

Abstract
Background:
Inflammatory response after myocardial infarction (MI) is essential for cardiac healing, while excessive and prolonged inflammation extends the infarction and promotes adverse cardiac remodeling. Understanding the mechanistic insight of these tightly controlled inflammatory processes has a significant impact on the post-MI recovery and therapy. Here, we uncover the critical role of small GTPase RhoE in the post-MI recovery and its clinical implication.
Methods:
Three genetic mouse lines are used. They are global RhoE knockout, cardiomyocyte-specific RhoE heterozygous and cardiomyocyte-specific RhoE overexpression mice. A set of molecular signaling experiments including bimolecular fluorescence complementation (BiFC), immunoprecipitation, electrophoretic mobility shift assay and mRNA microarray analysis are conducted. A permanent ligation of the left anterior descending artery (LAD) is performed followed by the assessments of cardiac function, inflammation and survival in the first week post-MI. Finally, we examine the correlation of the expression levels of RhoE in MI patient heart and the patient prognosis.
Results:
RhoE deficiency turns on a group of pro-inflammatory gene expressions in mouse heart. Mice with cardiomyocyte-specific haploinsufficiency exhibit excessive inflammatory response with deleterious cardiac function after MI. A profound increase in NF-κB activity is detected in the mutant heart as well as the isolated cardiomyocytes. We further find that the expression of RhoE is upregulated in response to MI. Mechanistically, RhoE interacts with p65 and p50 individually in cytosol and blocks their nuclear translocation. RhoE also occupies the dimerization domain of p65 and subsequently disrupts the heterodimerization between p65 and p50. Cardiac RhoE overexpression inhibits NF-κB activity, restrains post-MI inflammation and improves cardiac function and survival. Consistently, we find that the expression level of RhoE is elevated in MI patient heart, and the patients with higher expression level of RhoE exhibit a better prognosis in cardiac function recovery.
Conclusions:
The study uncovers RhoE as a new fine-tuning factor modulating MI-induced inflammation and promoting injured heart recovery. RhoE may serve as a new potential biomarker for the assessment of MI patient prognosis. Manipulation of RhoE could be as a potential therapeutic approach for MI and other inflammatory diseases.
Keywords: Myocardial infarction, Inflammation, NF-κB, Rho-GTP-binding proteins
Introduction
Myocardial infarction (MI) is a severe clinical condition caused by coronary artery thrombotic occlusion.1 Cardiac repair following MI can be divided into three distinct but overlapping phases: the inflammatory phase, the proliferative phase, and the maturation phase.2 The inflammatory phase is the most critical and dangerous stage since most of the adverse cardiac events occur in this period. Acute inflammation is developed in this phase to clear necrotic myocardium and to lay foundation for the upcoming healing processes. A well-balanced and tightly-controlled inflammation significantly impacts each stage of post-MI healing and cardiac remodeling.3, 4 Excessive or prolonged inflammation, for example, causes cytotoxic effects, which extends the ischemic injuries and delays the healing process.3, 5 Multiple anti-inflammatory strategies have been tested in experimental animals, and expected beneficial effects of reducing infarct size and preventing adverse cardiac remodeling have been observed.4 However, translations from bench and animal findings to clinical therapies are not satisfactory,6 suggesting that regulation of inflammation in MI patients is more complicated than we thought. Recent emerging clinical studies indicate that selectively modulating key inflammatory regulators provides more benefits to MI patients than globally suppressing downstream inflammatory factors.7–9
The transcription factor NF-κB is a pivot regulator upstream of the inflammatory response and other critical biological processes.10, 11 A low basal transcriptional activity of NF-κB is required in normal physiology, while aberrant NF-κB activation has been linked to various inflammatory diseases.12, 13 NF-кB activity is tightly regulated by IκB proteins. IκBα sequesters the inactive NF-кB complex in cytosol. Upon activation by stimuli, such as ischemia, bacteria and cytokines, IκBα is phosphorylated and subsequently degraded, allowing the NF-кB complex to translocate into the nucleus for regulation of target genes.14, 15 Many proteins, miRNAs, lncRNAs and small molecules regulate NF-κB activity through directly or indirectly modulating IκBα proteolysis. 16–19 However, few inhibitory factors directly target the NF-κB complex in cytosol.
RhoE/Rnd3 is a small GTPase that functions to regulate actin cytoskeleton organization and cell migration.20, 21 We have previously demonstrated that RhoE is essential for cardiac physiology, including ventricular remodeling, calcium handling and cardiac angiogenesis.22–24 Here we report that RhoE functions as a negative inflammatory mediator in myocardial infarction by specifically suppressing NF-кB in cytosol. Genetic manipulations of RhoE expression level in mouse heart determine the severity of inflammation and the outcomes after MI. Importantly, we found that RhoE expression level in the heart of MI patients positively correlated with their prognosis, indicating RhoE as a potential therapeutic target for MI and other inflammatory diseases.
Methods
The data, analytic methods, and study materials will be made available to other researchers for the purposes of reproducing the results or replicating the procedure. The detail methods and any associated references are provided in the online data supplement.
Human heart tissues and animal usage.
Human study protocol was approved by the Fuwai Cardiovascular Hospital. The procurement of human heart tissue usage for the study was obtained with written patient-informed consent and approval by the Ethics Committee of Fuwai Cardiovascular Hospital (approval number: 2013-496). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Texas A&M University Health Science Center Institute of Biosciences and Technology.
Statistics.
For two group comparisons, unpaired, 2-tailed student’s t-test was used in the animal and human study. For any multiple group comparisons, one-way ANOVA followed by Bonferroni test was performed. All values were presented as means ± SD; n refers to the sample size. A value of P < 0.05 was considered statistically significant.
Results
Expression level of RhoE is closely associated with inflammation in heart
We previously found that RhoE is downregulated in human failing heart and that RhoE is critical for cardiac calcium homeostasis and responsive angiogenesis.22–24 To further explore the cardiac functions of RhoE, we examined possible pathways that RhoE may be involved in. Since RhoE general knockout led to mouse embryo lethality at E11.0,22 we conducted mRNA microarray analysis in RhoE-null embryonic E10.5 mouse heart. Among the affected pathways (Fig. S1), significant upregulation of a large set of pro-inflammatory factors was observed, including cytokines/chemokines and their modulators, members of tumor necrosis factor (TNF) superfamily, interferons, immunoreceptors, matrix metalloproteinases (MMPs), etc. (Fig. S2A). Since cardiac immune cells have not been developed at mouse E10.5,25 elevation of these pro-inflammatory factors in heart is unexpected and intriguing, which led us to study the possible regulation of cardiac inflammation by RhoE.
It is well documented that myocardial infarction triggers active inflammatory responses, which are then gradually resolved after the acute inflammatory phase. When examining RhoE expression dynamics in wild-type mouse heart in the first week post-MI, we observed significant increases in RhoE protein expression (Fig. S2B). RhoE protein level continued to rise and reached the highest level at day 3 post-MI, and gradually decreased after then. The data showed the concurrence between the dynamic response of RhoE expression and the natural progression of post-MI inflammation.
