

Abstract
Background
Vaccines against all stages of the malaria parasite are in development, mainly for Plasmodium falciparum, which causes the most serious form of malaria. Pre‐erythrocytic vaccines act to prevent or delay a malaria attack by attacking the sporozoite and liver stages before the parasite reaches the bloodstream.
Objectives
To assess the efficacy and safety of pre‐erythrocytic malaria vaccines against any type of human malaria.
Search methods
In March 2006, we searched the Cochrane Infectious Diseases Group Specialized Register, CENTRAL (The Cochrane Library 2006, Issue 1), MEDLINE, EMBASE, LILACS, and the Science Citation Index. We also searched conference proceedings and reference lists of articles, and contacted organizations and researchers in the field.
Selection criteria
Randomized controlled trials comparing pre‐erythrocytic vaccines with placebo, control vaccine, or routine antimalarial control measures in people of any age receiving an artificial challenge or natural exposure to malaria infection.
Data collection and analysis
Both authors independently assessed trial quality and extracted data. Results of meta‐analyses were expressed as risk ratios with 95% confidence intervals (CI) using an intention‐to‐treat analysis.
Main results
Nine safety and efficacy trials, and two safety trials, with over 3000 participants were included. In semi‐immune children, RTS,S vaccine reduced clinical episodes of malaria by 26% (95% CI 13% to 37%) and severe malaria by 58% (95% CI 15% to 79%) for up to 18 months. Prevalence of parasitaemia was also reduced by 26% (95% CI 11% to 38%) at six months after immunization. RTS,S also reduced clinical malaria episodes by 63% (95% CI 18% to 83%) in semi‐immune adult men in the second year of follow up after a booster dose. No severe adverse events were judged to be related to RTS,S vaccine, although the frequencies of injection site pain, swelling, arm motion limitation, headache, and malaise were increased in the vaccine groups. There was no evidence for effect of the CS‐NANP vaccines (307 participants, 3 trials), CS102 peptide vaccine (14 participants, 1 trial), or the ME‐TRAP vaccine (372 participants, 1 trial).
Authors' conclusions
RTS,S vaccine was effective in preventing a significant number of clinical malaria episodes, including good protection against severe malaria in children for 18 months. No severe adverse events were attributable to the vaccine. Progression of this vaccine towards licensing is justified while efforts to increase its efficacy continue. The other vaccines do not look promising and further research is a priority.
23 April 2019
No update planned
Intervention not in general use or been superseded
This intervention is no longer available.
Plain language summary
Vaccines for preventing malaria in the pre‐erythrocytic phase
Malaria is a parasitic disease spread by mosquitoes. It affects millions of people worldwide and causes significant illness and mortality. The symptoms of uncomplicated malaria include fever, headache, muscle pain, and vomiting; and children commonly present also with rapid breathing, cough, and convulsions. Severe malaria leads to unconsciousness and death. Uncomplicated malaria can almost always be cured with appropriate drugs, given soon after symptoms appear, but in small children in particular, progression and death can come within 48 hours. The hope − bolstered by several decades of increasingly promising research − remains that one or more vaccines to prevent malaria will augment the existing malaria control tools. The expectation is that successful vaccines will decrease malaria incidence, but because of the complexity of the organism and other factors, protection will not be complete. The malaria parasite develops through several phases in the human body that evoke different immunologic responses, and vaccines for all phases are under development. This review looks at vaccines targeted at the 'pre‐erythrocytic' phase of the parasite's life, the phase before the parasites first enter the bloodstream from the liver. Trials of four types of vaccine against P. falciparum, the most important human malaria species, were available for this review. One of these (the RTS,S vaccine) significantly reduced the number of episodes of clinical malaria and severe malaria in children, while the other three vaccines were not effective under the conditions of the trials. No severe adverse events observed following the RTS,S vaccination were judged to be related to vaccination, though minor adverse events like headache, swelling, and malaise were.
Background
Malaria is a severe and debilitating disease caused by the parasitic protozoan Plasmodium, which is transmitted by many species of anopheline mosquitoes. Four Plasmodium species infect humans. Plasmodium falciparum is the most widespread and also the most serious and potentially fatal form. Recent estimates of the annual number of clinical malaria cases worldwide range from 214 to 397 million (WHO 2002; Breman 2004), although a higher estimate of 515 million (range 300 to 660 million) clinical cases of P. falciparum in 2002 has been proposed (Snow 2005). Estimates of annual mortality (nearly all from P. falciparum malaria) are thought to be around 1.1 million (WHO 2002; Breman 2004). Malaria deaths are believed to account for 3% of the world's total Disability Adjusted Life Years (DALYs) lost and 10% of DALYs in Africa (Breman 2004). Malaria also significantly increases the risk of childhood death from other causes (Snow 2004). Almost half of the world's population live in areas where they are exposed to risk of malaria (Hay 2004); increasing numbers of visitors to endemic areas are also at risk.
Despite continued efforts to control malaria, it remains a major health problem in many regions of the world. The number of drugs remaining effective is limited, and new ways to prevent the disease are urgently needed. Currently the major methods used to prevent malaria in endemic areas are impregnated mosquito nets and indoor residual spraying. Vaccines are widely considered a necessary component for the complete success of malaria control, but it is likely that vaccines will need to be used in conjunction with these other methods rather than replacing them completely. Early optimism for vaccines was tempered as the problems caused by genetic (hence, antigenic) variability of the parasite and the difficulty of generating high levels of durable immunity emerged. Recently, hope has been renewed by the development of several new vaccine candidates and delivery systems, as well as new formulations and adjuvants for previously existing candidates (Ballou 2004; Moorthy 2004a). Vaccines currently under evaluation include recombinant proteins, synthetic peptides (including multiple antigen peptides), DNA vaccines, inactivated whole parasites, and vaccines comprising mixtures of a large variety of potential antigens. All vaccines discussed in this review are candidate vaccines only, since no malaria vaccines are currently licensed in any country.
To be effective, a malaria vaccine could either prevent infection altogether or mitigate against severe disease and death in those who become infected despite vaccination. Four stages of the malaria parasite's life cycle have been the targets of vaccine development efforts. The first two stages are often grouped as 'pre‐erythrocytic stages' (ie before the parasite invades the human red blood cells): these are the sporozoites inoculated by the mosquito into the human bloodstream; and the parasites developing inside human liver cells. The other two targets are the stage when the parasite is invading or growing in the red blood cells (blood, merozoite, or erythrocytic stage); and the gametocyte stage, when the parasites emerge from red blood cells and fuse to form a zygote inside the mosquito vector (gametocyte, gamete, or sexual stage). Vaccines based on the pre‐erythrocytic stages usually aim to completely prevent infection, while blood‐stage vaccines aim to reduce (and preferably eliminate) the parasite load once a person has been infected. Gametocyte vaccines would prevent the parasite being transmitted to others through mosquitoes. Ideally, a vaccine effective at all these parasite stages is desirable (Richie 2002).
Given the complexity and wide range of malaria vaccines under development, we have chosen to consider them in three categories: pre‐erythrocytic vaccines (the subject of this review); SPf66 vaccine; and blood‐stage vaccines. Future Cochrane Reviews may consider transmission‐blocking and multi‐stage vaccines when these are tested. The SPf66 vaccine was the first to be tested extensively (Graves 2006a). SPf66 was ineffective in Africa (five trials) and Asia (one trial). It had marginal efficacy in South America (four trials). It is no longer being tested and development towards commercialization is not taking place. However, it is possible that new formulations of SPf66 or combinations with other antigens will be developed in the future.
One blood‐stage vaccine has advanced to Phase 2 trials, but it showed limited efficacy (Graves 2006b). This is currently a highly active area of research and new blood‐stage vaccine trials will be described in future updates of the blood‐stage vaccine Cochrane Review as they become available.
Pre‐erythrocytic vaccines
This review includes the randomized trials conducted to date on the efficacy of four types of pre‐erythrocytic vaccines: CS‐NANP; CS102; RTS,S; and ME‐TRAP.
The CS‐NANP‐based pre‐erythrocytic vaccines were the first to be tested, beginning in the 1980s. The vaccines used in the first trials comprised three different formulations of the four amino acid B‐cell epitope NANP, which is present as multiple repeats in the circumsporozoite protein covering the surface of the sporozoites of P. falciparum. The number of NANP repeats in these vaccines varied from three to 19, and three different carrier proteins were used.
The CS102 vaccine is also based on the sporozoite CS protein, but it does not include the NANP epitope. It is a synthetic peptide consisting of a stretch of 102 amino acids containing T‐epitopes from the C‐terminal end of the molecule.
The RTS,S recombinant vaccine also includes the NANP epitope. It contains 19 NANP repeats plus the C terminus of the CS protein fused to hepatitis B surface antigen (HBsAg), expressed together with unfused HBsAg in yeast. The resulting construct is formulated with the adjuvant ASO2/A. Thus the vaccine contains a large portion of the CS protein in addition to the NANP region, as well as the hepatitis B carrier.
The ME‐TRAP vaccine is entirely different from the other pre‐erythrocytic malaria vaccines. It is a DNA vaccine that uses the prime‐boost approach to immunization. It uses a malaria DNA sequence known as ME (multiple epitope)‐TRAP (thrombospondin‐related protein). The ME string contains 15 T‐cell epitopes, 14 of which stimulate CD8 T‐cells and the other of which stimulates CD4 T‐cells, plus two B‐cell epitopes from six pre‐erythrocytic antigens of P. falciparum. It also contains two non‐malarial CD4 T‐cell epitopes and is fused in frame to the TRAP sequence. This sequence is given first as DNA (two doses) followed by one dose of the same DNA sequence in the viral vector MVA (modified vaccinia virus Ankara). Phase 1 studies with this vaccine (excluded from this review) were very promising (McConkey 2003; Bejon 2005).
In addition to the vaccine candidates included in this review, many excluded studies describe Phase 1 trials carried out with other P. falciparum pre‐erythrocytic candidates, including other recombinant CS/hepatitis B vaccines (Walther 2005), sporozoite DNA vaccines (Le 1999), and multiple‐antigen peptides (Nardin 2000; Nardin 2001). Vaccines consisting of killed irradiated P. falciparum sporozoites are also being evaluated (Hoffman 2002). Those that progress to efficacy trials will be included in future updates of this review.
Objectives
To assess the efficacy and safety of pre‐erythrocytic malaria vaccines against any type of human malaria.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials.
Types of participants
People of any age.
Types of interventions
Intervention
Recombinant, synthetic peptide, parasite‐derived, or other vaccines containing antigens only from pre‐erythrocytic stages of any species of malaria parasite tested in humans in experimental or natural challenge trials. This currently includes the following vaccines: CS‐NANP; CS 102; RTS,S; and ME‐TRAP.
Control
Placebo or control vaccine, or routine antimalarial control measures.
Types of outcome measures
Primary
New malaria infection: Plasmodium appearance in blood sample.
Clinical malaria episodes.
Secondary
Severe malaria.
Prevalence of parasitaemia.
Parasite density: Plasmodium count from blood sample.
Fever episodes.
Anaemia.
Cerebral malaria.
Admission to hospital.
Admission to hospital with diagnosis of malaria.
Death.
Adverse events (local and systemic).
Search methods for identification of studies
We have attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and in progress).
Databases
We searched the following databases using the search terms and strategy described in Appendix 1: Cochrane Infectious Diseases Group Specialized Register (March 2006); Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library (2006, Issue 1); MEDLINE (1966 to March 2006); EMBASE (1980 to March 2006); LILACS (1982 to March 2006); and Science Citation Index (SCI; 1981 to March 2006).
