

Abstract
The architecture and bioactivity of natural products frequently serves as an embarkation point for exploration of biologically-relevant chemical space. Total synthesis followed by derivative synthesis has historically enabled a deeper understanding of structure-activity relationships. However, synthetic strategies toward a natural product are not always guided by hypotheses regarding structural features required for bioactivity. Here we report an approach to natural product total synthesis that we term ‘pharmacophore-directed retrosynthesis’. A hypothesized, pharmacophore of a natural product is selected as an early synthetic target and this dictates the retrosynthetic analysis. In an ideal application, sequential increases in structural complexity of this minimal structure enables development of an SAR profile throughout the course of the total synthesis effort. This approach enables the identification of simpler congeners retaining bioactivity at a much earlier stage of a synthetic effort as demonstrated herein for the spongiane diterpenoid, gracilin A, leading to simplified derivatives with potent neuroprotective and immunosuppressive activity.
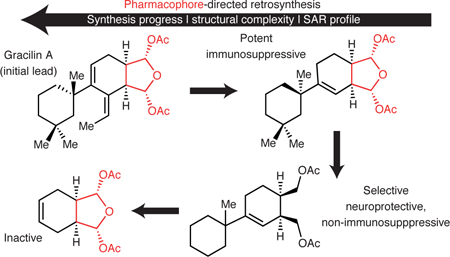
Graphical Abstract
The impactful and enduring role that natural products have played in improving the quality and duration of life for both humans and animals cannot be overstated. For example, rapamycin and its congeners have received FDA approval for various ailments and this natural product continues to provide insights into basic cell biology.1 Synthetic chemists are at the forefront of harvesting the full potential of natural products through synthetic efforts including classic total synthesis. Toward this goal, strategies to synthesize natural products have evolved significantly in recent years as more emphasis is placed on biological function.2 In Danishefsky’s ‘diverted total synthesis’ (DTS), a synthetic sequence is developed from simple building blocks employing classical retrosynthetic analysis and then various advanced intermediates previously employed in the synthesis effort are diverted toward simplified derivatives for biological analysis (Fig. 1a).3 In a strategy not necessarily directed toward total synthesis, Wender’s ‘function-oriented synthesis’ seeks to develop hypotheses regarding pharmacophores based on structural and computational analysis of distinct natural products and typically larger SAR data sets enabling the design and synthesis of simplified derivatives bearing a common pharmacophore.4 Alternatively, Schreiber’s ‘diversity oriented synthesis’ (DOS) of natural product-like libraries5 and the derived ‘biology-oriented synthesis’ (BIOS) by Waldmann6 seeks to synthesize collections of compounds based on structural features found in natural products. Finally, Myers recently developed a convergent building block strategy for rapid access to a diverse array of structurally related scaffolds such as macrolides related to erythromycin for the discovery of novel antibiotics7 that was subsequently termed ‘analogue-oriented synthesis’ by Vanderwal.8
Figure 1. ‘Pharmacophore–Directed Retrosynthesis’ (PDR) applied to gracilin A and comparison to other synthetic strategies harvesting the rich information content of natural products.

a, Strategies toward simplified, bioactive small molecules using natural products as starting points including diversity-oriented synthesis (DOS) directed toward libraries of natural product-inspired compounds. b, PDR seeks to identify or hypothesize the pharmacophore of a natural product, dictating the retrosynthesis by ensuring these features are present in multiple intermediates toward the natural product target.
The simultaneous alignment of total synthesis efforts with structure-function relationship studies has not been fully realized to the extent possible and in particular with novel natural products for which minimal SAR information exists. Several truncated natural products,9 which were found to possess similar bioactivity to the parent natural product, are known (e.g. eribulin mesylate from halichondrin10), however these derivatives were typically identified following completion of a total synthesis.11 In an example from our laboratory, a des-methyl, des-amino variant of the protein translation initiation inhibitor pateamine A was designed and synthesized following our total synthesis and found to have nearly equipotent activity to the natural product.12 This led to a retrospective question of whether such a derivative may have been accessed in route to the natural product and led us to consider the following question. Can the total synthesis of natural products, in particular with limited SAR or unknown or unconfirmed cellular targets, be more closely aligned to proposed biological activity during the retrosynthetic planning stages?
Herein, we describe a type of retrosynthetic analysis that seeks to more closely align total synthesis efforts with concurrent biological studies. The strategy opens the potential to identify simplified versions of the natural product with similar potency or potentially new functions in the course of a total synthesis effort. We term this strategy ‘pharmacophore-directed retrosynthesis’ (PDR) to emphasize the importance of considering hypothesized pharmacophores at the retrosynthetic planning stage of a total synthesis effort. While this approach increases the challenges of natural product total synthesis beyond important, contemporary goals including atom-economy,13 step and redox efficiency,14 and protecting group avoidance,15 it has the potential to greatly accelerate harvesting of the vast information content of natural products for basic cell biology and medicine. In PDR, we build on Wender’s notion of bringing function to the forefront of a synthetic endeavor, cf. function-oriented synthesis, but employ the logic of retrosynthesis16 to target simplified intermediates that importantly possess the proposed minimal structural features required for bioactivity, or pharmacophore, in route to the natural product.17 In applying PDR, a key first step is the identification of a proposed pharmacophore that may be based on (i) structural analysis with chemical intuition; (ii) existing SAR from isolated natural product congeners; (iii) activity of structurally related compounds; or (iv) anticipated reactivity. A retrosynthesis is then devised that ensures the proposed pharmacophore is present in multiple intermediates, with increasing complexity, ultimately leading to the natural product. We selected a member of the spongiane diterpenoid family, gracilin A (1),18 to initiate assessment of the utility of PDR toward exploring the recently described immunosuppressive19 and neuroprotective activity20 given the limited SAR information and no prior synthetic work (Fig. 2a).21 In the retrosynthetic analysis, as complexity is increased toward the natural product, several intermediates possessing the proposed pharmacophore are specifically targeted thus enabling SAR to be gathered as the total synthesis progresses (e.g. 8 → 7 → 6 → 1, Fig. 2b). It should be noted that several hypotheses regarding the pharmacophore of a particular natural product could be posited for PDR leading to alternate retrosynthetic strategies.
