

Abstract
Background:
4–9% of prostate cancers harbor homozygous deletions of the androgen-induced tumor suppressor gene, PLZF (ZBTB16). PLZF loss induces an in vitro phenotype of castration resistance and enzalutamide resistance. The association of low expression of PLZF and clinical outcomes is unclear.
Methods:
We assessed PLZF mRNA expression in patients diagnosed with primary prostate cancer during prospective follow-up of the Health Professionals Follow-up Study (HPFS; n=254) and the Physicians’ Health Study (PHS; n=150), as well as in The Cancer Genome Atlas (n=333). We measured PTEN status (using copy numbers and immunohistochemistry) and transcriptional activation of the mitogen-activated protein kinase (MAPK) pathway. Patients from HPFS and PHS were followed for metastases and prostate cancer-specific mortality (median, 15.3 years; 113 lethal events).
Results:
PLZF mRNA expression was lower in tumors with PLZF deletions. There was a strong, positive association between intratumoral androgen receptor signaling and PLZF expression. PLZF expression was also lower in tumors with PTEN loss. Low PLZF expression was associated with higher MAPK signaling. Patients in the lowest quartile of PLZF expression compared to those in the highest quartile were more likely to develop lethal prostate cancer, independent of clinicopathologic features, Gleason score, and androgen receptor signaling (odds ratio, 3.17; 95% CI, 1.32–7.60).
Conclusions:
Low expression of the tumor suppressor gene PLZF is associated with a worse prognosis in primary prostate cancer.
Impact:
Suppression of PLZF as a consequence of androgen deprivation may be undesirable. PLZF should be tested as a predictive marker for resistance to androgen deprivation therapy.
Keywords: PLZF, tumor suppressor, prostate cancer, lethality, androgen deprivation therapy
Introduction
Approximately two-thirds of those patients who die of prostate cancer initially present with localized disease (1). The mechanisms by which localized prostate cancers progress to a lethal disease are incompletely understood. One likely contributory mechanism is the loss of or alterations in tumor suppressor genes (e.g., PTEN, p53, RB1). We previously showed that loss of the tumor suppressor gene promyelocytic leukemia zinc finger (PLZF), also known as zinc finger and BTB domain containing 16 (ZBTB16), induces an in-vitro phenotype of castration and enzalutamide-resistant prostate cancer (CRPC) (2).
Intriguingly, PLZF is positively regulated by androgen signaling (2, 3). Androgen deprivation therapy (ADT), the standard of care for patients with advanced prostate cancer, may thus inhibit the tumor suppressor PLZF and in turn may activate deleterious pathways including MAPK signaling (2). Whether PLZF suppression with ADT indeed leads to worse clinical outcomes might depend on patient and tumor characteristics, such as the baseline expression of PLZF. Somatic deletions within the PLZF gene, potentially altering PLZF expression, occur in primary and metastatic prostate cancers, with 4–9% of patients reported to harbor focal homozygous deletions (4, 5). Additionally, in a preclinical study, activation of the PI3 kinase/Akt/mTORC1 pathway through PTEN loss suppressed PLZF expression (6). Whether and how these molecular changes impact outcomes in patients is unknown.
We hypothesized that PLZF expression in patient samples differs according to somatic copy number variation in the PLZF gene, PTEN status, and the androgen receptor (AR) activity in the tumor. We further hypothesized that low baseline expression of PLZF is associated with a higher risk of lethal prostate cancer. We studied large patient cohorts to validate regulators and effectors of PLZF and to evaluate the prognosis of low PLZF expression.
Methods
Study populations
Patients with primary prostate cancer were included from extreme case-control studies nested within the Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS), as well as from The Cancer Genome Atlas (TCGA). To allow for additional direct comparisons between primary and metastatic, presumably ADT-treated tumors, we additionally studied the Taylor et al. single-institutional cohort of a spectrum of prostate cancers with genomic profiling (7).
The HPFS and PHS prostate cancer cohorts are comprised of men who developed prostate cancer during prospective follow-up of two well-characterized cohort studies. The HPFS is an ongoing cohort study of initially 51,529 male health professionals, aged 40–75 years, who have been followed since 1986 (8). The PHS started in 1982 as randomized-controlled trials of aspirin and multivitamins among initially 29,067 male physicians, aged 40–84 years; participants were later followed as a prospective cohort (9, 10). Self-reported incident prostate cancers were verified with review of medical and pathology records. Patients have been followed for metastases and death causes through specific questionnaires, contact to treating physicians, and review of medical records and death certificates (>98% complete for mortality). We retrieved formalin-fixed paraffin-embedded primary cancer tissue for our biorepository. We here focus on patients in a nested extreme case-control study (n = 404; 92% prostatectomy) that oversampled patients who developed metastases or died from prostate cancer (lethal cancer) and those with prediagnostic blood samples (11).
The TCGA primary prostate cancer cohort included patients with previously untreated prostate cancer from clinical research sites and academic medical centers (4). Fresh-frozen prostatectomy specimens underwent comprehensive genomic profiling. We restricted our study to the published subset of cases with satisfactory RNA quality (n = 333) (4).
Histologic and genomic profiling
Tumors in all cohorts underwent histopathologic review, which included centralized re-grading by genitourinary pathologists in HPFS, PHS, and TCGA (4, 12). In HPFS and PHS, high-density tumor areas (>80%) on histopathologic review were selected for transcriptome profiling. In TCGA, tumor cellularity varied on pathology review, with 61% of samples having a tumor content of >60% cellularity; samples were retained if nucleic acid yield was sufficient. Taylor et al. required >70% tumor cell content (7).
Whole-transcriptome profiling including PLZF was performed in all cohorts. TCGA used RNA sequencing with the Illumina TruSeq RNA protocol and the Illumina HiSeq platform (4). In HPFS and PHS, the Affymetrix GeneChip Human Gene 1.0 ST array was used (Gene Expression Omnibus: GSE62872) (13). Taylor et al. used the Affymetrix Human Exon 1.0 ST array (7).
PLZF and PTEN copy number variations were assessed in TCGA and Taylor et al. As reported previously (4), tumor genome-wide copy number estimates in TCGA were normalized against non-cancer normal samples and segmented using circular binary segmentation, effectively filtering out germline variants, and focal alterations were identified using GISTIC. We also retrieved the overall proportion of genes with copy number alterations among all genes (fraction genome altered) in TCGA (4). Details for Taylor et al. are described elsewhere (7). PTEN status was assessed in HPFS and PHS using a genomically-validated immunohistochemical assay on tissue microarrays constructed from the dominant tumor nodule or the nodule with the highest Gleason score (14). Additionally, tissue microarrays from HPFS and PHS were assessed for percent nuclei positive for the cell proliferation marker Ki-67, as previously described (15).
Statistical analysis
Our analysis plan was geared at characterizing tumors with low PLZF, defined as the lowest quartile of mRNA expression in each cohort. In the cross-sectional analysis of PLZF regulators, we estimated differences in PLZF mRNA, as expressed in standard deviations [SD], using linear regression. We assessed whether PLZF copy number variation, PTEN copy number variation or PTEN status by immunohistochemistry (complete loss vs. any expression) (14), and the z score of a well-described, parsimonious mRNA signature of AR signaling (16) are associated with PLZF mRNA expression. We chose this signature due to its association with AR protein expression (4), and we repeated analyses using other well-described signatures (17, 18). We also evaluated the association of Gleason score (with coding in grade groups: 5–6, 3+4, 4+3, 8, 9–10, and ordinal coding) and fraction genome altered (linear) and PLZF expression. In models for PTEN loss and PLZF expression, we additionally adjusted for age and Gleason score, even though Gleason score could be considered as an intermediate in this association. Finally, we compared PLZF expression between primary and metastatic samples from a single cohort (7).
To validate downstream effects of low PLZF, we assessed the association of PLZF expression and proportion of Ki-67 positivity (continuous, after quantile normalization across tissue microarrays and logarithmic scaling) in the HPFS and PHS cohorts. To validate the association of PLZF and activation of MAPK signaling, we used principal components analysis to combine the 267 genes of the MAPK signaling pathway, as curated by the KEGG database (Molecular Signatures Database, version 6.1, pathway M10792) (19). The directionality was determined by comparison with a sum of z scores of the 267 genes; higher levels of the first principal component correlated positively with z (r = 0.73). We tested for differences in the first principal component by PLZF expression, using linear regression, in TCGA and HPFS and PHS combined.
In longitudinal analyses in HPFS and PHS, we assessed the association of PLZF expression at cancer diagnosis (continuous and binary as above) and lethality, contrasting lethal disease (development of metastases or prostate cancer-specific death) versus nonlethal disease (no evidence of metastases at >8 years of follow up). We used logistic regression to estimate age-adjusted and multivariable-adjusted odds ratios (ORs). HPFS and PHS were initially analyzed separately and then combined for multivariable models that adjusted for age at cancer diagnosis (continuous), calendar year of cancer diagnosis (categorical, pre-prostate specific antigen [PSA] era, 1982–1988; peri-PSA era, 1989–1993; PSA era, 1994–2005), AR signature (continuous), and additionally for PTEN loss (binary). Since Ki-67 and stage at diagnosis, and possibly Gleason score, are probable intermediates between PLZF expression and lethal disease, we did not include them in our models designed to assess an etiologic factor. Models including PTEN or Ki-67 were restricted to patients with non-missing data.
Tests were two-sided and all confidence intervals (CIs) are presented at a 95% level. The institutional review boards at Harvard T.H. Chan School of Public Health and Partners Healthcare approved the research.
Results
Characteristics of the study populations
Baseline characteristics of patients in HPFS, PHS, and TCGA are presented in Table 1. From 254 patients in HPFS, 81 developed metastases or died from prostate cancer (lethal disease) over long-term follow up in the extreme case-control subset (median, 15.0 years). In PHS, among 150 patients, 32 developed lethal disease (median follow up, 15.8 years). In total, we included 113 lethal cases from both studies. Additional tissue biomarker data for Ki-67 positivity and PTEN loss was available for 257 and 260 patients, respectively, from HPFS and PHS. Baseline characteristics of the patients in the Taylor et al. cohort, including mRNA data on 131 primary tumors and 14 metastases, have been published elsewhere (4, 7).
Table 1.
Baseline characteristics at cancer diagnosis of men with prostate cancer and tumor transcriptome profiling in The Cancer Genome Atlas (TCGA), the Health Professionals Follow-up Study (HPFS), and the Physicians’ Health Study (PHS), by PLZF mRNA expression (low: first quartile; normal: all higher quartiles).
| TCGA | HPFS | PHS | ||||
|---|---|---|---|---|---|---|
| PLZF expressiona | Low | Normal | Low | Normal | Low | Normal |
| n | 84 | 249 | 68 | 186 | 33 | 117 |
| Age, median (range) | 64 (46–74) | 61(43–76) | 65 (47–76) | 66 (49–80) | 65 (55–79) | 66 (51–81) |
| Gleason score in grade | ||||||
| groups, n (%) | ||||||
| 5–6 | 9 (11) | 56 (22) | 4 (6) | 20 (11) | 3 (9) | 30 (26) |
| 3+4 | 30 (36) | 72 (29) | 20 (29) | 71 (38) | 11 (33) | 37 (32) |
| 4+3 | 23 (27) | 55 (22) | 22 (32) | 52 (28) | 10 (30) | 18 (15) |
| 8 | 10 (12) | 35 (14) | 8 (12) | 13 (7) | 5 (15) | 17 (15) |
| 9–10 | 12 (14) | 31 (12) | 14 (21) | 30 (16) | 4 (12) | 15 (13) |
| Clinical stage, n (%) | ||||||
| T1/T2 | 84 (100) | 249 (100) | 56 (85) | 158 (86) | 29 (88) | 107 (93) |
| T3 | 5 (8) | 16 (9) | 3 (9) | 3 (3) | ||
| T4/N1/M1 | 5 (8) | 9 (5) | 1 (3) | 5 (4) | ||
| PSA,b n (%) | ||||||
| <4 | 7 (15) | 12 (9) | 7 (12) | 17 (11) | 5 (19) | 12 (12) |
| 4–10 | 30 (65) | 78 (55) | 27 (47) | 90 (58) | 15 (58) | 64 (62) |
| 10–20 | 7 (15) | 30 (21) | 13 (22) | 30 (19) | 3 (12) | 17 (17) |
| >20 | 2 (4) | 21 (15) | 11 (19) | 17 (11) | 3 (12) | 10 (10) |
| Missing | 38 | 108 | 10 | 32 | 7 | 14 |
| Tissue source, n (%) | ||||||
| Prostatectomy | 84 (100) | 249 (100) | 64 (94) | 172 (92) | 29 (88) | 104 (89) |
| TURPc | 4 (6) | 14 (8) | 4 (12) | 13 (11) | ||
| PLZF copy number | Not available | Not available | ||||
| Gaind | 3 (4) | 16 (6) | ||||
| Diploid | 63 (75) | 215 (86) | ||||
| Heterozygous deletion | 11 (13) | 11 (4) | ||||
| Homozygous deletion | 7 (8) | 7 (3) | ||||
| PTEN status | ||||||
| Intact/Diploid | 43 (51) | 197 (79) | 32 (68) | 97 (82) | 11 (50) | 55 (79) |
| Loss/Any deletion | 41 (49) | 52 (21) | 15 (32) | 21 (18) | 11 (50) | 15 (21) |
Categorized as: low, first quartile of mRNA expression; normal, all higher quartiles combined.
Serum prostate specific antigen, in ng/ml.
Transurethral resection of the prostate. Includes one lymph node sample in a patient from PHS.
Includes one amplification event in the mRNA expression category “normal”.
Regulators of PLZF expression
First, we sought to establish whether differences in PLZF copy number were associated with differences in PLZF mRNA expression (Figure 1.A). In TCGA, compared to tumors with diploid PLZF, those with heterozygous deletions had 0.18 standard deviations [SD] lower PLZF (95% CI, –0.29 to 0.67). Those with homozygous deletions had 0.65 SD lower PLZF expression (95% CI, 0.07 to 1.23; ptrend = 0.022). Gains/amplifications did not have different expression levels compared to diploid PLZF (difference, –0.21 SD; 95% CI, –0.67 to 0.26). For comparison, we assessed if alterations of PLZF expression were non-specifically driven by genome instability; however, fraction genome altered was not correlated with PLZF expression (r = 0.01; 95% CI, –0.09 to 0.12).
Figure 1.
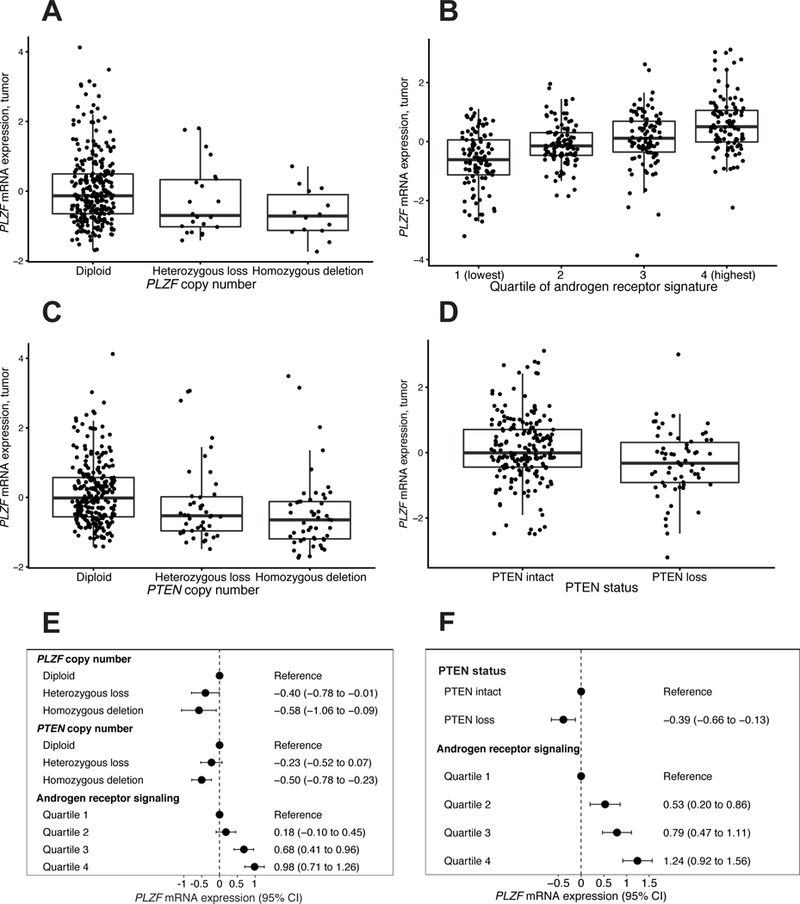
Regulators of PLZF mRNA expression. A, PLZF copy number and PLZF mRNA expression in TCGA (n = 333). B, Androgen receptor signature and PLZF mRNA expression in HPFS and PHS combined (n = 404). C, PTEN copy number and PLZF mRNA expression in TCGA (n = 333). D, PTEN by immunohistochemistry and PLZF mRNA expression in HPFS and PHS (n = 260). In panels A–E, horizontal lines indicate the medians; boxes reach from the first to the third quartiles; whiskers extend 1.5 times the interquartile range. E–F, Regulators of PLZF expression in multivariable models in the TCGA primary prostate cancer cohort (E) and in HPFS and PHS combined (F). All units of tumor PLZF expression are standard deviations.
Next, we sought to validate the influence of AR signaling on PLZF expression, described in vitro, across the clinical spectrum of prostate cancer. Higher expression of a well-described transcriptome signature of AR signaling (16) was positively correlated with higher PLZF expression both in TCGA (r = 0.41; 95% CI, 0.32 to 0.50) as well as in the combined HPFS and PHS cohorts (r = 0.52; 95% CI, 0.45 to 0.59; Fig. 1.B). Results were similar with other signatures of AR signaling (17, 18). In line with these observations, in a smaller cohort of primary and metastatic tumors (7), PLZF expression was lower among metastatic prostate cancers, patients who had presumably been treated with ADT, compared to primary tumors (difference, –0.71 SD; 95% CI, – 0.23 to –1.19). Finally, we aimed to validate the directionality of the association between AR signaling and PLZF expression, using the TCGA cohort. Patients with PLZF deletions, compared to those with wild-type PLZF, did not have lower expression of the AR signature (difference, 1.29 SD; 95% CI, –3.65 to 6.22; ptrend = 0.13), supporting the expectation that AR signaling has a stronger impact on PLZF expression than vice versa.
A preclinical study had suggested that PTEN/PI3K signaling affects PLZF expression (6). Validating these observations, we found that PLZF expression differed by PTEN status. In TCGA, compared to tumors with intact PTEN, those with heterozygous deletions had 0.34 SD lower PLZF (95% CI, 0.01 to 0.67) while those with homozygous deletions had 0.46 SD lower PLZF (95% CI, 0.13 to 0.79; ptrend < 0.001; Figure 1.C). In the combined HPFS and PHS cohorts, PLZF mRNA expression was 0.44 SD lower among tumors with PTEN loss, compared to those with intact PTEN (95% CI, 0.15 to 0.73; Fig. 1.D).
To assess if differences in PLZF expression were merely attributable to differing Gleason scores of these tumors, we compared PLZF expression between low-grade and high-grade tumors. Gleason grade groups were not strongly associated with PLZF expression in TCGA (difference in PLZF between Gleason 3+3 and 9–10, – 0.06 SD; 95% CI, –0.44 to 0.32; ptrend = 0.87) nor in the combined HPFS and PHS cohorts (difference, –0.35 SD; 95% CI, –0.72 to 0.00; ptrend = 0.06). PLZF copy number, PTEN copy number, and the AR signature were independent predictors of PLZF expression in TCGA (Figure 1.E). Validation in HPFS and PHS using PTEN status by immunohistochemistry, where data on copy number alterations was unavailable, yielded similar results for both PTEN and the AR signature as predictors of PLZF expression (Fig. 1.F). Further adjustment for Gleason score did not alter the associations.
Consequences of low PLZF expression
Given our previous observation that shRNA knockdown of PLZF induced MAPK signaling in vitro (2), we aimed to assess if primary prostate cancers with low PLZF had higher proliferation indices in general, as quantified through Ki-67 levels, or specifically more activation of the MAPK pathway, as quantified through a transcriptome signature. Tumors in the lowest quartile of PLZF expression did not have higher Ki-67 levels compared to those in the highest quartile (difference, –0.07 SD; 95% CI, –0.43 to 0.29; ptrend = 0.44). In contrast, among tumors in the lowest quartile of PLZF expression in TCGA, the MAPK score was 0.32 SD higher than in the highest quartile of PLZF (95% CI, 0.02 to 0.62; ptrend = 0.006). In the combined HPFS and PHS cohorts, the MAPK scores did not show a linear trend across quartiles of PLZF mRNA (ptrend = 0.45); however, tumors with the lowest quartile of PLZF level had higher MAPK scores than tumors with higher PLZF expression (difference, 0.45 SD; 95% CI, 0.19 to 0.73; Figure 2.A). Differences were not attenuated in either cohort when additionally adjusting for AR signaling.
Figure 2.
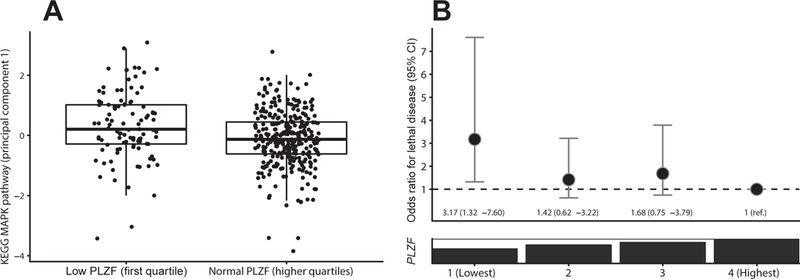
Downstream consequences of low PLZF in HPFS and PHS. A, PLZF mRNA expression and a signature of MAPK signaling (n = 404). Horizontal lines indicate the medians; boxes reach from the first to the third quartiles; whiskers extend 1.5 times the interquartile range. B, PLZF mRNA expression and risk of lethal disease over long-term follow-up in a multivariable model adjusting for patient, histologic, and genomic covariates.
Finally, we determined the clinical outcome of patients with low PLZF (Table 2). Patients from HPFS in the lowest quartile of PLZF expression, compared to those with higher expression, had an approximately two-fold higher odds of developing lethal disease over long-term follow up (age-adjusted OR, 1.92; 95% CI, 1.07 to 3.45). In the independent validation cohort PHS, the age-adjusted OR was 3.19 (95% CI, 1.22 to 8.36). Combining both cohorts and adjusting for further patient and tumor characteristics including Gleason score and AR signaling, PLZF expression was independently associated with lethal disease (OR for lowest vs. highest quartile, 3.17; 95% CI, 1.32 to 7.60; ptrend = 0.021; Figure 2.B). The association was also essentially unchanged when additionally adjusting for PTEN status in the subset of patients with available PTEN data (Table 2).
Table 2.