Conference proceedings
We checked the proceedings of the annual meetings of the American Society for Tropical Medicine and Hygiene for 2002 to 2004, the conference proceedings for the MIM Malaria Pan‐Africa Conference, 18 to 22 November 2002, Arusha, Tanzania, and the third Pan‐African Malaria Conference, 22 to 24 June 1998, Nairobi, Kenya. We also accessed the proceedings of the Global Vaccine Research Forum, 7 to 10 June 2004, Montreux, Switzerland, and 12 to 15 June 2005, Bahia, Brazil, organized by the WHO Initiative for Vaccine Research.
Researchers and organizations
In October 2005, we contacted the following researchers working in the field: A Saul; B Genton; B Greenwood; T Smith; A Thomas; and S Hoffman. We also contacted R Rabinovich and the websites of the Malaria Vaccine Initiative at Program for Appropriate Technology in Health (PATH) and the Malaria Vaccine Technology Roadmap in January 2006. Other web sources searched in September 2005 included the European Malaria Vaccine Initiative, the European Malaria Vaccine Consortium, and the African Malaria Network Trust. We also accessed the portfolio of candidate malaria vaccines currently in development from the WHO Initiative for Vaccine Research (WHO 2005).
Reference lists
We checked the reference lists of all studies identified by the above methods.
Data collection and analysis
Selection of studies
Two people independently applied the inclusion criteria to all identified trials (one author and an Editor of the Cochrane Infectious Diseases Group, or both authors). Differences were discussed until consensus was reached.
Data extraction and management
Both authors independently extracted data on number of each outcome and number of participants from the trials using a pre‐specified form. Differences were resolved by discussion. Data details were checked with the trial authors for Alonso 2005a and Alonso 2005b.
Assessment of risk of bias in included studies
Both authors independently assessed the trials for four dimensions of quality using a pre‐specified form: method of generation of allocation sequence and allocation concealment (adequate, inadequate, not done, or unclear as defined by Jüni 2001); blinding (described who was blinded, eg participants, investigators, and outcome assessors); and completion of follow up (proportion of those randomized who completed all doses and who completed follow up, if stated). Differences were resolved by discussion.
Data synthesis
We analysed the data using Review Manager 5. Results for dichotomous data were expressed as risk ratios (RR) of an outcome occurring in the vaccine group compared to the control group. The risk ratio may be converted to an estimate of vaccine efficacy: efficacy = (1 ‐ RR) x 100%. Similarly, the 95% confidence interval (CI) for the vaccine efficacy may be obtained by substituting the upper and lower 95% confidence intervals of the risk ratio into the formula. Continuous results (parasite density) were expressed as mean difference, using the geometric mean parasite density in positive blood samples.
If trials continued after a booster dose, we separated the analysis of these results accordingly. Trials were also subgrouped according to the age of the participants (children versus adults) representing different immune status or transmission conditions, or both, and according to the type of challenge (experimental or natural).
This review used an intention‐to‐treat analysis, that is, the denominators were the numbers randomized into each arm. Many trials included adjusted incidence rates, such as by bed net use or performed time‐to‐event analysis. This meta‐analysis uses only unadjusted incidence rates based on the number of participants in each arm of the trial.
Results
Description of studies
Nine safety and efficacy trials, and two safety trials, including more than 3000 participants aged between three months and 45 years met the inclusion criteria; see the 'Characteristics of included studies' for detailed information on these trials. In two instances, more than one trial is reported in the same publication (Alonso 2005a and Alonso 2005b; Bojang 2005a and Bojang 2005b). Nineteen trials were excluded for reasons listed in the 'Characteristics of excluded studies'.
The included efficacy trials comprised three of CS‐NANP vaccines (Guiguemde 1990; Brown 1994; Sherwood 1996); one of CS‐102 vaccine (Genton 2005); four of RTS,S vaccine (Kester 2001; Bojang 2001; Alonso 2005a; Alonso 2005b); and one of ME‐TRAP vaccine (Moorthy 2004b). Two randomized controlled trials of safety of RTS,S in a total of 225 children have also been included (Bojang 2005a; Bojang 2005b).
1. CS‐NANP vaccines
Two different constructs containing the NANP epitope of the CS protein were used in the three trials of CS‐NANP vaccines. Guiguemde 1990 used three synthetic NANP repeats conjugated to tetanus toxoid; the control was tetanus toxoid. Brown 1994 and Sherwood 1996 used a recombinant vaccine (R32toxA) containing 30 NANP and 2 NVDP repeats conjugated to the toxin A of Pseudomonas aeruginosa with alum. The controls were tetanus/diphtheria toxoid and hepatitis B vaccine, respectively.
Guiguemde 1990 and Sherwood 1996 were conducted in Africa, and Brown 1994 was conducted in Asia. All the CS‐NANP trials were conducted in situations of natural challenge. Guiguemde 1990 was carried out with 123 infants (three to five months old) in a highly malaria endemic area. The two trials of R32toxA were in adult males only (199 in Brown 1994; 76 in Sherwood 1996). In Sherwood 1996, all participants were given a treatment course of quinine/doxycycline before each vaccination.
Guiguemde 1990 used active case detection. Brown 1994 used a combination of passive and active (bi‐weekly) case detection, while Sherwood 1996 did surveillance by daily home visitation.
In the CS‐NANP trials in endemic areas, data were reported on clinical and parasitaemic episodes. Clinical malaria was defined as symptoms plus parasitaemia in Guiguemde 1990 and Sherwood 1996. Brown 1994 was conducted in a less endemic area and the outcome measure was parasitaemia. Both Brown 1994 and Sherwood 1996 also used time to infection as an outcome measure and employed survival analysis methods. For this review, the incidence of the first episode of either parasitaemia (if given) or clinical malaria (passive and active detection combined) in each group has been used for analysis. Data from Guiguemde 1990 were reported as the cumulative incidence of parasitaemia over the course of the study. The total number of incident cases was not reported, although some children may have been positive at more than one survey.
2. CS102 vaccine
CS102 is a synthetic peptide vaccine containing T epitopes from the C‐terminal end of the CS protein. Genton 2005, a small trial of CS102 in malaria‐naive volunteers in Switzerland with 16 adult participants, allocated 10 participants to vaccine and six to control (adjuvant alone). The trial assessed efficacy using polymerase chain reaction (PCR) to detect parasites in the blood after artificial challenge by infected mosquitoes.
3. RTS,S vaccine
RTS,S is a recombinant product containing 19 NANP repeats as well as another portion of the CS protein, conjugated to the hepatitis B surface antigen and formulated with AS02A adjuvant. Four RTS,S efficacy trials (Kester 2001; Bojang 2001; Alonso 2005a; Alonso 2005b) and two safety trials (Bojang 2005a; Bojang 2005b) are included in this review. One trial of RTS,S in 46 non‐immune adults used experimental challenge with infected mosquitoes (Kester 2001). The control group received hepatitis B vaccine. After promising results, a field trial in 306 semi‐immune adult males in The Gambia used natural challenge (Bojang 2001). The control group received rabies vaccine. All participants in this trial were given a course of chemotherapy to clear parasites before the third dose of vaccine. A booster dose of vaccine was given to 158 of the participants in the second year.
Safety trials of RTS,S in two cohorts of children (first six to 11 years, then one to four years) were conducted in The Gambia (Bojang 2005a; Bojang 2005b). There were 90 children in the first trial and 135 in the second; in both cases the control groups were given rabies vaccine. The first efficacy trials of RTS,S in children were in two cohorts of one‐ to four‐year olds in Mozambique (Alonso 2005a; Alonso 2005b). Cohort one (1605 children) focused on assessing protection from clinical malaria (Alonso 2005a), while cohort two (417 children) assessed mainly new infections (Alonso 2005b). The control vaccines given in both trials were as follows: children under 24 months were given pneumococcal conjugate vaccine (first and third doses) and Haemophilus influenzae b vaccine (second dose); and children over 24 months were given hepatitis B vaccine in three doses. All children in cohort two were treated with amodiaquine and sulfadoxine‐pyrimethamine four weeks before the start of surveillance to clear any parasites.
Results of the RTS,S Mozambique trials were reported separately for two periods of follow up − six months and 18 months − starting two weeks after the third dose. In this review, the results at six and 18 months are reported separately for some outcome measures (eg prevalence).
Bojang 2001 conducted follow up by daily surveillance for 15 weeks in the first year and nine weeks in the second year. In Mozambique, cohort one was followed mainly by passive surveillance, although monthly home visits were also done (Alonso 2005a). Cohort two was followed more intensely by active surveillance for new infections in addition to passive surveillance through health facilities (Alonso 2005b). Follow up for adverse events was reported for the combined cohorts and tabulated under the trial name Alonso 2005ab.
4. ME‐TRAP vaccine
ME‐TRAP is a DNA vaccine given in a prime‐boost sequence: DNA representing multiple pre‐erythrocytic antigen epitopes is given first in two doses followed by DNA inserted in the viral vector MVA (modified vaccinia Ankara). One ME‐TRAP trial was conducted with 372 adult males in The Gambia using rabies vaccine in the control group (Moorthy 2004b). All participants were treated with sulfadoxine‐pyrimethamine two weeks before the third dose. The follow‐up period was 11 weeks after the final dose.
Risk of bias in included studies
1. CS‐NANP vaccines
The method of randomization was not described in any of the three trials, and allocation concealment was described only in Brown 1994. All trials were described as double blind. The withdrawal rate was low (< 10%) in the Sherwood 1996 trial, but between 10% and 20% in Guiguemde 1990 and Brown 1994. The withdrawal rate did not appear to differ between the vaccine and control groups.
2. CS102 vaccines
Randomization and allocation concealment were both adequate in Genton 2005, and investigators, participants, and outcome assessors were blinded. Two out of 16 participants in the trial did not receive the challenge infection due to previously undisclosed medical histories.
3. RTS,S vaccines
The RTS,S trials were generally good quality. Both randomization and allocation concealment were adequate in all RTS,S trials except Kester 2001 in which allocation concealment was not used, and all included groups were described as double blind or blinded participants, investigators, and outcome assessors, at least initially. The RTS,S Mozambique trials (Alonso 2005a and Alonso 2005b) technically became single blind after the code was broken and results were reported to investigators after six months follow up. However, only the study statistician held the code, which was not available to other investigators, participants, or assessors, and no further immunizations were given. Therefore, it is unlikely that bias was introduced in the single‐blind phase.
In Kester 2001, only 52% of those starting the immunization series were subsequently challenged: 50% of the vaccine groups and 58% of the placebo group. There was a relatively high dropout rate in both arms of Bojang 2001: 14% of the vaccine group and 22% of the control group dropped out or were excluded between the first dose and the follow‐up period in the first year. Fifty‐two per cent of original participants took part in the second year of the trial (48% of the vaccine group and 56% of the control group). More than 94% of participants in each of the two trials from The Gambia completed the short follow up (Bojang 2005a; Bojang 2005b). In Mozambique, follow up was better in Alonso 2005a (cohort one) where 93% received three doses and 86% completed the first six months follow up, than in Alonso 2005b (cohort two) where the proportions were 92% and 72%, respectively. Some participants not present in the double‐blind, six‐month, follow‐up phase were included in the subsequent single‐blind follow up from six to 18 months; over 90% of those entering the single‐blind phase completed the follow up in both cohorts.