Figure 2. Select members of the gracilin A family and application of PDR to gracilin A.

a, Naturally–occurring spongiane diterpenoids bearing a common bis-acetoxy furanose moiety (red) selected as the hypothetical pharmacophore. b, PDR applied to gracilin A (1) leads to increasingly complex intermediates (i.e. 8, 7, 6) throughout the course of total synthesis from key intermediate 9.
The gracilins, including the rare nor–diterpene gracilin A,18 were originally isolated and characterized from the Mediterranean sponge Spongionella gracilis.22 These diterpenes are structurally unique owing to the unusual diacetoxy furanose found in most members. The cytotoxic activity of gracilins B and G–I isolated from Spongionella pulchella against a diverse panel of 12 human cancer cell lines has been reported, but these compounds did not progress further in preclinical evaluation.23 The absolute configuration of gracilin A was never established, while its relative configuration was based on X–ray analysis of the keto derivative of 9,11–dihydrogracilin A (3).24 Gracilin A was reported to be a potent inhibitor of phospholipase A2 (PLA2) with a 69% inactivation efficiency.25 We previously reported that gracilin A was mildly cytotoxic against K562 and PBMC cells, with IC50 values of 0.6 and 0.8 μM respectively, and inhibited EGF–R by 70% at 100 μM concentration.22 We also recently posited that gracilin A mimics the effects of cyclosporin A (CsA) through interaction with cyclophilin A (CypA)19 and also improves Alzheimer’s Disease (AD) hallmarks in vitro and in vivo.20, 26, 27 These results prompted the current study to employ PDR to unravel and ideally differentiate the structural requirements for immunosuppressive and neuroprotective effects observed with gracilin A.
The cyclophilins (Cyp) are highly conserved peptidyl-prolyl, cis-trans isomerases (PPIase) involved in protein folding and trafficking28, 29, 30 and are found in multiple cellular compartments.31CypA, the cytoplasmic isoform, has several roles in cell metabolism and energy homeostasis 32 with an enhanced expression in inflammation and cancer.33 The use of small molecules that selectively block the inflammation-related functions of CypA, is an important pharmacological strategy leading to effective immunosuppressive agents.33 On the other hand, Cyclophilin D (CypD), the mitochondrial isoform, translocates to form the mitochondrial permeability transition pore (mPTP) and its activity correlates with the mitochondrial dysfunction observed in AD leading to neuronal death.34, CypD inhibitors devoid of immunosuppressive activity through lack of binding to CypA but possessing the desired mitochondrial effect and appropriate blood-brain barrier (BBB) permeability could provide an approach to address AD.
Herein, we demonstrate a proof-of-principle study of PDR through application to the spongiane diterpene, gracilin A, that has led to useful lead compounds for both immunosuppression and neuroprotection. This study revealed simplified gracilin A derivatives with high affinity for CypA and others that demonstrated significant selectivity for CypD over CypA demonstrating their potential as neuroprotective agents devoid of immunosuppressive effects.
Results and discussion
Pharmacophore-directed retrosynthesis applied to gracilin A.
The bis-acetoxy furanose moiety was selected as the pharmacophore of gracilin A based on several lines of evidence. Studies of the reactivity of macfarlandin E, which possesses a 1,4-dicarbonyl masked as a bis-acyloxy furanose, with lysine derivatives provided evidence for pyrrole formation through a Paal-Knorr process35 and mounting evidence suggests this is possible with a number of other bis-acetoxy furanose-containing natural products.36 Furthermore, a computational study demonstrated the potential of the bis-acyloxy furanoses of gracilin A and aplysulphurin-1 to bind divalent cations such as Ca2+ pointing to the potential importance of this moiety for bioactivity.37 Finally, it is interesting to note that several bioactive members of the spongiane diterpenoid family possess a bis-acyloxy furanose including the structurally related gracilin L (2),22 9, 11 dihydrogracilin A (3), dendrillin (4),38 and tetrahydroaplysulphurin-1 (5).39 These considerations guided application of PDR to gracilin A and imposed a requirement that multiple intermediates along the synthetic route would bear or be converted to the proposed pharmacophore, namely the bis-acetoxy furanose (e.g. derivatives 6-8, Fig. 2b). In this way, structure-activity relationship (SAR) studies could be conducted throughout the course of the total synthesis. We recognized that keto lactone 9, accessible in gram quantities through our recently described Diels-Alder-lactonization organocascade,40 would serve as a key intermediate to study the importance of the C8-exocyclic alkene (e.g. derivative 6), the C9-appended cyclohexyl moiety (e.g. derivative 7). The importance of the bicyclic core of gracilin A would be ascertained by synthesis of the highly simplified monocyclic furanose 8 (Fig. 2b).
Sequential synthesis of increasingly complex gracilin A derivatives.