RhoE deficiency in heart promotes post-MI inflammation
To investigate the correlation between RhoE expression and the inflammatory response in heart, we generated RhoE loxP mice and then bred the mice with αMHC-Cre (Myh6-Cre) mice to induce cardiomyocyte-specific RhoE knockout (Fig. S3). The homozygous RhoE knockout led to embryonic lethality, while RhoE haploinsufficient mice (RhoEflox/+;αMHC-Cre) were viable without detectable abnormalities and cardiac dysfunction. A decrease in RhoE protein level was confirmed in RhoE haploinsufficient mouse heart (Fig. 1A). We used these RhoE-deficient mice for the study and RhoE loxP (RhoEflox/+) mice as a control. MI operation was performed and cardiac inflammation was then analyzed at day 3 post-MI. We observed robust increases in neutrophil and macrophage infiltration in the RhoEflox/+;αMHC-Cre mouse heart compared to the RhoEflox/+ control mouse heart (Fig. 1, B and C). RhoE deficiency also promoted production of the pro-inflammatory cytokines and MMPs (Fig. 1D). Consistent with the excessive inflammation in heart, enlarged infarct size, deteriorated cardiac function, and increased mortality were also exhibited in RhoE deficient mice in the first week post-MI (Fig. 1, E-G). These results indicate the critical role of RhoE in cardiomyocytes in regulation of post-MI inflammation.
Figure 1. Cardiac RhoE deficiency induces excessive post-MI inflammation.
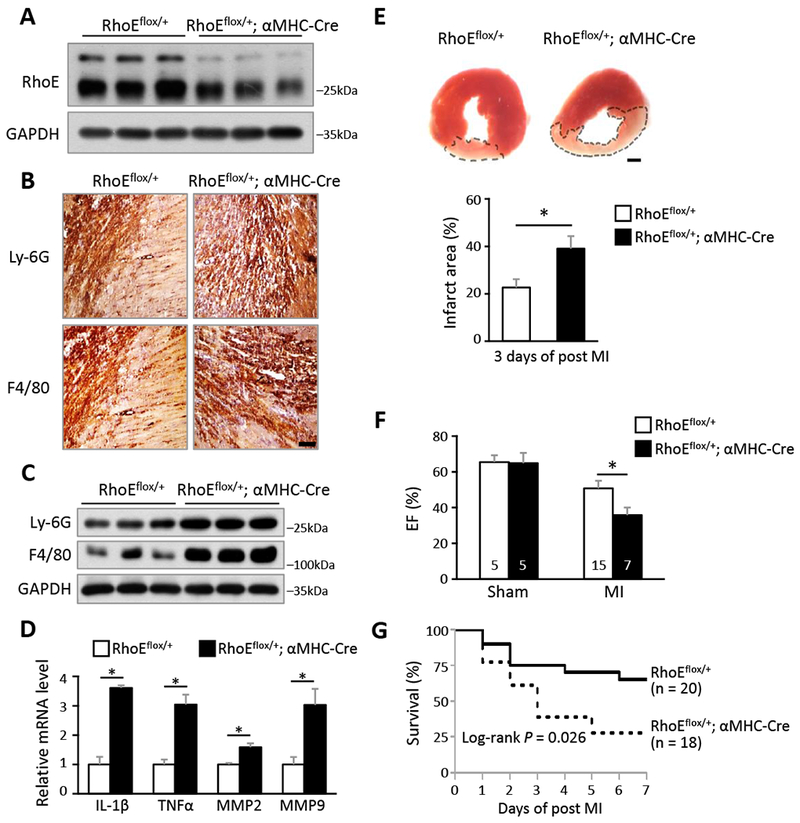
(A) Immunoblot for RhoE in 3 pairs of RhoEflox/+ and RhoEflox/+;αMHC-Cre mouse hearts. (B) IHC staining for neutrophil marker (Ly-6G) and macrophage marker (F4/80) in RhoEflox/+ and RhoEflox/+;αMHC-Cre mouse hearts on day 3 post MI. Scale bar: 0.2 mm. (C) Immunoblot for Ly-6G and F4/80 in whole heart lysates from the above mice. (D) qRT-PCR for IL-1β, TNFα, MMP2 and MMP9 in whole heart lysates from above mice. (E) TTC staining for infarct area (top panel, outlined by dashed line). Scale bar: 1.0 mm. Infarct area was quantified to left ventricle area in 3 pairs of mice (bottom panel). (F) Ejection fraction of sham and MI mice on day 3 post MI. (G) Kaplan-Meier survival curves of mice in the first week post MI. n: number of mice. *: P < 0.05.
RhoE negatively regulates NF-κB activation
NF-κB is a master upstream regulator of inflammatory response. Overactivated NF-κB triggers excessive inflammation during the cardiac healing process after MI, resulting in detrimental outcomes of MI.26–28 We assessed NF-κB activation in the post-MI mouse heart by evaluating nuclear p65 and p50 protein levels, and detected a profound increase in nuclear p65 and p50 levels in the RhoE-deficient mouse heart compared to the control mouse heart (Fig. 2A). To confirm if RhoE deficiency can directly induce NF-κB activation in mouse cardiomyocytes, we compared nuclear p65 and p50 protein levels in the cardiomyocytes isolated from RhoE haploinsufficient mice versus control. We observed a significant increase in p65 protein level and a moderate increase in p50 protein level in RhoE-deficient cardiomyocytes (Fig. 2B, lane 1 and 2). TNFα stimulation further promotes NF-κB activation in RhoE deficient cardiomyocytes (Fig. 2B, lane 3 and 4). Together, these results suggest that RhoE negatively regulates NF-κB activation in mouse cardiomyocytes.
Figure 2. RhoE negatively regulates NF-κB activation in vivo and in vitro.
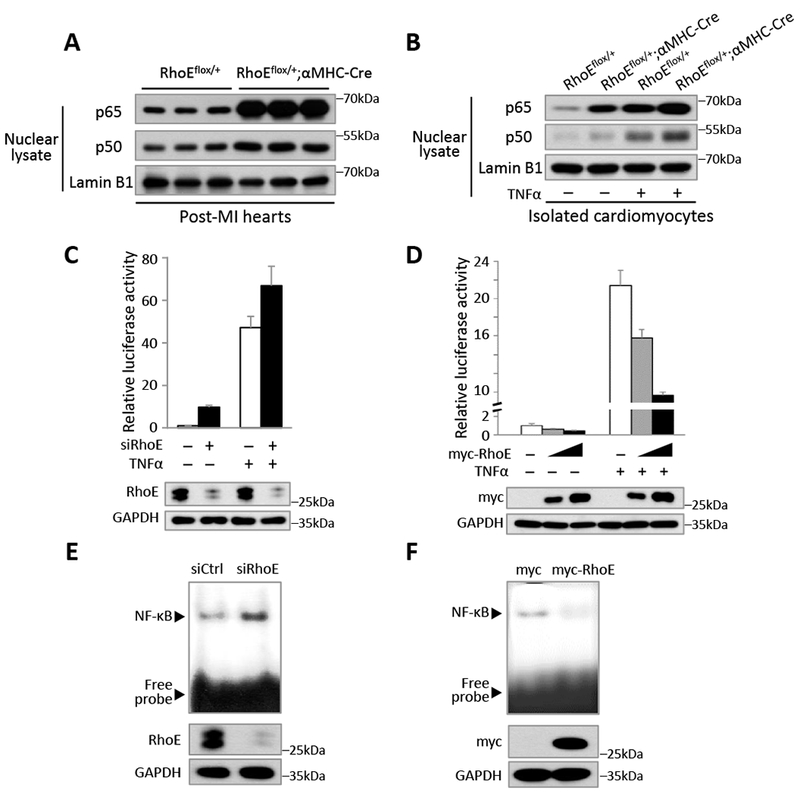
(A) Immunoblot for nuclear p65 and p50 in RhoEflox/+ and RhoEflox/+;αMHC-Cre mouse hearts on day 3 post MI. (B) Immunoblot for nuclear p65 and p50 in isolated cardiomyocytes. The cells were either untreated or stimulated with 40 ng/ml of TNFα for 15 minutes. (C-D) NF-κB-dependent reporter luciferase assay in C2C12 cells transfected with control siRNA or RhoE-specific siRNA (C), and in C2C12 cells transfected with empty vector or myc-RhoE expression construct (D). The cells were either untreated or stimulated with 40 ng/ml of TNFα for 4 hours. RhoE expression levels were assessed by immunoblot. (E-F) EMSA assay for NF-κB activity in C2C12 cells transfected with control siRNA or siRhoE (E), or in C2C12 cells transfected with empty vector or myc-RhoE expression construct (F).