4. ME‐TRAP vaccine
The one trial of ME‐TRAP was of good quality (Moorthy 2004b); it was double blind with adequate allocation concealment. Among 372 randomized participants, 86% received three doses and 80% completed follow up.
Effects of interventions
1. CS‐NANP vaccines (3 trials)
There was no evidence for effectiveness of CS‐NANP vaccines in the three trials. The combined risk ratio for reduction of new infections in the three trials was 1.05 (95% CI 0.82 to 1.35; 307 participants, Analysis 1.1).
1.1. Analysis.
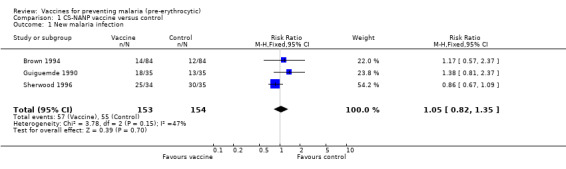
Comparison 1 CS‐NANP vaccine versus control, Outcome 1 New malaria infection.
2. CS102 vaccines (1 trial)
All 14 participants in this small trial of non‐immune individuals had malaria infection as detected by PCR (8 participants in the vaccine group, and 6 in the control group; Genton 2005). However, participants in the vaccine group had a significant reduction in the frequency of nausea (RR 0.13, 95% CI 0.02 to 0.78; 14 participants, Analysis 2.1). There were also non‐significant reductions in the frequency of fever (14 participants, Analysis 2.2) and malaise (14 participants, Analysis 2.3).
2.1. Analysis.
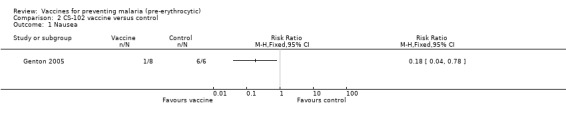
Comparison 2 CS‐102 vaccine versus control, Outcome 1 Nausea.
2.2. Analysis.
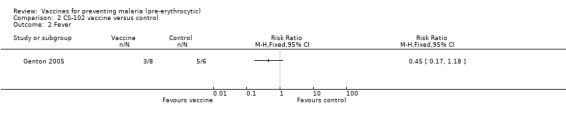
Comparison 2 CS‐102 vaccine versus control, Outcome 2 Fever.
2.3. Analysis.
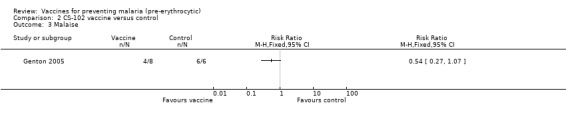
Comparison 2 CS‐102 vaccine versus control, Outcome 3 Malaise.
3. RTS,S vaccines (4 safety/efficacy trials and 2 safety trials)
RTS,S protected strongly against malaria infection in one trial in non‐immune people using experimental challenge (RR 0.53, 95% CI 0.34 to 0.83; 24 participants, Analysis 3.1; Kester 2001). However, this was a small trial with no allocation concealment and a large number of dropouts between immunization and challenge. When tested against natural challenge, in an intention‐to‐treat analysis, RTS,S did not significantly prevent new infections within the follow‐up period (RR 0.96, 95% CI 0.87 to 1.06; 723 participants, Analysis 3.1). No evidence was seen of effect by age group (330 adults, 417 children, Analysis 3.2).
3.1. Analysis.
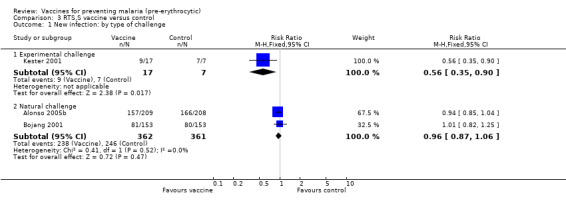
Comparison 3 RTS,S vaccine versus control, Outcome 1 New infection: by type of challenge.
3.2. Analysis.
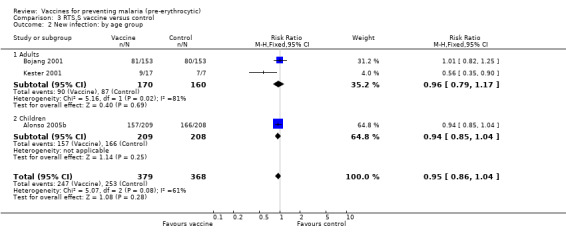
Comparison 3 RTS,S vaccine versus control, Outcome 2 New infection: by age group.
RTS,S vaccine significantly reduced the incidence of clinical episodes of malaria in children, up to 18 months after immunization (RR 0.74, 95% CI 0.63 to 0.87; 1605 participants, Analysis 3.3). This represents efficacy of 26% (95% CI 13% to 37%). The same effect was not seen in clinical malaria in semi‐immune adults in The Gambia in the first season after immunization (306 participants, Analysis 3.3). However, after a booster dose, there was a large reduction in clinical malaria episodes in the second year of follow up (RR 0.37, 95% CI 0.17 to 0.82; 158 participants, Analysis 3.4). This corresponds to a vaccine efficacy of 63% (95% CI 18% to 83%).
3.3. Analysis.

Comparison 3 RTS,S vaccine versus control, Outcome 3 Clinical malaria episodes.
3.4. Analysis.
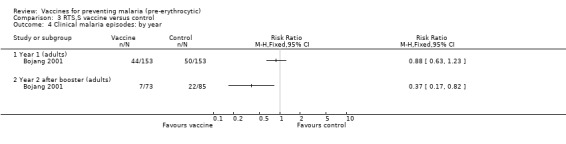
Comparison 3 RTS,S vaccine versus control, Outcome 4 Clinical malaria episodes: by year.
In the Mozambique trial (Alonso 2005a), there was a significant reduction in episodes of severe malaria in children by 58% (95% CI 15% to 79%), derived from the risk ratio of 0.42 (95% CI 0.21 to 0.85; 1605 participants, Analysis 3.5). The number of cases of malaria requiring admission to hospital was also reduced by 32% (RR 0.68, 95% CI 0.46 to 0.99; 1605 participants, Analysis 3.6). No significant effects were seen on admission to hospital for any cause (1605 participants, Analysis 3.7). However, these efficacy estimates for severe malaria and hospital admission were based on exploratory analyses of outcomes not intended as primary outcome measures in the trial design.
3.5. Analysis.
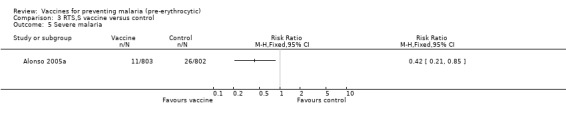
Comparison 3 RTS,S vaccine versus control, Outcome 5 Severe malaria.
3.6. Analysis.
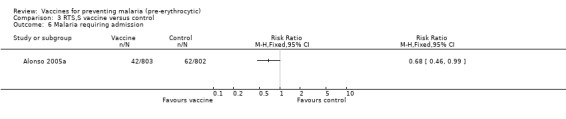
Comparison 3 RTS,S vaccine versus control, Outcome 6 Malaria requiring admission.
3.7. Analysis.

Comparison 3 RTS,S vaccine versus control, Outcome 7 Admission to hospital for any cause.
Outcome measures detected by a cross‐sectional survey (prevalence and anaemia) were assessed after six months and 18 months of follow up. Prevalence of parasitaemia was reduced significantly by 26% at six months (RR 0.74, 95% CI 0.62 to 0.89; 2022 participants, Analysis 3.8) and by 20% at 18 months (RR 0.80, 95% CI 0.65 to 0.99; 1794 participants, Analysis 3.8). No effect was seen on anaemia at either survey (Analysis 3.9), but the prevalence of anaemia was low in these study populations.
3.8. Analysis.
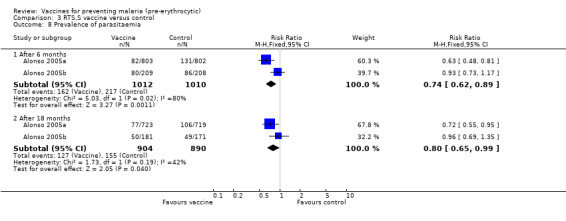
Comparison 3 RTS,S vaccine versus control, Outcome 8 Prevalence of parasitaemia.
3.9. Analysis.
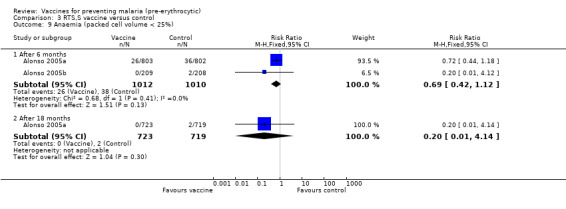
Comparison 3 RTS,S vaccine versus control, Outcome 9 Anaemia (packed cell volume < 25%).
Adverse events were assessed in adults in the USA and The Gambia, and in children in The Gambia and Mozambique (Analysis 3.10). In The Gambia (Bojang 2005a; Bojang 2005b), adverse events were graded on a one to four scale, but the results given represent the numbers of children with any adverse event, not necessarily those with grade three or four reaction. Also in each of these trials, the results from all three vaccine dose groups have been combined. In Mozambique, adverse events were reported for both cohorts one and two combined (cited here as Alonso 2005ab). Severe adverse events are reported by length of follow up: zero to six months; and six to 18 months.
3.10. Analysis.
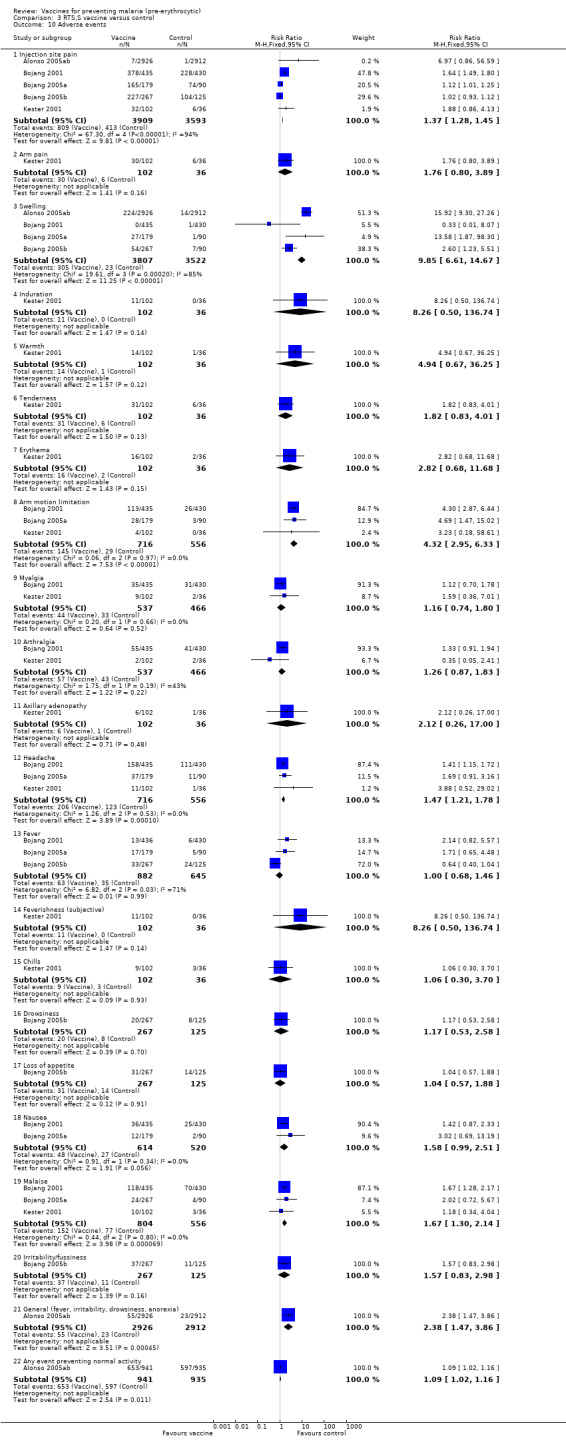
Comparison 3 RTS,S vaccine versus control, Outcome 10 Adverse events.
