72 | 3.73 | 0.44 | |||
| SS peak | 1.61 | 76 | 3.78 | ||||||
| SS trough | 0.84 | 64 | 3.97 | ||||||
| 12 | Postdose | 100 | 1.23 | 80 | 3.08 | 0.32 | |||
| SS peak | 2.53 | 87 | 2.90 | ||||||
| SS trough | 1.43 | 82 | 3.24 | ||||||
| 13 | Postdose | 50 | 0.91 | 67 | 3.04 | 0.50 | |||
| SS peak | 1.99 | 78 | 2.71 | ||||||
| SS trough | 1.22 | 70 | 2.89 | ||||||
| 12 | LEV | Postdose | 250 | 3.65 | 58 | 4.01 | N/A | 3.58 | |
| 13 | Postdose | 600 | 10.7 | 68 | 2.51 | ||||
| 11 | Postdose | 250 | 4.25 | 52 | 2.10 | ||||
| 2 and 3 | BRV | 0.46 | |||||||
| LEV | 4.02 |
BRV, brivaracetam; conc., concentration; ID, subject identifier; LEV, levetiracetam; N/A, not available; SS, steady state.
The relationship between SO and BRV or LEV plasma concentration is shown in Figure 3, and the IC 50 values are shown in Table 2. The SO data followed the pharmacokinetic‐brain target occupancy relationship between drug plasma concentration and SO. The combined IC 50 estimate across Cohorts 2 and 3 was 0.46 μg/mL for BRV and 4.02 μg/mL for LEV, that is, BRV has 8.7‐fold higher affinity for SV2A than LEV.
Figure 3.
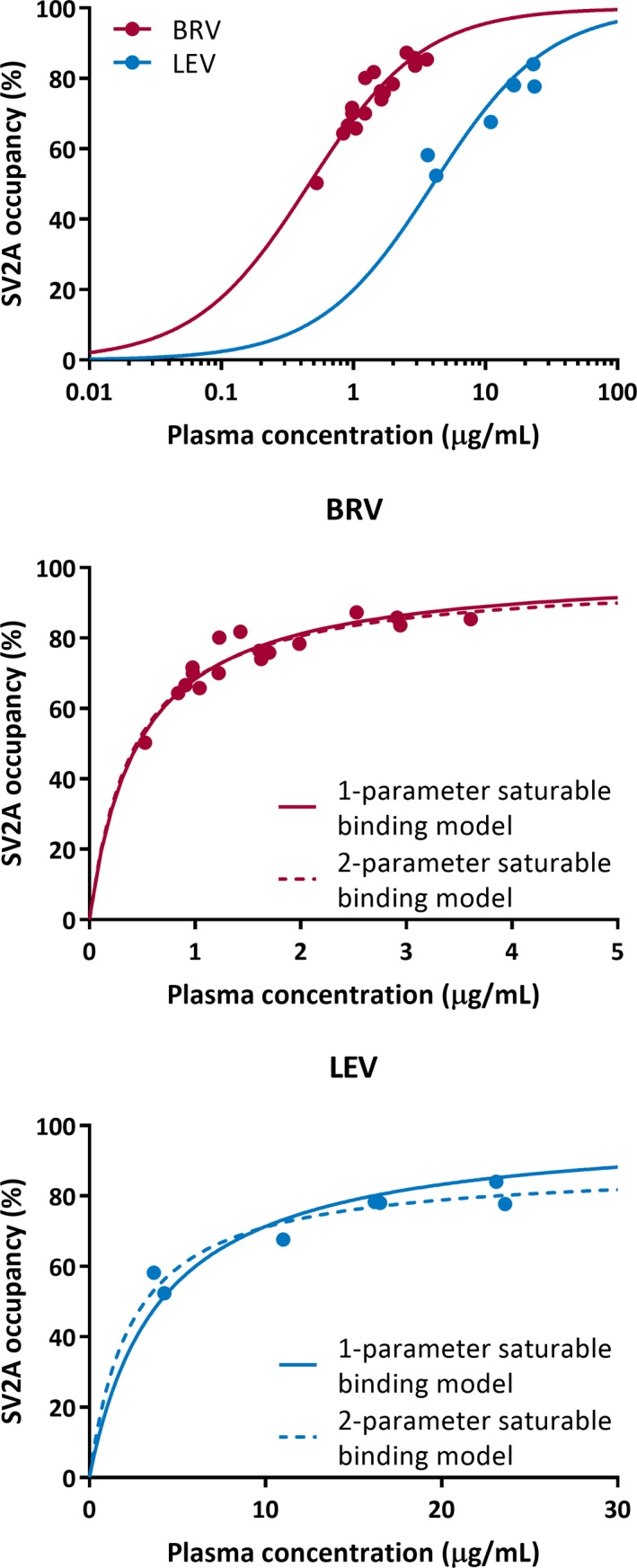
The relationship between the measured drug plasma concentration during each positron emission tomography measurement and SV2A occupancy is shown for all subjects in Cohorts 2 and 3. (Top) One‐parameter saturable binding fits are shown for SV2A occupancy versus brivaracetam (BRV) concentration (red) or levetiracetam (LEV) concentration (blue). Model comparison is shown between one‐parameter saturable binding fit (solid line) to estimate IC 50, and two‐parameter saturable binding fit (dotted line) to estimate IC 50 and maximal occupancy (E max) for BRV (middle) and LEV (bottom)
The BRV SS peak occupancy was 4%‐12% higher than the acute postdose occupancy. The SS trough occupancy was 5%‐12% lower than the SS peak occupancy. These data demonstrate high reproducibility of the occupancy measures with 11C‐UCB‐J, and the calculated SOs are fully explained by the pharmacokinetic‐brain target occupancy relationship between drug plasma concentration and SO.
The IC 50 of BRV was also estimated on a subjectwise basis using the postdose, SS peak, and SS trough occupancy and drug concentration data in the four subjects in Cohort 3 (Table 2). The average IC 50 across these subjects was 0.45 ± 0.09 μg/mL, which was in excellent agreement with the estimate of combined data from both cohorts.