Next, we investigated the regulatory role of RhoE in NF-κB transcriptional activity. We conducted NF-κB-dependent luciferase reporter assay. Knockdown of RhoE induced about 10-fold increase in NF-κB reporter activity (Fig. 2C). Further increase was detected under TNFα stimulation. In contrast, overexpression of RhoE repressed luciferase activity (Fig. 2D). Furthermore, the inhibitory role of RhoE on NF-κB activity was confirmed by electrophoretic mobility shift assay (EMSA). Knockdown of RhoE enhanced NF-κB binding to the κB site-containing probe (Fig. 2E), while overexpression of RhoE weakened NF-κB the DNA binding affinity (Fig. 2F). Collectively, these results suggest that RhoE is a suppressor of NF-κB.
RhoE binds to p65 and p50 in cytosol to inhibit NF-κB nuclear translocation
To explore RhoE regulatory mechanisms of NF-κB, we examined the relationship between RhoE and the NF-κB components: p65 and p50. GST pull-down assay was conducted and the result showed strong binding of GST-RhoE recombinant protein to His-p65 and His-p50, respectively (Fig. 3, A and B). Co-immunoprecipitations revealed physical interactions of endogenous RhoE with endogenous p65 and p50, respectively (Fig. 3C). To verify the binding specificity of RhoE with p65 and p50, we also examined the interaction of RhoE with IκBα, and the interaction of p65 or p50 with other GTPases, such as RhoA and Rnd1 (Fig. S4 A-E). No interactions were detected among these proteins, indicating the high specificity of RhoE binding to p65 and p50. To further illustrate protein-protein interactions among RhoE, p65 and p50, we conducted the bimolecular fluorescence complementation (BiFC) assay. BiFC is an imaging technology used to visualize protein-protein interaction within a cell. The assay is based on re-formation of a functional fluorescent protein when two non-fluorescent fragments are brought together by two interacting proteins. The result provides location and intensity of the interaction between two proteins.29 Here, we fused RhoE to N-terminus of fluorescent protein Venus (termed VN-RhoE); and p65 or p50 to C-terminus of fluorescent protein Venus (termed VC-p65 or VC-p50). No fluorescent signal was observed when two non-interactive proteins were co-expressed in C2C12 cells, which was presented as a negative control (Fig. 3D, left panel). BiFC assay also did not detect any positive interaction of constitutively active RhoA (Q63L) with p65 and p50 (Fig. S4F). However, clear fluorescent signals were exhibited when VN-RhoE and VC-p65, or VN-RhoE and VC-p50 expression vectors were co-transfected, confirming the physical interactions of RhoE with p65 and p50 (Fig. 3D, right panel). Importantly, the fluorescent signal was solely detected in cell cytosol, suggesting that RhoE interacts with p65 and p50 only in cytosol to suppress NF-κB activation.
Figure 3. RhoE interacts with p65 and p50 individually in the cytosol.
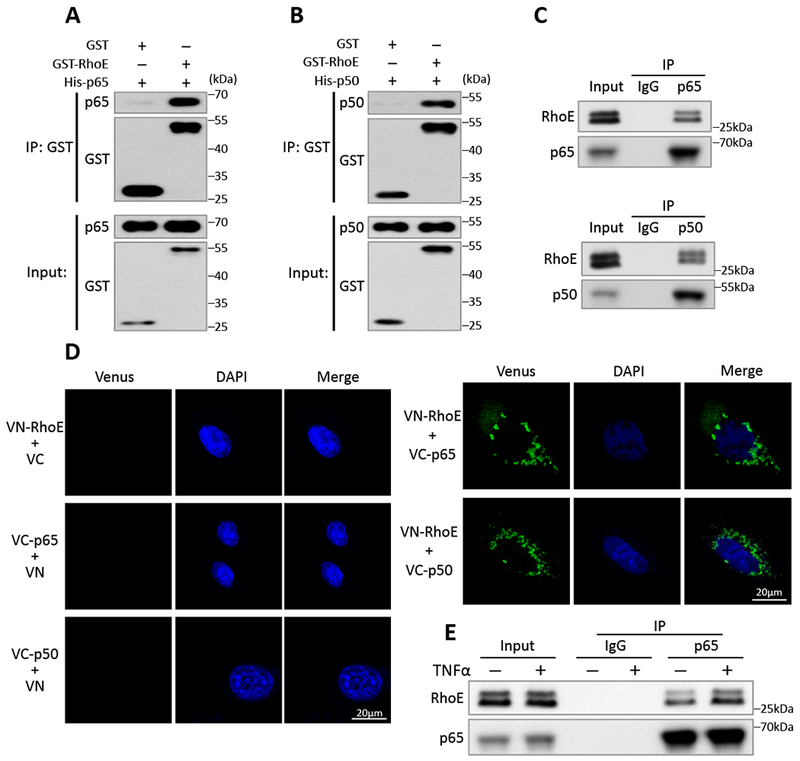
(A-B) In vitro GST pull-down assay of GST and GST-RhoE fusion proteins with His-p65 fusion protein (A) or His-p50 fusion protein (B). (C) Co-IP assay of endogenous p65 (top panel) or endogenous p50 (bottom panel) with endogenous RhoE in C2C12 cells. (D) BiFC assay exhibited negative controls (left panel), and interactions of RhoE (fused with N-terminus of Venus, termed VN-RhoE) with p65 and p50 (fused with C-terminus of Venus, termed VC-p65 or VC-p50) in C2C12 cells (right panel). (E) Co-IP assay between endogenous p65 and endogenous RhoE in C2C12 cells. The cells were either untreated or stimulated with 40 ng/ml of TNFα for 15 minutes.
NF-κB activity is tightly regulated and remains at a low basal level to maintain normal biological functions. Dynamic repressive mechanisms are essential to prevent NF-κB overactivation. Since we have shown that RhoE represses NF-κB activation under both normal and stimulated conditions (Fig. 2), we further tested if RhoE interacted with NF-κB in a dynamic manner. We used co-IP assay to compare the RhoE-p65 interaction before and after TNFα treatment, and found that TNFα stimulation rapidly led to increased interaction between RhoE and p65, suggesting a dynamic response of RhoE-mediated NF-κB inhibition (Fig. 3E).
