

Abstract
Catalytic carbonyl-olefin metathesis reactions have recently been developed as a powerful tool for carbon–carbon bond formation. However, currently available synthetic protocols rely exclusively on aryl ketone substrates while the corresponding aliphatic analogs remain elusive. We herein report the development of Lewis acid-catalyzed carbonyl-olefin ring-closing metathesis reactions for aliphatic ketones. Mechanistic investigations are consistent with a distinct mode of activation relying on the in situ formation of a homobimetallic singly bridged iron(III)-dimer as the postulated active catalytic species. These “superelectrophiles” function as more powerful Lewis acid catalysts that form upon association of individual iron(III)-monomers. While this mode of Lewis acid activation has previously been postulated to exist, it has not yet been applied in a catalytic setting. The insights presented are expected to enable further advancement in Lewis acid catalysis by building upon the activation principle of “superelectrophiles” and to broaden the current scope of catalytic carbonyl-olefin metathesis reactions.
Graphical Abstract

INTRODUCTION
Carbonyl-olefin metathesis reactions are at the forefront of current research as a result of their potential for direct carbon–carbon bond formation between carbonyl and olefin functionalities. Distinct approaches for carbonyl-olefin metathesis have been advanced relying on stepwise oxetane formation and subsequent fragmentation,1 the use of molybdenum alkylidenes as reagents,2 or bicyclic hydrazine catalysts that enable the first catalytic protocol to affect this transformation.3 Additionally, Lewis acid-catalyzed protocols have recently been developed as a viable reaction design for carbonyl-olefin metathesis.4–6 Upon binding to a Lewis acid catalyst, such as FeCl3, the carbonyl functionality is activated to perform a [2+2]-cycloaddition forming an intermediate oxetane, which subsequently undergoes a retro-[2+2]-cycloaddition resulting in the desired metathesis product. While successful protocols for intramolecular and intermolecular Lewis acid-catalyzed carbonyl-olefin metathesis have been reported following this reaction design principle,7,8 all current procedures rely exclusively on aryl carbonyls as substrates while the reaction of aliphatic ketones remains elusive.4–8 We report herein the development of a catalytic carbonyl-olefin ring-closing metathesis of aliphatic ketones that proceeds in yields of up to 94% and tolerates a variety of functional groups. Mechanistic investigations reveal a distinct mode of activation for aliphatic ketones relative to their aromatic analogs in which the formation of a homobimetallic singly bridged iron(III)-dimer as the active catalytic species enables the formation of a significantly enhanced electrophilic moiety.
In our initial efforts to develop an FeCl3-catalyzed carbony-olefin metathesis reaction, we had investigated both aryl ketone 1 and aliphatic ketone 4 in their ability to undergo the desired transformation and observed profound differences in reactivity.5a While phenyl ketone 1 leads to carbonyl-olefin metathesis product 2 in 99% yield relying on 5 mol % FeCl3 as the Lewis acid catalyst (Figure 1), the corresponding methyl ketone 4 failed to undergo the desired transformation under otherwise identical reaction conditions. As part of our mechanistic investigations toward FeCl3-catalyzed carbonyl-olefin metathesis reactions,5c we identified three main challenges associated with aliphatic ketones as substrates. (1) In the carbonyl-olefin metathesis reactions of aryl ketones, catalyst turnover is enabled due to the favored binding of the substrate 8 to FeCl3 by 2.4 kcal/mol in comparison to acetone 6 (Figure 2A). However, aliphatic ketones (7) bind less strongly to the FeCl3 catalyst by ~1 kcal/mol as compared to their aromatic analogs (8) thereby requiring stronger Lewis acids to efficiently activate these less reactive substrates for carbonyl-olefin metathesis (Figure 2A). Additionally, competitive binding of the Lewis acid with the acetone byproduct 3 is more prominent for aliphatic ketones and catalyst inhibition becomes a concern. (2) Our mechanistic investigations identified the aromatic moiety of aryl ketones as a required structural component that plays a crucial role in transition state stabilization5c by redistributing electron density (8, Figure 2B). (3) Recent investigations of Brønsted acid-catalyzed oxygen atom transfer reactions have shown that alternate oxetane fragmentation pathways exist competing with carbonyl-olefin metathesis reactions.9 Specifically, acidic protons in the α-position to the carbonyl functionality can engage in distinct oxetane fragmentation pathways under acid-catalyzed conditions forming the corresponding unsaturated alcohols 10 via elimination (Figure 2C).10 On the basis of the identified challenges, we expected aliphatic ketones to require activation by a Lewis acid far exceeding the strength of FeCl3. Additionally, we switched from β-ketoester 4 bearing an acidic α-proton to methyl ketone 11 incorporating an α-quaternary carbon as a substrate for the evaluation of distinct Lewis acids to avoid competing oxetane fragmentation pathways (Table 1).
Figure 1.

Comparison of aryl ketones and aliphatic ketones in FeCl3-catalyzed carbonyl-olefin metathesis.
Figure 2.

Challenges associated with aliphatic ketones in catalytic carbonyl-olefin metathesis reactions.
Table 1.