Further evaluation of model fitting (ie, assuming maximum 100% occupancy or fitting the maximum occupancy E max) was performed with the combined occupancy and plasma concentration data from both cohorts for both BRV and LEV (Figure 3). The extra‐sum‐of‐squares F test was used to determine statistical significance between the two model fits for each drug, with α = 0.05 and a critical F value of F 2 − 1, N − 2, where N is the number of occupancy points for BRV or LEV (Table 2), and 1 and 2 correspond to the number of parameters in the one‐ and two‐parameter models. For BRV, the calculated F statistic was less than the critical F value, so the assumption of 100% maximal occupancy was not rejected. For LEV, the calculated F statistic (7.09) was greater than the critical F value (6.61); from the two‐parameter fit, the estimated E max was 88.4%, which was statistically different from 100%.
5. DISCUSSION
The results of the current study provide the first direct clinical evidence that BRV enters brain faster than LEV, consistent with previous in vitro and in vivo preclinical data. Although brain entry rate is an important contributing factor in the treatment of some forms of acute seizures requiring rapid therapeutic intervention, the therapeutic benefit of the faster brain entry of BRV has yet to be confirmed in clinical studies. In addition, BRV has 8.7‐fold higher SV2A affinity in humans than LEV. Also, SO by BRV was consistent between acute and SS conditions.
The tracer displacement half‐time analysis utilized in this study follows our previous study in nonhuman primates12 and shows that BRV enters the brain approximately ninefold faster than LEV. The corrected half‐times for 100 mg BRV and 1500 mg LEV in this clinical study are similar to the 3‐minute and 23‐ minute half‐times observed in the nonhuman primate study at doses intended to result in equivalent plasma levels in humans. Despite neither BRV nor LEV being indicated for SE, recent studies have suggested that SE patients can be treated with BRV, with loading doses similar to those in this study.24, 25 A further recent study has reported that a human dose of at least 2 mg/kg and up to 5 mg/kg is necessary for efficacy in SE, suggesting that doses slightly above 100 mg BRV intravenously could be in the efficacious range.26 The tracer displacement half‐time is an underestimation of the drug brain entry half‐time, because of the time taken for tracer clearance from tissue. The clearance of tracer from tissue is expected to be the same for both treatments and does not impact the observation that BRV brain entry is faster than for LEV. More advanced modeling, including direct estimation of drug brain entry rates, requires incorporation of the drug plasma input function and can be explored to more accurately estimate the brain entrance rates of BRV and LEV.