Significantly higher proportions of participants in the malaria vaccine groups reported injection site pain, swelling, arm motion limitation, headache, and malaise after vaccination compared with the control groups (Analysis 3.10). The frequency of adverse events was stated to be similar in hepatitis B surface antigen‐positive and antigen‐negative participants (Bojang 2001), although five hepatitis B virus (HBV) chronic carriers developed elevated alanine amino‐tranferase concentrations after vaccination and were not given further vaccinations. After a fourth booster dose, three of 79 vaccine recipients in the Bojang 2001 trial developed severe injection site pain compared with none in the control group. In Bojang 2005a, one child in the vaccine group developed grade‐two axillary lymphadenopathy after the first vaccine dose; it resolved within four days. In both safety trials from The Gambia, symptoms rated as grade‐three severity were infrequent and resolved or decreased within 24 hours. No severe adverse events believed to be related to vaccination were reported. In Alonso 2005ab, a high frequency of severe events was reported especially during the first six months (RR 0.72, 95% CI 0.61 to 0.85; 1876 participants, Analysis 3.11), but they were less frequent in the malaria vaccine compared with the control groups, and none was judged to be related to vaccination.
3.11. Analysis.
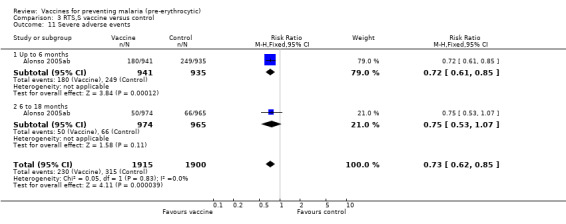
Comparison 3 RTS,S vaccine versus control, Outcome 11 Severe adverse events.
4. ME‐TRAP vaccine (1 trial)
There was no evidence for effectiveness of ME‐TRAP vaccine in preventing new infections (296 participants, Analysis 4.1) or clinical malaria episodes (296 participants, Analysis 4.2). Nor did the vaccine reduce density of parasites (171 cases, Analysis 4.3) or increase mean packed cell volume (a measure of anaemia) in semi‐immune adult males (296 participants, Analysis 4.4) in The Gambia (Moorthy 2004b).
4.1. Analysis.
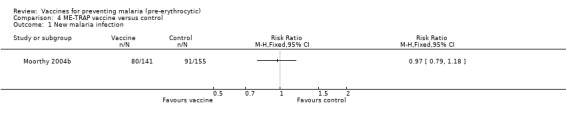
Comparison 4 ME‐TRAP vaccine versus control, Outcome 1 New malaria infection.
4.2. Analysis.
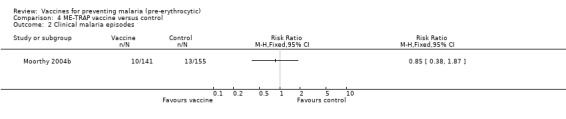
Comparison 4 ME‐TRAP vaccine versus control, Outcome 2 Clinical malaria episodes.
4.3. Analysis.
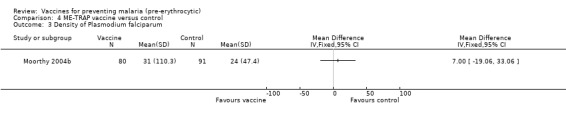
Comparison 4 ME‐TRAP vaccine versus control, Outcome 3 Density of Plasmodium falciparum.
4.4. Analysis.
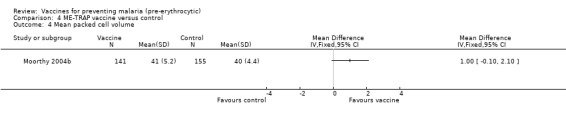
Comparison 4 ME‐TRAP vaccine versus control, Outcome 4 Mean packed cell volume.
Adverse events were significantly more frequent in vaccine than control groups (Analysis 4.5). The frequencies of all types of local and systemic events, except objective fever and nausea, were greater in the vaccine than control groups. However, no serious adverse events were reported.
4.5. Analysis.
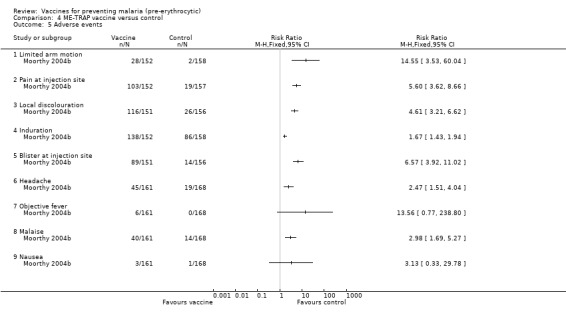
Comparison 4 ME‐TRAP vaccine versus control, Outcome 5 Adverse events.
Discussion
Four different types of pre‐erythrocytic vaccine are described in this review. The RTS,S vaccine has shown significant efficacy against both experimental challenge (in non‐immunes) and natural challenge (in participants living in endemic areas) with malaria. Children in Mozambique were protected by RTS,S against episodes of clinical malaria and severe malaria in trials in for up to 18 months after immunization. Protection against clinical malaria in these children was estimated at 26% (95% CI 13% to 37%). It is encouraging that RTS,S also showed significant protection against severe malaria in children, estimated at 58% (95% CI 15 to 79%). However the efficacy estimate for severe malaria in the Mozambique RTS,S trial was based on exploratory analyses and more precise estimates are needed. Further trials of RTS,S are in the planning stages. Although no evidence was found for efficacy of RTS,S against clinical malaria in adults in The Gambia in the first year of follow up, efficacy was 63% (95% CI 18% to 93%) in the second year after immunization, after a booster dose. The results from the Mozambique efficacy trial described here are generally consistent with the efficacy results presented in the published papers (Alonso 2005a; Alonso 2005b), which were based on time‐to‐event analysis and hazard ratios. These estimated that the efficacy of the vaccine (defined as 1 ‐ hazard ratio) was 29.9% against clinical episodes and 57.7% against severe malaria. However for first infection, this review's finding of lack of significant efficacy (1 ‐ risk ratio) against new infection is different from the published paper's estimate, which was 45%, based on a time‐to‐infection analysis.
Of the three other types of vaccine reviewed, the CS‐NANP vaccine has been tested in three relatively small trials under natural challenge, and CS102 has only been tested in one very small good quality trial in non‐immune participants using experimental challenge. Evidence is insufficient to evaluate the efficacy of either of these vaccines, although with current evidence they do not appear promising. The ME‐TRAP vaccine has also only been tested in one randomized trial, although it was a good quality, large trial in an endemic area.
There are no significant safety issues with RTS,S vaccines, although the frequency of local and systemic adverse events is increased compared to control. No severe adverse events were judged to be vaccine related.
If further trials of RTS,S show similar results to those already reported, the level of protection against severe malaria justifies speedy progression of this vaccine towards licensing for routine use as well as further development of the vaccine for greater efficacy. In addition to exploring efficacy of other pre‐erythrocytic antigens and whole irradiated parasites, methods of improving the immunogenicity and effectiveness of the RTS,S vaccine and its combination with DNA vaccines or antigens from other malaria stages are the highest research priorities. In addition, since infants less than one year old are a high‐risk population and the future target group for malaria vaccines, RTS,S and other vaccines must be tested in infants.
Authors' conclusions
Implications for practice.
The RTS,S vaccine showed extremely promising results, especially with regard to prevention of severe malaria in children and duration of protection of 18 months. The frequency of local and systemic adverse events is increased by RTS,S vaccine, but no severe adverse events were judged to be vaccine related. If further trials show similar results to those already reported, these results justify speedy progression of this vaccine towards licensing for routine use as well as further development of the vaccine for greater efficacy. Progression towards licensing should be accompanied by development of deployment strategies, including funding for the countries most in need of the vaccine.
Implications for research.
The CS‐NANP epitope alone appears to be ineffective in a vaccine. Research on vaccines that combine the NANP epitope with other antigens may be more productive. CS102 and ME‐TRAP have not been evaluated sufficiently to make a judgement about their ultimate value. Methods of improving the immunogenicity and effectiveness of the RTS,S vaccine and its combination with DNA vaccines or antigens from other malaria stages are research priorities, as is testing candidate vaccines in infants. Future trials should be designed to detect effects on clinical and severe malaria.
What's new
| Date | Event | Description |
|---|---|---|
| 21 July 2008 | Amended | Converted to new review format with minor editing. |
History
Protocol first published: Issue 4, 2006 Review first published: Issue 4, 2006
| Date | Event | Description |
|---|---|---|
| 15 August 2006 | New citation required and conclusions have changed | 2006, Issue 4: The original Cochrane Review of malaria vaccines (Graves 2003) has been divided into three parts for pre‐erythrocytic vaccines, SPf66 vaccine, and blood‐stage vaccines. This review includes the trials of pre‐erythrocytic vaccines. The CS‐NANP and RTS,S trials from Graves 2003 are included; one trial of CS‐NANP vaccine from Nigeria has been reclassified as a multi‐stage vaccine (CS/5.1) and will be included in a future Cochrane Review for multi‐stage vaccines. Six new trials have been added to this review: one of a new vaccine CS102; one of a new vaccine ME‐TRAP; two safety trials of RTS,S; and two efficacy trials of RTS,S. With the addition of new field trials, it became apparent that there was heterogeneity between trials that used experimental and natural challenge, and these have been subgrouped in this review. The text of the review has been extensively updated and revised, and meta‐analysis is now done with risk ratio as the default statistic. |
Acknowledgements
This document is an output from a project funded by the UK Department for International Development (DFID) for the benefit of developing countries. The views expressed are not necessarily those of DFID.
Appendices
Appendix 1. Search methods: detailed search strategies
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb | Science Citation Index |
| 1 | malaria | malaria | malaria | malaria | malaria | malaria |
| 2 | Plasmodium | Plasmodium | Plasmodium | Plasmodium | Plasmodium | Plasmodium |
| 3 | 1 or 2 | 1 or 2 | 1 or 2 | 1 or 2 | 1 or 2 | 1 or 2 |
| 4 | vaccin* | vaccin* | vaccin* | vaccin* | vaccin* | vaccin* |
| 5 | 3 and 4 | 3 and 4 | 3 and 4 | 3 and 4 | 3 and 4 | 3 and 4 |
| 6 | — | MALARIA VACCINES | MALARIA VACCINES | MALARIA VACCINES | — | — |
| 7 | — | 5 or 6 | 5 or 6 | 5 or 6 | — | — |
| 8 | — | — | Limit 7 to human | Limit 7 to human | — | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Higgins 2005); upper case: MeSH or EMTREE heading; lower case: free text term.
Data and analyses
Comparison 1. CS‐NANP vaccine versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 New malaria infection | 3 | 307 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.82, 1.35] |
Comparison 2. CS‐102 vaccine versus control.