Lewis Acid Evaluation in the Carbonyl-Olefin Metathesis of Aliphatic Ketone 11*
 | ||||||
|---|---|---|---|---|---|---|
| entry | Lewis acid | mol% | solvent | time (h) | yield 12 (%) | conv. (%) |
| 1 | AlCl3 | 5 | DCE | 24 | 0 | 0 |
| 2 | TiCl4 | 5 | DCE | 24 | 0 | 36 |
| 3 | GaCI3 | 5 | DCE | 16 | 21 | 82a |
| 4 | BF3·OEt2 | 5 | DCE | 24 | 24 | 51 |
| 5 | SnCl4 | 5 | DCE | 16 | 30 | 70a |
| 6 | EASC | 100 | DCE | 15 min | 30 | 100a |
| 7 | FeCl3 | 5 | DCE | 15 min | 44 | 48 |
| 8 | FeCI3 | 10 | DCE | 3 | 68 | 70 |
| 9 | FeCI3 | 10 | DCE | 24 | 74 | 78 |
| 10 | FeCI3 | 5 | DCM | 24 | 37 | 40 |
| 11 | FeCl3 | 5 | toluene | 24 | 0 | 25 |
| 12 | HCI | 5 | DCE | 24 | 0 | 7 |
| 13 | TfOH | 5 | DCE | 24 | 0 | 3 |
| 14 | H2SO4 | 5 | DCE | 24 | 0 | 93a |
Conditions: All reactions were performed using 0.16 mmol of ketone 11 and Lewis acid in solvent (0.05M) at 23 °C. EASC = ethyl aluminum sesquichloride.
Substrate decomposition was observed
RESULTS AND DISCUSSION
Our initial efforts focused on the evaluation of more powerful Lewis acids compared to FeCl3 upon conversion with aliphatic ketone 11. However, using substoichiometric amounts of the strong Lewis acid, AlCl3, no formation of the desired metathesis product 12 was observed (entry 1, Table 1). Nevertheless, the use of stoichiometric amounts of the strong Lewis acid ethyl aluminum sesquichloride (EASC) resulted in the formation of 12 in 30% yield and with complete consumption of substrate 11 (entry 6, Table 1). Control reactions with weaker Lewis acids, including SnCl4 and GaCl3, also resulted in the formation of the desired cyclopentene 12, albeit in low yields of 30% and 21%, respectively, and increased decomposition of starting material (entries 3 and 5, Table 1). Similarly, BF3·Et2O formed the desired product in 24% while TiCl4 failed to provide cyclopentene 12 under otherwise identical reaction conditions. Based on this range of reactivities observed with Lewis acids varying in strength, we investigated varying amounts of FeCl3 upon conversion with methyl ketone 11. Surprisingly, formation of metathesis product 12 was observed in yields of up to 44% with an abbreviated reaction time of 15 min, in comparison to 16 to 24 h with other Lewis acids (entry 7, Table 1). Increasing catalyst loading to 10 mol % FeCl3 proved beneficial and resulted in improved yields of 68% within 15 min (entry 8, Table 1). Further efforts identified conducting the reaction for 3 h as optimal, resulting in 74% yield of metathesis product 12 and 78% conversion of starting material 11 (entry 9, Table 1). In ensuing attempts to further optimize this transformation, we observed a decreased yield of 37% when the reaction was conducted in dichloromethane as solvent under otherwise identical reaction conditions (entry 10, Table 1). Furthermore, no formation of the desired metathesis product 12 was detected using toluene as the reaction solvent. Both of these results are in stark contrast to carbonyl-olefin metathesis reactions of aryl ketones which tolerate chlorinated hydrocarbon solvents as well as aromatic solvents.5a,c Less than stoichiometric amounts of Brønsted acids including HCl, TfOH, and H2SO4 did not result in the formation of the desired carbonyl-olefin metathesis product 12 (entries 12–14, Table 1). Interestingly, the results obtained relying on FeCl3 as optimal Lewis acid catalyst did not corroborate our preceding theoretical investigations focused on enthalpies for Lewis acid activation that predict the need for a more potent Lewis acid to effectively activate aliphatic ketones (Figure 2).
We next explored the effect of varying alkene substitution in the iron(III)-catalyzed carbonyl-olefin metathesis reaction of aliphatic ketones. While ketone 11a bearing a prenyl fragment resulted in the formation of metathesis product 12 in 74% yield, the corresponding styrenyl analogs 11b-11f initially showed little or no reactivity under identical reaction conditions. Subsequent studies revealed that the lack of reactivity can be attributed to competing product inhibition of the aryl aldehyde byproduct formed (see Supporting Information for details). Therefore, we focused on the evaluation of reagents capable of sequestering the corresponding aldehyde byproducts to avoid catalyst inhibition.5f Ultimately, allyltrimethylsilane was identified as the most promising additive enabling catalytic carbonyl-olefin metathesis of electronically differentiated styrene derivatives in up to 78% yield (Table 2).
Table 2.
Evaluation of Olefin Substitution in the Carbonyl-Olefin Metathesis of Aliphatic Ketones 11a–f
Conditions: All reactions were performed using 0.05 mmol of ketone 11a–f and 0.05 mmol of FeCl3 in dichloroethane as solvent (0.05 M) at 23 °C for 16–24 h.
Addition of 5.0 equiv of allyltrimethylsilane.
The optimized reaction conditions developed for the catalytic carbonyl-olefin metathesis of aliphatic ketones proved efficient for a variety of substrates incorporating distinct substituents and functional groups (Tables 3 and 4). Substrates including electron-deficient aryl residues in the β-position resulted in good to excellent yields of 63–93% of the desired metathesis products (13–17, Table 3). Electron-rich aryl-moieties bearing methyl, isopropyl, tert-butyl or methoxy substituents provided up to 94% yield of the desired products (18−21, Table 3). Additionally, substrates with methyl or chloro substituents in the ortho-position led to diminished yields of 63% and 50%, respectively (25 and 26), while meta-substituted substrates provided good yields ranging from 64 to 76% (22−24, Table 3). Interestingly, a thiophene-containing substrate was well tolerated under the reaction conditions resulting in 54% yield of the desired product 27. In addition to methyl ketones, distinct aliphatic substituents including ethyl and isobutyl carbonyls were tolerated under the optimized reactions conditions (30, 31, and 32, Table 4) although in slightly lower yields of 34−62%, presumably due to the increased steric bulk near the reactive carbonyl. Substrates bearing additional alkene functionalities, such as cinnamyl and geranyl substituents, proved compatible and resulted in the corresponding products 37 and 38 in 60% and 31% yield, respectively. Various substituents in the α-position to the carbonyl were tolerated under the reaction conditions. Cyclopentene 36 bearing two aliphatic moieties was obtained in 56% yield while cyclopropyl derivative 35 was formed in 58% isolated yield. Substrates bearing the β-ketoester moiety (44, 45, and 46, Table 4) yielded lower amounts of the metathesis products or did not undergo metathesis at all. Cyclohexyl carbonyl derivatives were efficient under the optimized reaction conditions leading to the formation of the corresponding bicyclic scaffolds in up to 78% yield (39−43). Interestingly, a cycloheptyl-derived substrate did not result in the formation of the desired metathesis product, but led to the isolation of oxetane 4711 in 52% yield, providing additional support for the reaction mechanism.
Table 3.
Substrate Scope of Aryl Variation*
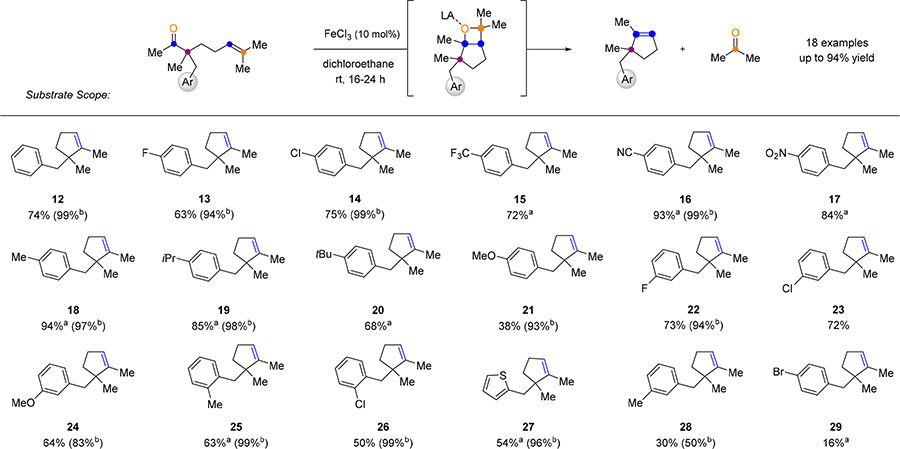
|
Conditions: All reactions were performed using 10 mol % of Lewis acid, FeCl3, in DCE (0.05M) at 23 °C for 16–24 h or a80 °C for 3 h. bYields are based on recovered starting material. c50 mol % FeCl3 in DCE (0.05M) at 0 to 23 °C.
Table 4.
Substrate Scope of Distinct Functional Group Incorporation
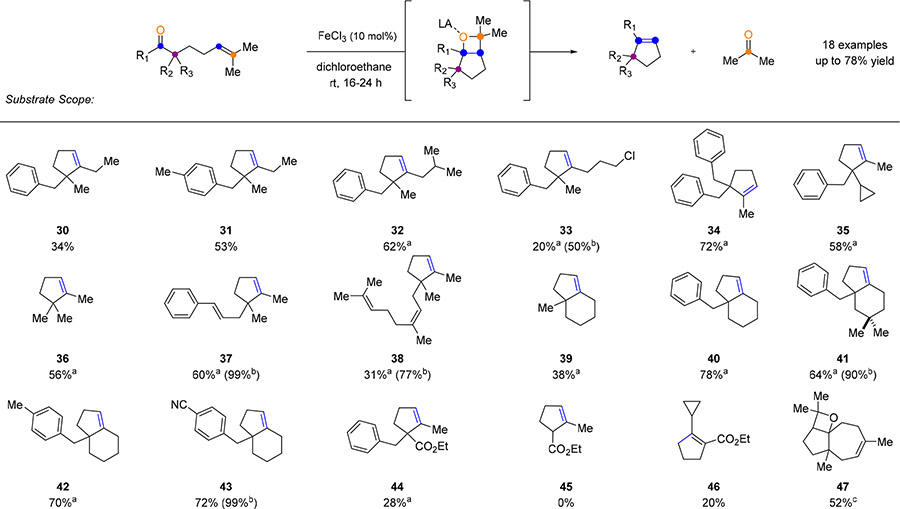
|
Conditions: All reactions were performed using 10 mol % of Lewis acid, FeCl3, in DCE (0.05M) at 23 °C for 16–24 h or a80 °C for 3 h. bYields are based on recovered starting material. c50 mol % FeCl3 in DCE (0.05 M) at 0 to 23 °C.
Mechanistic Investigations.
Over the course of our studies toward optimizing catalytic carbonyl-olefin metathesis reactions of aliphatic ketones, we note three key differences in reactivity compared to aryl ketone substrates. (1) FeCl3-catalyzed carbonyl-olefin metathesis of aliphatic ketones proceeded best in dichloroethane as solvent resulting in optimal yields of 74% (entry 6, Table 5). No product was observed relying on toluene as solvent and diminished yields of 37% under otherwise identical conditions were obtained in dichloromethane (entries 4 and 5, Table 5). In comparison, product formation in high yields ranging from 65% to 99% is observed when the corresponding aryl ketone 48 is reacted with 5 mol % of FeCl3 in these solvents. (2) Higher catalyst loadings of 10 mol % FeCl3 were shown to result in higher yields of the desired carbonyl-olefin metathesis product 12 while the analogous transformation of aryl ketones 48 proceeds in high yields with as little as 1 mol % FeCl3. 3) Additionally, subsequent gas-phase simulations of the reaction path for FeCl3-catalyzed carbonyl-olefin metathesis of aliphatic ketone 11 revealed activation barriers of >25 kcal/mol, which are too high for the reaction to proceed. Comparatively, the corresponding activation barrier for aryl ketones was found to be 14.5 kcal/mol (Table 5).
Table 5.
Evaluation of Solvent and Catalyst Loading for Aryl (48) vs Aliphatic (11) Ketone Substrates
 | ||||
|---|---|---|---|---|
| entry | R | FeCl3 (X mol %) | solvent | yield 49 or 12 (%) |
| 1 | Ph | 5 | tolunce | 65 |
| 2 | Ph | 5 | dichloromethane | 78 |
| 3 | Ph | 5 | dichloromethane | 99 |
| 4 | Me | 10 | tolunce | 0 |
| 5 | Me | 10 | dichloromethane | 37 |
| 6 | Me | 10 | dichloromethane | 74 |
Conditions: All reactions were performed using 0.16 mmol % of ketone 48 or 11 and Lewis acid in solvent (0.05 M) at 23 °C.
Intrigued by the high reactivity of FeCl3 and unique solvent dependence observed in the carbonyl-olefin metathesis of aliphatic ketone 11, we initiated kinetic studies to obtain further insight into the controlling features of this transformation (Figure 3). Mechanistic investigations of the catalytic carbonyl-olefin metathesis reaction of aryl ketone 1 had previously revealed a zero-order dependence on substrate concentration and first-order dependence in FeCl3.5c This indicated that the catalyst resting state has monomeric FeCl3 bound to the substrate in Lewis acid–base complex 50 and undergoes a classic activation mode with a single Lewis acid monomer (Figure 3). Similarly, carbonyl-olefin metathesis of aryl ketone 48 bearing an α-quaternary center was also found to have first-order dependence on FeCl3 concentration and zero order in substrate. Importantly, kinetic evaluation of the analogous aliphatic ketone 11 also displayed zero order with respect to substrate, which is consistent with a catalyst resting state of monomeric FeCl3 bound to substrate (52, Figure 3). However, ketone 11 was shown to proceed with second-order kinetics in FeCl3,12,13 implying that a different mode of Lewis acid activation is operative for aliphatic ketones (Figure 3). Specifically, these kinetic results are consistent with a hypothesis that two equivalents of FeCl3 are involved in the rate-determining step of carbonyl-olefin metathesis of aliphatic ketones while only one equivalent of FeCl3 is involved in the analogous reaction of aryl ketones.
Figure 3.

Kinetic investigations of the catalytic carbonyl-olefin metathesis reaction with two independent methods relying on “initial rates” based on the decay of substrate and the “Normalized time scale method”. (A) Rate order results for aryl ketones. (B) Rate order results for aliphatic ketones. (C) Catalytic resting states following kinetic investigation.
It has previously been postulated that individual Lewis acid monomers 55 could associate to form singly bridged dimers 54 that retain an open coordination site rendering them as stronger Lewis acids than their corresponding monomers (Figure 4A). This strategy was described as a “highly desirable”14 reaction design principle in Lewis acid catalysis as early as the 1960s as an approach to generate stronger Lewis acids by taking advantage of their inherent tendency to associate into “superelectrophiles”. Polarization of singly bridged dimers 54 could induce subsequent ionization into doubly electron-deficient ion pairs 53. Importantly, both singly bridged dimers 54 and ion pairs 53 fall under Olah’s definition of superelectrophiles15 by exhibiting reactivity that substantially exceeds that of their corresponding monomer 55. While the synthetic realization and application of heterobimetallic superelectrophiles that result from the association of Lewis acids comprised of two different metals has led to important developments in organometallic chemistry,16 the related homobimetallic case is still considered uncommon.17 Consequently, the principle of homobimetallic association of Lewis acids has remained unexplored in catalysis. Isolated reports of homobimetallic association of Lewis acids have been postulated to be operative but exclusively in stoichiometric reaction settings. The observation of second-order rate dependence in GaCl3-mediated Friedel–Crafts alkylations18 led Brown and co-workers to suggest an activation mode based on Lewis acid superelectrophiles (59, Figure 4B) but dimers 54 or ion pairs 53 could not be differentiated. Later, Evans suggested carbonyl activation by homobimetallic ion pairs 60 in stoichiometric Et2AlCl-mediated Diels–Alder reactions to account for unique reactivity observed with Al-based Lewis acids (Figure 4B).19–21
Figure 4.

(A) Lewis acid activation modes (M = metal) and postulated activation mode: “superelectrophiles”. (B) Homobimetallic association of Lewis acids in Friedel–Crafts alkylation and Diels–Alder reactions.
On the basis of this literature precedent and our results obtained in the kinetic investigations, we considered two distinct activation modes for aliphatic ketones in catalytic carbonyl-olefin metathesis reactions relying on singly bridged FeCl3 dimer (61) or ion pair (62) (Figure 5). In the neutral pathway, the first equivalent of FeCl3 binds aliphatic ketone 11 to form the catalyst resting state 52. Coordination of a second equivalent of FeCl3 generates singly bridged homobimetallic dimer 61 (Figure 5) as a Lewis acid superelectrophile. The resulting increase in substrate polarization leads to efficient activation of the substrate for carbonyl-olefin metathesis. Alternatively, homobimetallic dimer 61 can undergo solvent-assisted polarization to result in ion pair 62, which similarly represents a Lewis acid super-electrophile capable of activating the substrate for carbonyl-olefin metathesis.
Figure 5.

Proposed superelectrophiles for aliphatic ketones in catalytic carbonyl-olefin metathesis reactions.
In order to gain initial support for the formation of Lewis acid superelectrophiles and differentiate between neutral singly bridged dimers or ion pairs as active catalytic species, we conducted infrared spectroscopic measurements that relate Lewis acid-carbonyl activation to Lewis acid strength based on the change in absorption frequency observed.22,23 A new signal with an absorption frequency of 1642 cm−1 is observed when ketone 63 is treated with equimolar amounts of FeCl3, consistent with single carbonyl activation upon coordination of FeCl3 to form complex 64 (Figure 6A). Addition of a second equivalent of FeCl3 resulted in a third signal with a lower absorption frequency at 1615 cm−1, suggesting increased carbonyl activation of 63 by a stronger Lewis acid. Both the singly bridged FeCl3 dimer (61) and the corresponding ion pair (62) are expected to lower carbonyl absorption frequencies upon coordination and are consistent with calculations of the expected differences in shifts of absorption frequencies that match the experimentally observed shifts (ΔC=Omeasured vs ΔC=Ocalc.: 27 vs 20.0 cm−1) (Figure 6B).
Figure 6.
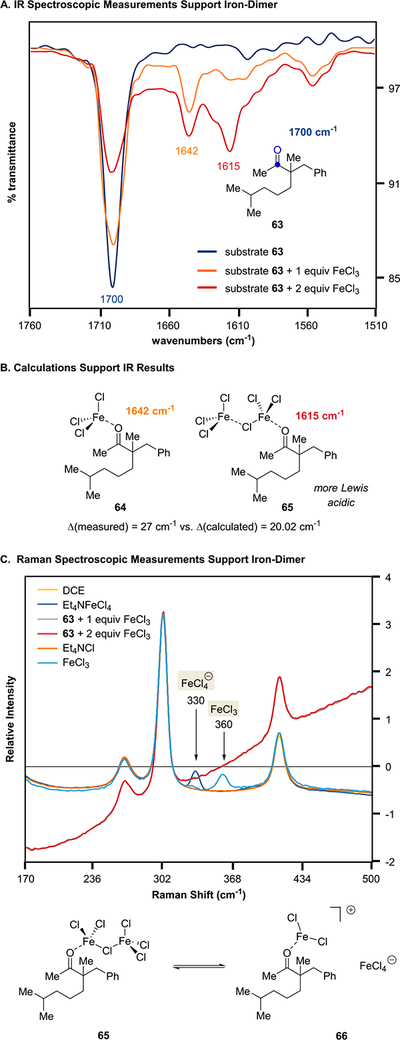
(A) Infrared spectroscopic measurements of 63. (B) Calculations for IR data. (C) Raman spectroscopic data of 63.
Experiments relying on Raman spectroscopy show no formation of FeCl4− (330 cm−1)25,26 upon addition of FeCl3 to aliphatic ketone 63 in dichloroethane (Figure 6C), indicating that the ion pair 66 is not present. Electron paramagnetic resonance (EPR) experiments were performed to gain further support for the catalyst resting state in this transformation and distinguish between singly bridged dimers or ion pairs as active catalytic species. Increasing amounts of aliphatic ketone 63 were added to a solution of FeCl3 in dichloroethane while the concentration of FeCl3 remained the same (Figure 7). High spin EPR spectra with g = 4.29 were obtained for all ratios of 63 bound to FeCl3 as expected for iron(III) species. The addition of excess aliphatic ketone 63 leads to an increase in signal strength, consistent with an iron(III)-bound complex 64 as the major species in solution and supports the catalyst resting state of monomeric FeCl3 bound to substrate. In comparison, the singly bridged homo-dimer 65 is expected to be favored with an excess of FeCl3 relative to ketone 63. While the homo-dimer 65 is EPR silent, the corresponding monomeric complex 64 is EPR active in accordance with the observed signal. Importantly, no signal is observed for the FeCl4− anion, which is EPR-active,24 providing additional support for a neutral reaction pathway.
Figure 7.

Electron paramagnetic resonance spectroscopic measurements of 63.
These combined mechanistic investigations and substrate evaluation in the carbonyl-olefin metathesis reaction of aliphatic ketones support a carbonyl-activation mode based on iron(III) homo-dimers as superelectrophiles. To reinforce this hypothesis, we examined the possibility of generating even more reactive dimers functioning as Lewis acidic superelectrophiles which could lead to increased yields of the desired metathesis products. The composition of GaCl3 and GaBr3 mixtures in nonpolar solvents has previously been studied by Černý and co-workers.27 Specifically, halogen exchange between both Lewis acids was shown to occur and the predominant species at equimolar composition are Ga2Cl3Br3 and Ga2Cl4Br2. We postulated that two distinct iron-derived Lewis acids bearing substituents differing in electronegativity, specifically FeCl3 and FeBr3, would also undergo halogen exchange in solution to form a stronger Lewis acidic superelectrophile in situ, which could result in increased yields of the desired metathesis product (Figure 8). When 10 mol % of a 1:1 ratio of FeCl3 and FeBr3 was used with ketone 11, the desired carbonyl-olefin metathesis product was obtained in a slightly higher yield when compared to the FeCl3 homo-dimer (82% yield vs 74% yield). This result is consistent with the hypothesis that stronger superelectrophiles can be generated from dissimilar iron-based Lewis acids and result in increased substrate activation (see Supporting Information for additional experimental and spectroscopic details on this experiment).
Figure 8.

(A) Proposed homobimetallic superelectrophile utilizing different Lewis acids. (B) Generation of stronger superelectrophiles with MCl3 and MBr3 to increase reactivity.
As a next step, quantum chemical simulations were performed to probe the mechanism of catalytic carbonyl-olefin metathesis reaction of aliphatic ketones.28,29 These simulations were performed using density functional theory at the B97-D level including implicit solvation, see Supporting Information for more details. Figure 9 shows the most favorable reaction pathway found for the monomeric FeCl3–substrate complex (shown in gray) and the corresponding singly bridged homo-dimer (shown in blue). Both pathways consist of concerted, asynchronous ring-closing (B) and ring-opening (D) steps involving an oxetane intermediate C, which were operative in the carbonyl-olefin metathesis of aryl ketones. The overall barriers, however, show that the initial step of the reaction is enthalpically preferred by 3.5 kcal/mol when two equivalents of FeCl3 are bound to the substrate in a homo-dimeric manner. The most favored computed metathesis pathway is the Lewis acid activation of a Lewis acid, which ultimately leads to a superelectrophile functioning as a stronger catalyst consistent with contraction of the Fe–O bond by 0.05 Å in A compared to the monomeric FeCl3-complex (see Supporting Information for details). Likewise, the Fe–Cl distance of the Fe–Cl–Fe bridge is elongated by 0.12 Å, resulting in charge transfer from the first iron to the second iron as this bond is activated. In total, a single FeCl3 molecule is able to withdraw 0.33 electrons from the substrate, but two molecules of FeCl3 extract 0.45 units of charge. This increased charge withdrawal through this singly bridged dimer is needed for aliphatic substrates, but seemingly not for the corresponding aryl analogs due to the ability of aryl ketones to delocalize charge through conjugation. These simulations are therefore consistent with spectroscopic results, which display increased carbonyl activation when multiple units of Lewis acid are available and are also in accordance with the observed second-order rate dependence of FeCl3.
Figure 9.

Quantum chemical investigations. (A) Enthalpic profile and (B) reaction pathway of the computationally proposed reaction mechanism for carbonyl-olefin metathesis of aliphatic ketones using FeCl3.
While we have demonstrated the use of the carbonyl-olefin metathesis reaction on a wide range of aliphatic substrates, the significant changes in structure coupled with the competitive binding scenarios to the Lewis acid make it difficult to separate the individual features of the substrate that impact the reaction outcome. To avoid the complexity of connecting the substrate structure to more than one fundamental process, we hypothesized that analysis of a modular model substrate class could offer more informative insight into the substrate sensitives to a singular reaction event. The diversity of the benzylic-derived substrates (Table 3) offers the requisite changes to both the remote electronic and steric environments (Figure 10) for a training set but also incorporates sufficient overlapping features for analysis. The relative rate values were measured for 12 substrates under uniform reaction conditions and, as expected, do not mirror the trend in the associated isolated yields, suggesting other events contribute to this outcome. Considering the substituents under evaluation, we employed the corresponding benzoic acids on the basis of the Sigman group’s recent effort to develop enhanced parameter sets using these simple surrogates.30 Computation optimizations were performed at the M06–2X/def2-TZVP level of theory wherein NBO charges, IR vibrations, and Sterimol values were collected to probe structural effects.31 These parameters were correlated to ΔΔG‡, calculated using the equation ΔΔG‡ = −RT ln(krel), by using linear regression fitting to quantitatively analyze the substituent effects on reaction rate. A two parameter model was sufficient to describe the structural effects of this subset of substrates affecting the rate of reaction (Figure 10A). Note that a reasonable linear correlation can only be obtained after the removal of the 4-Br substrate (R2 = 0.46, if included). Electron density measured through the NBOC=O charge was found to be the most significant reactivity discriminant, in which increasing the electron donating ability of the substituent on the aromatic ring increased the rate of reaction. Steric effects were of little consequence to rate and were only pronounced when substituents larger than fluorine were introduced at the 3-position, which slow the rate of reaction substantially. Reasonable steric bulk at the 3-position is not taken into account by NBOC=O therefore displaying two outliers (Figure 10B). This data is in agreement with the hypothesis that a stronger Lewis acid initially accelerates the reaction rate to allow for the less reactive aliphatic substrates to undergo metathesis.
Figure 10.

Evaluation of aromatic α-substituent features dictating rate in the reaction of aliphatic ketones. (A) MLR model using benzoic acids as simple probes prioritizes NBOC=O and sterimol L as key contributing factors in the benzylic series. (B) Univariate correlation between NBOC=O and measured ΔΔG‡. Red points represent outliers with large substituents at the 3-position that perform worse than expected based on electron density effects alone.
On the basis of these combined results, we propose separate activation modes for Lewis acid-catalyzed carbonyl-olefin metathesis reactions of aryl and aliphatic ketones. When considering aryl ketones, the catalyst resting state 51, in which monomeric FeCl332,33 is bound to substrate, is sufficiently activated to undergo carbonyl-olefin metathesis. This is consistent with the observed first-order kinetics in FeCl3 and our computational investigations that identified the aryl moiety as an essential structural component for transition state stabilization by delocalizing electron density to facilitate formation of the oxetane intermediate.5c In comparison, aliphatic ketones are devoid of this stabilization and thus the catalyst remains in its resting state 52 until a second equivalent of FeCl3 binds to form singly bridged homo-dimer 61. This homobimetallic association of a second equivalent of FeCl3 generates a stronger Lewis acid that functions as a superelectrophile capable of lowering the energy of the transition state to provide sufficient activation for carbonyl-olefin metathesis (Figure 11). The resulting increase in substrate polarization leads to formation of intermediate oxetane 70, which upon subsequent fragmentation results in the desired metathesis product 12 and acetone byproduct 3.
Figure 11.

(A) Mechanistic hypothesis presenting reactive activation mode for aliphatic ketones requiring second coordination of FeCl3 to undergo metathesis. (B) Substrate activation of aryl ketone substrates by monomeric FeCl3.5c
Studies by Wong and Brown33a have shown that Lewis acids, such as GaCl3, are stabilized in hydrocarbon solvents. Specifically, GaCl3–Cl–R adducts are formed and the stability of these decreases with increased branching of the hydrocarbon solvents. Consequently, FeCl3–DCE adducts are expected to be less stable than FeCl3–DCM adducts which is consistent with DCE being the superior solvent in carbonyl-olefin metathesis reactions of aliphatic ketones. Additionally, DCE has a higher capability to stabilize charge due to its increased dielectric constant compared to DCM, which also aids the transition state stabilization of carbonyl-olefin metathesis reactions of aliphatic ketones (see Supporting Information for details).
CONCLUSION
Lewis acid activation through bimetallic association was postulated 60 years ago as a viable avenue to generate stronger Lewis acid catalysts. While this concept has been advanced in the context of heterobimetallic association, the corresponding homobimetallic association was considered to be “of little or no synthetic consequence”.17 The results presented herein show that the concept of Lewis acid activation by association can be expanded to include homobimetallic interactions as a desirable reactivity mode. We demonstrate the synthetic realization and importance that homobimetallic association has in Lewis acid catalysis by accessing superelectrophiles in situ to give rise to more potent catalytic species. These proposed superelectrophilic singly bridged iron(III) homo-dimers lead to more reactive Lewis acid-complexes that are capable of activating previously unreactive substrates for the catalytic carbonyl-olefin metathesis reactions of aliphatic ketones.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH/National Institute of General Medical Sciences (R01-GM118644 to C.S.S. and R35GM128830 to P.M.Z.), the National Science Foundation (CHE-1763436 to M.S.S), the Alfred P. Sloan Foundation and the David and Lucile Packard Foundation (fellowships to C.S.S.). H.A., C.C.M., and J.R.L. thank the National Science Foundation for predoctoral fellowships (DGE 1256260). Computational resources were provided by the Center for High Performance Computing at the University of Utah and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (ACI-1548562). We are very grateful to Prof. Nathaniel Szymczak, Prof. Adam Matzger, Prof. Nicolai Lehnert, Andrew Hunt, and Corey White (University of Michigan) for help conducting IR, Raman, and EPR spectroscopic measurements as described in this paper and accompanying Supporting Information. Prof. James Devery (Loyola University Chicago) is gratefully acknowledged for helpful suggestions regarding the kinetic measurements described in this paper.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11840.
General methods; detailed experimental studies; computational experiments; 1H and 13C NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).For carbonyl-olefin metathesis reactions proceeding via oxetane photoadducts, see:; (a) Jones G II; Schwartz SB; Marton MTJ Chem. Soc., Chem. Commun 1973, 11, 374–375. [Google Scholar]; (b) Jones G II; Acquadro MA; Carmody MAJ Chem. Soc., Chem. Commun 1975, 0, 206–207. [Google Scholar]; (c) Carless HAJ; Trivedi HSJ Chem. Soc., Chem. Commun 1979, 8, 382–383. [Google Scholar]; (d) D’Auria M; Racioppi R; Viggiani L Photochem. Photobiol. Sci 2010, 9, 1134–1138. [DOI] [PubMed] [Google Scholar]; (e) Pérez-Ruiz R; Gil S; Miranda MA J. Org. Chem 2005, 70, 1376–1381. [DOI] [PubMed] [Google Scholar]; (f) Pérez-Ruiz R; Miranda MA; Alle R; Meerholz K; Griesbeck AG Photochem. Photobiol. Sci 2006, 5, 51–55. [DOI] [PubMed] [Google Scholar]; (g) Valiulin RA; Kutateladze AG Org. Lett 2009, 11, 3886–3889. [DOI] [PubMed] [Google Scholar]; (h) Valiulin RA; Arisco TM; Kutateladze AG J. Org. Chem 2011, 76, 1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Valiulin RA; Arisco TM; Kutateladze AG J. Org. Chem 2013, 78, 2012–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fu GC; Grubbs RH J. Am. Chem. Soc 1993, 115, 3800–3801. [Google Scholar]
- (3).For organocatalytic approaches, see:; (a) Griffith AK; Vanos CM; Lambert TH J. Am. Chem. Soc 2012, 134, 18581–18584. [DOI] [PubMed] [Google Scholar]; (b) Hong X; Liang Y; Griffith AK; Lambert TH; Houk KN Chem. Sci 2014, 5, 471–475. [Google Scholar]
- (4).For Brønsted and Lewis acid mediated carbonyl-olefin metathesis reactions, see:; (a) Schopov I; Jossifov C Makromol. Chem., Rapid Commun 1983, 4, 659–662. [Google Scholar]; (b) Soicke A; Slavov N; Neudörfl J-M; Schmalz H-G Synlett 2011, 2011, 2487–2490. [Google Scholar]; (c) van Schaik H-P; Vijn R-J; Bickelhaupt F Angew. Chem., Int. Ed. Engl 1994, 33, 1611–1612. [Google Scholar]; (d) Jossifov C; Kalinova R; Demonceau A Chim. Oggi 2008, 26, 85–87. [Google Scholar]
- (5).(a) Ludwig JR; Zimmerman PM; Gianino JB; Schindler CS Nature 2016, 533, 374–379. [DOI] [PubMed] [Google Scholar]; (b) McAtee CM; Riehl PS; Schindler CS J. Am. Chem. Soc 2017, 139, 2960–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ludwig JR; Phan S; McAtee CM; Zimmerman PM; Devery JJ III; Schindler CS J. Am. Chem. Soc 2017, 139, 10832–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Groso EJ; Golonka AN; Harding RA; Alexander BW; Sodano TM; Schindler CS ACS Catal. 2018, 8, 2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Albright H; Vonesh HL; Becker MR; Alexander BW; Ludwig JR; Wiscons RA; Schindler CS Org. Lett 2018, 20, 4954–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ma L; Li W; Xi H; Bai X; Ma E; Yan X; Li Z Angew. Chem., Int. Ed 2016, 55, 10410–10413. [DOI] [PubMed] [Google Scholar]; For example, a single literature report of an aluminum-mediated fragmentation of aliphatic oxetanes exists resulting in 30% yield of the corresponding olefin product: Intramolecular and intermolecular Lewis acid catalyzed ene reactions using ketones as enophiles.; Jackson AC; Goldman BE; Snider BB J. Org. Chem 1984, 49, 3988–3994. [Google Scholar]; In comparison, boron trifluoride and Brønsted acid-mediated fragmentation of oxetanes bearing aromatic moieties are more prominent.For an example, see:; Carless HAJ; Trivedi HSJ Chem. Soc., Chem. Commun 1979, 8, 382–383. [Google Scholar]
- (6).For a review on Lewis acid-catalyzed carbonyl-olefin metathesis reactions, see:; Ludwig JR; Schindler CS Synlett 2017, 28, 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For approaches relying on carbocation-based Lewis acids as catalysts in carbonyl-olefin metathesis, see:; (a) Bah J; Franzén J; Naidu VR Eur. J. Org. Chem. 2015, 2015, 1834–1839. [Google Scholar]; (b) Tran UPN; Oss G; Pace DP; Ho J; Nguyen TV Chem. Sci 2018, 9, 5145–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For an approach relying on Brønsted acid-catalyzed carbonyl-olefin metathesis reactions inside a supramolecular host, see:; (a) Catti L; Tiefenbacher K Angew. Chem., Int. Ed 2018, 57, 14589–14592. [DOI] [PubMed] [Google Scholar]; For an iodine-catalyzed approach, see:; (b) Nguyen TV; Tran UPN; Oss G; Breugst M; Detmar E; Liyanto K ChemRxiv. DOI: 10.26434/chemrxiv.7057913. [DOI] [Google Scholar]
- (9).Ludwig JR; Watson RB; Nasrallah DJ; Gianino JB; Zimmerman PM; Wiscons R; Schindler CS Science 2018, 361, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Unpublished results.