We recognized that in applying PDR, synthetic progress with concurrent biological assays of intermediates could best be achieved in stages based on increasing complexity of intermediates in the synthetic sequence as outlined in Figs. 3a–c, 4a. A final stage of diverted total synthesis could enable further refinement of the SAR profile through ‘gap filling’ with particular targeted derivatives to answer more specific questions building on information gathered during the initial stages. As applied to gracilin A, we first targeted the simple bis-acetoxy furanose 8 as the minimal pharmacophore that was readily obtained through a two-step oxidation/hydrogenation sequence to deliver the racemic syn-substituted acetoxy furanose (±)-8a along with the anti-diastereomer (±)-8b (dr 1.7:1). In a second stage, derivatives devoid of the cyclohexyl substituent and exocyclic alkene were targeted, namely bicyclic bis-acetoxy furanose 7a,b (Fig. 3b). The bicyclic lactone endo-15a (3:1, dr; 94% ee) was obtained from a Diels-Alder-lactonization organocascade employing diene 12 and acryloyl chloride (11) with tetramisole as Lewis base promoter. The required endo-diastereomer 15a could be isolated in 58% yield and subsequent reduction with LiAlH4 followed by desilylation delivered the ketodiol 16. Swern oxidation led to a dialdehyde which was directly subjected to acid-promoted acetylation to give the unstable keto bis-acetoxy furanoses 17 as a mixture of syn/anti diastereomers. A subsequent reduction with NaBH4 and dehydration with SOCl2 delivered the bicyclic bis-acetoxy furanoses 7a,b as a mixture of alkene regioisomers. The low yields obtained in this and subsequent 4-step sequences leading to the bis-acetoxy furanoses were primarily a result of incomplete or non-simultaneous Swern oxidation of the diols leading to regioisomeric mono-acetoxy furanoses.
Figure 3. Pharmacophore-directed retrosynthesis applied to gracilin A:

a, Synthesis of a highly simplified analogue bearing the proposed pharmacophore. b, Assembly of the cis–fused, 6,5–bicyclic core 15a of gracilin A through an enantioselective, organocatalytic Diels–Alder/lactonization cascade and manipulation to a simplified bicyclic analog 7. c, Annulation of the natural and a simplified cyclohexyl moiety. (inset: ORTEP representation of the single crystal X-ray structure of (–)-21b)
Figure 4. Synthesis of gracilin A derivatives toward SAR profile gap filling.

a, Alternate oxidation states of the original bis-acetoxy furanose. b, Select gracilin A derivatives in the enantiomeric series.
We next targeted derivatives devoid of the exocyclic alkene but bearing the cyclohexyl substituent (Fig. 3c). Desilylation of endo-Diels-Alder adduct 15a, gave bicyclic lactone (–)-9 which was subjected to an allylzinc reagent derived from cyclohexenyl choride 19 employing conditions we previously employed in our synthesis of spongiolactone.41 This delivered the tricyclic adducts 21a as a 1:1 mixture of diastereomers at the generated quaternary carbon but with high facial selectivity leading to a single epimer at the tertiary alcohol center. The contra-steric addition of the allylzinc reagent to the concave face of the ketone (–)-9 was unexpected but likely due to stereoelectronic effects leading directly to a chair rather than a boat conformation and was verified by extensive NMR studies (see Supplementary, Sect. C for details). In the case of addition of the gem-dimethyl cyclohexenyl zinc reagent derived from (±)-19, the stereochemistry was further verified by single crystal X-ray analysis of adduct (–)-21b. However, this diastereoselectivity is inconsequential since the tertiary alcohol in (–)-21b is subsequently dehydrated. Hydrogenation reduced the cyclohexene of (–)-21b and a 3-step process delivered the hydroxy bis-acetoxy furanose 22 as a 1:1 mixture of diastereomers. Dehydration with Martin’s sulfurane gave the alkenes 23 as a mixture of 4 diastereomers due to the alkene regioisomers produced. The four diastereomers were separable by preparative chiral HPLC enabling biological analysis of each stereoisomer.
Gaps in the SAR profile were also back-filled following initial assays described below (Fig. 4). This entailed application of aspects of diverted total synthesis to synthesize derivatives devoid of the cyclohexyl moiety (i.e. 24), variations in the oxidation state and substituents of the tetrahydrofuran and cyclohexyl moiety (i.e. 26-27), and also exploration of the enantiomeric series (i.e. 28-29).
PDR enables sequential increases in complexity with correlation to immunosuppressive activity.
As a preliminary biological screen, the initial gracilin A derivatives synthesized were analyzed by surface plasmon resonance (SPR) with immobilized CypA.42, 43 Biological testing of gracilin A derivatives was performed in sets as the various stages of PDR were accomplished (Figs. 3a–c, 4a,b). Association curves (KD) were measured at various concentrations in comparison to gracilin A and CsA and the initially synthesized highly simplified derivatives, monocyclic (±)-8 and bicyclic 7 bis-acetoxy furanoses, exhibited no affinity for CypA up to 10 μM (Fig. 5). Upon binding to CypA, CsA and gracilin A modulate interleukin 2 release (IL-2) through the calcineurin pathway,19 thus the effects of these simplified derivatives on IL-2 production was also measured. Concanavalin A (ConA) was used to activate human T lymphocytes and induce IL-2 release.44 T lymphocytes were pre-treated for 2-hour with different non-toxic concentrations of compounds and then activated for 48 hours in the presence of ConA (50 µg/mL). After this time, levels of IL-2 released to the medium were measured by ELISA kit. As expected, the greatly simplified monocyclic (±)-8a,b and bicyclic furanoses 7a,b did not inhibit IL-2 production nor exhibit T-lymphocyte toxicity at concentrations up to 10 μM (MTT assay).
Figure 5. Immunosuppressive activity of gracilin A derivatives.

Kinetic equilibrium dissociation constants (KD, μM) for binding of gracilin A derivatives to CypA as measured by SPR (n=4, Mean +/− SEM), interleukin–2 release inhibition (IL-2 (IC50, n=10, mean, CI)) by ELISA, and cell viability of human T lymphocytes (Viability (EC50, n=3, mean, CI)) as determined by an MTT assay. CI (95% confidence interval); R2 values (0.91–0.98) were in acceptable range (see Supplementary, Sect. B for details). Inhibition of calcineurin (CN) phosphatase activity at 1 µM expressed in percentage of control cells (n=4, mean +/− SEM). (>10 = not active up to 10 µM).