It is well established that in vitro affinity of SV2A ligands strongly correlates with their in vivo anticonvulsant potency across seizure models.27 The SO measurements demonstrated that BRV has 8.7‐fold higher SV2A affinity than LEV. These results are consistent with in vitro studies demonstrating 20‐fold higher SV2A affinity in cells and human brain tissue and 10‐fold higher potency in in vivo models of epilepsy.8, 25, 28 Consistently, clinical effective plasma concentrations of BRV are lower than for LEV.
The V ND values from the Lassen plots were similar for BRV and LEV. Occupancy of SV2A by BRV was well described by the one‐parameter model, and the two‐parameter model did not outperform the one‐parameter model. This means that 100% of the tracer‐specific binding could be blocked by BRV. For LEV, there was an improvement of fit when including E max in the model, although the number of data points was very limited. To assess whether the LEV E max is lower than 100%, a more extensive occupancy study applying a larger dose range is required. In addition, the occupancy analysis assumes that drug levels in plasma are in equilibrium with those in the brain. It is possible that LEV concentrations in brain were not in equilibrium with plasma concentrations, especially given the slower entry rate of LEV into the brain. Thus, this lack of equilibrium could contribute to the slight lack of fit to the one‐parameter model.
The relationship between SO and seizure protection in audiogenic mice as a function of time after single‐dose administration was first explored with LEV using an ex vivo binding approach. The postadministration time needed to achieve maximal SO was well correlated with the time needed for maximal protection against seizures.29 Interestingly, after single oral dosing to audiogenic mice, BRV provided maximal protection against seizures 30 minutes postdose, when maximal plasma concentrations were also achieved, whereas peak anticonvulsant activity of LEV was delayed by almost 1 hour with respect to peak plasma levels.12 Further pharmacokinetic measurements also indicated that BRV showed a faster entry into the rodent brain than LEV, which explained faster onset of action against seizures in audiogenic mice.12
LEV is approved in adults with epilepsy at doses from 1000 to 3000 mg/d (maximum twice daily single dose = 1500 mg). BRV is approved in adults with epilepsy at doses from 50 to 200 mg/d (maximum twice daily single dose = 100 mg). Despite the small difference in SO between both 100 mg and 200 mg BRV and 1500 mg LEV in this study, there was considerable overlap in the measured occupancies at these doses. Variability in the occupancy between subjects was well explained by plasma drug concentrations, and each drug could be fitted to a single saturation occupancy curve across the range of plasma concentrations. The difference in SO between the 4‐hour postdose scans and the SS peak and trough occupancies for BRV are explained by the differences in drug plasma concentration, and the data fit the same pharmacokinetic‐brain target occupancy relationship.
More rapid penetration of BRV versus LEV provides the potential for more rapid onset of action30 and therefore could be important in acute unremitting seizures requiring prompt therapeutic intervention. A clinical study investigating the efficacy and safety of BRV as treatment for increased seizure activity in an epilepsy monitoring unit setting (EP0087; http://www.clinicaltrials.gov NCT03021018) has been completed and concluded that intravenous lorazepam, intravenous 100 mg brivaracetam, and intravenous 200 mg brivaracetam showed similar efficacy in acute seizures.31 The therapeutic benefit of the faster brain entry of BRV has yet to be confirmed; however, brain‐entry rate is an important contributing factor for assessing the efficacy of drugs and their place in clinical practice.
In conclusion, BRV approximately ninefold more rapidly achieves SO than LEV. At SS trough conditions, SO levels at 100 and 200 mg BRV were comparable to postdose 1500 mg LEV occupancy. These studies indicate that BRV may have utility in the treatment of acute seizures, and further clinical studies are required to confirm such efficacy in patients.