In cytosol, p50 and p65 form the NF-кB complex with the inhibitory protein IκBα to remain inactive. IκBα is phosphorylated and degraded by the ubiquitin proteasome system (UPS) upon NF-кB stimulation. The dimerized p50 and p65 then translocate into nucleus to regulate target genes.16, 30–32 We examined these regulatory mechanisms to see how RhoE involved in the inhibitory regulation of NF-кB in cytosol. We found that silencing or forcing RhoE expression did not affect the phosphorylation and expression level of IκBα (Fig. 4, A and B). The knockdown of RhoE by the siRNA specific for RhoE (siRhoE) or overexpression of RhoE also had no effect on either total expression levels of p65 and p50, or p65 phosphorylation level at Ser536 site (Fig. 4, A and B), suggesting that RhoE exerts its inhibitory role neither upstream of IκBα, nor through affecting the degradation and phosphorylation of p65 and p50.
Figure 4. RhoE inhibits NF-κB nuclear translocation.
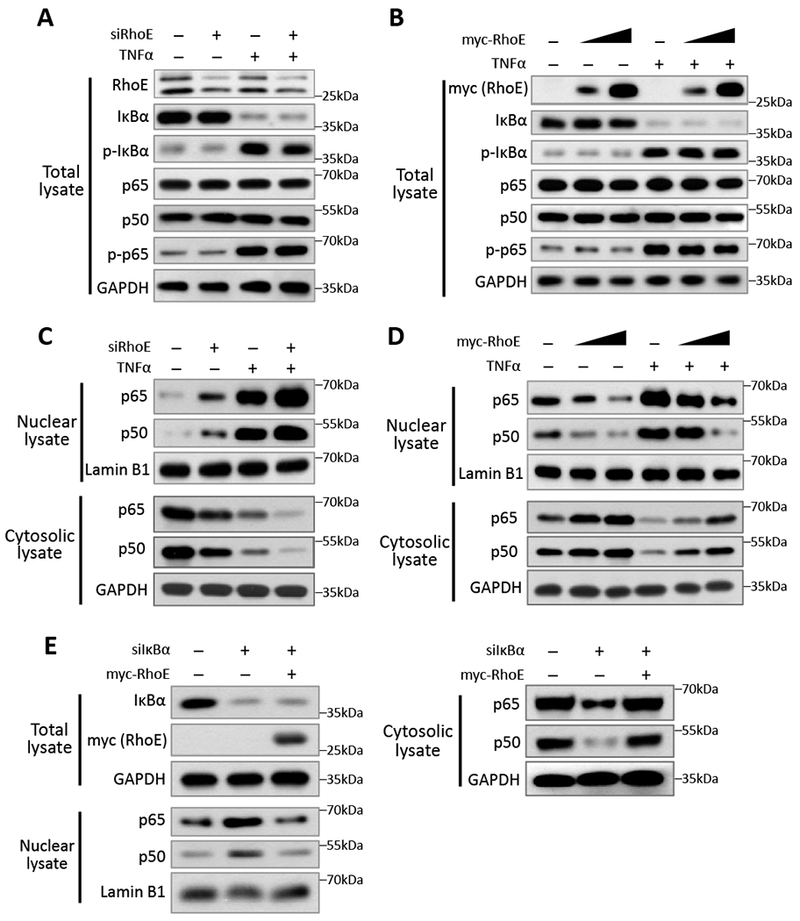
(A-B) Immunoblot for total levels of IκBα, phospho-IκBα (Ser32), p65, p50 and phospho-p65 (Ser536) in C2C12 cells with RhoE knockdown (A), or with RhoE overexpression (B). In TNFα treatment groups, cells were treated with 40 ng/ml of TNFα for 15 minutes. (C-D) Nuclear and cytosolic lysates extracted from above cells were immunoblotted for p65 and p50. (E) C2C12 cells were transfected with control siRNA or IκBα-specific siRNA together with empty vector or myc-RhoE expression construct. Total lysates were immunoblotted for IκBα and myc-RhoE. Nuclear and cytosolic lysates were immunoblotted for p65 and p50.
Nuclear translocation of the NF-κB complex is an essential step for NF-κB activation. We measured nuclear p65 and p50 protein levels to determine if RhoE affects their nuclear translocation. RhoE knockdown significantly increased nuclear p65 and p50 protein level in both resting and TNFα-stimulated conditions (Fig. 4C), while overexpression of RhoE decreased nuclear p65 and p50 protein levels (Fig. 4D), suggesting that RhoE sequesters NF-κB in cytosol and prevents its nuclear translocation. To visualize this regulatory process, we labelled p65 and p50 with GFP and monitored their nuclear translocation in RhoE knockdown or overexpression conditions. The result confirmed the inhibitory effect of RhoE on NF-κB nuclear translocation (Fig. S5).
IκBα is the most critical factor that inhibits NF-κB nuclear translocation.33 Since we have shown that RhoE did not directly change IκBα phosphorylation and total protein level, indicating that RhoE did not affect the upstream regulators of IκBα. We next wondered if RhoE-mediated NF-κB inhibition was directly through IκBα. We knocked down IκBα by siIκBα. As expected, the knockdown led to increased nuclear p65 and p50 levels (Fig. 4E, lane 2). However, under the IκBα knockdown condition, forced expression of RhoE maintained its inhibitory effect in preventing p65 and p50 nuclear translocation (Fig. 4E, lane 3), further indicating that RhoE targets downstream of IκBα to inhibit NF-κB signaling.
Previous studies have demonstrated that RhoA is closely involved in pathological cardiac hypertrophy and ischemic injury,34, 35 while RhoE competitively antagonizes RhoA and subsequently inhibits RhoA/ROCK signaling.36, 37 We therefore determined RhoA impact on RhoE-mediated NF-κB inhibition by overexpression of active RhoA isoform RhoA Q63L and the application of RhoA inhibitor C3 under RhoE overexpression and downregulation, respectively. NF-κB activity was assessed by NF-κB-dependent luciferase assay and the Western blot detecting NF-κB nuclear translocation (Fig. S6). As expected, overexpression of RhoE decreased NF-κB activity, while activation of RhoA significantly increased NF-κB activity. However, activation of RhoA failed to diminish RhoE-mediated NF-κB inhibition (Fig. S6, A and B). We then validated the result by a loss-of function approach using RhoA inhibitor C3 under RhoE knockdown condition. We found that inhibition of RhoA cannot completely block RhoE deficiency-induced NF-κB activation although a moderate decrease of NF-κB activity was detected (Fig. S6, C and D). Together, these data strongly suggest that the activation of RhoA has a minimal effect on RhoE-mediated NF-κB regulation.
RhoE impedes the heterodimerization between p65 and p50
Next, we tried to identify the interaction regions of p50 and p65 with RhoE. A series of p50 and p65 truncated mutants were generated for co-IP assay. We found that both N-terminus and C-terminus of p50 bound to RhoE (Fig. S7), indicating that RhoE associated with p50 via multiple interaction sites. For p65, RhoE only bound to its N-terminus (1-286 aa) but not to its C-terminus (286-551 aa) (Fig. 5, A and B). The N-terminus of p65 contains two functional domains, N-terminal domain (NTD, 1-186 aa), responsible for DNA binding activity; and dimerization domain (DD, 187-286 aa), required for homo- or hetero-dimerization of p65 with other NF-κB subunits.38 We found that dimerization domain, but not N-terminal domain of p65, was capable of binding to RhoE (Fig. 5B). The result was further confirmed by the BiFC assay, which RhoE only interacted with dimerization domain-containing p65 fragments to generate fluorescent signals (Fig. 5C).
Figure 5. RhoE impedes heterodimerization between p65 and p50.