Gracilin A derivative 23 bearing the pendant cyclohexyl moiety, began to display immunosuppressive activity (Fig. 5). The initial diastereomeric mixture of tertiary alcohols 22 derived from cyclohexenyl zinc addition were inactive. However, upon dehydration to introduce the tri-substituted alkenes, immunosuppressive activity was observed with only a 2-fold decrease in KD relative to gracilin A (2.53 ± 0.40 μM) for alkene 23b bearing the 10S configuration (5.83 ± 3.33 μM) with moderate IL-2 inhibition (~19%). The interplay between alkene regioisomers (Δ8,9 vs. Δ9,11) and the C10 configuration was revealed through derivatives 23a and 23d that exhibited nearly a 1000-fold increase in KD toward CypA compared to gracilin A, 5.34 ± 1.68 nM and 7.57 ± 1.61 nM, respectively while isomers 23b and 23c were either significantly less active or completely inactive, respectively. Derivatives 29a and 23a displayed IC50 values for IL-2 inhibition of 0.12 and 0.15 µM, respectively whereas the C10 epimers of 23a, derivatives 23b and 23d had higher values of 0.30 and 0.36 µM, respectively, while the R-stereoisomer 23c exhibited a value of >10 µM again pointing to the importance of the C10 stereocenter for IL-2 inhibition. The discrepancy between binding (KD) to CypA as measured by SPR and the cellular inhibition of IL-2 expression may point to differential cell permeability or reflect differential binding to calcineurin, the presumed target of the CypA-gracilin A complex, impacted by the absence of the C8 exocyclic ethylidene. In addition, the conformation of the cyclophilins is highly dependent on the pH of the assay. This assay was performed at low pH (4.5) to demonstrate the activity of gracilin derivatives which also lowers the KD for CsA compared to literature values (see Supplementary Sect. B, Discussion for details). To address this discrepancy, we also performed an enzymatic assay to measure calcineurin inhibition by gracilin A derivatives (at 1 μM) and as expected the most potent derivatives in the SPR assay showed comparable inhibition (22–31%) to CsA (23% at 1 μM). Despite differences in CypA binding affinity, these derivatives have comparable activity in both IL-2 and calcineurin assays which supports the hypothesis that these derivatives lead to differential binding to calcineurin by the respective CypA-gracilin derivative complexes. These data taken together suggest that conformational preferences about the C9-C10 bond play a pivotal role in the immunosuppressive activity of the gracilin family.
To fill gaps in the SAR profile regarding immunosuppressive activity, we turned to application of diverted total synthesis and lastly targeted additional simplified derivatives (Fig. 5). The greatly simplified bicyclic mono-acetoxy ketone 24 and the bicyclic lactone 28, differing from the highly active bis-acetoxy furanose 23a only by the oxidation state of the tetrahydrofuran, were completely inactive. Epimerization of exo-15b enabled access to the enantiomeric series leading to mono-acetoxy furanoses 29a,b epimeric at the quaternary C10 center. Affinity to CypA (KD ~3 μM) for both diastereomers 29a,b was comparable to gracilin A, however differential inhibition of IL-2 production (IC50 = 0.12 vs >10 µM) was observed for both diastereomers while remaining relatively non-toxic to T-cells (EC50 = 1–4 µM). The importance of the quaternary carbon stereochemistry and alkene regisomer on IL-2 inhibition, which directly impacts the conformation about the C9-C10 bond, is again highlighted by this enantiomeric series with the natural (S) configuration imparting the greatest activity.
Neuroprotective activity of gracilin A derivatives.
Given the neuroprotective effects previously observed for gracilin A, the derivatives synthesized through application of PDR for immunosuppressive effects were also studied for potential neuroprotective activity and of particular interest was possible selectivity for CypD versus CypA PPiase activity (Fig. 6), a key property required for neuroprotective lead compounds devoid of immunosuppressive effects. All assays were performed at the indicated concentrations (0.001, 0.01, 0.1 or 1.0 µM) based on cytotoxicity determined with SHSY5Y cells. Particularly, compounds which displayed some toxicity against this cell line at 1 µM were tested at lower concentrations. Treatment of SH-SY5Y cells with a known potent oxidant such as H2O2 produces oxidative damage with the consequent increase in ROS release. ROS generation provokes mitochondrial dysfunction, increases cell death and affects the cellular antioxidant systems such as the glutathione (GSH) cycle.45 Therefore, four parameters were measured to evaluate the neuroprotective effects of the simplified gracilin A derivatives: cell viability, mitochondrial membrane potential (ΔΨm), ROS release, and glutathione levels after cellular challenge with H2O2 (see Supplementary, Sect. C for details). Several highly simplified gracilin A derivatives, namely monoacetoxy furanose 27a and the highly simplified diacetate 27b both derived from 21a, the inseparable, tertiary alcohols 22 (1:1, dr), and the enantiomeric mono-acetoxy furanose 29a, but not its diastereomer 29b, and even the simplest bis-acetoxy furanose (±)-8a,b (1.7:1, syn/anti) displayed significant neuroprotective effects. In view of these results, the compounds with greater activity in oxidative stress (OS) assays (27a, 27b, 29a, 22 and 23c) were chosen to test their ability to block the opening of the mitochondrial permeability transition pore (mPTP). The simplest derivatives 8a, b and 7a, b displayed lower activities and only at one of the concentrations tested, so these compounds were not subjected to the following assays. In PPIase activity assays, selectivity for CypD over CypA was observed for derivatives 27a (~3-fold) and 29a (~18-fold) with the very simple diacetate 27b displaying the greatest differential CypD activity since it was inactive against CypA (up to 10 μM) but displayed activity against CypD (IC50 = 0.48 μM). In contrast, tertiary alcohols 22 had opposite selectivity for CypA over CypD (~4-fold). Our results suggest that the antioxidant effect of these gracilin A derivatives is mitochondrial-related, similar to gracilin A, mediated through interaction with CypD. These compounds protect cells from oxidative damage induced by H2O2 improving mitochondrial functioning measured by MTT and TMRM assays and increased GSH levels. Moreover, they block mitochondrial pore opening and induce CypD activity inhibition. Therefore, gracilin A derivatives hold potential as neuroprotective lead compounds that are devoid of immunosuppressive effects.