Comparison 3. RTS,S vaccine versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 New infection: by type of challenge | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Experimental challenge | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.35, 0.90] |
| 1.2 Natural challenge | 2 | 723 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.87, 1.06] |
| 2 New infection: by age group | 3 | 747 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.86, 1.04] |
| 2.1 Adults | 2 | 330 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.79, 1.17] |
| 2.2 Children | 1 | 417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.85, 1.04] |
| 3 Clinical malaria episodes | 2 | 1911 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.66, 0.88] |
| 3.1 Adults | 1 | 306 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.63, 1.23] |
| 3.2 Children | 1 | 1605 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.63, 0.87] |
| 4 Clinical malaria episodes: by year | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Year 1 (adults) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Year 2 after booster (adults) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Severe malaria | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Malaria requiring admission | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Admission to hospital for any cause | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8 Prevalence of parasitaemia | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 After 6 months | 2 | 2022 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.62, 0.89] |
| 8.2 After 18 months | 2 | 1794 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.65, 0.99] |
| 9 Anaemia (packed cell volume < 25%) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 After 6 months | 2 | 2022 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.42, 1.12] |
| 9.2 After 18 months | 1 | 1442 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.01, 4.14] |
| 10 Adverse events | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Injection site pain | 5 | 7502 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [1.28, 1.45] |
| 10.2 Arm pain | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.76 [0.80, 3.89] |
| 10.3 Swelling | 4 | 7329 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.85 [6.61, 14.67] |
| 10.4 Induration | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.26 [0.50, 136.74] |
| 10.5 Warmth | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.94 [0.67, 36.25] |
| 10.6 Tenderness | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.82 [0.83, 4.01] |
| 10.7 Erythema | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.82 [0.68, 11.68] |
| 10.8 Arm motion limitation | 3 | 1272 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.32 [2.95, 6.33] |
| 10.9 Myalgia | 2 | 1003 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.74, 1.80] |
| 10.10 Arthralgia | 2 | 1003 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.87, 1.83] |
| 10.11 Axillary adenopathy | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.12 [0.26, 17.00] |
| 10.12 Headache | 3 | 1272 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [1.21, 1.78] |
| 10.13 Fever | 3 | 1527 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.68, 1.46] |
| 10.14 Feverishness (subjective) | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.26 [0.50, 136.74] |
| 10.15 Chills | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.30, 3.70] |
| 10.16 Drowsiness | 1 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.53, 2.58] |
| 10.17 Loss of appetite | 1 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.57, 1.88] |
| 10.18 Nausea | 2 | 1134 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.99, 2.51] |
| 10.19 Malaise | 3 | 1360 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [1.30, 2.14] |
| 10.20 Irritability/fussiness | 1 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [0.83, 2.98] |
| 10.21 General (fever, irritability, drowsiness, anorexia) | 1 | 5838 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.47, 3.86] |
| 10.22 Any event preventing normal activity | 1 | 1876 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [1.02, 1.16] |
| 11 Severe adverse events | 1 | 3815 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.62, 0.85] |
| 11.1 Up to 6 months | 1 | 1876 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.61, 0.85] |
| 11.2 6 to 18 months | 1 | 1939 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.53, 1.07] |
Comparison 4. ME‐TRAP vaccine versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 New malaria infection | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Clinical malaria episodes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Density of Plasmodium falciparum | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Mean packed cell volume | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 Limited arm motion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Pain at injection site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Local discolouration | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Induration | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.5 Blister at injection site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.6 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.7 Objective fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.8 Malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.9 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alonso 2005a.
| Methods | Randomized controlled trial Generation of allocation sequence: computer generated in blocks of 6 Allocation concealment: central randomization was done at GlaxoSmithKline and the code released to the investigators after completion of follow up; opaque masked and coded syringes were used Blinding: investigator, participants, and outcome assessors blinded for first 6 months; investigators were not blinded during next 12 months Inclusion of all randomized participants: 1493/1605 (93.0%) of those randomized received 3 doses; 1380/1605 (86.0%) completed 6‐month follow up (92.4% of those who received 3 doses); 1442 entered single‐blind phase of whom 1319 (91.5%) completed follow up Length of follow up: 18 months after third dose |
|
| Participants | Number: 1605 children Inclusion criteria: aged 1 to 4 years; resident in study area; full immunization with Expanded Programme of Immunization (EPI) vaccines; parental consent Exclusion criteria: history of allergic disease; packed cell volume ≤ 25%; weight for height ≤ 3 Z score; clinically significant chronic or acute disease; abnormal haematology or biochemistry variables |
|
| Interventions | 1. RTS,S vaccine: 3 doses, 25 µg in 250 µL AS02A adjuvant, intramuscularly in deltoid (alternating arms) at 0, 1, and 2 months 2. Pneumococcal conjugate vaccine (under 24‐months old; first and third doses) plus Hib vaccine (second dose) or hepatitis B vaccine (over 24‐months old; 3 doses) | |
| Outcomes | 1. Time to first clinical episode of symptomatic Plasmodium falciparum malaria (case definition: child presenting with temperature > 37.5 °C and parasitaemia > 2500/µL) 2. Clinical episodes of malaria 3. Malaria needing admission: (P. falciparum sole cause of illness or important contributing factor) 4. All‐cause admission 5. Severe malaria (derived from World Health Organization definition: asexual P. falciparum parasitaemia; no other more probable cause of illness; plus composite of severe malaria anaemia (packed cell volume < 15%), cerebral malaria (Blantyre coma score < 2), and severe disease of other body systems (multiple seizures, prostration, hypoglycaemia, clinically suspected acidosis, or circulatory collapse) 6. Prevalence of parasitaemia 7. Prevalence of anaemia (packed cell volume < 25%) 8. Geometric mean parasite density in first clinical episode 9. Geometric mean parasite density in parasitaemic children at 6.5 months 10. Geometric mean titre to CS protein and hepatitis B surface antigen (HBsAg) 11. Seropositivity rates for anti‐CS antibody (> 0.5 international units/mL) and anti‐hepatitis B surface antigen (HBsAg) antibody (≥ 10 international units/mL) 12. Adverse events | |
| Notes | Location: within 10 km radius of Manhica, Mozambique where the entomological inoculation rate in 2002 was 38 infective bites/year Method of surveillance: passive surveillance of illness and adverse events through health centre staffed 24/7; observation for 1 h after vaccination and once/day at home for 3 days after each dose for adverse events; home visits once per month starting 60 days after third dose to check residence and document unreported adverse events; complete blood count done 1 month after dose 3; creatinine, alanine aminotransferase (ALT), and bilirubin at months 1 and 6.5 after dose 3; cross‐sectional surveys with blood slide and axillary temperature taken at 6.5 months and 18 months after dose 3 This trial reported in same publication as Alonso 2005b |
|
Alonso 2005ab.
| Methods | Study reference created for the reporting of adverse event data, which were reported jointly in the two trial reports (see Alonso 2005a and Alonso 2005b) | |
| Participants | Not applicable | |
| Interventions | Not applicable | |
| Outcomes | Not applicable | |
| Notes | Not applicable | |
Alonso 2005b.
| Methods | Randomized controlled trial Generation of allocation sequence: computer generated in blocks of 6 Allocation concealment: central randomization was done at GlaxoSmithKline and the code released to the investigators after completion of follow up; opaque and masked coded syringes were used Blinding: investigator, participants, and outcome assessors blinded for first 6 months; investigators were not blinded during next 12 months Inclusion of all randomized participants: 383/417 (91.8%) of those randomized received 3 doses; 299/417 (71.7%) completed 6 months follow up (78.1% of those who received 3 doses); 352 entered single‐blind phase of whom 320 (90.9%) completed follow up Length of follow up: 18 months after third dose |
|
| Participants | Number: 417 children Inclusion criteria: age 1 to 4 years; resident in study area; full Expanded Programme of Immunization (EPI) immunization; parent's consent Exclusion criteria: history of allergic disease; packed cell volume ≤ 25%; weight for height ≤ 3 Z score; clinically significant chronic or acute disease; abnormal haematology or biochemistry variables |
|
| Interventions | 1. RTS,S vaccine: 3 doses, 25 µg in 250 µL AS02A adjuvant, intramuscularly in deltoid (alternating arms) at 0, 1, and 2 months
2. 7‐valent pneumococcal conjugate vaccine (< 24‐months old; doses 1 and 3) plus Hib vaccine (dose 2) or hepatitis B vaccine (> 24‐months old; 3 doses) 4 weeks before start of surveillance, presumptive treatment with amodiaquine and sulfadoxine‐pyrimethamine was given; those children positive 2 weeks later were treated with second‐line drug and excluded from follow up |
|
| Outcomes | 1. Time to first infection with Plasmodium falciparum malaria (case definition: presenting with temperature > 37.5 °C and parasitaemia > 2500/µL) 2. Clinical episodes of malaria 3. Malaria needing admission (P. falciparum sole cause of illness or important contributing factor) 4. All‐cause admission 5. Severe malaria (derived from World Health Organization definition: asexual P. falciparum parasitaemia; no other more probable cause of illness; plus composite of severe malaria anaemia (packed cell volume < 5%), cerebral malaria (Blantyre coma score < 2), and severe disease of other body systems (multiple seizures, prostration, hypoglycaemia, clinically suspected acidosis, or circulatory collapse) 6. Prevalence of parasitaemia 7. Prevalence of anaemia (packed cell volume < 25%) 8. Geometric mean parasite density in first clinical episode 9. Geometric mean parasite density in parasitaemic children at 6.5 months 10. Geometric mean titre to CS protein and hepatitis B surface antigen (HBsAg) 11. Seropositivity for anti‐CS antibody (> 0.5 international units/mL) and anti‐hepatitis B surface antigen (HBsAg) antibody (≥ 10 international units/mL) 12. Adverse events | |
| Notes | Location: Ilha Josina, a lowland area 55 km north of Manhica, Mozambique with pronounced seasonality of transmission and more intense transmission than in Manhica Method of surveillance: active surveillance for infection by morbidity questionnaire, axillary temperature, and blood slides at home visits starting 2 weeks after dose 3, every 2 weeks for 2.5 months then monthly for 2 months; observation for 1 h after vaccination and once/day at home for 3 days after each dose for adverse events; complete blood count done 1 month after dose 3; creatinine, alanine aminotransferase (ALT), and bilirubin at months 1 and 6.5 after dose 3; cross‐sectional surveys with blood slide and axillary temperature taken at 6.5 months and 18 months after dose 3 This trial reported in same publication as Alonso 2005a |
|
Bojang 2001.
| Methods | Randomized controlled trial Generation of allocation sequence: externally generated list; in village blocks Allocation concealment: each individual's vaccine doses were packaged in sealed boxes labelled with a unique randomization number; vaccines given by nurses with no other role in study Blinding: double blind Inclusion of all randomized participants: 264/306 (86.3%) received 3 doses; 250/306 (81.7%) completed follow up to year 1 (94.7% of those who received 3 doses); 158 received dose 4 and were followed up in second year Length of follow up: 15 weeks in 1998; and 9 weeks in 1999 transmission season |
|
| Participants | Number: 306 males Inclusion criteria: age 18 to 45 years; no clinically significant disease. Exclusion criteria: known allergy to any vaccine or sulfadoxine‐pyrimethamine; chronic clinically significant pulmonary, cardiovascular, hepatic, or renal functional abnormality; history of splenectomy; packed cell volume < 30%; any blood transfusions within last month; any immunosuppressive drugs, previous vaccine with MPL‐ or QS‐21‐containing vaccine; participation in another trial; any other vaccine or immunoglobulin within last 2 weeks; known human immunodeficiency virus (HIV) infection |
|
| Interventions | 1. RTS,S vaccine: 3 doses (50 µg per 0.5 mL dose) on days 0, 28, and 150; dose 4 given in following year (1999)
2. Rabies human diploid cell vaccine Sulfadoxine‐pyrimethamine (3 tablets) was given to all participants 2 weeks before dose 3 |
|
| Outcomes | 1. Time to first asexual Plasmodium falciparum infection 2. Symptomatic malaria (case definition: presence of P. falciparum at any density with at least one of the following: fever (axillary temp ≥ 37.5 °C; history of fever in last 24 h); malaise; chills; headache; myalgia; arthralgia; and nausea, with no other obvious cause) 3. Malaria infection 4. Packed cell volume 5. Anti‐circumsporozoite protein antibodies 6. T‐cell responses to RTS,S and hepatitis B surface antigen 7. Adverse events | |
| Notes | Location: 6 villages in Upper River Division, The Gambia, where malaria occurs during the rainy season (July to November) with greatest incidence in October to November Entomological inoculation rate: 1 to 50 infective bites per person per year Method of surveillance: daily home visits for symptom surveillance and weekly blood slide; passive surveillance |
|
Bojang 2005a.
| Methods | Randomized controlled trial Generation of allocation sequence: random selection from list of eligible participants sent to external statistician at GlaxoSmithKline Biologicals, in ratio of 2 vaccine to 1 placebo per dose level Allocation concealment: masked identical syringes prepared by staff otherwise uninvolved in trial Blinding: investigators, participants, and evaluators blinded Inclusion of all randomized participants: 85/90 (94%) completed follow up Length of follow up: 30 days after last dose |
|
| Participants | Number: 90 children Inclusion criteria: age 6 to 11 years; no clinically significant chronic or acute disease (chronic hepatitis B carriers were not excluded) Exclusion criteria: known allergy to any vaccine; severe malnutrition (weight for height < 3 Z scores); haematocrit < 30% |
|
| Interventions | 1. RTS,S/AS02A vaccine: 10 µg in 0.1 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
2. RTS,S/AS02A vaccine: 25 µg in 0.25 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
3. RTS,S/AS02A vaccine: 50 µg in 0.5 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
4. Rabies human diploid cell vaccine (Merieux HDCV): single‐dose vial with diluent; 3 doses 0, 1, and 3 month intervals plus dose 4 given after trial completion (RTS,S groups were also offered 3 doses of rabies vaccine after trial completion) Increasing doses of vaccine were given in a dose‐escalating fashion, ie dose groups were staggered at 10 day intervals |
|
| Outcomes | 1. Injection site pain 2. Swelling > 50 mm and persisting > 24 h 3. Limitation of arm motion 4. Fever 5. Headache 6. Malaise 7. Nausea 8. Haemoglobin, haematocrit, white blood cell count, and platelets on days 14, 60, and 104 9. Creatinine and alanine aminotransferase (ALT) on days 14, 60, and 104 10. Antibody to circumsporozoite protein repeat epitopes 11. Circulating hepatitis B surface antigen 12. Anti‐hepatitis B surface antigen (HbSAg) antibodies | |
| Notes | Location: village of Dampha Kunda, near Basse, Upper River Division, The Gambia; highly seasonal malaria with peak in October to November Method of surveillance: home visits daily for 3 days or until symptoms resolved; case reporting on standardized forms by study nurse in village or at clinic in Basse This trial reported in same publication as Bojang 2005a |
|
Bojang 2005b.
| Methods | Randomized controlled trial Generation of allocation sequence: random selection from list of eligible participants sent to external statistician at GlaxoSmithKline Biologicals, in ratio 2 vaccine to 1 placebo per dose level Allocation concealment: masked identical syringes prepared by staff otherwise uninvolved in trial Blinding: investigators, participants, and evaluators blinded Inclusion of all randomized participants: 130/135 (96.3%) completed follow up Length of follow up: 30 days after last dose |
|
| Participants | Number: 135 children Inclusion criteria: age 1 to 5 years; no clinically significant chronic or acute disease (chronic hepatitis B carriers were not excluded) Exclusion criteria: known allergy to any vaccine; severe malnutrition (weight for height < 3 Z scores); haematocrit < 30% |
|
| Interventions | 1. RTS,S/AS02A vaccine: 10 µg in 0.1 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
2. RTS,S/AS02A vaccine: 25 µg in 0.25 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
3. RTS,S/AS02A vaccine: 50 µg in 0.5 mL adjuvant; 3 doses at 0, 1, and 3 month intervals
4. Rabies human diploid cell vaccine (Merieux HDCV): single‐dose vial with diluent; 3 doses at 0, 1, and 3 month intervals plus dose 4 given after trial completion; (vaccine groups were also offered 3 doses of rabies vaccine after trial completion) Increasing doses of vaccine were given in a dose‐escalating fashion, ie dose groups were staggered at 10 day intervals |
|
| Outcomes | 1. Injection site pain 2. Swelling > 20 mm 3. Fever 4. Drowsiness 5. Loss of appetite 6. Irritability/fussiness 7. Haemoglobin, haematocrit, white blood cell count, and platelets on days 14, 60, and 104 8. Creatinine and alanine aminotransferase (ALT) on days 14, 60, and 104 9. Antibody to circumsporozoite protein repeat epitopes 10. Circulating hepatitis B surface antigen (HBsAG) antigen 11. Anti‐HbSAg antibodies | |
| Notes | Location: village of Dampha Kunda, near Basse, Upper River Division, The Gambia; highly seasonal malaria with peak in October to November Method of surveillance: home visits daily for 3 days or until symptoms resolved; case reporting on standardized forms by study nurse in village or at clinic in Basse This trial reported in same publication as Bojang 2005b |
|
Brown 1994.
| Methods | Randomized controlled trial Generation of allocation sequence: not stated Allocation concealment: coded vials of similar appearance for vaccine and control; prepared externally by the Swiss Serum and Vaccine Institute and provided to the investigators Blinding: double blind Inclusion of all randomized participants: 84% completed the study Length of follow up: 4 months after last dose |
|
| Participants | Number: 199 male Thai soldiers; malaria‐naive (44) or malaria‐experienced (155) Inclusion criteria: age 18 to 45 years Exclusion criteria: significant cardiac, hepatic, renal, or immunological disease; recent surgery; human immunodeficiency virus (HIV) antibody positivity; use of immunosuppressive drugs; anaemia (haemoglobin < 10 g/dL); diabetes; history of significant allergy |
|
| Interventions | 1. R32Tox‐A vaccine: 3 doses (320 µg per 0.4 mL dose, adsorbed onto aluminium hydroxide) at 8 and 16 weeks 2. Tetanus/diphtheria toxoids: 10 and 1 Lf units, respectively, in first dose and phosphate buffered saline in subsequent doses | |
| Outcomes | 1. Malaria cases (case definition: positive slide) 2. Time to malaria diagnosis 3. Polymerase chain reaction (PCR)‐detected CS sequences from cases 4. Anti‐R32LR IgG and IgM levels 5. Anti‐toxin A antibody 6. Adverse events | |
| Notes | Location: Ubon Ratchatani Province on Thai‐Cambodian border Participants were vaccinated in a non‐endemic area and then deployed in camps in endemic areas Method of surveillance: bi‐weekly active and passive case detection |
|
Genton 2005.
| Methods | Randomized controlled trial Generation of allocation sequence: by computer, in blocks of 8 (5 vaccine and 3 placebo), by independent statistician Allocation concealment: syringes prepared and labelled with randomization number by independent pharmacist Blinding: investigators, participants, and outcome assessors blinded Inclusion of all randomized participants: 14/16 (87.5%) completed follow up Length of follow up: 21 days after challenge |
|
| Participants | Number: 16 adults Inclusion criteria: age 18 to 45 years; either gender; live near Lausanne, Switzerland; scored ≥ 10/12 correct on test of understanding Exclusion criteria: history of malaria; possible exposure to malaria within the previous 6 months; positive serology for Plasmodium falciparum in indirect fluorescent antibody test; history of severe reactions or allergy to mosquito bites, artemether‐lumefantrine (Riamet), or vaccines; pregnancy or lactation; confirmed or suspected immunodeficient condition; chronic or active neurological, gastrointestinal, cardiovascular, or respiratory disease; haemoglobinopathies; history of > 2 hospitalizations for invasive bacterial infections; requirement of any chronic medication; suspected or known current alcohol or illegal drug abuse (excluding cannabis); any other significant finding which, in the opinion of the investigator, would significantly increase the risk of having an adverse outcome from participating in this protocol or of dropping out of the study; body mass index < 18 kg/m2 or > 32 kg/m2; evidence of past or present psychiatric condition; seropositivity for human immunodeficiency virus (HIV), hepatitis C or B (other than antibody to hepatitis B surface antigen (HBsAg)); 10‐year risk of coronary heart disease > 10%; clinically significant deviation from normal range in biochemistry or haematology blood tests or in urinalysis |
|
| Interventions | 1. Pf CS102: 2 doses (30 µg each dose) in Montanide ISA 720 adjuvant as 0.5 mL intramuscular injection on days 0 and 60 2. Adjuvant only, on same schedule | |
| Outcomes | 1. Time between experimental challenge and detection of blood‐stage parasites in thick blood film 2. Time between experimental challenge and polymerase chain reaction (PCR) detection of blood stage parasites 3. Adverse events 4. Humoral and cell‐mediated immune responses before and after sporozoite challenge | |
| Notes | Location: Lausanne, Switzerland Challenge by bite of 5 sporozoite‐infected mosquitoes in Nijmegen, the Netherlands, 2 weeks after dose 2 Methods of surveillance: for adverse events, used diary card for self report of symptoms up to day 4, and solicited and unsolicited events assessed on days 4 and 14 after each dose; for efficacy, participants were seen once per day from day 3 to 5 after challenge, twice per day from day 6 to 15, and once per day from day 15 to 21 |
|
Guiguemde 1990.
| Methods | Randomized controlled trial Generation of allocation sequence: stated to be randomized, but method not clear Allocation concealment: unclear Blinding: double blind Inclusion of all randomized participants: 109/123 (88.6%) infants completed the study Length of follow up: 5 months |
|
| Participants | Number: 123 infants Inclusion criteria: age 3 to 5 months; weight > 3 kg; good general health; parents' consent Exclusion criteria: fever ≥ 38 °C; positive blood slide for Plasmodium falciparum |
|
| Interventions | 1. (NANP)3‐tetanus toxoid vaccine: 3 doses (subcutaneous, 100 µg) 2. Tetanus toxoid (TT) and NANP)3‐TT vaccine: 2 doses TT and 1 dose vaccine 3. Tetanus toxoid (TT): 3 doses, 1 month apart (Expanded Programme of Immunization (EPI) schedule) | |
| Outcomes | 1. Infection with malaria 2. Cases of malaria (case definition: rectal temperature ≥ 38 °C with P. falciparum density ≥ 10,000/µL and no other aetiology for the fever) 3. Anaemia (packed cell volume) 4. Proportion with seroconversion to NANP at day 75 5. Proportion with "efficacious seroconversion" (4‐fold elevation in titre) at day 75 6. Absence of parasites at end of immunization 7. Adverse events | |
| Notes | Location: Vallee du Ko, north of Bobo‐Dioulasso, Burkina Faso; area of permanent malaria transmission with maxima in July and November | |
Kester 2001.
| Methods | Randomized controlled trial Generation of allocation sequence: random (method not stated) for included trial subgroups, balanced for sex and antibodies to hepatitis B surface antigen (HBsAg) Allocation concealment: not used Blinding: included subgroups were double blind Inclusion of all randomized participants: 46 participants were immunized; 24 of these were subsequently challenged Length of follow up: 60 days after last dose |
|
| Participants | Number: 46 malaria‐naive adults Inclusion criteria: age 18 to 45 years; written informed consent Exclusion criteria: splenectomy; any cardiovascular, hepatic, or renal abnormalities; allergy to any antimalarial drugs; immunodeficiency; pregnancy; conditions that would increase risk of adverse outcome from malaria |
|
| Interventions | 1. RTS,S/SBAS2 vaccine: 3 doses in groups; C = 50 µg vaccine; D = 25 µg vaccine; E = 10 µg vaccine
2. Engerix hepatitis B vaccine (group F) Doses at 0, 1, and 9 months |
|
| Outcomes | 1. Incidence of malaria 2. Time to malaria infection 3. Adverse events | |
| Notes | Location: USA Artificial challenge with sporozoite infected mosquitoes, 2 to 4 weeks after last vaccine dose |
|
Moorthy 2004b.
| Methods | Randomized controlled trial Generation of allocation sequence: generated by Data Safety and Monitoring Board (assume by computer); block randomization to avoid imbalances within villages and overall Allocation concealment: opaque sealed envelopes Blinding: investigators, participants, and assessors blinded (including separate assessors for reactogenicity and outcomes) Inclusion of all randomized participants: 320/372 (86.0%) received 3 doses, 296/372 (79.6%) completed follow up (92.5% of those receiving 3 doses) Length of follow up: 11 weeks after dose 3 |
|
| Participants | Number: 372 males Inclusion criteria: age 15 to 45 years; living in study villages Exclusion criteria: any chronic illness detected by clinical evaluation; alanine aminotransferase (ALT) > 42 international units/L; creatinine > 130 µmol/L; packed cell volume < 30%; positive antibody ELISA to human immunodeficiency virus (HIV) 1 or HIV2; simultaneous participation in another clinical trial; blood transfusion in month prior to vaccination; previous experimental malaria vaccination; administration of another vaccine within 2 weeks of vaccination; previous rabies vaccination; allergy to any previous vaccine or to sulfadoxine‐pyrimethamine; history of splenectomy; any treatment with immunosuppressive drugs |
|
| Interventions | 1. DNA ME‐TRAP vaccine: 2 mg on days 0 and 21 (2 intramuscular injections each time, 1 into each deltoid muscle) and MVA ME‐TRAP 1.5 x 10^8 plaque forming units on day 42 (4 intradermal injections, 2 in each deltoid)
2. Rabies vaccine (Chiron Behring): 3 doses on same schedule and routes of administration Sulfadoxine‐pyrimethamine given 2 weeks before dose 3 (4 weeks before beginning of surveillance) |
|
| Outcomes | 1. Time to first infection with Plasmodium falciparum 2. Clinical malaria (case definition: asexual P. falciparum at any level plus temperature ≥ 37.5 °C plus any of headache, myalgia, arthralgia, malaise, nausea, dizziness, or abdominal pain) 3. P. falciparum parasitaemia 4. Packed cell volume 5. Adverse events | |
| Notes | Location: North Bank Division, The Gambia; 13 villages near alluvial flood plain Malaria incidence in adult men was 72% over 11 weeks of high transmission season in a low‐transmission year Entomological inoculation rate: estimated at 10 to 20 per year Method of surveillance: twice weekly home visits and minimum weekly blood slides; passive case detection by study nurses residing in study villages; observation for adverse events for 1 h after vaccination and at home visits on days 1, 2, and 7 |
|
Sherwood 1996.
| Methods | Randomized controlled trial Generation of allocation sequence: participants were in self‐selected pairs, assignment in each pair was random (method not stated) Allocation concealment: unclear Blinding: double blind Inclusion of all randomized participants: 69/76 (90.8%) completed the study Length of follow up: 6 months after last vaccination |
|
| Participants | Number: 76 males in 38 pairs sleeping adjacently in houses in 5 villages Inclusion criteria: age 18 to 30 years; willing to reside in study area for 12 months and use no antimalarial prophylaxis or bed net during study; human immunodeficiency virus (HIV) negative Exclusion criteria: evidence of cardiac, pulmonary, renal, or immunologic disease; antibody to HIV |
|
| Interventions | 1. Recombinant R32LRToxA vaccine ((NANP)15‐(NVDP)2‐LR covalently linked to Pseudomonas aeruginosa toxin A): 3 doses at 1, 8, and 24 weeks; each dose was 175 µg peptide and 225 µg toxin A
2. Recombinant hepatitis B vaccine (Engerix B) In both groups: parasitaemia cleared with quinine sulfate (650 mg 3 times per day for 3 days) and doxycycline (100 mg 2 times per day for 7 days) before each vaccination, and after last vaccination quinine sulfate (as above) and doxycycline (100 mg 2 times per day for 28 days) to eliminate liver stages |
|
| Outcomes | 1. Number of symptomatic Plasmodium falciparum cases after 1, 2, or 3 doses (case definition: positive blood slide together with fever, chills, sweats, headache, cough, or diarrhoea) 2. Number of fever cases 3. Number of days until P. falciparum positive blood slide 4. Density of P. falciparum 5. Prevalence of P. falciparum, P. vivax, and P. malariae 6. Levels of anti‐R32LR antibody by ELISA 7. Levels of antisporozoite antibody by indirect fluorescent antibody 8. Lymphocyte proliferation to R32 LR 9. Adverse events | |
| Notes | Location: near Saradidi, Western Kenya; malaria incidence 90% over 4 months Method of surveillance: scheduled blood slides taken weekly; slides made at daily home visits if symptoms reported |
|
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bejon 2005 | Nonrandomized review and methodology study |
| Doherty 1999 | Nonrandomized study |
| Edelman 2002 | Nonrandomized study |
| Epstein 2004 | Nonrandomized study |
| Fries 1992 | Nonrandomized study |
| Gordon 1990 | Nonrandomized study |
| Gordon 1995 | Nonrandomized study |
| Heppner 1996 | Nonrandomized study |
| Hoffman 1994 | 2 groups with very small numbers of participants, and only 1 group randomized |
| Hoffman 2002 | Nonrandomized study |
| Le 1999 | Nonrandomized open label dose‐finding safety and immunogenicity study |
| McConkey 2003 | Nonrandomized immunogenicity study |
| Moorthy 2003 | Nonrandomized safety and immunogenicity study; no placebo group |
| Moorthy 2004c | Quasi‐randomized safety and immunogenicity study; no placebo group |
| Nardin 2000 | Nonrandomized immunogenicity study |
| Nardin 2001 | Nonrandomized immunogenicity study |
| Roggero 1999 | Nonrandomized study |
| Stoute 1998 | Participants were randomized between different doses of the vaccine, but not randomized between vaccine and control groups |
| Walther 2005 | Nonrandomized study |
Contributions of authors
Patricia Graves wrote the protocol, extracted data, and drafted the review. Hellen Gelband extracted data and co‐wrote the review.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
External sources
Department for International Development, UK.
Declarations of interest
None known.
Unchanged
References
References to studies included in this review
Alonso 2005a {published data only}
- Alonso P, Sacarlal J, Aponte JJ, Leach A, Macete E, Milman J, et al. Efficacy of the RTS,S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: randomised controlled trial. Lancet 2004;364(9443):1411‐20. [DOI] [PubMed] [Google Scholar]
- Alonso PL, Sacarlal J, Aponte JJ, Leach A, Macete E, Aide P, et al. Duration of protection with RTS,S ASO2A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single‐blind extended follow‐up of a randomised controlled trial. Lancet 2005;366(9502):2012‐8. [DOI] [PubMed] [Google Scholar]
Alonso 2005ab {published data only}
- See Alonso 2005a, 2005b.