- (11).Demole E; Enggist P; Borer MC Helv. Chim. Acta 1971, 54, 1845–1864. [Google Scholar]
- (12).Baxter RD; Sale D; Engle KM; Yu J; Blackmond DG J. Am. Chem. Soc 2012, 134, 4600–4606. [DOI] [PubMed] [Google Scholar]
- (13).Burés J Angew. Chem., Int. Ed 2016, 55, 2028–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Negishi E Chem. - Eur. J 1999, 5, 411–420. [Google Scholar]
- (15).(a) Olah GA Angew. Chem., Int. Ed. Engl 1993, 32, 767–788. [Google Scholar]; (b) Olah GA; Klumpp DA Superelectrophiles and Their Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, 2007. [Google Scholar]; (c) Shambayati S; Crowe WE; Schreiber SL Angew. Chem., Int. Ed. Engl 1990, 29, 256–272. [Google Scholar]
- (16).In comparison, the corresponding heterobimetallic association of two different metal-derived Lewis acids have been widely investigated to induce distinct reactivity, such as ate complexation and transmetalation. For examples, see:; (a) Sugasawa T; Toyoda T; Adachi M; Sasakura KJ Am. Chem. Soc 1978, 100, 4842–4852. [Google Scholar]; (b) Douglas AW; Abramson NL; Houpis IN; Karady S; Molina A; Xavier LC; Yasuda N Tetrahedron Lett. 1994, 35, 6807–6810. [Google Scholar]
- (17).Negishi E Pure Appl. Chem 1981, 53, 2333–2356. [Google Scholar]
- (18).DeHaan FP; Brown HC J. Am. Chem. Soc 1969, 91, 4844–4850. [Google Scholar]
- (19).Evans DA; Chapman KT; Bisaha JJ Am. Chem. Soc 1988, 110, 1238–1256. [Google Scholar]
- (20).Castellino S; Dwight WJ J. Am. Chem. Soc 1993, 115, 2986–2987. [Google Scholar]
- (21).Lam Y.-h.; Cheong PH-Y; Blasco Mata JM; Stanway SJ; Gouverneur V; Houk KN J. Am. Chem. Soc 2009, 131, 1947–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cook D Can. J. Chem 1963, 41, 522–526. [Google Scholar]
- (23).Hilt G; Pünner F; Möbus J; Naseri V; Bohn MA Eur. J. Org. Chem 2011, 2011, 5962–5966. [Google Scholar]
- (24).Hertel GR; Clark HB J. Phys. Chem 1961, 65, 1930–1932. [Google Scholar]
- (25).Sitze MS; Schreiter ER; Patterson EV; Freeman RG Inorg. Chem 2001, 40, 2298–2304. [DOI] [PubMed] [Google Scholar]
- (26).Wyrzykowski D; Maniecki T; Pattek-Janczyk A; Stanek J; Warnke Z Thermochim. Acta 2005, 435, 92–98. [Google Scholar]
- (27).Cerny Z; Machacek J; Fusek J; Casensky B; Kriz O; Tuck DG J. Organomet. Chem 1993, 456, 25–30. [Google Scholar]
- (28).Zimmerman PM J. Comput. Chem 2015, 36, 601–611. [DOI] [PubMed] [Google Scholar]
- (29).Pendleton IM; Pérez-Temprano MH; Sanford MS; Zimmerman PM J. Am. Chem. Soc 2016, 138, 6049–6060. [DOI] [PubMed] [Google Scholar]
- (30).Santiago CB; Milo A; Sigman MS J. Am. Chem. Soc 2016, 138, 13424–13430. [DOI] [PubMed] [Google Scholar]
- (31).For reviews on this approach, see:; (a) Santiago CB; Guo J-Y; Sigman MS Chem. Sci 2018, 9, 2398–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sigman MS; Harper KC; Bess EN; Milo A Acc. Chem. Res 2016, 49, 1292–1301. [DOI] [PubMed] [Google Scholar]
- (32).Asakura K; Nomura M; Kuroda H Bull. Chem. Soc. Jpn 1985, 58, 1543–1550. [Google Scholar]
- (33).Interactions of GaCl3 with chlorinated solvents are well investigated and suggest that GaCl3 is monomeric in chlorinated hydrocarbons. For details, see:; (a) Wong R; Brown HC J. Inorg. Nucl. Chem. 1955, 1, 402–410. [Google Scholar]; (b) DeHaan FP; Brown HC J. Am. Chem. Soc 1969, 91, 4844–4850. [Google Scholar]; (c) DeHaan FP; Gibby MG; Aebersold DR J. Am. Chem. Soc 1969, 91, 4860–4863. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.