DISCLOSURE OF CONFLICT OF INTEREST
S.J.F., S.R., M.N., S.H., H.G., R.P., D.M., N.B.N., and Y.H. have no conflicts of interest to report. R.P.M., J.M., S.K., J.‐M.N., H.K., S.D., C.O., P.Ma., P.Mu., and A.S. are current employees of UCB Pharma. R.M.K. and J.H. were employees of UCB Pharma at the time this work was conducted; J.H. is currently a full‐time employee of Alkahest. R.E.C. has received research support from Astellas Pharma, Astra‐Zeneca, Bristol‐Myers Squibb, Lilly, Pfizer, Taisho Pharmaceutical, and UCB Pharma, outside of the submitted work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
DATA SHARING
Qualified researchers whose proposed use of the data has been approved by an independent review panel will be given access to anonymized individual participant data and redacted study documents. Additional information is available at http://www.clinicalstudydatarequest.com.
Supporting information
ACKNOWLEDGMENTS
Research support was provided by UCB Pharma. The authors thank the participants for their participation in the study. The authors also thank the staff of the Yale PET Center for their expert assistance. S.J.F. was supported by an International Postdoc grant from the Swedish Research Council. UCB Pharma provided the radiolabeling precursor and the reference material for 11C‐UCB‐J. This study was also made possible by CTSA grant number UL1 TR000142 from the National Center for Advancing Translational Sciences, a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH. The authors acknowledge Barbara Pelgrims, PhD (UCB Pharma, Brussels, Belgium) for publication coordination.
Finnema SJ, Rossano S, Naganawa M, et al. A single‐center, open‐label positron emission tomography study to evaluate brivaracetam and levetiracetam synaptic vesicle glycoprotein 2A binding in healthy volunteers. Epilepsia. 2019;60:958–967. 10.1111/epi.14701
REFERENCES
- 1. Moshe SL, Perucca E, Ryvlin P, Tomson T. Epilepsy: new advances. Lancet. 2015;385:884–98. [DOI] [PubMed] [Google Scholar]
- 2. Hauser WA, Kurland LT. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:958–66. [DOI] [PubMed] [Google Scholar]
- 3. Picot MC, Baldy‐Moulinier M, Daures JP, Dujols P, Crespel A. The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population‐based study in a Western European country. Epilepsia. 2008;49:1230–8. [DOI] [PubMed] [Google Scholar]
- 4. Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9. [DOI] [PubMed] [Google Scholar]
- 5. Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the National General Practice Study of Epilepsy. Lancet. 1995;346:140–4. [DOI] [PubMed] [Google Scholar]
- 6. Gillard M, Chatelain P, Fuks B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur J Pharmacol. 2006;536:102–8. [DOI] [PubMed] [Google Scholar]
- 7. Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101:9861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gillard M, Fuks B, Leclercq K, Matagne A. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti‐convulsant properties. Eur J Pharmacol. 2011;664:36–44. [DOI] [PubMed] [Google Scholar]
- 9. Bajjalieh SM, Peterson K, Shinghal R, Scheller RH. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science. 1992;257:1271–3. [DOI] [PubMed] [Google Scholar]
- 10. Feany MB, Lee S, Edwards RH, Buckley KM. The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell. 1992;70:861–7. [DOI] [PubMed] [Google Scholar]
- 11. Klitgaard H, Matagne A, Nicolas JM, et al. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. 2016;57:538–48. [DOI] [PubMed] [Google Scholar]
- 12. Nicolas JM, Hannestad J, Holden D, et al. Brivaracetam, a selective high‐affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia. 2016;57:201–9. [DOI] [PubMed] [Google Scholar]
- 13. Mercier J, Provins L, Valade A. Discovery and development of SV2A PET tracers: potential for imaging synaptic density and clinical applications. Drug Discov Today Technol. 2017;25:45–52. [DOI] [PubMed] [Google Scholar]
- 14. Mercier J, Archen L, Bollu V, et al. Discovery of heterocyclic nonacetamide synaptic vesicle protein 2A (SV2A) ligands with single‐digit nanomolar potency: opening avenues towards the first SV2A positron emission tomography (PET) ligands. Chem Med Chem. 2014;9:693–8. [DOI] [PubMed] [Google Scholar]
- 15. Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain. Sci Transl Med. 2016;8:348ra396. [DOI] [PubMed] [Google Scholar]
- 16. Nabulsi NB, Mercier J, Holden D, et al. Synthesis and preclinical evaluation of 11C‐UCB‐J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med. 2016;57:777–84. [DOI] [PubMed] [Google Scholar]
- 17. Carson RE, Channing MA, Blasberg RG, et al. Comparison of bolus and infusion methods for receptor quantitation: application to [18F]cyclofoxy and positron emission tomography. J Cereb Blood Flow Metab. 1993;13:24–42. [DOI] [PubMed] [Google Scholar]
- 18. Jin X, Mulnix T, Gallezot JD, Carson RE. Evaluation of motion correction methods in human brain PET imaging—a simulation study based on human motion data. Med Phys. 2013;40:102503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holmes CJ, Hoge R, Collins L, Woods R, Toga AW, Evans AC. Enhancement of MR images using registration for signal averaging. J Comput Assist Tomogr. 1998;22:324–33. [DOI] [PubMed] [Google Scholar]
- 20. Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. Neuroimage. 2002;15:273–89. [DOI] [PubMed] [Google Scholar]
- 21. Finnema SJ, Nabulsi NB, Mercier J, et al. Kinetic evaluation and test‐retest reproducibility of [11C]UCB‐J, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2A in humans. J Cereb Blood Flow Metab. 2018;38:2041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koole M, van Aalst J, Devrome M, et al. Quantifying SV2A density and drug occupancy in the human brain using [(11)C]UCB‐J PET imaging and subcortical white matter as reference tissue. Eur J Nucl Med Mol Imaging. 2018;46(2):396–406. [DOI] [PubMed] [Google Scholar]
- 23. Cunningham VJ, Rabiner EA, Slifstein M, Laruelle M, Gunn RN. Measuring drug occupancy in the absence of a reference region: the Lassen plot re‐visited. J Cereb Blood Flow Metab. 2010;30:46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strzelczyk A, Steinig I, Willems LM, et al. Treatment of refractory and super‐refractory status epilepticus with brivaracetam: a cohort study from two German university hospitals. Epilepsy Behav. 2017;70:177–81. [DOI] [PubMed] [Google Scholar]
- 25. Kalss G, Rohracher A, Leitinger M, et al. Intravenous brivaracetam in status epilepticus: a retrospective single‐center study. Epilepsia. 2018;59(Suppl 2):228–33. [DOI] [PubMed] [Google Scholar]
- 26. Aicua‐Rapun I, Andre P, Rossetti AO, Decosterd LA, Buclin T, Novy J. Intravenous brivaracetam in status epilepticus: correlation between loading dose, plasma levels and clinical response. Epilepsy Res. 2019;149:88–91. [DOI] [PubMed] [Google Scholar]
- 27. Kaminski RM, Matagne A, Leclercq K, et al. SV2A protein is a broad‐spectrum anticonvulsant target: functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology. 2008;54:715–20. [DOI] [PubMed] [Google Scholar]
- 28. Matagne A, Margineanu DG, Kenda B, Michel P, Klitgaard H. Anti‐convulsive and anti‐epileptic properties of brivaracetam (ucb 34714), a high‐affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. 2008;154:1662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaminski RM, Gillard M, Klitgaard H. Targeting SV2A for discovery of antiepileptic drugs In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado‐Escueta AV. eds. Jasper's Basic Mechanisms of the Epilepsies. Bethesda, MD: Oxford University Press; 2012;1441–54. [PubMed] [Google Scholar]
- 30. Klein P, Diaz A, Gasalla T, Whitesides J. A review of the pharmacology and clinical efficacy of brivaracetam. Clin Pharmacol. 2018;10:958–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Szaflarski J, Sadek A, Greve B, Williams P, Varner J, Moseley B. Efficacy and safety of IV brivaracetam as a treatment for increased seizure activity in an epilepsy monitoring unit. Paper presented at: American Epilepsy Society 72nd Annual Meeting; November 30–December 4, 2018, New Orleans, LA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials