

Abstract
One of the functions of Human Serum Albumin (HSA) is binding and transport of fatty acids. This ability could be altered by the presence of several blood components such as toxins or peptides – which in turn alters the functionality of the protein. We aim at characterizing HSA and its fatty acid binding in native serum environment. Native ligand binding and deviations from normal function can be monitored by electron paramagnetic resonance (EPR) spectroscopy using spin labeled fatty acids (FAs). Blood serum from healthy individuals is used to examine healthy HSA in its natural physiological conditions at different loading ratios of protein to FAs. Among the EPR spectroscopic parameters (like hyperfine coupling, line shape, rotational correlation time and population of different binding sites) the rotational correlation time is found to differ significantly between binding sites of the protein, especially at loading ratios of four FAs per HSA. Although differences are observed between individual samples, a general trend regarding the dynamics of healthy HSA at different loading ratios could be obtained and compared to a reference of purified commercially available HSA in buffer.
Keywords: HSA in blood, serum test, EPR spectroscopy, fatty acid binding
1. Introduction
Human Serum Albumin (HSA) as a carrier protein with high concentration in serum (∼60 %) is responsible for binding and blood circulation/delivery of peptides, drugs, other endogenous and exogenous components, and is the main carrier for fatty acids (FAs). It is an important protein for detoxification and affects pharmacokinetics of many drugs.1,2,3 Among the numerous binding partners, the protein has a unique ability to bind FAs – 99 % of the non‐esterified FAs are bound to HSA under physiological conditions.4, 5, 6 Although low density serum proteins are also capable of binding to FAs, they have especially high affinities for very long‐chain FAs (containing 20–24 carbon atoms) at an FA excess of (i.e FA/HSA loading ratios higher than) four.7 Thus, intermediate length FAs are to the largest degree bound to HSA.
HSA concentration levels in blood of cancer patients reflect their nutritional status and may serve as prognostic factor in inflammation or in several malignant conditions.3 Independently of HSA concentration in blood, peptides are able to bind to HSA and change its binding properties to FAs due to allosteric modifications of the protein structure. Endotoxines released by malignant tumours8 can compete with fatty acids for the proper binding site.9,10 Binding of such metabolites to HSA can modify its structure and thus lead to an altered binding capacity of HSA for FAs.11
Electron paramagnetic Resonance (EPR) is a powerful tool which can monitor such structural and therefore functional modifications.12, 13, 14, 15, 16 EPR spectroscopy as a magnetic resonance method detects spins of unpaired electron, hence (unlike in nuclear magnetic resonance (NMR) spectroscopy) one needs to introduce paramagnetic species into the sample. Here, the labeled stearic acid (16‐doxyl stearic acid, 16‐DSA) is our main probe molecule of choice and is used as a spin probe throughout this study. Although other labeling positions along the methylene‐backbone of the stearic acid are available and could be used, it is shown that 16‐DSA displays the widest difference in spectral parameters (hyperfine coupling and correlation time as described upon binding to Bovine Serum Albumin (BSA)).17,18 The vast majority of our studies (and other reports in literature) specifically study HSA and BSA in in vitro situations (mostly buffered solutions). With the exception of a few studies that used empirical spectral parameters8,9,19,20 to analyze paramagnetic FA binding to HSA in actual blood samples, no systematic understanding of the effects, differences, possibilities and caveats for albumin studies that ensue when one exposes FA ligands to HSA (and all other proteins and small molecules contained in blood plasma) has been achieved. In this study, by systematically comparing EPR spectroscopic measurements and their rigorous spectral simulations on varying HSA to FA ratios of purified HSA in buffered solution with blood serum samples of healthy adults, we aim at understanding FA binding behavior of HSA in its natural physiological medium and how it differs from its behavior in purified form in buffer medium.
To achieve this purpose on this rather complex set of samples, we reduce the spectral complexity by focusing our continuous wave (CW) EPR spectroscopic measurements and their analyses on two parameters: the hyperfine coupling tensor element A′zz, which is a scale of the immediate environmental effects on the spin probe (like polarity/hydrophobicity of the direct spin probe environment or the presence of H‐bonding) and the rotational correlation time , which by definition is the time for a particle/system to rotate by one radian and is a simple measure of spin probe mobility. It reveals information about system dynamics and it is size dependent; the larger or heavier the system, the slower the rotation. HSA has two classes of binding sites for FAs: one bearing a high affinity for FA (strong binding, hereafter called site (A)) and another one with intermediate affinity (more loosely bound, site (B)) toward FAs.21,22 The population of these sites could be obtained by spectral simulation.
Previous studies have reported that allosteric changes of HSA may be detected in cancer and potentially may serve as a tumor marker.8,19,20,23,24 The long‐term goal in our studies is to use EPR spectroscopy on EPR‐active FAs that bind to HSA to discriminate between blood sample of “healthy” and “diseased” groups of patients based on its spectral features. Specifically, one may ask whether there are loading ratios of HSA to FA that allow to distinguish HSA in blood serum of two or more groups of people. When there are spectral differences, which kinds of structural modifications do they reflect? And as a long term goal, could HSA be used as a biomarker, e. g. for different types of cancer?
Herein, we make the first step in the quest to obtain answers to these questions that are more universal than mere clinical case studies. We would like to base the understanding of HSA‐FA binding on the above mentioned spectral parameters and their differences when comparing HSA in buffered aqueous solution with that in blood serum of healthy adults. By contrasting/comparing the blood‐sample based spectral analyses with the purified HSA samples, we can characterize the scope and limitations of such measurements in blood serum of reference patients quantitatively.
2. Results and Discussion
All measurements and simulations were performed for ratios of 1 : 1 to 1 : 7 (HSA:16‐DSA), but up to a ratio of 1 : 3, no significant changes between simulation parameters (A′zz, and population of each class of binding sites) were found. Therefore, we focus on data and discussion of the loading ratios of 1 : 3 to 1 : 7 in the main text. As an example, the experimental and simulated spectra of all loading ratios of a healthy individual (sample numbered 1 in control group) are given in Figure 1(a). As we have reported earlier12,13,15,16 the EPR spectra at these loading ratios of HSA:FA are comprised of three spectral fractions, as shown in Figure 1(b): (A) strongly bound and immobilized FAs that have high rotational correlation times ( ≥10 ns); (B) FA that are intermediately‐strong bound ( ≥1 ns); and (C) FAs that float relatively free in solution ( ≥0.1 ns): The rigorously simulated EPR spectra thus always consist of these three fractions in different weights, of course the and hyperfine coupling values were always allowed to vary during the simulation to a small degree.
Figure 1.
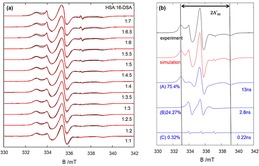
(a) Experimental (black) and simulated (red) EPR spectra of a healthy individual (“patient 1”) at different loading ratios. (b) Deconvolution by simulation of the spectrum of the same sample at loading ratio of 1 : 4. The component (A), 16‐DSA strongly bound to HSA, makes up 75.4 % of the experimental spectrum, while (B) the intermediate affinity binding sites give 24.27 % contribution and the free component (C) has only a 0.32 % share of the total spectrum. The corresponding (in ns) is also given.
Throughout the paper we mostly discuss the spectral parameters related to high and intermediate affinity binding sites (A) and (B). Since the spectral features (A′zz and ) of non bound (free) fatty acids (C) remain almost identical during the simulations, only the spectral contributions of free 16‐DSA component in the different samples are given.
We compare the results obtained with patient HSA in blood serum with reference samples of commercially available and purified HSA in buffer solution to test for the impact of serum with all its soluble components on the behavior of HSA and FA binding.
2.1. Bound Fraction of FAs
Inspecting the EPR spectra of a series of loading ratios (for the same sample, see Figure 1(a)), it already becomes apparent that the change in population (spectral weight) of strong and intermediate binding sites, as well as changes in their line shape and an estimation of the free spin probe can be qualitatively extracted. These properties can then be quantified by spectral simulations as shown in Figure 1(b).
Table 1 reports the median of the simulation parameters of 16‐DSA bound to blood sample HSA of our group of 24 individuals and the simulation values for purified HSA in buffer at different loading ratios.
Table 1.
Median values of the simulation parameters (2A′zz, ) for all 24 measured blood samples and purified HSA. Binding sites with strong and intermediate immobilization are denoted by (A) and (B), respectively. Hyperfine couplings and rotational correlation times are given in MHz and ns. The Median Absolute Deviation (MAD) is given in parenthesis in blue.
| (2A′zz, ) (A) | (2A′zz, ) (B) | (2A′zz, ) (A) | (2A′zz, ) (B) | |
|---|---|---|---|---|
| 1 : 3 | (98.2(1.1),12.6(0.9)) | (90.7(0.0),2.6(0.4)) | (97.0,12.8) | (91.5,3.20) |
| 1 : 4 1 : 5 1 : 6 1 : 7 | (95.7(2.6),13.4(2.7)) (98.7(1.9),11.7(2.7)) (97.1(0.8),12.5(0.6)) (97.8(0.9),12.5(1.1)) | (88.6(0.0),4.2(0.8)) (90.7(0.2),2.5(0.2)) (91.5(0.9),3.6(0.2)) (91.5(0.0),3.6(0.3)) | (97.0,12.8) (98.0,9.20) (98.80,8.60) (97.8,8.94) | (91.5,3.20) (91.5,2.40) (91.5,2.70) (91.5,2.27) |
When comparing the blood sample values with those in purified HSA solutions, one finds that variations in A′zz and in purified HSA are not as pronounced as in blood samples. In purified HSA, faster dynamics are observed with increasing the FA loading ratio while the blood samples do not show this behavior. There is no significant change in A′zz for both types of samples.