PLZF expression and odds ratios (OR) for lethal prostate cancer (with 95% confidence intervals) in patients from the Health Professionals Follow-up Study (HPFS) and the Physicians’ Health Study (PHS).
| Quartile of PLZF |
1st (lowest) |
2nd |
3rd |
4th (highest) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Cases |
Lethal Non-lethal |
Lethal Non-lethal |
Lethal Non-lethal |
Lethal Non-lethal |
|||||
| 40 | 61 | 22 | 79 | 27 | 74 | 24 | 77 | ||
| Model | OR | 95% CI | OR | 95% CI | OR | 95% CI | OR | 95% CI | ptrenda |
| A Age-adjusted | 2.43 | 1.30–4.53 | 1.00 | 0.51–1.96 | 1.24 | 0.65–2.38 | 1.00 | (ref.) | 0.011 |
| B A+clinicalb | 2.53 | 1.20–5.33 | 1.30 | 0.58–2.89 | 1.56 | 0.70–3.47 | 1.00 | (ref.) | 0.026 |
| C B+ARc | 3.17 | 1.32–7.60 | 1.42 | 0.62–3.22 | 1.68 | 0.75–3.79 | 1.00 | (ref.) | 0.021 |
| D C in PTEN subsetd | 3.39 | 1.06–10.9 | 1.62 | 0.55–4.73 | 1.39 | 0.46–4.23 | 1.00 | (ref.) | 0.041 |
| E C+PTENd | 3.51 | 1.03–12.0 | 1.64 | 0.55–4.84 | 1.42 | 0.46–4.39 | 1.00 | (ref.) | 0.046 |
Trend for linear trend across quartiles
Demographics and clinical factors: age, calendar year, Gleason score
Hieronymus et al. (2006) AR signature (16)
PTEN status was available only in a subset of 253 patients. Models D and E are both estimated in this subset of patients. Model D includes the same predictors as model C. Model E additionally adjusts for PTEN status. To gauge the change in estimates due to adjusting for PTEN status, compare results from model E to results from model D.
Discussion
In this study, we assessed a tumor suppressor that is androgen induced and in turn inhibited by ADT. We showed that prostate tumors with PLZF and PTEN deletions have lower PLZF expression levels, and we validated across a spectrum of prostate cancers that PLZF expression is tightly coupled to the activity of AR signaling. Likely partially mediated through activation of MAPK signaling, low PLZF expression was associated with inferior prognosis over long-term follow-up, independent of Gleason score, AR signaling, and PTEN loss.
Deletions of PLZF are among the more frequent copy number alterations in presumptive driver genes in prostate cancer, as independently demonstrated in three large genomic landscape studies of primary and metastatic disease (4, 5, 20). We demonstrated here that tumors with PLZF deletions have lower PLZF mRNA expression. More importantly, key signaling pathways in prostate cancer are important regulators of PLZF expression beyond alterations in PLZF copy numbers. PLZF can experimentally be induced by androgens, and there is marked androgen-induced AR recruitment to PLZF enhancer regions, as we and others have previously shown in vitro (2, 3). Clinically, tumors from our three cohorts with low AR signaling activity had considerably lower PLZF expression (Fig 1.B). We also demonstrated that tumors with loss of PTEN also have lower PLZF expression (Fig. 1.C–D), in line with mechanistic work suggesting FOXO3a as an Akt-regulated mediator of PLZF regulation (6). In light of feedback regulation between PI3K and AR signaling (21), we verified that the association of PTEN loss and PLZF expression was not merely due to differences in AR signaling. We also verified that low expression of the tumor suppressor gene PLZF was not simply driven by genome instability. Collectively, PLZF copy number loss, PTEN loss, and low AR signaling activity were statistically independent predictors of low PLZF expression (Fig. 1.E–F).
We also demonstrated that PLZF expression was lower in presumably ADT-treated patients with metastases at the time of genomic profiling in the cohort described by Taylor, Schultz (7), compared to primary tumors. These results are in line with a smaller cross-sectional study of PLZF immunohistochemistry, showing lower expression in metastases and higher grade-tumors, albeit without controlling for key regulators of PLZF expression (22). Of note, our large study with centrally re-reviewed histology had precise estimates that excluded any meaningful differences in average PLZF expression across groups of Gleason grades, suggesting that low PLZF expression is specifically influenced by PTEN status and AR signaling but not by unspecific tumor dedifferentiation. Additionally, it has been suggested that PLZF itself, interacting with KLK4, in turn inhibits AR through a feedback loop (23). We did not find tumors with PLZF copy number alteration differed in AR signaling; however, these results were imprecise and do not exclude the presence of such a feedback loop.
To determine the clinical relevance of low PLZF expression, we harnessed prospectively collected long-term outcome data on metastases and cause-specific death among men diagnosed with primary prostate cancer in HPFS and PHS. In both independent studies, patients with low PLZF had a higher risk of lethal disease (Fig. 2.B). Importantly, we accounted for patient and tumor characteristics, such that the elevated risk of lethality among patients with low PLZF is not merely due to PTEN loss or tumors arising in a low-AR signaling environment. As one potential mechanism linking low PLZF to lethal disease, we validated our preclinical observation that PLZF is a repressor of the MAPK signaling pathway with binding sites in the MAPK pathway regulators DDIT3, MKNK2, JUND, JUN, and RRAS (2). We did indeed observe higher expression of a MAPK signature in tumors with low PLZF (Fig. 2.A). Even if emergence of MAPK signaling has been described in low-AR signaling states (24), the difference in MAPK expression was not due to low AR signaling in our cohorts. Numerous additional downstream effects of PLZF beyond the scope of our study have been described, such as posttranslational modification of MYC and MTORC1 inhibition (25–28). Further study would be needed to tease out which pathways connect PLZF expression and lethality.
Our results beg the question as to whether ADT through its suppression of PLZF may paradoxically accelerate ADT resistance and tumor progression (Fig. 3). Constitutive AR signaling and experimental alteration of androgen levels in model systems tightly control PLZF, and as our results demonstrate, tumors with low PLZF have inferior clinical outcomes. These observations strongly support that ADT-driven PLZF downregulation is one potential mechanism that contributes to castration resistance. It is possible that PLZF levels before ADT treatment are predictive of clinical outcomes, suggesting that tumors with low pre-ADT PLZF are particularly susceptible to ADT-induced PLZF suppression. We assessed PLZF expression in primary tumors, in general many years before ADT initiation. This is an imperfect measure of PLZF expression at the time of ADT initiation, probably resulting in nondifferential misclassification and underestimation of differences in outcomes by PLZF expression. Ideally, future work would analyze randomized-controlled trials of ADT in high-risk patients and quantify PLZF in tumor tissue before ADT initiation to assess it as a predictive biomarker of ADT resistance. Further, it remains to be shown if PLZF mRNA levels as assessed through transcriptome profiling or PLZF protein levels as assessed through immunohistochemistry (2, 22) are better suited for predicting clinical outcomes. A second corollary of our previous (2) and current studies is that assessing for low PLZF expression may aid in enriching clinical trials of MAPK inhibitors with patients who are more likely to benefit.
Figure 3.
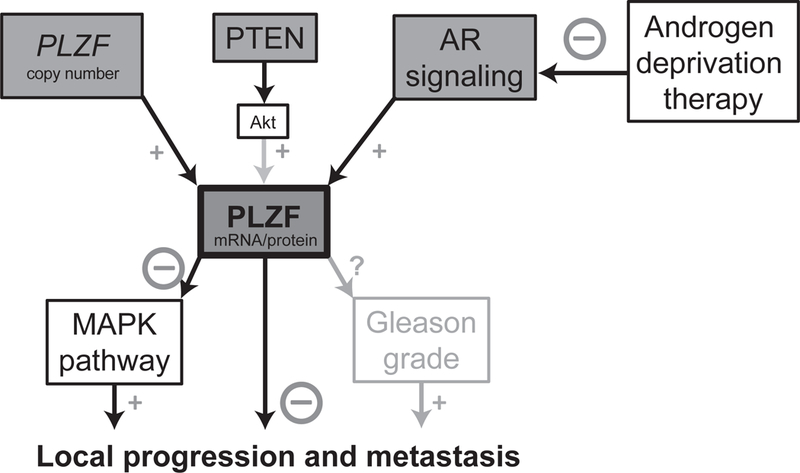
Schematic overview of PLZF regulation and downstream consequences.
Taken together, PLZF is not only one of the most commonly deleted putative driver genes in prostate cancer, but also tightly coupled in its expression levels with key mechanisms of prostate cancer progression, AR signaling activity and PTEN loss. Our results from large patient populations with prospective follow-up highlight the clinical implications of low PLZF and contribute to our understanding of the potentially undesirable effects of ADT in prostate cancer treatment. Assessing PLZF levels before ADT may aid in predicting ADT resistance and in biomarker-based patient stratification for MAPK inhibitors trials in high-risk prostate cancer.
Acknowledgements:
We would like to thank the participants and staff of the Health Professionals Follow-up Study and the Physicians’ Health Study for their valuable contributions. In particular, we would like to recognize the contributions of Liza Gazeeva, Siobhan Saint-Surin, Robert Sheahan, and Betsy Frost-Hawes.We would like to thank the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data.
Funding: The Health Professionals Follow-up Study is supported by the National Institutes of Health (U01 CA167552). The Physicians’ Health Study was supported by the National Institutes of Health (CA097193, CA34944, CA40360, HL26490, HL34595). P.W.K. was supported by the Department of Defense (DOD-W81XWH-14–1-0515). This research was funded in part by the Dana-Farber/Harvard Cancer Center Specialized Programs of Research Excellence program in Prostate Cancer 5P50 CA090381 and the NIH/NCI Cancer Center Support Grant P30 CA008748. K.H.S., W.A., and L.A.M. are Prostate Cancer Foundation Young Investigators.