Alonso 2005b {published data only}
- Alonso P, Sacarlal J, Aponte JJ, Leach A, Macete E, Milman J, et al. Efficacy of the RTS,S/AS02A vaccine against Plasmodium falciparum infection and diseases in young African children: randomised controlled trial. Lancet 2004;364(9443):1411‐20. [DOI] [PubMed] [Google Scholar]
- Alonso PL, Sacarlal J, Aponte JJ, Leach A, Macete E, Aide P, et al. Duration of protection with RTS,S ASO2A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single‐blind extended follow‐up of a randomised controlled trial. Lancet 2005;366(9502):2012‐8. [DOI] [PubMed] [Google Scholar]
Bojang 2001 {published data only}
- Alloueche A, Milligan P, Conway DJ, Pinder M, Bojang K, Doherty T, et al. Protective efficacy of the RTS,S/ASO2 Plasmodium falciparum malaria vaccine is not strain specific. American Journal of Tropical Medicine and Hygiene 2003;68(1):97‐101. [PubMed] [Google Scholar]
- Bojang KA, Milligan PJ, Pinder M, Vigneron L, Alloueche A, Kester KE, et al. Efficacy of RTS,S/AS02 malaria vaccine against Plasmodium falciparum infection in semi‐immune adult men in The Gambia: a randomised trial. Lancet 2001;358(9297):1927‐34. [DOI] [PubMed] [Google Scholar]
Bojang 2005a {published data only}
- Bojang KA, Olodude F, Pinder M, Ofori‐Anyinam O, Vigneron L, Fitzpatrick S, et al. Safety and immunogenicity of RTS,S/ASO2A candidate malaria vaccine in Gambian children. Vaccine 2005;23(32):4148‐57. [DOI] [PubMed] [Google Scholar]
Bojang 2005b {published data only}
- Bojang KA, Olodude F, Pinder M, Ofori‐Anyinam O, Vigneron L, Fitspatrick S, et al. Safety and immunogenicity of RTS,S/ASO2A candidate malaria vaccine in Gambian children. Vaccine 2005;23(32):4148‐57. [DOI] [PubMed] [Google Scholar]
Brown 1994 {published data only}
- Brown AE, Singharaj P, Webster HK, Pipithkul J, Gordon DM, Boslego JW, et al. Safety, immunogenicity and limited efficacy study of a recombinant Plasmodium falciparum circumsporozoite vaccine in Thai soldiers. Vaccine 1994;12(2):102‐7. [DOI] [PubMed] [Google Scholar]
Genton 2005 {published and unpublished data}
- Genton B, D'Acremont V, Lurati F, Verhage D, Hermsen R, Audran R, et al. Randomized, double‐blind placebo‐controlled Phase IIa trial to test efficacy of the malaria vaccine PfCS102 to protect non‐immune volunteers against falciparum challenge. Acta Tropica 2005;Suppl 95:84. [Google Scholar]
Guiguemde 1990 {published data only}
- Guiguemde TR, Sturchler D, Ouedraogo JB, Drabo M, Etlinger H, Douchet C, et al. Vaccination against malaria: initial trial with an ant‐sporozoite vaccine, (NANP)3‐TT (RO 40‐2361) in Africa (Bobo‐Dioulasso, Burkina Faso) [Vaccination contre le paludisme: premier essai avec un vaccin antisporozoite, le (NANP)3‐TT (RO40‐2361) en Afrique (Bobo‐Dioulasso, Burkina Faso)]. Bulletin de la Societe de Pathologie Exotique 1990;83(2):217‐27. [PubMed] [Google Scholar]
Kester 2001 {published data only}
- Kester KE, McKinney DA, Tornieporth N, Ockenhouse C, Heppner DG, Hall T, et al. Efficacy of recombinant circumsporozoite protein vaccine regimens against experimental Plasmodium falciparum malaria. Journal of Infectious Diseases 2001;183(4):640‐7. [DOI] [PubMed] [Google Scholar]
Moorthy 2004b {published data only}
- Moorthy VS, Imoukhuede EB, Milligan P, Bojang K, Keating S, Kaye P, et al. A randomised, double‐blind controlled vaccine efficacy trial of DNA/MVA ME‐TRAP against malaria infection in Gambian adults. PLOS Medicine 2004;1(2):128‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sherwood 1996 {published data only}
- Sherwood JA, Copeland RS, Taylor KA, Abok K, Oloo AJ, Were JB, et al. Plasmodium falciparum circumsporozoite vaccine immunogenicity and efficacy trial with natural challenge quantitation in an area of endemic human malaria in Kenya. Vaccine 1996;14(8):817‐27. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bejon 2005 {published data only}
- Bejon P, Andrews L, Andersen RF, Dunachie S, Webster D, Walther M, et al. Calculation of liver‐to‐blood inocula, parasite growth rates, and preerythrocytic vaccine efficacy, from serial polymerase chain reaction studies of volunteers challenged with malaria sporozoites. Journal of Infectious Diseases 2005;191(4):619‐26. [DOI] [PubMed] [Google Scholar]
Doherty 1999 {published data only}
- Doherty JF, Pinder M, Tornieporth N, Carton C, Vigneron L, Milligan P, et al. A phase I safety and immunogenicity trial with the candidate malaria vaccine RTS,S/SBAS2 in semi‐immune adults in the Gambia. American Journal of Tropical Medicine and Hygiene 1999;61(6):865‐8. [DOI] [PubMed] [Google Scholar]
Edelman 2002 {published data only}
- Edelman R, Wasserman SS, Kublin JG, Bodison SA, Nardin EH, Oliveira GA, et al. Immediate‐type hypersensitivity and other clinical reactions in volunteers immunized with a synthetic multi‐antigen peptide vaccine (PfCS‐MAP1NYU) against Plasmodium falciparum sporozoites. Vaccine 2002;21(3‐4):269‐80. [DOI] [PubMed] [Google Scholar]
Epstein 2004 {published data only}
- Epstein JE, Charoenvit Y, Kester K, Wang R, Newcomer R, Fitzpatrick S, et al. Safety, tolerability and antibody responses in humans after sequential immunization with a PfCSP DNA vaccine followed by the recombinant protein vaccine RTS,S/ASO2A. Vaccine 2004;22(13‐4):1592‐603. [DOI] [PubMed] [Google Scholar]
Fries 1992 {published data only}
- Fries LF, Gordon DM, Schneider I, Beier J, Long GW, Gross M, et al. Safety, immunogenicity and efficacy of a Plasmodium falciparum vaccine comprising a circumsporozoite repeat region peptide conjugated to Pseudomonas aeruginosa toxin A. Infection and Immunity 1992;60(5):1834‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gordon 1990 {published data only}
- Gordon DM, Cosgriff TM, Schneider I, Wasserman GF, Majarian WR, Hollingdale MR, et al. Safety and immunogenicity of a Plasmodium vivax sporozoite vaccine. American Journal of Tropical Medicine and Hygiene 1990;42(6):527‐31. [DOI] [PubMed] [Google Scholar]
Gordon 1995 {published data only}
- Gordon DM, McGovern TW, Krzych U, Cohen JC, Schneider I, LaChance R, et al. Safety, immunogenicity and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein‐hepatitis B surface antigen subunit vaccine. Journal of Infectious Diseases 1995;171(6):1576‐85. [DOI] [PubMed] [Google Scholar]
Heppner 1996 {published data only}
- Heppner DG, Gordon DM, Gross M, Wellde B, Leitner W, Krzych U, et al. Safety, immunogenicity and efficacy of Plasmodium falciparum repeatless circumsporozoite protein vaccine encapsulated in liposomes. Journal of Infectious Diseases 1996;174(2):361‐6. [DOI] [PubMed] [Google Scholar]
Hoffman 1994 {published data only}
- Hoffman SL, Edelman R, Bryan JP, Schneider I, Davis J, Sedegah M, et al. Safety, immunogenicity, and efficacy of a malaria sporozoite vaccine administered with monophosphoryl lipid A, cell wall skeleton of mycobacteria, and squalene as adjuvant. American Journal of Tropical Medicine and Hygiene 1994;51(5):603‐12. [DOI] [PubMed] [Google Scholar]
Hoffman 2002 {published data only}
- Hoffman SL, Goh ML, Luke TC, Schneider I, Le TP, Doolan DL, et al. Protection of humans against malaria by immunization with radiation‐attenuated Plasmodium falciparum sporozoites. Journal of Infectious Diseases 2002;185(8):1155‐64. [DOI] [PubMed] [Google Scholar]
Le 1999 {published data only}
- Le TP, Coonan KM, Hedstrom RC, Charoenvit Y, Sedegah M, Epstein JE, et al. Safety, tolerability and humoral immune responses after intramuscular administration of a malaria DNA vaccine to healthy adult volunteers. Vaccine 2000;18(18):1893‐901. [DOI] [PubMed] [Google Scholar]
McConkey 2003 {published data only}
- McConkey SJ, Reece WHH, Moorthy VS, Webster D, Dunachie S, Butcher G, et al. Enhanced T‐cell immunogenicity of plasmid DNA vaccines boosted by recombinant modified vaccinia virus Ankara in humans. Nature Medicine 2003;9(6):729‐35. [DOI] [PubMed] [Google Scholar]
Moorthy 2003 {published data only}
- Moorthy VS, Pinder M, Reece WH, Watkins K, Atabani S, et al. Safety and immunogenicity of DNA modified vaccinia virus Ankara malaria vaccination in African adults. Journal of Infectious Diseases 2003;188(8):1239‐44. [DOI] [PubMed] [Google Scholar]
Moorthy 2004c {published data only}
- Moorthy VS, Imoukhuede EB, Keating S, Pinder M, Webster D, Skinner MA, et al. Phase 1 evaluation of 3 highly immunogenic prime‐boost regimens, including a 12‐month reboosting vaccination, for malaria vaccination in Gambian men. Journal of Infectious Diseases 2004;189(12):2213‐9. [DOI] [PubMed] [Google Scholar]
Nardin 2000 {published data only}
- Nardin EH, Oliveira GA, Calvo‐Calle JM, Castro ZR, Nussenzweig RS, Schmeckpeper B, et al. Synthetic malaria peptide vaccine elicits high levels of antibodies in vaccinees of defined HLA genotypes. Journal of Infectious Diseases 2000;182(5):1486‐96. [DOI] [PubMed] [Google Scholar]
Nardin 2001 {published data only}
- Nardin EH, Calvo‐Calle JM, Oliveira GA, Nussenzweig RS, Schneider M, Tiercy JM, et al. A totally synthetic polyoxime malaria vaccine containing Plasmodium falciparum B cell and universal T cell epitopes elicits immune responses in volunteers of diverse HLA types. Journal of Immunology 2001;166(1):481‐9. [DOI] [PubMed] [Google Scholar]
Roggero 1999 {published data only}
- Roggero MA, Weilenmann C, Bonelo A, Audran R, Renggli J, Spertini F, et al. Plasmodium falciparium CS C‐terminal fragment: preclinical evaluation and phase I clinical studies. Parassitologia 1999;41(1‐3):421‐4. [PubMed] [Google Scholar]
Stoute 1998 {published data only}
- Nussenzweig R, Zavala F. A malaria vaccine based on a sporozoite antigen. New England Journal of Medicine 1997;336(2):128‐30. [DOI] [PubMed] [Google Scholar]
- Stoute JA, Kester KE, Krzych U, Wellde BT, Hall T, White K, et al. Long‐term efficacy and immune response following immunization with the RTS,S malaria vaccine. Journal of Infectious Diseases 1998;178(4):1139‐44. [DOI] [PubMed] [Google Scholar]
- Stoute JA, Slaoui M, Heppner DG, Momin P, Kester KE, Desmons P, et al. A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS,S malaria vaccine evaluation group. New England Journal of Medicine 1997;336(2):86‐91. [DOI] [PubMed] [Google Scholar]
Walther 2005 {published data only}
- Walther M, Dunachie S, Keating S, Vuola JM, Berthoud T, Schmidt A, et al. Safety, immunogenicity and efficacy of a pre‐erythrocytic malaria candidate vaccine, ICC‐1132 formulated in Seppic ISA 720. Vaccine 2005;23(7):857‐864. [DOI] [PubMed] [Google Scholar]
Additional references
Ballou 2004
- Ballou WR, Arrevalo‐Herrera M, Carucci D, Richie TL, Corradin G, Diggs C, et al. Update on the clinical development of candidate malaria vaccines. American Journal of Tropical Medicine and Hygiene 2004;71 Suppl 2:239‐47. [PubMed] [Google Scholar]
Breman 2004
- Breman JG, Alilio MS, Mills A. Conquering the intolerable burden of malaria: what's new, what's needed: a summary. American Journal of Tropical Medicine and Hygiene 2004;71 Suppl 2:1‐15. [PubMed] [Google Scholar]
Graves 2006a
- Graves P, Gelband H. Vaccines for preventing malaria (SPf66). Cochrane Database of Systematic Reviews 2006, Issue 2. [DOI: 10.1002/14651858.CD005966] [DOI] [PMC free article] [PubMed] [Google Scholar]
Graves 2006b
- Graves PM, Gelband H. Vaccines for preventing malaria (blood‐stage). Cochrane Database of Systematic Reviews 2006, Issue 4. [DOI: 10.1002/14651858.CD006199] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hay 2004
- Hay SI, Guerra CA, Tatem AJ, Noor AM, Snow RW. The global distribution and population at risk of malaria: past, present, and future. Lancet Infectious Diseases 2004;4(6):327‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2005
- Higgins J, Green S, editors. Highly sensitive search strategies for identifying reports of randomized controlled trials in MEDLINE. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]; Appendix 5b. www.cochrane.org/resources/handbook/hbook.htm (accessed 1 September 2005).
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moorthy 2004a
- Moorthy VS, Good MF, Hill AV. Malaria vaccine developments. Nature 2004;363(9403):150‐6. [DOI] [PubMed] [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Richie 2002
- Richie TL, Saul A. Progress and challenges for malaria vaccines. Nature 2002;415(6872):694‐701. [DOI] [PubMed] [Google Scholar]
Snow 2004
- Snow RW, Korenromp E, Gouws E. Pediatric mortality in Africa: Plasmodium falciparum as a cause or risk?. American Journal of Tropical Medicine and Hygiene 2004;71 Suppl 2:16‐24. [PubMed] [Google Scholar]
Snow 2005
- Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005;434(7030):214‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2002
- World Health Organization. The world health report : 2002 : Reducing the risks, promoting healthy life. Geneva: World Health Organization, 2002. [Google Scholar]
WHO 2005
- World Health Organization. Initiative for Vaccine Research. Portfolio of candidate malaria vaccines currently in development: March 2005. www.who.int/vaccine_research/documents/en/malaria_table.pdf (accessed 3 May 2006).