We could also monitor the difference between binding sites (A) and (B) in terms of their hyperfine coupling and rotational correlation time. The data obtained by simulation of experimental spectra were collected and are summarized in form of a box plot (see Figure 2) to check to which extent the spectral differences are significant.
Figure 2.
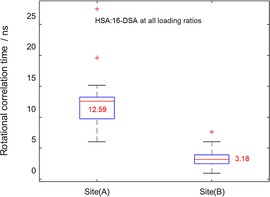
Boxplot of the rotational correlation times for all loading ratios of HSA: 16‐DSA in terms of their binding affinities. As appears from the median values (red lines and corresponding values in each box), the high affinity binding site (A) has a slower dynamic of about four times, compared to the intermediate binding sites (B). Red crosses are outliers in each series.
Box plots concisely contain information about median values (red line), spread of the data (length of box) and outliers (which are shown by red crosses). The readout values from Figure 2 reveal that FAs in binding sites (A) are about four times slower in their rotational motion than those in class (B) sites, which in turn enables us to fully separate the motional regimes of FAs in these two sites by plotting their τc values against both their respective hyperfine couplings or their population (in percent contribution to the overall simulation). Such plots for the 1 : 3 loading ratio samples of all 24 examined individuals are shown in Figure 3, as an example.
Figure 3.
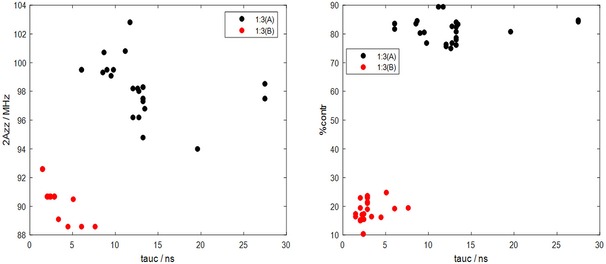
Correlations between spectral feature 2A′zz vs. (tauc) in the left panel and population of binding site (%contr.) vs. in the right panel are shown. A deconvoluted/resolved dynamics at low loading ratio 1 : 3 of two classes of binding sites (A) in red black dots and (B) in red dots can be seen. The two motional regimes of two binding sites are such visible clearly separated.
2.2. Non Bound (Free) FAs
High amounts of non‐bound 16‐DSA (free component) even at low loading ratios have been considered as disease signatures in HSA‐cancer related studies.8,9, 19, 20 In our case of the reported healthy adult “control group” samples, this amount has increased when increasing the HSA:FA ratio consecutively from 1 : 3 to 1 : 7. This is expected, however the median values of free component of the spectra never exceed one percent, regardless of the ratio (Figure 4).
Figure 4.
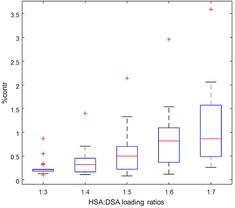
Free component contributions at different loading ratios are depicted in a boxplot. Although a linear increasing trend is observed by moving toward higher loading ratios, the medians (red lines) of contributions stay below one percent for healthy individuals. The red crosses are outliers in each series.
This remarkably shows that the FA : HSA loading ratio series in “healthy” individual samples are in fact different from previously reported disease‐related blood serum samples and reflect a behavior similar to the reference series of purified HSA, which is discussed in the following.
2.3. Comparisons with Purified HSA
To observe the difference of HSA behavior in its native blood serum and the commercially available purified HSA, we prepared HSA at the average concentration of control group samples (∼4.0 g/dl). The results of such comparisons, for one of the samples (“patient 7”), are given in Figure 5. Obviously, even higher amounts of free component (C) could be seen in purified HSA sample than in the control group which suggests either a stronger binding of FA to HSA in blood so it could not be easily released (in contrast to pure HSA), which is in agreement with the fact that HSA is the first binding target of FAs with high probability. Also, there could be nonspecific binding sites for FAs in serum, as there are other proteins in biological medium to which FAs could bind, although with much lower probability and which in previous studies, if existent, never had such an increasing effect on FA binding.
Figure 5.
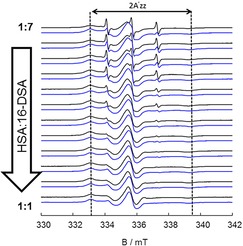
The EPR spectra of a healthy individual (blue) and purified HSA (black) are compared. Deviations from purified HSA (in black) could be observed in terms of high amount of free component and variation in population of binding sites (A) and (B). With an exception to1 : 4 loading ratio, no big changes in 2A′zz could be seen.
When inspecting the EPR spectra and their spectral simulations as summarized with the data in Table 1, we find that in case of the blood samples, the 1 : 4 loading ratio has the smallest hyperfine couplings and slowest rotational motion, both for binding to (A) and (B) sites. This indicates that at this ratio, the probes are mostly bound to the more high affinity binding sites with lowest polarity (lowest A′zz) and highest immobilization, either tightly bound (A‐sites) or with faster residual orientational motion (B‐sites). When the fatty acid loading is increased even further, hyperfine couplings grow larger again and movement of the spin probes becomes faster as lower affinity binding sites are increasingly occupied and the dynamic equilibrium between bound and free states shifts towards non‐bound states ever more.
The difference between A′zz (or ΔA′zz) of binding sites (A) and (B) is about 6–8 MHz in blood samples while it is about 6 MHz in purified protein. Also, in both, purified or serum HSA samples, the (A) binding sites have larger A′zz than the corresponding (B) sites. As the fatty acid spin probes contain nitroxyl groups as persistent radical moieties (NO.), one can identify the parameters which may increase the hyperfine couplings. In a simplified picture, one can describe organic NO.‐radicals using two resonance structure; a zwitterion structure with partial charges on the nitrogen and oxygen atoms, in which the unpaired electron spin population is formally situated on N, and a neutral resonance structure in which the unpaired electron formally resides on the oxygen atom The more polar the environment is, the more of the zwitterionic resonance structure contributes to the overall situation – formally – and the larger the hyperfine couplings become. Shorter N−O bond length and the presence of negative charges close to NO. moiety and the deviation of N−O from planarity (the so called out of plane −OOP‐angle) can be named as other factors which increase the hyperfine coupling values.25, 26, 27, 28, 29, 30, 31
At physiological pH, HSA is negatively charged (∼19e)14,32,33 and it is a well‐established fact that hydrophilic negative head‐groups and hydrophobic moieties are required for binding to the fatty acid binding sites of HSA. The high affinity binding site of FAs to the protein are the ones who have hydrogen bond and /or electrostatic interactions with the ligand carboxylate head group.21,34,35 From an EPR viewpoint the lower affinity binding sites (B) in purified HSA have a 2–3 MHz larger A′zz value as compared to those in blood samples. This observed lower polarity in the B‐sites in blood samples might either be due to small but non‐negligible binding of FAs to other, slightly less polar blood sample components (proteins), or due to actual stronger hydration (increasing polarity) in B‐sites in blood samples, which would indicate slight differences in HSA hydration in serum. At this point, we cannot exclude either of these explanations. In A‐sites, the hydration/polarity does not seem to differ between HSA in blood and in buffered solution, yet the FA rotational motion in the blood samples is always slower for both sites in blood samples, which could also stem from stronger hydration and hence H‐bond‐anchoring of FAs in the binding sites (probably at the carboxylate head group end, remote from the NO.‐group at the chain end) or might indicate interaction of secondary structure elements of HSA in blood samples with other blood serum components.
In the correlation plots between the two spectral parameters and the relative weights of the two FA species (A and B) obtained in the simulations (cf. Figure 3), one can clearly see that the spectral components (reflecting actual FAs in the two binding situations (A) and (B)) occupy a rather distinct A′zz vs. parameter space, which in itself is rather well defined. For future studies, this might by a very valuable piece of information that may help in detection and characterization of blood samples from patients with various diseases.
In the case of cancer it has been demonstrated, that bioactive proteins released from tumor cells may bind to albumin and thus lead to changes in binding properties of HSA8,19 . Hence, deviations from the occupied value space in A′zz vs. and/or spectral contribution vs. can differ from those of healthy adults of this study as, e. g. the motional behavior of the disease‐altered samples or the hyperfine coupling etc. may vary. In terms of the significance test, a P‐value <0.01 were obtained for reliability of correlation times data. The same is true in case of hyperfine couplings (see Figure S1).