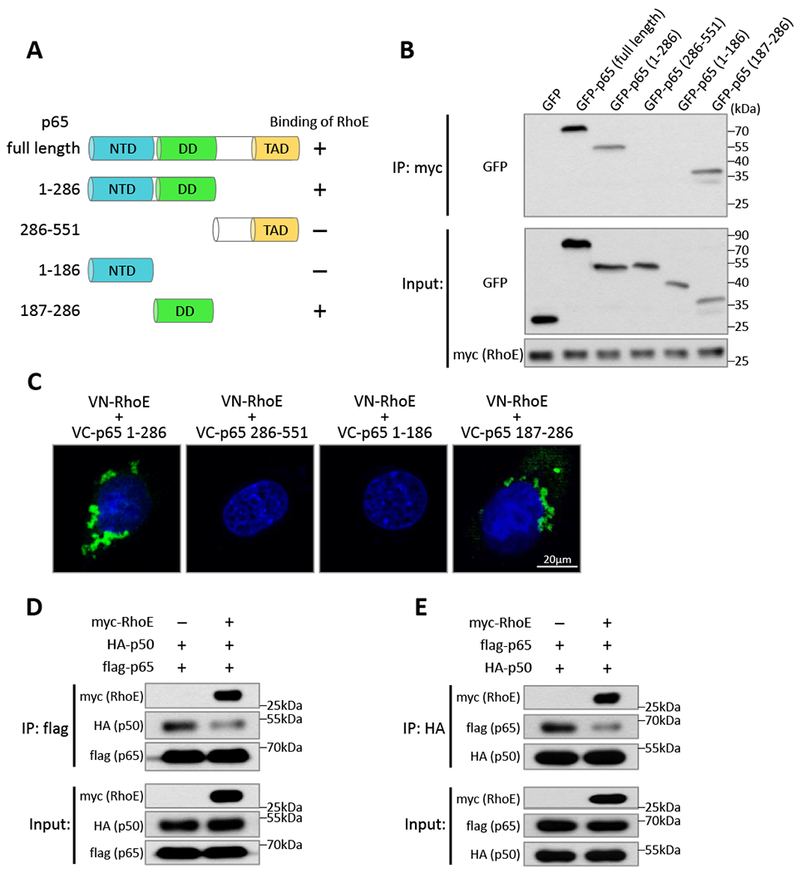
(A) Schematic of p65 domains and truncated mutants. NTD: N-terminal domain; DD: dimerization domain; TAD: transactivation domain. (B) Co-IP assay in HEK293T cells co-transfected with myc-RhoE expression construct and GFP-tagged p65 full length or truncated mutants. (C) BiFC assay in C2C12 cells co-transfected with VN-RhoE and VC-p65 truncated mutants. (D-E) HEK293T cells were co-transfected of HA-p50 and flag-p65 expression constructs together with empty vector or myc-RhoE expression construct. Interaction between p65 and p50 was analyzed by co-IP assay using anti-flag (D) or anti-HA (E) antibodies as the immunoprecipitation antibody.
Dimerization domain of p65 is essential for its dimerization with other NF-κB subunits, such as p50, to execute NF-κB activity.38 We wondered if the binding of RhoE to dimerization domain of p65 impedes the heterodimerization between p65 and p50. Co-IP assay was conducted to evaluate formation of the p65/p50 heterodimer. We found that enforcing RhoE expression significantly blocks the heterodimerization between p65 and p50 (Fig. 5, D and E), suggesting that RhoE also inhibits NF-κB activity by interfering p65/p50 heterodimer formation.
Enforcing RhoE expression in heart diminishes post-MI inflammation
While loss of RhoE in animal heart resulted in overactivated NF-κB and excessive inflammation after MI, we examined if overexpression of RhoE could temper the severity of post-MI inflammation. Transgenic mice with cardiomyocyte-specific RhoE overexpression (RhoE-TG) were previously generated in our lab23 and nearly 4-fold increase in RhoE protein level was achieved compared to wild-type mouse (Fig. 6A). Less nuclear p65 and p50 proteins were detected in the RhoE-TG cardiomyocytes compared to the wild-type cardiomyocytes in both baseline and TNFα-treated condition (Fig. 6B), which further confirmed the regulatory mechanism of RhoE on NF-κB in cardiomyocytes. Consistently, MI-induced NF-κB activation was significantly attenuated in the RhoE-TG heart compared to the wild-type heart (Fig. 6C).
Figure 6. Cardiac RhoE overexpression diminishes post-MI inflammation.
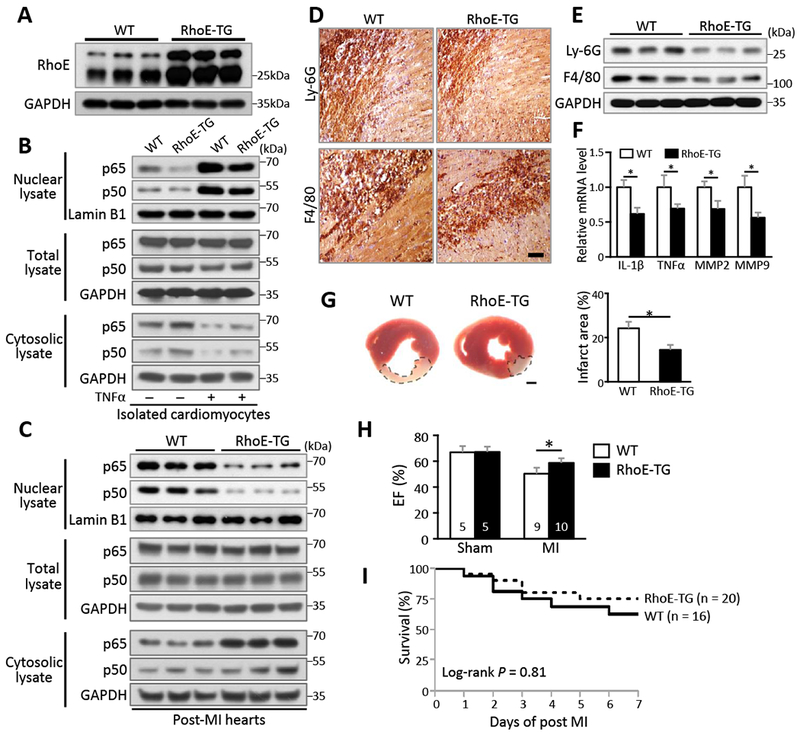
(A) Immunoblot for RhoE in the hearts of 3 pairs of WT and RhoE-TG mice. (B) Immunoblot for nuclear p65 and p50 in isolated cardiomyocytes. The cells were either untreated or stimulated with 40 ng/ml of TNFα for 15 minutes. (C-F) WT and RhoE-TG mouse hearts were used for the following assays on day 3 post MI: (C) Immunoblot for p65 and p50. (D) IHC staining and (E) immunoblot for Ly-6G and F4/80. Scale bar: 0.2 mm. (F) qRT-PCR for IL-1β, TNFα, MMP2 and MMP9. (G) TTC staining for infarct area (highlighted by dashed lines, left panel). Scale bar: 1.0 mm. Infarct area was quantified to left ventricle area in 3 pairs of experimental mice (right panel). (H) Ejection fraction of sham and MI group mice on day 3 post MI. (I) Kaplan-Meier survival curves of mice in the first week post MI. n: number of mice. *: P < 0.05.
Cardiac inflammation in RhoE-TG mice was also assessed at day 3 post-MI. Moderate neutrophil and macrophage infiltration with less production of pro-inflammatory cytokines and MMPs were observed in the RhoE-TG heart compared to the wild-type heart (Fig. 6, D-F). RhoE-TG mice showed reduced infarct size, improved cardiac function and survival (Fig. 6, G-I). Together, these results have provided physiological evidence to support RhoE as an endogenous NF-кB suppressor to balance post-MI inflammation.
RhoE also plays a cardioprotective role against MI injury in female mice
While the clinical epidemiology shows a higher prevalence of MI patients in man population than in women, it is equally important to know if the regulatory role of RhoE occurs in female mice. To our knowledge, no report suggests any functional disparity of RhoE between genders in both mice and humans. To fill in this knowledge deficiency, we repeated the animal experiments in RhoE deficient and RhoE transgenic female mice. The results were consistent with the observations detected in the male mice. We found that the female RhoEflox/+;αMHC-Cre mice exhibited excessive post-MI inflammation and enlarged infarct size with deleterious cardiac function after MI. A significant increase in NF-κB activity was detected in the RhoE deficient heart (Fig. S8). Meanwhile, cardiac specific-overexpression of RhoE protected the heart from MI injury and improved cardiac function (Fig. S9).
RhoE expression level in human heart positively associates with MI patient prognosis
Finally, to explore the relationship of RhoE expression levels and the MI patient prognosis, we assessed RhoE expression levels in the left ventricular tissues from ten MI patients who underwent left ventricular aneurysm resection. Demographics of the patients was summarized in the supplement table 1. These patients were divided into good or poor prognosis group based on the improvement of their postoperative (3-month) cardiac function. The patients in good prognosis group showed two- or more fold increases in ejection fraction (EF) after the operation; while the patients in poor prognosis group remained EF at 35% or lower without significant postoperative improvement in cardiac function (Fig. 7A). There was no statistically significant difference in ages between the good (58.8 ± 4.6 years) and the poor prognosis group (56.2 ± 8.9 years). We found that RhoE protein level was significantly higher in the heart from the good prognosis patients than the heart from the poor prognosis patients (Fig. 7, B-C). Meanwhile, less neutrophil and macrophage infiltration was detected in the heart from the good prognosis patients compared to the heart from the poor prognosis patients (Fig. 7, D and E). The data are consistent with the animal study. Interestingly, RhoE expression level in the heart was increased in both groups of MI patients compared to the non-MI subjects (Fig. 7, B-C), a similar pattern exhibited in MI mouse heart (Fig. S2B), suggesting a shared dynamic response of RhoE in human beings and mice.
Figure 7. Upregulation of RhoE associates with good prognosis of MI patients.
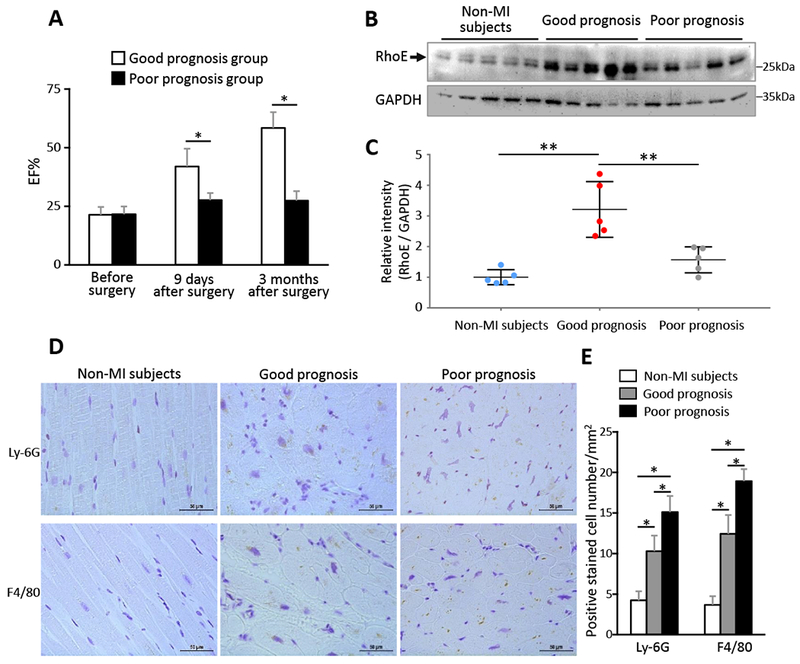
(A) Patients with MI-induced left ventricle aneurysm received aneurysm resection surgery, ejection fraction was measured by echocardiography before surgery, at 9 days and 3 months after surgery. n= 5 in each group. (B) Immunoblot for RhoE in myocardium from non-MI subjects and the patients in (A) before aneurysm resection surgery. (C) Relative intensity of RhoE/GAPDH in (B). n = 5 in each group. (D) IHC staining for Ly-6G and F4/80 in myocardium sections from the above patients before aneurysm resection surgery. Scale bar: 50 μm. (E) Quantification of Ly-6G and F4/80 positive cells. Each group contains 3 subjects and 24 sections. *: P < 0.05; **: P < 0.01.
Discussion
Tight control and timely resolution of post-MI inflammation is one of the most critical mechanisms in post-MI injury recovery.39 The present study has provided experimental evidence that RhoE protects heart from excessive inflammatory injuries during the acute healing processes after MI. RhoE negatively regulates post-MI inflammation by directly targeting the key inflammatory regulator NF-κB. We have uncovered two novel mechanisms elucidating how RhoE inhibits NF-κB activation: blocking NF-κB nuclear translocation, and interrupting p65/p50 heterodimerization (Fig. 8).
Figure 8. Schematic representing of RhoE-mediated negative regulation of inflammation.
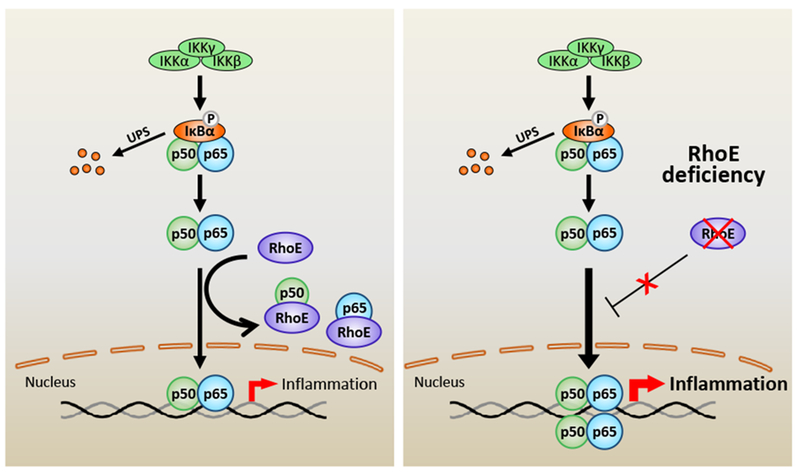
NF-κB is the pivot transcriptional regulator of inflammatory responses. Classical NF-κB pathway is primarily activated by IκB kinase (IKK)-mediated phosphorylation and degradation of IκBα. Activated p65/p50 complex is then translocated into nucleus to promote expression of target genes. RhoE functions as a novel NF-κB suppressor. RhoE physically binds to p65 and p50 individually in cytosol to inhibit their nuclear translocation. RhoE-p65 and RhoE-p50 interactions also impede formation of the p65/p50 heterodimer. Under RhoE deficiency condition, the inhibitory role of RhoE on NF-κB is diminished, which promotes the heterodimerization of p65 and p50, and the nuclear translocation of the p65/p50 complex, resulting in overactivated NF-κB and excessive inflammation.