Figure 6. Activity of gracilin A derivatives as neuroprotective agents.

Cell viability against H2O2 toxicity, mitochondrial membrane potential restoration (TMRM), % inhibition of reactive oxygen species (ROS) release, % increase cellular glutathione (GSH) levels, mPTP blockage, and IC50 values for binding to CypD and CypA. The lowest effective concentration of each derivative is indicated in parentheses (μM). Values are calculated as percentage of untreated control cells in comparison to H2O2 treated cells with values: H2O2 Toxicity 55.6 ± 4.4, TMRM 75.9 ± 3, ROS 140.3 ± 4.4, GSH 82.9 ± 1.6, mPTP blockage: 76.7 ± 2.8 (n=4, Mean +/− SEM). Cyp D and A activity expressed as IC50 values (µM). CI (95% confidence interval); R2 values (0.91–0.99) were in acceptable range (see Supplementary, Sect. C for details); derivatives exhibiting neuroprotective effects while also showing differential activity of CypA vs CypD are highlighted (red boxes; n=4, Mean +/− SEM)) (ND = not determined).
Summary of PDR and structure-activity relationships gleaned from application of PDR to gracilin A.
The total synthesis of natural products continues to be an important endeavor for discovery of novel synthetic strategies and methods in addition to exploration of biologically relevant chemical space. The described ‘pharmacophore-directed retrosynthesis’ approach brings biological function into the retrosynthetic planning stages to target multiple, simplified derivatives bearing a hypothesized pharmacophore in route to the natural product. PDR can be considered to be a subset of Wender’s function-oriented synthesis and will of course not be applicable to every natural product, i.e. those where the majority of the structure is required for bioactivity. A caveat to this approach is that a balance must be found between synthetic convergency and targeting of bioactivity during the total synthesis. However, the application of PDR may only be limited by the creativity and intuition of synthetic chemists building on minimal SAR. For example, SAR of isolated natural product congeners and other lines of evidence may direct one or more hypotheses regarding the proposed pharmacophore. Indeed, application of PDR can generate alternative strategies for a given natural product through alternative hypothesized pharmacophores in a similar way to consideration of various strategic bond disconnections in classic retrosynthesis. In the present study, the first SAR profiles of gracilin A derivatives were secured providing proof-of-principle studies of PDR and support the notion of the bis-acetoxy furanose as a pharmacophore for immunosuppressive activity but not necessarily for neuroprotective activity (Fig. 7).
Figure 7. Structure–activity relationship (SAR) profile of gracilin A for both immunosuppressive and neuroprotective activity enabled through application of PDR.

a, SAR profile in immunosuppressive assays. b, SAR profile in neuroprotective assays.
Evidence was gathered for our initial hypothesis invoking the bis-acetoxy furanose of gracilin A as the pharmacophore, as relates to immunosuppressive activity, given that derivatives with this moiety (e.g. 23a and 23d) displayed the greatest activity (KD ~5–7 nM), while those lacking this moiety were inactive (e.g. (–)-28). Importantly, PDR disclosed that the C8-ethylidene is not required to elicit potent immunosuppressive activity and with the availability of natural gracilin A as a comparator, it was unnecessary to complete a total synthesis.
While derivatives of CsA were previously investigated as potential neuroprotective agents through inhibition of CypD, they lacked effectiveness due to their high molecular weights and low BBB permeability.46, 47, 48 Several gracilin A derivatives (e.g. 27a,b, and 29a) accessed through this study, that interestingly includes the enantiomeric series, displayed significant neuroprotective activity, importantly while also displaying selectivity for CypD vs CypA inhibition. These gracilin derivatives serve as lead compounds for neurodegenerative diseases and other CypD-mediated diseases including atherosclerosis or autoimmune diseases.49, 50
Although an ideal application of PDR will generally be challenging to implement, in particular when reactive functionality precludes a completely linear synthetic strategy as dictated by PDR and as in the present case of the gracilins, we expect that pharmacophore hypotheses brought into retrosynthetic planning will enable greater SAR information to be gathered in route to a natural product. It is also anticipated that application of PDR will provide an avenue for hypothesis-driven, natural product total synthesis efforts while simultaneously accelerating the exploration of natural product chemical space through total synthesis efforts premised on PDR.
Methods
Gram-scale Diels-Alder-Lactonization Organocascade providing γ-lactones (–)-15a and (+)-15b, the bicyclic core of gracilin A derivatives.
To an oven-dried, 250-mL round-bottomed flask equipped with a magnetic stir bar was added silyloxydiene alcohol 1251 (2.14 g, 10.0 mmol, 1.0 equiv), (S)-(–)-TM·HCl (2.40 g, 10.0 mmol, 1.0 equiv), 2,6-lutidine (3.5 mL, 30.0 mmol, 3.0 equiv) and anhydrous CH2Cl2 (100 mL, to make the final concentration of silyloxydiene alcohol 0.1 M) at ambient temperature (23 ºC). With vigorous stirring, 11 (1.05 mL, 13.0 mmol, 1.3 equiv) in CH2Cl2 (9.0 mL) was added over a period of 5 h by syringe pump addition. After stirring for an additional 13 h, the reaction mixture was filtered through a short pad of SiO2 and the filtrate was concentrated by rotary evaporation. Purification by automated flash chromatography (5→50% EtOAc/hexanes) afforded bicyclic γ-lactones (–)-15a (1.55 g, 58% yield, 94% ee) and (+)-15b (0.51 g, 19% yield, 94% ee). See Supporting for full characterization of 15a and 15b.