Such a distinction between different motional regimes between two binding sites could be clearly observed for all other loading ratios as well (cf. Figures S2 and S3). However, for both binding sites in particular there are variations in their relative population with increasing FA loading. It should be noted, though, that some samples in the healthy individual group displayed spectral behavior very similar to that of purified HSA (Figure S5). For loading ratios of 1 : 3, 1 : 5 to 1 : 7 we found no strong changes in A′zz (0.084 MHz) for both sites (A) and (B). At a loading ratio of 1 : 4, a difference of ∼2.9 MHz could be observed in A′zz of the intermediately‐strong binding sites (B). Also, comparing with the median of correlation times in the blood samples of the healthy group, we find that the binding site (A) in the purified HSA rotates faster as in real medium of about 2 ns, which looks reasonable due to the presence of several proteins/ components in a real medium (cf. Table 1), as explained above.
2.4. Binding Models of HSA
Considering that HSA as a receptor has multiple independent binding sites for FAs as ligands and using the receptor‐ligand terminology in binding with FAs, there are a few models which are able to describe binding cases like HSA. Among several available models for ligand binding, the first one was released in 1949 by Scatchard36 who proposed a “ligand to one binding site” model in a graphical representation (the so called Scatchard plot, biochemistry textbook knowledge by now). He grouped binding sites into classes with equal affinity for the ligand. By plotting the ratio of ligand bound to ligand free vs. ligand free fractions one could obtain a linear graph for systems, and extract the binding parameters (binding constant and number of binding sites). Efforts were made to improve data interpretation37 or generalize Scatchard's model for systems with multiple interacting binding sites.38, 39, 40, 41, 42 In case of HSA, Karush showed the flexibility of HSA binding sites and that they can adopt different conformations with almost the same energy, under physiological conditions which is in contradiction with the main assumption of the Scatchard model. The Karush model thus is in better agreement with experimental data regarding binding of FAs to HSA.43,44
Taken together, it is well known that Scatchard plots are an oversimplification of the receptor‐ligand binding mechanism. Yet, by detecting deviations from linearity one can find deviations from simple, non‐cooperative FA binding. These deviations may e. g. point to the presence of multiple binding sites, non‐specific binding and cooperativity. An upward curved plot suggests for an either negative cooperativity or non‐specific binding while a concave downward plot may be due to positive cooperativity or instability of the ligand.45 When inspecting the Scatchard plots of HSA from blood samples and purified HSA, one observes rather slight deviations from linearity for both cases. The purified HSA has less propensity for binding FAs with increasing FA ratio, therefore it reveals a negative cooperativity case which could be observed for some of the samples in healthy adult group as well. On the other hand, there are cases which show a bound state even at 1 : 7 loading ratio which reflects the presence of non‐specific binding in addition to negative cooperativity in these samples. Scatchard plots for two samples of the healthy individuals group (numbered as 14 and 17) in comparison with purified HSA are depicted in Figure S4.
3. Conclusions
Aiming at ultimately understanding the differences between healthy and diseased HSA bound FA behavior in detail, we examined blood serum samples of healthy adult individuals and their fatty acid binding to HSA. The population of strongly and intermediately immobilized fatty acids, the hyperfine coupling value A′zz of the bound state, the population of non‐bound FAs and rotational correlation times were obtained by rigorous spectral simulations at each loading ratio of HSA to FAs. Monitoring different spectroscopic variables showed the sets of hyperfine couplings, rotational correlation times, and spectral contributions for both binding site types at specific FA loading ratios can be correlated and may serve as discriminant parameter sets in future studies. This is especially promising for a loading ratio of HSA to FAs of 1 : 4.
Although the blood samples investigated here are from a group of 24 healthy individuals, both spectral features A′zz and reveal deviations from corresponding values for buffered solutions of commercially available, purified HSA. Such variations reflect the impact of a natural, physiological medium, which contains several components, and other factors like charges on FAs. Also in a ligand‐receptor terminology, negative cooperativity was observed for most of the samples in the healthy adult group as well as (very slightly) in pure buffered HSA samples.
Since we deal with large amounts of data, we need to use statistical methods, which are also sensitive to deviations from general trends (outlier points). As native blood samples in fact are rather complex mixtures of small and macromolecular components, issues like nutritional habits or unidentified ailments may significantly impact the results. By using groups of individuals that are large enough, we can here show that a robust description of “healthy” behavior concerning FA binding is possible in the vast parameter space spanned by EPR spectroscopy and FA binding to HSA. These parameters are kept as simple as possible to avoid complexity, so that we have used an isotropic model to describe the rotational correlation times of bound FAs and the hyperfine coupling tensor element A′zz as a measure of local polarity and hydrophilicity and finally the relative spectral weight of the two binding sites (A) and (B).
Based on these findings and comparing with the well understood reference data set of pure HSA in buffered solution, we are able to obtain in‐depth insights into a HSA in native serum and we have established an open, clear, and reproducible EPR protocol and EPR‐based parameters to analyze HSA in serum samples of humans in general. Thus, for potential further studies of HSA in patients with diseases, we are able to compare their HSA binding properties with those of the healthy individuals examined here.