RhoE, a member of Rho family GTPases, has a unique characteristic in that it lacks GTPase activity and stays in the GTP-bound state. Therefore, it is not regulated by GTP/GDP cycling, but by expression level and protein modifications such as prenylation and phosphorylation.40 The initial function of RhoE is linked to cell actin cytoskeleton dynamics, cell migration and proliferation through inhibition of Rho kinase 1 (ROCK1).41 Recent studies have revealed much diversified ROCK1-independent functions of RhoE, including regulation of calcium homeostasis, angiogenesis, cell proliferation, tumorigenesis, and cancer metastasis.22–24, 40, 42–44 Importantly, we recently observed a profound decrease of RhoE expression in human failing heart, suggesting a translational significance of understanding RhoE’s physiological function in heart.23
In this study, we demonstrated a protective role of RhoE in acute MI. RhoE expression is rapidly increased during the acute inflammatory phase in animal heart after cardiac ischemia. MI-induced RhoE upregulation is also detected in MI patients, suggesting a conserved mechanism. Using a loss-of-function approach of cardiomyocyte-specific RhoE haploinsufficient mice, we determined the inhibitory role of RhoE in MI-induced cardiac inflammation. RhoE deficiency in mouse heart leads to excessive inflammation, resulting in detrimental MI injuries. In contrast, overexpression of RhoE in mouse heart prevents excessive inflammatory response, improves cardiac function and post-MI survival. We would like to indicate that while these beneficial effects were significant, the improved cardiac contractility and survival were moderate since only one transgenic RhoE mouse line was used to prove a concept. Future study should explore if the RhoE-induced cardiac protection is dosage-dependent. Interestingly, MI patients with higher cardiac RhoE expression showed significantly improved cardiac function after aneurysm resection operation, while those with lower cardiac RhoE expression exhibited poor cardiac outcomes. Thus, upregulation of RhoE in response to MI is essential and beneficial during the healing processes of MI. The expression level of RhoE, therefore, might be a valuable biomarker for the assessment of MI patient prognosis. An increase in RhoE expression level could be a potential therapeutic strategy for MI treatment.
NF-κB signaling is closely involved in many critical physiological processes, and the interest of discovering selective and effective NF-κB suppressors has never faded. The prevailing mechanism is that IκB proteins function as the major inhibitors to sequester the NF-κB complex in cytosol. Most NF-κB regulators exert their effects through directly or indirectly targeting on IκB proteins. Very few factors are identified to directly act on p65 and p50, and keep them in cytosol.28, 45 In this study, we discover RhoE as a new regulator to sequester NF-κB in cytosol by directly interacting with p65 and p50 in cytosol. A series of protein dissection experiments reveal that RhoE binds to p65 dimerization domain, which is close to p65 nuclear localization signal (NLS) site. RhoE also binds to p50 at multiple sites. Together, the experimental data support the following model, which the binding of RhoE to p65 and p50 might mask the NLS, and subsequently prevent the nuclear translocation process (Fig. 8). Interestingly, recent studies show that nuclear NF-κB level is also affected by nuclear degradation of p6531, 46 and nuclear export of p65.30, 47 The RhoE-mediated NF-κB inhibition seems not through these mechanisms since RhoE does not affect the protein levels of p65 and p50; and the interactions of RhoE with p65 and p50 occur only in cytosol.
Assembly of individual subunits of Rel family into homo- and hetero-dimer is a critical step in the formation of a functional NF-κB complex.48 The p65/p50 heterodimer has been proved to be the most stable and abundant NF-κB complex. Heterodimerization of p65 and p50 enables p65/p50 heterodimer to bind to DNA and to execute transactivation activity. There are limited studies about the regulation of p65/p50 dimerization. In this study, another important discovery is that RhoE can interrupt p65/p50 dimerization process. RhoE binds to the dimerization domain of p65 and therefore occupies the dimerization surface of p65, preventing p65 forming a complex with other NF-κB subunits such as p50. Indeed, a decrease in p65/p50 heterodimer level is observed when we force the expression of RhoE.
Two early studies reported that ROCK1, a downstream effector of RhoE, can promote NF-κB activation by increasing the phosphorylation level of p65 at Ser536.49, 50 Interestingly, we do not observe the difference of p65 phosphorylation level at Ser536 by the changes of RhoE expression, suggesting the regulatory role of RhoE in NF-κB activity is not through its antagonism of ROCK1.
Collectively, this study reveals a novel function of RhoE as a fine-tuning factor in the regulation of NF-κB activation, and demonstrates the importance of RhoE precise abundance in post-MI recovery. As an inhibitory factor, RhoE directly acts on p65 and p50, providing an additional layer to secure NF-κB signaling network. Importantly, our animal and clinical observations suggest that the expression level of RhoE is dynamic and rapid in response to myocardial infarction. We propose that this responsive upregulation of RhoE is an endogenous protective mechanism against MI-induced overactive inflammation through titrating NF-κB activity. Given the pivotal role of NF-κB in cardiovascular and other inflammatory diseases, this discovery may render RhoE as a new therapeutic candidate.
Given the complexity in regulation of inflammatory response, many important questions remain to be answered in future studies. For example, we only used the mice with cardiomyocyte-specific RhoE deficiency or overexpression. While the alternation of RhoE expression in cardiomyocytes has shown a significant impact on post-MI inflammation, verification of the mechanism in different tissues/cells, such as the immune system/cells, with different stimuli is intriguing. We also only explored the relationship between RhoE and IκBα; we do not know if RhoE interacts with IKK complexes, the further upstream of NF-κB signaling. Whether RhoE has a similar regulatory role in the non-canonical NF-κB pathway is another interesting extension in the study of lymphoid organogenesis. Further investigations will provide insight for these fascinating questions.
Supplementary Material
Clinical Perspective.
What is new?
Small GTPase RhoE is an inflammatory response modulator participating in post-MI cardiac healing processes.
RhoE functions as an inhibitor of NF-κB in an IκBα-independent manner.
The associated molecular mechanism is that RhoE directly binds to p65 and p50 individually, impedes their dimerization, and subsequently blocks the two proteins nuclear translocation.
RhoE deficiency causes excessive inflammatory response in infarcted animal heart, resulting in enlarged infarct size, decreased contractility, and increased mortality.
The expression of RhoE can be dynamically induced in response to MI in both animal and human hearts.
MI patients with higher induction of RhoE exhibit a better prognosis.
What are the clinical implications?
The discovery of RhoE as a new member in NF-кB regulatory network adds an additional regulatory layer to the negative regulatory machinery of NF-kB signaling, and provides a potential therapeutic target for MI and other inflammatory diseases.
The RhoE abundance in the heart might be used for the assessment of MI patient prognosis.
Acknowledgments
Sources of Funding
This study was supported by the following funding resources: the China Scholarship Council 201206270109 (Y.D.); the American Heart Association Predoctoral Fellowship (T.Y.); the National Natural Science Foundation of China (X. Yue); the National Natural Scientific Foundation of China 81460042 and 81160020 (J. G.); and the NIH’s National Heart, Lung, and Blood Institute R01HL102314, R01HL123953 and R01HL141215 (J.C.).
Footnotes
The authors have declared that no conflict of interest exists.
Disclosures
None.