Binding experiments: Surface activation, ligand immobilization and binding.
A Biacore X SPR biosensor with Control Software and BIAevaluation software version 3.0 from Biacore (GE Healthcare, Uppsala, Sweden) was used to check the binding between gracilin A derivatives and CypA. Sensor surface activation and ligand immobilization were performed by using HBS-EP as running buffer at a flow rate of 5 μL/min and 25 °C. CM5 sensor chips were used as surface where Cyp A was immobilized as ligand. The CM5 chip is a glass slide coated with a thin layer of gold with a matrix of carboxymethylated dextran covalently attached. The CM5 chip was activated using an amine coupling kit. Following manufacture instructions, a mixture (1:1, v/v) of EDC and NHS was applied for 2 min over the sensor chip. After activation, the ligand, 100 μg/mL of active human CypA protein dissolved in sodium acetate 10 mM at pH 4.5 was added to be immobilized over a CM5 sensor chip. Finally, ethanolamine-HCl was injected to deactivate the remaining active esters and to avoid non-specific binding. Next, analytes (CsA as positive control or synthetic compounds) were added to evaluate the binding with CypA. Once analytes were tested and interaction was observed, individual binding curves were analyzed by determining the kinetic constants of analytes-CypA binding, namely, the observed rate constant (Kobs), the association rate constant (Kass), the dissociation rate constant (Kdiss), and the kinetic equilibrium dissociation constant (KD). At equilibrium, by definition, Kdiss/Kass= KD. The pseudo-first-order association rate constants Kobs (s−1) were determined for each compound concentration by using the 1:1 Langmuir association model of BiaEvaluation software (BiaCore, Uppsala, Sweden). Then a representation of Kobs against the corresponding concentration of each compound was done. These plots follow a linear correlation coefficient. From the equation of these representations, Kass, M−1 s−1, gradient of the plot, and Kdiss, s −1, intercept of the plot was obtained. Within these two values, the kinetic equilibrium dissociation constant KD for each analyte-CypA binding was obtained.
The duration of the sample injection was 2 min at 10 μL/min flow rate. Next, dissociation of bound molecules in HBS-EP buffer flow was studied. The bound drugs were removed from the chip surface before the next injection by adding 1 M Glycine-HCl at pH 2.5 for 1 min. The association phase was used to quantify the compound-CypA interactions. All experiments were performed four times.
Determination of intracellular ROS levels.
Intracellular levels of reactive oxygen species (ROS) were determined with carboxy-H2DCFDA (5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate). This dye diffuses through the cellular membrane and is converted by cellular esterases to carboxy-H2DCFH (non-fluorescent). When carboxy-H2DCFH is oxidized by ROS, it becomes fluorescent.52 Cells were seeded at a density of 2.5×105 cells/mL in 96-well plates and allowed to attach for 24h. Following treatment with compounds at various concentrations (1 µM→1 nM) and 150 µM H2O2 for 6h, SH-SY5Y cells were washed twice with serum-free culture medium. Then, carboxy-H2DCFDA 20 µM dissolved in serum-free culture medium was added to each well and the cells were incubated for 1h at 37 ºC. After this incubation, the medium with the fluorescent dye was replaced with PBS and the plate was incubated for 30 min at 37 ºC. Fluorescence was read at 527 nm, with an excitation wavelength of 495 nm. All experiments were performed four times.
Mitochondrial membrane permeability transition pore (mPTP) measurement.
The blockage of mPTP by compounds was determined with the MitoProbe Transition Assay Kit following manufacturer’s instructions. Briefly, SH-SY5Y human neuroblastoma cells were re-suspended in pre-warmed PBS/Ca+2 buffer at a final concentration of 1×106 cells/mL. Cells were loaded with 0.01 µM Calcein-AM and incubated at 37ºC for 15 min. Then, 0.4 mM CoCl2 and compounds at selected concentrations were added and incubated for 15 min at 37ºC. Cyclosporine A (CsA) at 0.2 µM was used as positive control. After this incubation, cells were centrifuged and re-suspended in 100 µL of PBS. Just before analyzing, 1 mM tert-butyl hydroperoxide (TBHP) was added to the samples to induce pore opening. Fluorescence intensity was measured at 488 nm excitation and 517 nm emission wavelengths by flow cytometry using the ImageStreamMKII (Amnis Corporation, Merck-Millipore) and INSPIRE® software. The fluorescence of 10,000 events was analyzed with IDEAS® Application vs 6.0 (Amnis Corporation, Merck-Millipore). Experiments were carried out four times.
Supplementary Material
Acknowledgement.
Support from NIH (R37 GM052964 to D.R.), the Robert A. Welch Foundation (AA-1280 to D.R.), FEDER cofunded-grants: from CONSELLERIA DE Cultura, EDUCACION e ordenación Universitaria Xunta de Galicia, 2017 GRC GI-1682 (ED431C 2017/01), from CDTI and Technological Funds, supported by Ministerio de Economía, Industria y Competitividad, AGL2014-58210-R, AGL2016-78728-R (AEI/FEDER, UE)(LB), ISCIII/PI16/01830 (AA) and RTC-2016-5507-2, ITC-20161072, and from European Union POCTEP 0161-Nanoeaters −1-E-1, Interreg AlertoxNet EAPA-317-2016, and H2020 778069-EMERTOX (LB) and from the European Union’s Seventh Framework Programme managed by REA–Research Executive Agency (FP7/2007–2013 under grant agreement 312184 PHARMASEA to L.B. and M.J.) is gratefully acknowledged. Drs. Nattamai Bhuvanesh and Joe Reibenspies (Center for X-ray Analysis, TAMU) secured X-ray data and Dr. Bill Russell (Laboratory for Biological Mass Spectrometry, TAMU) provided mass data. Correspondence and requests for materials should be directed to D. Romo (chemistry) and L. Botana (biology).
Footnotes
Data Availability.
Crystallographic data for the structure reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition number CCDC 1557733 ((–)-21b). A copy of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
Additional information
Full synthetic details for all new, bioactive gracilin derivatives and full characterization including 1H (1D and 2D) and 13C NMR spectra (Supplementary Sections A and C). Further details of the immunosuppressive (Cyp A binding, IL-2 release inhibition and cellular viability) and neuroprotective assays (cellular viability, TMRM assay, ROS release, GSH content, mPTP opening and PPiase activity; Supplementary Section B).
Competing interests
The authors declare no competing interests.
References
- 1.Li J, Kim Sang G. & Blenis J Rapamycin: One Drug, Many Effects. Cell Metab 19, 373–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trauner D Finding function and form. Nat. Prod. Rep 31, 411–413 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Wilson RM & Danishefsky SJ Small Molecule Natural Products in the Discovery of Therapeutic Agents: The Synthesis Connection. J. Org. Chem 71, 8329–8351 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Wender PA Toward the ideal synthesis and molecular function through synthesis-informed design. Nat. Prod. Rep 31, 433–440 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Schreiber SL Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 287, 1964–1969 (2000). [DOI] [PubMed] [Google Scholar]
- 6.van Hattum H & Waldmann H Biology-Oriented Synthesis: Harnessing the Power of Evolution. J. Am. Chem. Soc 136, 11853–11859 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Seiple IB, Zhang Z, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, Kitamura Y, Zhou X, Condakes ML, Szczypiński FT, Green WD & Myers AG A platform for the discovery of new macrolide antibiotics. Nature 533, 338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Könst ZA, Szklarski AR, Pellegrino S, Michalak SE, Meyer M, Zanette C, Cencic R, Nam S, Voora VK, Horne DA, Pelletier J, Mobley DL, Yusupova G, Yusupov M & Vanderwal CD Synthesis facilitates an understanding of the structural basis for translation inhibition by the lissoclimides. Nat. Chem 9, 1140 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bathula SR, Akondi SM, Mainkar PS & Chandrasekhar S “Pruning of biomolecules and natural products (PBNP)”: an innovative paradigm in drug discovery. Org. Biomol. Chem 13, 6432–6448 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Yu MJ, Zheng W & Seletsky BM From micrograms to grams: scale-up synthesis of eribulin mesylate. Nat. Prod. Rep 30, 1158–1164 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Crane EA & Gademann K Capturing Biological Activity in Natural Product Fragments by Chemical Synthesis. Angew. Chem. Int. Ed 55, 3882–3902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romo D, Choi NS, Li S, Buchler I, Shi Z & Liu JO Evidence for Separate Binding and Scaffolding Domains in the Immunosuppressive and Antitumor Marine Natural Product, Pateamine A: Design, Synthesis, and Activity Studies Leading to a Potent Simplified Derivative. J. Am. Chem. Soc 126, 10582–10588 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Trost B The atom economy--a search for synthetic efficiency. Science 254, 1471–1477 (1991). [DOI] [PubMed] [Google Scholar]
- 14.Newhouse T, Baran PS & Hoffmann RW The economies of synthesis. Chem. Soc. Rev 38, 3010–3021 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young IS & Baran PS Protecting-group-free synthesis as an opportunity for invention 1, 193 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Corey EJ C. X-M The Logic of Chemical Synthesis John Wiley & Sons, 1995. [Google Scholar]
- 17.Czakó B, Kürti L, Mammoto A, Ingber DE & Corey EJ Discovery of Potent and Practical Antiangiogenic Agents Inspired by Cortistatin A. J. Am. Chem. Soc 131, 9014–9019 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Mayol L, Piccialli V & Sica D Gracilin A, an unique: nor-diterpene metabolite from the marine sponge spongionella gracilis. Tetrahedron Lett 26, 1357–1360 (1985). [Google Scholar]
- 19.Sanchez JA, Alfonso A, Leiros M, Alonso E, Rateb ME, Jaspars M, Houssen WE, Ebel R, Tabudravu J & Botana LM Identification of Spongionella compounds as cyclosporine A mimics. Pharmacol. Res 107, 407–414 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Leiros M, Alonso E, Rateb ME, Houssen WE, Ebel R, Jaspars M, Alfonso A & Botana LM Gracilins: Spongionella-derived promising compounds for Alzheimer disease. Neuropharmacology 93, 285–293 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Corey EJ & Letavic MA Enantioselective Total Synthesis of Gracilins B and C Using Catalytic Asymmetric Diels-Alder Methodology. J. Am. Chem. Soc 117, 9616–9617 (1995). [Google Scholar]
- 22.Rateb ME, Houssen WE, Schumacher M, Harrison WT, Diederich M, Ebel R & Jaspars M Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod 72, 1471–1476 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Rueda A L. A; Fernandez R; Cabanas C; Garcia-Fernandez LF; Reyes F; Cuevas C Gracilins G-I, Cytotoxic Bisnorditerpenes from Spongionella pulchella, and the Anti-Adhesive Properties of Gracilin B. Lett. Drug Des. Disc 3, 753–760 (2006). [Google Scholar]
- 24.Puliti R, Fontana A, Cimino G, Mattia CA & Mazzarella L Structure of a keto derivative of 9,11-dihydrogracilin A. Acta Crystallographica Section C 49, 1373–1376 (1993). [Google Scholar]
- 25.Potts BC, Faulkner DJ & Jacobs RS Phospholipase A2 inhibitors from marine organisms. J. Nat. Prod 55, 1701–1717 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Leiros M, Alonso E, Sanchez JA, Rateb ME, Ebel R, Houssen WE, Jaspars M, Alfonso A & Botana LM Mitigation of ROS insults by Streptomyces secondary metabolites in primary cortical neurons. ACS Chem. Neurosci 5, 71–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leiros M, Sanchez JA, Alonso E, Rateb ME, Houssen WE, Ebel R, Jaspars M, Alfonso A & Botana LM Spongionella secondary metabolites protect mitochondrial function in cortical neurons against oxidative stress. Mar. Drugs 12, 700–718 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kofron JL, Kuzmic P, Kishore V, Colon-Bonilla E & Rich DH Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30, 6127–6134 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Walsh CT, Zydowsky LD & McKeon FD Cyclosporin A, the cyclophilin class of peptidylprolyl isomerases, and blockade of T cell signal transduction. J. Biol. Chem 267, 13115–13118 (1992). [PubMed] [Google Scholar]
- 30.Ferreira PA & Orry A From Drosophila to humans: reflections on the roles of the prolyl isomerases and chaperones, cyclophilins, in cell function and disease. J. Neurogenet 26, 132–143 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J & Kim SS An overview of cyclophilins in human cancers. J. Int. Med. Res 38, 1561–1574 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Hogan PG, Chen L, Nardone J & Rao A Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17, 2205–2232 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Nigro P, Pompilio G & Capogrossi MC Cyclophilin A: a key player for human disease. Cell Death Dis 4, e888 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Picone P, Nuzzo D, Caruana L, Scafidi V & Di Carlo M Mitochondrial dysfunction: different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev 2014, 780179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schnermann MJ, Beaudry CM, Egorova AV, Polishchuk RS, Sütterlin C & Overman LE Golgi-modifying properties of macfarlandin E and the synthesis and evaluation of its 2,7-dioxabicyclo[3.2.1]octan-3-one core. Proc. Natl. Acad. Sci. U.S.A 107, 6158–6163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kornienko A & La Clair JJ Covalent modification of biological targets with natural products through Paal-Knorr pyrrole formation. Nat. Prod. Rep 34, 1051–1060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nirmal N, Praba GO & Velmurugan D Modeling studies on phospholipase A₂-inhibitor complexes. Indian J. Biochem. Biophys 45, 256–262 (2008). [PubMed] [Google Scholar]
- 38.Baker BJ, Kopitzke RW, Yoshida WY & McClintock JB Chemical and Ecological Studies of the Antarctic Sponge Dendrilla membranosa. J. Nat. Prod 58, 1459–1462 (1995). [Google Scholar]
- 39.Buckleton JS, Bergquist PR, Cambie RC, Clark GR, Karuso P & Rickard CEF Structure of tetrahydroaplysulphurin-1. Acta Crystallogr. C 43, 2430–2432 (1987). [Google Scholar]
- 40.Abbasov ME, Hudson BM, Tantillo DJ & Romo D Stereodivergent, Diels–Alder-initiated organocascades employing α,β-unsaturated acylammonium salts: scope, mechanism, and application. Chem. Sci 8, 1511–1524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harvey NL, Krysiak J, Chamni S, Cho SW, Sieber SA & Romo D Synthesis of (±)-Spongiolactone Enabling Discovery of a More Potent Derivative. Chem. Eur. J 21, 1425–1428 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Alfonso A, Pazos MJ, Fernandez-Araujo A, Tobio A, Alfonso C, Vieytes MR & Botana LM Surface plasmon resonance biosensor method for palytoxin detection based on Na+,K+-ATPase affinity. Toxins 6, 96–107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanchez JA, Alfonso A, Leiros M, Alonso E, Rateb ME, Jaspars M, Houssen WE, Ebel R & Botana LM Spongionella Secondary Metabolites Regulate Store Operated Calcium Entry Modulating Mitochondrial Functioning in SH-SY5Y Neuroblastoma Cells. Cell Physiol. Biochem 37, 779–792 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Damsker JM, Bukrinsky MI & Constant SL Preferential chemotaxis of activated human CD4+ T cells by extracellular cyclophilin A. J. Leukocyte Biol 82, 613–618 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreira PI, Zhu X, Wang X, Lee H. g., Nunomura A, Petersen RB, Perry G & Smith MA Mitochondria: A therapeutic target in neurodegeneration. Biochim. Biophys. Acta, Mol. Basis Dis 1802, 212–220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azzolin L, Antolini N, Calderan A, Ruzza P, Sciacovelli M, Marin O, Mammi S, Bernardi P & Rasola A Antamanide, a derivative of Amanita phalloides, is a novel inhibitor of the mitochondrial permeability transition pore. PLoS One 6, e16280 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo HX, Wang F, Yu KQ, Chen J, Bai DL, Chen KX, Shen X & Jiang HL Novel cyclophilin D inhibitors derived from quinoxaline exhibit highly inhibitory activity against rat mitochondrial swelling and Ca2+ uptake/ release. Acta Pharmacol Sin 26, 1201–1211 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao VK, Carlson EA & Yan SS Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 1842, 1267–1272 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dawar FU, Tu J, Khattak MN, Mei J & Lin L Cyclophilin A: A Key Factor in Virus Replication and Potential Target for Anti-viral Therapy. Curr Issues Mol Biol 21, 1–20 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Satoh K Cyclophilin A in cardiovascular homeostasis and diseases. Tohoku J Exp Med 235, 1–15 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Abbasov ME, Hudson BM, Tantillo DJ & Romo D Acylammonium Salts as Dienophiles in Diels–Alder/Lactonization Organocascades. J. Am. Chem. Soc 136, 4492–4495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halliwell B & Whiteman M Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol 142, 231–255 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.