Experimental Section
Blood Samples
Experimental Blood samples were obtained of a total number of 24 healthy individuals presenting at the Alfried Krupp Krankenhaus in Essen for elective surgical hernia repair. Samples were collected according to Standardized Operation Procedures. The time range between sample collection and following centrifugation was 15 minutes. After clotting, serum was separated by centrifugation (1000 g) for 10 minutes at room temperature, removed, and then stored at −20 °C.
Albumin Concentration
The HSA concentration (g per dl) was measured by titration of a standard solution of the anionic dye bromcresol green, which is known to bind to cationic albumin at pH 4.1, to the albumin solution until a color change (blue‐green complex) could be observed photometrically. Norm values are 3.5–5.0 g/dl.
All individuals provided their consent for participation in this study. The informed consent was obtained according to an ethical permission (Ethics commission permit of the medical association Nordrhein, Germany, Nr. 2015076).
CW EPR Spectroscopy
All CW EPR spectra were recorded on a Magnettech MiniScope MS400 benchtop CW‐EPR spectrometer operating at X‐Band frequencies (9.43 GHz) at 298 K. A microwave power of 3.16 mW with a modulation frequency of 100 KHz, modulation amplitude of 0.1 mT and 4096 points were used throughout the measurements. The respective final spectrum which is subjected to analysis is accumulated ten times, each for 60 seconds. Since at this frequency only the z‐component of the hyperfine coupling tensor is well resolved, determining the overall width of the spectrum, we only focus on variations of this parameter during the discussions.
Sample Preparation
16‐DSA (16‐DOXYL stearic acid) as a spin probe was purchased from Sigma Aldrich and prepared as a 26 mM stock solution, by dissolving the proper amounts in 0.1 M KOH solution. It is well known that under physiological conditions, up to seven FAs can be taken by HSA, therefore loading ratios of HSA:16‐DSA were prepared from 1 : 1 to 1 : 7 in 0.5 steps (in total 12 preparations per sample). Concentrations of 200 μM of HSA from blood samples (24 samples of healthy adults with known HSA concentrations) was used to mix with corresponding concentrations of 16‐DSA in DPBS buffer. Glycerol (87 % from Thermo Fisher Scientific) was used as cryoprotectant for comparability with further pulse EPR measurements which necessitates cryogenic temperatures (typically 50 K). As we have pointed out before,12,14,16 addition of glycerol increases the viscosity of solutions, so the actual blood medium conditions could be simulated somewhat more realistically. Overall, the final sample volume was always set to 200 μL.
Due to the amphiphilicity of 16‐DSA, ethanol is used in most of the previous studies, while it is shown that the binding affinity of HSA for 16‐DSA is drastically decreased in the presence of ethanol. Moreover, ethanol above a certain concentration in the sample can cause HSA denaturation.46 Examining the pH of the samples showed that they all range between pH 7.4 to 8.0, in which HSA is in its native form, so the natural conformation and environment of the protein is completely preserved.14
To know if the obtained results are reproducible on the one hand and to ascertain stability of the blood samples on the other hand, we chose three samples at random at a loading ratio of 1 : 4 and repeated the measurements after three months. During this time interval samples were kept at −80 °C and were thawed before reexamining them. The results are given in Figure S5. Obviously the samples were not degraded and reproduced the same EPR spectra.
Computational Section
EPR spectra were simulated based on the spin Hamiltonian, using the eayspin software package (version 5.1.2 and 5.2.11)47 in MATLAB version 8.6 (R2015b48). The principal values of the g‐tensor were chosen according to the values reported for DOXYL‐labeled stearic acids in interaction with BSA.11,18 Margins of confidence/error in simulations are based on the RMS‐deviations and were found to be around 10 % for loading ratios up to 1 : 4 and about 20 % for higher loading ratios (1 : 5 to 1 : 7). A three component system was considered for simulations ( details are shown in the Discussion section). To reduce complexity but to still have one value as a measure of rotational mobility, the reported rotational correlation times are calculated based on an isotropic rotational model.49,50
Statistical analyses of the data were also performed in MATLAB. All quantitative data are given in terms of their median values to monitor the central tendency of the set of observations and the median absolute deviation (MAD) which both are considered robust statistics against outliers.51,52 To compare between two sets of independent samples, the two‐tailed t‐test was performed, so the reliability of data within 95 % range of significance were tested.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Stefanie Weber for technical support and gratefully acknowledge financial support from the state of Saxony‐Anhalt (European Regional Development – ERDF grant ZS/2016/06/79740).
H. H. Haeri, B. Schunk, J. Tomaszewski, H. Schimm, M. J. Gelos, D. Hinderberger, ChemistryOpen 2019, 8, 650.
References
- 1. Levitt D., Levitt M., Int. J. Gen. Med. 2016, 9, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Merlot A. M., Kalinowski D. S., Richardson D. R., Front. Plant Physiol. 2014, 5, 1–7. [Google Scholar]
- 3. Fanali G., DiMasi A., Trezza V., Marino V., Fasano M., Ascenzi P., Mol. Aspects Med. 2012, 33, 209–290. [DOI] [PubMed] [Google Scholar]
- 4. Spector A. A., Methods Enzymol. 1986, 320–339. [Google Scholar]
- 5. Spector A. A., J. Lipid Res. 1975, 165–179. [PubMed] [Google Scholar]
- 6. Goodman D. S., Shafrir E., J. Am. Chem. Soc. 1959, 81, 364–370. [Google Scholar]
- 7. Shafri E., Gatt S., Khasis S., Biochim. Biophys. Acta. 1965, 98, 365–371. [DOI] [PubMed] [Google Scholar]
- 8. Lowenthal M. S., Mehta A. I., Frogale K., Bandle R. W., Araujo R. P., Hood B. L., Veenstra T. D., Conrads T. P., Goldsmith P., Fishman D., E. F. Petricoin III , Liotta L. A., Clin. Chem. 2005, 51, 1933–1945. [DOI] [PubMed] [Google Scholar]
- 9. Gurachevsky A., Muravskaya E., Gurachevskaya T., Smirnova L., Muravsky V., Cancer Invest. 2007, 25, 378–383. [DOI] [PubMed] [Google Scholar]
- 10. Graner M. W., Likhacheva A., Davis J., Raymond A., Brandenberger J., Romanoski A., Thompson S., Akporiaye E., Katsanis E., Cancer Res. 2004, 64, 8085–8092. [DOI] [PubMed] [Google Scholar]
- 11. Pavicevic A. A., Popovic-Bijelic A. D., Mojovic M. D., Susnjar S. V., Bacic G. G., J. Phys. Chem. 2014, 118, 10898–10905. [DOI] [PubMed] [Google Scholar]
- 12. Reichenwallner J., Hinderberger D., Biochim. Biophys. Acta, 2013, 1830, 5382–5393. [DOI] [PubMed] [Google Scholar]
- 13. Junk M. J. N., Spiess H. W., Hinderberger D., Angew. Chem. Int. Ed. 2010, 49, 8755–8759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8937–8941. [Google Scholar]
- 14. Reichenwallner J., Oehmichen M. T., Schmelzer C. E. H., Hauenschild T., Kerth A., Hinderberger D., Magnetochemistry 2018, 4, 47–82. [Google Scholar]
- 15. Arabi S. H., Aghelnejad B., Schwieger C., Meister A., Kerth A., Hinderberger D., Biomater. Sci. 2018, 6, 478–492. [DOI] [PubMed] [Google Scholar]
- 16. Reichenwallner J., Thomas A., Steinbach T., Eisermann J., Schmelzer C. E. H., Wurm F., Hinderberger D., Biomacromolecules 2019, 20, 1181–1131. [DOI] [PubMed] [Google Scholar]
- 17. Ge M., Rananavare S. B., Freed J. H., Biochim. Biophys. Acta 1990, 1036, 228–236. [DOI] [PubMed] [Google Scholar]
- 18. Earle K. A., Budil D. E., Freed J. H., J. Phys. Chem. 1993, 97, 13289–13297. [Google Scholar]
- 19. Kazmierczak S. C ., Gurachevsky A., Matthes G., Muravsky V., Clin. Chem. 2006, 52, 2129–2134. [DOI] [PubMed] [Google Scholar]
- 20. Gelos M., Hinderberger D., Welsing E., Belting J., Schnurr K., Mann B., Int. J. Colorectal Dis. 2010, 25, 119–127. [DOI] [PubMed] [Google Scholar]
- 21. Fasano M., Curry S., Galliano E. Terreno„ M., Fanali G., Narciso P., Notari S., Ascenzi P., IUBMB Life. 2005, 57, 787–796. [DOI] [PubMed] [Google Scholar]
- 22. Simard J. R., Zunszain P. A., Hamilton J. A., Curry S., Mol. Biol. 2006, 361, 336–351. [DOI] [PubMed] [Google Scholar]
- 23. Seidel P., Gurachevsky A., Muravsky V., Schnurr K., Seibt G., Matthes G., Z. Med. Phys. 2005, 15, 265–72. [DOI] [PubMed] [Google Scholar]
- 24. Gurachevsky A., Kazmierczak S., Jörres A., Muravsky V., Clin. Chem. Lab. Med. 2008, 46, 1203–1210. [DOI] [PubMed] [Google Scholar]