References
- 1.Anderson JL and Morrow DA. Acute Myocardial Infarction. N Engl J Med. 2017;376:2053–2064. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–1939. [DOI] [PubMed] [Google Scholar]
- 3.Prabhu SD and Frangogiannis NG. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ Res. 2016;119:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. 2014;11:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liehn EA, Postea O, Curaj A and Marx N. Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol. 2011;58:2357–2362. [DOI] [PubMed] [Google Scholar]
- 6.Saxena A, Russo I and Frangogiannis NG. Inflammation as a therapeutic target in myocardial infarction: learning from past failures to meet future challenges. Transl Res. 2016;167:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seropian IM, Toldo S, Van Tassell BW and Abbate A. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63:1593–1603. [DOI] [PubMed] [Google Scholar]
- 8.Dinarello CA. Anti-inflammatory Agents: Present and Future. Cell. 2010;140:935–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruparelia N, Chai JT, Fisher EA and Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 2017;14:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, Lenardo MJ and Baltimore D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayden MS and Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. [DOI] [PubMed] [Google Scholar]
- 12.Tak PP and Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoesel B and Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huxford T, Huang DB, Malek S and Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–770. [DOI] [PubMed] [Google Scholar]
- 15.Karin M How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. [DOI] [PubMed] [Google Scholar]
- 16.Gilmore TD and Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. [DOI] [PubMed] [Google Scholar]
- 17.Ma X, Becker Buscaglia LE, Barker JR and Li Y. MicroRNAs in NF-kappaB signaling. J Mol Cell Biol. 2011;3:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, Lin L, Yao H, Su F, Li D, Zeng M and Song E. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370–381. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Li M, Su X, Qin L, Miao M, Yu C, Shen Y, Luo Q and Chen Q. Mycoepoxydiene induces apoptosis and inhibits TPA-induced invasion in human cholangiocarcinoma cells via blocking NF-kappaB pathway. Biochimie. 2014;101:183–191. [DOI] [PubMed] [Google Scholar]
- 20.Guasch RM, Scambler P, Jones GE and Ridley AJ. RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18:4761–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Wang T, Lin X, Yue X, Wang Q, Wang G, Fu Q, Ai X, Chiang DY, Miyake CY, Wehrens XH and Chang J. Genetic Deletion of Rnd3/RhoE Results in Mouse Heart Calcium Leakage Through Upregulation of Protein Kinase A Signaling. Circ Res. 2015;116:e1–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue X, Lin X, Yang T, Yang X, Yi X, Jiang X, Li X, Li T, Guo J, Dai Y, Shi J, Wei L, Youker KA, Torre-Amione G, Yu Y, Andrade KC and Chang J. Rnd3/RhoE Modulates Hypoxia-Inducible Factor 1alpha/Vascular Endothelial Growth Factor Signaling by Stabilizing Hypoxia-Inducible Factor 1alpha and Regulates Responsive Cardiac Angiogenesis. Hypertension. 2016;67:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yue X, Yang X, Lin X, Yang T, Yi X, Dai Y, Guo J, Li T, Shi J, Wei L, Fan GC, Chen C and Chang J. Rnd3 haploinsufficient mice are predisposed to hemodynamic stress and develop apoptotic cardiomyopathy with heart failure. Cell death & disease. 2014;5:e1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landreth KS. Critical windows in development of the rodent immune system. Hum Exp Toxicol. 2002;21:493–498. [DOI] [PubMed] [Google Scholar]
- 26.Jin JL, Lv RG, Guo J, Liu XH, Liang YW, Wei JR and Wang L. Improvement of Left Ventricular Remodelling by Inhibition of NF-kappaB in a Rat Model of Myocardial Infarction. Heart Lung Circ. 2016;25:1007–1012. [DOI] [PubMed] [Google Scholar]
- 27.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H and Sunagawa K. Blockade of NF-kappaB improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–1344. [DOI] [PubMed] [Google Scholar]
- 28.Ruland J Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. 2011;12:709–714. [DOI] [PubMed] [Google Scholar]
- 29.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Li HH, Fu J, Wang XF, Ren YB, Dong LW, Tang SH, Liu SQ, Wu MC and Wang HY. Oncoprotein p28 GANK binds to RelA and retains NF-kappaB in the cytoplasm through nuclear export. Cell Res. 2007;17:1020–1029. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Grusby MJ and Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–591. [DOI] [PubMed] [Google Scholar]
- 32.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. [DOI] [PubMed] [Google Scholar]
- 33.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. [DOI] [PubMed] [Google Scholar]
- 34.Lauriol J, Keith K, Jaffre F, Couvillon A, Saci A, Goonasekera SA, McCarthy JR, Kessinger CW, Wang J, Ke Q, Kang PM, Molkentin JD, Carpenter C and Kontaridis MI. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci Signal. 2014;7:ra100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiang SY, Vanhoutte D, Del Re DP, Purcell NH, Ling H, Banerjee I, Bossuyt J, Lang RA, Zheng Y, Matkovich SJ, Miyamoto S, Molkentin JD, Dorn GW 2nd and Brown JH. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest. 2011;121:3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riento K, Guasch RM, Garg R, Jin B and Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wennerberg K, Forget MA, Ellerbroek SM, Arthur WT, Burridge K, Settleman J, Der CJ and Hansen SH. Rnd proteins function as RhoA antagonists by activating p190 RhoGAP. Curr Biol. 2003;13:1106–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng C, Yin Q and Wu H. Structural studies of NF-kappaB signaling. Cell Res. 2011;21:183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kempf T, Zarbock A, Vestweber D and Wollert KC. Anti-inflammatory mechanisms and therapeutic opportunities in myocardial infarct healing. J Mol Med (Berl) 2012;90:361–369. [DOI] [PubMed] [Google Scholar]
- 40.Jie W, Andrade KC, Lin X, Yang X, Yue X and Chang J. Pathophysiological Functions of Rnd3/RhoE. Comprehensive Physiology. 2016;6:169–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chardin P Function and regulation of Rnd proteins. Nat Rev Mol Cell Biol. 2006;7:54–62. [DOI] [PubMed] [Google Scholar]
- 42.Lin X, Liu B, Yang X, Yue X, Diao L, Wang J and Chang J. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc Natl Acad Sci U S A. 2013;110:8236–8241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Lin X, Yang X, Dong H, Yue X, Andrade KC, Guo Z, Yang J, Wu L, Zhu X, Zhang S, Tian D, Wang J, Cai Q, Chen Q, Mao S, Chen Q and Chang J. Downregulation of RND3/RhoE in glioblastoma patients promotes tumorigenesis through augmentation of notch transcriptional complex activity. Cancer Med. 2015;4:1404–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu B, Dong H, Lin X, Yang X, Yue X, Yang J, Li Y, Wu L, Zhu X, Zhang S, Tian D, Wang J, Cai Q, Mao S, Chen Q and Chang J. RND3 promotes Snail 1 protein degradation and inhibits glioblastoma cell migration and invasion. Oncotarget. 2016;7:82411–82423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan F and Lenardo MJ. The nuclear signaling of NF-kappaB: current knowledge, new insights, and future perspectives. Cell Res. 2010;20:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maine GN, Mao X, Komarck CM and Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007;26:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao N, Asamitsu K, Hibi Y, Ueno T and Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann A, Natoli G and Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–6716. [DOI] [PubMed] [Google Scholar]
- 49.Rodriguez PL, Sahay S, Olabisi OO and Whitehead IP. ROCK I-mediated activation of NF-kappaB by RhoB. Cell Signal. 2007;19:2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anwar KN, Fazal F, Malik AB and Rahman A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol. 2004;173:6965–6972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.