Footnotes
Prior presentation: Presented in part at the Genitourinary Cancers Symposium, San Francisco, CA, 2018
Conflicts of interest: W. Abida reports receiving commercial research grants from AstraZeneca, Zenith Epigenetics, Clovis Oncology, and GlaxoSmithKline, honoraria from CARET, and is a consultant/advisory board member for Clovis Oncology, Janssen, and MORE Health. P.W. Kantoff reports ownership interest in Context Therapeutics LLC, DRGT, Placon, Seer Biosciences, and Tarveda Therapeutics, is a company board member for Context Therapeutics LLC, is a consultant/advisory board member for BIND Biosciences, Inc., BN Immunotherapeutics, DRGT, GE Healthcare, Janssen, Metamark, New England Research Institutes, Inc., OncoCellMDX, Placon, Progenity, Sanofi, Seer Biosciences, Tarveda Therapeutics, and Thermo Fisher, and serves on data safety monitoring boards for Genentech/Roche and Merck. No potential conflicts of interest were disclosed by the other authors.
References
- 1.ICECaP Working Group, Sweeney C, Nakabayashi M, Regan M, Xie W, Hayes J, et al. The Development of Intermediate Clinical Endpoints in Cancer of the Prostate (ICECaP). J Natl Cancer Inst. 2015;107:djv261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hsieh CL, Botta G, Gao S, Li T, Van Allen EM, Treacy DJ, et al. PLZF, a tumor suppressor genetically lost in metastatic castration-resistant prostate cancer, is a mediator of resistance to androgen deprivation therapy. Cancer Res. 2015;75:1944–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang F, Wang Z. Identification and characterization of PLZF as a prostatic androgen-responsive gene. Prostate. 2004;59:426–35. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao J, Zhu S, Zhou W, Li J, Liu C, Xuan H, et al. PLZF mediates the PTEN/AKT/FOXO3a signaling in suppression of prostate tumorigenesis. PLoS One. 2013;8:e77922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giovannucci E, Liu Y, Platz EA, Stampfer MJ, Willett WC. Risk factors for prostate cancer incidence and progression in the health professionals follow-up study. Int J Cancer. 2007;121:1571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaziano JM, Sesso HD, Christen WG, Bubes V, Smith JP, MacFadyen J, et al. Multivitamins in the prevention of cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2012;308:1871–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steering Committee of the Physicians’ Health Study Research Group. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med. 1989;321:129–35. [DOI] [PubMed] [Google Scholar]
- 11.Sinnott JA, Peisch SF, Tyekucheva S, Gerke T, Lis R, Rider JR, et al. Prognostic Utility of a New mRNA Expression Signature of Gleason Score. Clin Cancer Res. 2017;23:81–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stark (Rider) JR, Perner S, Stampfer MJ, Sinnott JA, Finn S, Eisenstein AS, et al. Gleason score and lethal prostate cancer: does 3 + 4 = 4 + 3? J Clin Oncol. 2009;27:3459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penney KL, Sinnott JA, Tyekucheva S, Gerke T, Shui IM, Kraft P, et al. Association of prostate cancer risk variants with gene expression in normal and tumor tissue. Cancer Epidemiol Biomarkers Prev. 2015;24:255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahearn TU, Pettersson A, Ebot EM, Gerke T, Graff RE, Morais CL, et al. A Prospective Investigation of PTEN Loss and ERG Expression in Lethal Prostate Cancer. J Natl Cancer Inst. 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rider JR, Fiorentino M, Kelly R, Gerke T, Jordahl K, Sinnott JA, et al. Tumor expression of adiponectin receptor 2 and lethal prostate cancer. Carcinogenesis. 2015;36:639–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–30. [DOI] [PubMed] [Google Scholar]
- 17.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, et al. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet. 2018;50:682–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao GQ, Unger P, Yang Q, Kinoshita Y, Singh K, McMahon L, et al. Loss of PLZF expression in prostate cancer by immunohistochemistry correlates with tumor aggressiveness and metastasis. PLoS One. 2015;10:e0121318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin Y, Qu S, Tesikova M, Wang L, Kristian A, Maelandsmo GM, et al. Molecular circuit involving KLK4 integrates androgen and mTOR signaling in prostate cancer. Proc Natl Acad Sci U S A. 2013;110:E2572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell. 2017;32:474–89 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Vogt PK. Posttranslational regulation of Myc by promyelocytic leukemia zinc finger protein. Int J Cancer. 2009;125:1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hobbs RM, Seandel M, Falciatori I, Rafii S, Pandolfi PP. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell. 2010;142:468–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McConnell MJ, Chevallier N, Berkofsky-Fessler W, Giltnane JM, Malani RB, Staudt LM, et al. Growth suppression by acute promyelocytic leukemia-associated protein PLZF is mediated by repression of c-myc expression. Mol Cell Biol. 2003;23:9375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suliman BA, Xu D, Williams BR. The promyelocytic leukemia zinc finger protein: two decades of molecular oncology. Front Oncol. 2012;2:74. [DOI] [PMC free article] [PubMed] [Google Scholar]