- 25. Steinhoff H. J. in: Supramolecular Structure and Function 8 (Eds.: G. Pifat-Mrzljak), Springer, Boston, MA. 2005, pp157-177. [Google Scholar]
- 26. Owenius R., Engstrom M., Lindgren M., J. Phys. Chem. A. 2001, 105, 10967–10977. [Google Scholar]
- 27. Improta R., Barone V., Chem. Rev. 2004, 104,1231-1253. [DOI] [PubMed] [Google Scholar]
- 28. Saracino G. A. A., Tedeschi A., D'Errico G., Improta R., Franco L., Ruzzi M., Corvaia C., Barone V., J. Phys. Chem. A 2002, 106, 10700–10706. [Google Scholar]
- 29. Cimino P., Pedone A., Stendardo E., Barone V., Phys. Chem. Chem. Phys. 2010, 12, 3741–3746. [DOI] [PubMed] [Google Scholar]
- 30. Hunold J., Wolf T., Wurm F. R., Hinderberger D., Chem. Commun. 2019, 55, 3414–3417. [DOI] [PubMed] [Google Scholar]
- 31. Kurzbach D., Junk M. J. N., Hinderberger D., Macromol. Rapid Commun. 2013, 34, 119–134. [DOI] [PubMed] [Google Scholar]
- 32. Alvarez B., Carballal S., Turell L., Radi R., Methods Enzymol. 2010, 473, 117–136. [DOI] [PubMed] [Google Scholar]
- 33. Baler K., Martin O. A., Carignano M. A., Ameer G. A., Vila J. A., Szleifer I., J. Phys. Chem. B, 2014, 118, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Curry S., Mandelkow H., Brick P., Franks N., Nat. Struct. Biol. 1998, 5,827-35. [DOI] [PubMed] [Google Scholar]
- 35. Bhattacharya A. A., Gruene T., Curry S., J. Mol. Biol. 2000, 303, 721–732. [DOI] [PubMed] [Google Scholar]
- 36. Scatchard G., Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar]
- 37. Rosenthal H. E., Anal. Chem. 1967, 20, 525–532. [DOI] [PubMed] [Google Scholar]
- 38. Klotz I. M., Walker F. M., Pivan R. B., J. Am. Chem. Soc. 1946, 68, 1486–1490. [DOI] [PubMed] [Google Scholar]
- 39. Arvidsson E. O., Green F. A., Laurell S., J. Biol. Chem. 1971, 246, 5373–5379. [PubMed] [Google Scholar]
- 40. Laiken N., Nementy G., Biochemistry, 1971, 10, 2101–2106. [DOI] [PubMed] [Google Scholar]
- 41. Hulme E. C., in Receptor – Ligand Interactions: A Practical Approach, Oxford University Press, New York, 1992, p. 458. [Google Scholar]
- 42. Klotz I. M., in Ligand – Receptor Energetics: A Guide for the Perplexed, Wiley, New York, 1997, p. 170. [Google Scholar]
- 43. Karush F., J. Am. Chem. Soc. 1950, 72, 2705–27013. [Google Scholar]
- 44. Karush F., J. Am. Chem. Soc. 1954, 76, 5536–5542. [Google Scholar]
- 45. Copeland R. A., in Enzymes, A Practical Introduction to Structure, Mechanism, and Data Analysis, Wiley-VCH, second edition: 2000, p. 416. [Google Scholar]
- 46. Akdogan Y., Hinderberger D., J. Phys. Chem. B. 2011, 115, 15422–15429. [DOI] [PubMed] [Google Scholar]
- 47. Stoll S., Schweiger A., J. Magn. Reson. 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- 48.MATLAB 2015b, The MathWorks, 2015.
- 49. Freed J. H., in Theory of slow tumbling ESR spectra for nitroxides. In Spin labeling: Theory and application. (Eds.: LJ. Berliner), Academic Press Inc. ; 1976, pp. 53–130. [Google Scholar]
- 50. Oppenheim S. F., Buettner G. R., Rodgers V. G. J., J. Membr. Sci. 1996, 118, 133–139. [Google Scholar]
- 51. Leys C., Ley C., Klein O., Bernard P., Licata L., J. Exp. Soc. Psychol. 2013, 49, 764–766. [Google Scholar]
- 52. Massart D. L., Meyers-Verbeke J. S., Capron X., Schlesier K., LC-GC 2005, 18, 215–218. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary