

SUMMARY
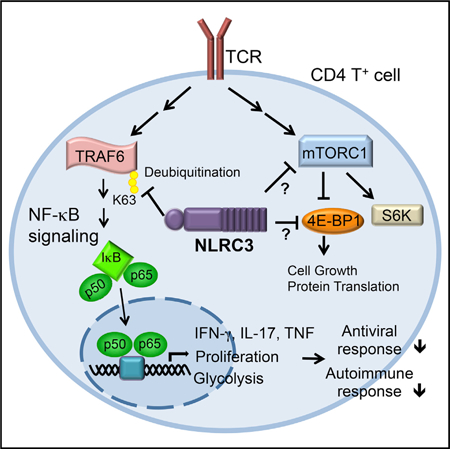
Appropriate immune responses require a fine balance between immune activation and attenuation. NLRC3, a non-inflammasome-forming member of the NLR innate immune receptor family, attenuates inflammation in myeloid cells and proliferation in epithelial cells. T lymphocytes express the highest amounts of Nlrc3 transcript where its physiologic relevance is unknown. We show that NLRC3 attenuated interferon-γ and TNF expression by CD4+ T cells and reduced T helper 1 (Th1) and Th17 cell proliferation. Nlrc3−/− mice exhibited increased and prolonged CD4+ T cell responses to lymphocytic choriomeningitis virus infection and worsened experimental autoimmune encephalomyelitis (EAE). These functions of NLRC3 were executed in a T-cell-intrinsic fashion: NLRC3 reduced K63-linked ubiquitination of TNF-receptor-associated factor 6 (TRAF6) to limit NF-κB activation, lowered phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), and diminished glycolysis and oxidative phosphorylation. This study reveals an unappreciated role for NLRC3 in attenuating CD4+ T cell signaling and metabolism.
Graphical Abstract
In Brief
NLRC3 limits inflammatory signaling in myeloid cells, but its role in T cells has been unclear. Uchimura et al. reveal that T cell expression of Nlrc3 restricts autoimmune and virus-specific CD4+ T cell responses by attenuating T cell signaling and metabolic pathways in CD4+ T cells.
INTRODUCTION
Pattern-recognition receptors (PRRs) are indispensable for innate immune responses. PRRs are receptors or sensors that detect pathogen-associated molecular patterns (PAMPs) from microbes and damage-associated molecular patterns (DAMPs) from cellular sources either directly or indirectly. In most cases, these molecules function to initiate innate immune and inflammatory signaling cascades (Brubaker et al., 2015). The nucleotide-binding domain and leucine-rich-repeat-containing (NLR) proteins are cytosolic PRRs and are known to sense or bind intracellular PAMPs and DAMPs. There are two groups of NLRs based on their functions: inflammasome-forming NLRs and non-inflammasome-forming NLRs (Allen, 2014; Guo et al., 2015). Some of the best-studied inflammasome-forming NLRs, including NLRP1, NLRP3, and NLRC4, regulate the activation of caspase-1, which is necessary for the processing of key pro-inflammatory cytokines that drive innate immune responses to PAMPs and DAMPs. Upon activation by PAMPs or cellular disturbances, inflammasome NLRs undergo a conformational change that allows the recruitment and binding of the ASC (apoptotic-speck-containing protein with a CARD) adaptor and procaspase-1 to form a fibril-like macromolecular structure (Lechtenberg et al., 2014; Lu et al., 2014). This leads to the catalytic cleavage and activation of caspase-1, which drives the subsequent catalytic cleavage and activation of IL-1 and IL-18. Non-inflammasome-forming NLRs—for example, CIITA, NLRC5, NOD1, and NOD2—demonstrate enhanced major histocompatibility gene expression and immune signaling. By contrast, an increasingly large group of NLRs negatively regulate innate immunity and inflammatory responses in the myeloid lineage. These include NLRP2 (Bruey et al., 2004), NLRP4 (Lin et al., 2016; Cui et al., 2012), NLRP6 (Anand et al., 2012), NLRP12 (Allen et al., 2012; Zaki et al., 2011), NLRX1 (Allen et al., 2011; Xia et al., 2011), NLRC5 (Cui et al., 2010), and NLRC3 (Schneider et al., 2012; Zhang et al., 2014; Karki et al., 2016), although some of these proteins have pleiotropic functions.
Murine NLRC3 reduces toll-like-receptor (TLR)-induced nuclear factor kappa B (NF-κB) activation through inhibition of the adaptor protein TNF-receptor-associated factor 6 (TRAF6) in peritoneal macrophages (Schneider et al., 2012). NLRC3 also reduces stimulator of interferon genes (STING)-dependent innate immune activation in response to cytosolic DNA and cyclic di-GMP by blocking STING and TANK-binding kinase 1 (TBK1) interaction required for type 1 interferon (IFN) production (Zhang et al., 2014). NLRC3 additionally plays a role in non-immune cells, where its association with PI3K blocks activation of the PI3K-dependent Akt kinase and inhibits mTOR pathways in colon epithelial cells (Karki et al., 2016). Although its function has been studied in myeloid and epithelial cells, Nlrc3 expression in these cells is extremely low. Paradoxically, both mouse Nlrc3 and human NLRC3 show the highest expression in T cells and secondary lymphoid organs (Conti et al., 2005); however, the biologic role of NLRC3 in primary T cells remains to be elucidated. Although PRRs in general have been extensively analyzed in the innate immune system, there are few reports addressing their roles in adaptive immune cells. Nlrc3 is unique among NLRs because of its high expression in lymphocytes.
Herein, we used Nlrc3−/− cells or mice to investigate the physiologic role of NLRC3 in T cell responses. We found that NLRC3 had a T-cell-intrinsic role in attenuating T cell proliferation and cytokine production upon T cell receptor (TCR) stimulation in part by reducing the immune signaling mediated by NF-κB, TRAF6, and 4E-BP1. Additionally, NLRC3 regulated cellular metabolism in a NF-κB-dependent fashion, marked by an attenuation of glycolysis, which is known to control both IFN-γ secretion and cellular proliferation (Peng et al., 2016). Nlrc3−/− mice displayed increased CD4+ T cell responses to lymphocytic choriomeningitis virus (LCMV) infection and enhanced CD4+ T cell autoreactivity against self-antigens. Cumulatively, we reveal an unappreciated role for NLRC3 in directly regulating adaptive immunity through T-cell-intrinsic mechanisms.
RESULTS
Nlrc3 Is Down-regulated Upon T Cell Activation and Suppresses CD4+ T Cell Activation
To explore the role of Nlrc3 in T cell function, we analyzed Nlrc3 expression in splenic CD4+ T cells and CD8+ T cells from wild-type (WT) mice after in vitro stimulation with anti-CD3 and anti-CD28 monoclonal antibodies. Nlrc3 expression in CD4+ T cells but not CD8+ T cells was decreased after stimulation (Figure 1A). CD4+ T cells differentiated under Th0, Th1 (IL-12), and Th17 (IL-6 and TGF-β) cell conditions also showed decreased Nlrc3 expression 24 and 48 hr after anti-CD3 and anti-CD28 stimulation (Figure 1B), suggesting that T cell activation reduces Nlrc3 expression in Th0, Th1, and Th17 CD4+ T cells. To define the functional role of NLRC3 in T cells, we evaluated its role during T cell development in the thymus and spleen under homeostatic conditions. Eight-week-old Nlrc3−/− mice had normal proportions of splenic and thymic T cell subsets, and cell numbers were similar to those in WT mice (Figures S1A–S1F). CD4+ or CD8+ T cells from WT and Nlrc3−/− mice were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin or with anti-CD3 and anti-CD28 antibodies and analyzed for surface expression of CD25 (IL-2 receptor) and CD69, which are upregulated early after T cell activation. Compared with WT CD4+ T cells, Nlrc3−/− CD4+ T cells expressed significantly greater percentages of CD25 (Figure 1C, top panels) and CD69 (Figure 1C, lower panels) 2–4 hr after PMA and ionomycin treatment, and the mean fluorescence intensity (MFI) for CD25 and CD69 was also increased for Nlrc3−/− CD4+ T cells (Figure 1C, right). Similar expression patterns were seen when T cells were stimulated with anti-CD3 and anti-CD28 antibodies (Figure 1D). In contrast to CD4+ T cells, Nlrc3−/− CD8+ T cells expressed similar or slightly reduced amounts of those markers in comparison with WT CD8+ T cells under the same stimulatory conditions (Figure S2). These findings suggest that NLRC3 suppresses the activation of CD4+ T cells.
Figure 1. Nlrc3 Is Down-regulated upon TCR Stimulation, Suppressing CD4+ T Cell Activation.

(A) Nlrc3 expression in splenic CD4+ T cells and CD8+ T cells from WT mice after activation by anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies.
(B) Nlrc3 expression in splenic CD4+ T cells from WT mice upon activation by anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies under Th0, Th1, and Th17 cell conditions.
(C) CD4+ T cells isolated from WT and Nlrc3−/− mice were stimulated with PMA (50 ng/mL) and ionomycin (1 μg/mL). Graphs show the percentage of CD4+CD25+ and CD4+CD69+ cells, MFI of CD25 among CD4+CD25+ cells, and MFI of CD69 among CD4+CD69+ cells after stimulation.
(D) Cells isolated from WT and Nlrc3−/− mice were stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies. Graphs show the percentage of CD4+CD25+ and CD4+CD69+ cells, MFI of CD25 among CD4+CD25+ cells, and MFI of CD69 among CD4+CD69+ cells.
Data are from one experiment representative of two or three experiments and are shown as mean ± SEM of triplicate samples. Statistical significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figures S1 and S2.
NLRC3 Negatively Regulates Cytokine Expression by Activated CD4+ T Cells
The expression data above suggest that NLRC3 might affect T cell activity. Significantly greater percentages of Nlrc3−/− CD4+ T cells than of WT cells were able to make IFN-γ or TNF 24 hr after anti-CD3 and anti-CD28 stimulation, and the amount of cytokine expressed per cell was also increased for Nlrc3−/− CD4+ T cells (Figures 2A and 2B), though the effect of Nlrc3 deletion on IL-2 expression was modest. In addition, the frequency of Nlrc3−/− cells co-expressing IFN-γ and TNF was nearly 4-fold higher than in WT T cells (Figure 2C). Nlrc3−/− CD4+ T cells could produce more IFN-γ, TNF, and IL-2 than WT cells, as shown by ELISA (Figure 2D). By contrast, CD8+ T cells purified from Nlrc3−/− mice showed no change or even slightly less expression of these cytokines than did WT CD8+ T cells. (Figures S3A–S3C). In contrast to TCR-induced signals, NLRC3 did not affect inflammatory cytokine production by T cells after stimulation with a TLR agonist or IL-1β (Figure S3D). Collectively, these results suggest that NLRC3 limits cytokine production by CD4+ T cells.
Figure 2. NLRC3 Negatively Regulates Cytokine Expression by Activated CD4+ T Cells.

CD4+ T cells purified from WT and Nlrc3−/− mice were stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies and incubated for 0, 4, 24, and 48 hr.
(A) Flow-cytometry analysis of CD4+IFN-γ+, CD4+TNF+, and CD4+IL-2+ cells.
(B) Percentages of IFN-γ+, TNF+, and IL-2+CD4+ T cells; MFI of IFN-γ expression among IFN-γ+CD4+ cells; MFI of TNF expression among TNF+CD4+ cells; and MFI of IL-2 expression among IL-2+CD4+ cells by intracellular staining.
(C) Representative results at the 24 hr time point of (B).
(D) IFN-γ, TNF, and IL-2 in supernatants of CD4+ T cell cultures as measured by ELISA.
Representative data from three experiments are presented as mean ± SEM. Statistical significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
NLRC3 Negatively Regulates Cell Proliferation in a T-Cell-Intrinsic Manner In Vitro
We next determined whether NLRC3 intrinsically regulates T cell proliferation or cell death. We labeled CD4+ or CD8+ T cells from WT and Nlrc3−/− mice with the fluorescent dye carboxyfluorescein succinimidyl ester (CFSE) and stimulated them with different concentrations of anti-CD3 and anti-CD28 antibodies to induce proliferation across 3–5 days. At lower concentrations of anti-CD3 (1 or 2.5 μg/mL), there were approximately twice as many proliferating Nlrc3−/− CD4+ T cells as WT cells (Figures 3A and 3B). At the highest concentration of anti-CD3 (5 μg/mL), Nlrc3−/− CD4+ T cells proliferated more than WT T cells early on, but this difference diminished by day 5 when >90% of the cells had proliferated in both groups (Figure 3C). More Nlrc3−/− CD4+ T cells than WT T cells accumulated in these cultures, consistent with their increased proliferation and survival (Figure 3D). A similar analysis was performed for CD4+ T cells under Th1- and Th17-cell-polarizing conditions. There were approximately 2- to 3-fold more proliferating Nlrc3−/− CD4+ T cells than WT cells at low and medium concentrations of anti-CD3 under Th1 (IL-12)-and Th17 (IL-6 and TGF-β)-cell-polarizing conditions (Figure 3E), though these differences diminished with the highest anti-CD3 concentration. These findings suggest that NLRC3 negatively regulates Th1 and Th17 cell proliferation when TCR stimulation is limited (Figure 3E). We found little impact of NLRC3 deficiency on naive CD25loCD44lo CD4+ T cell proliferation after anti-CD3 and anti-CD28 stimulation (Figure S4A), and Nlrc3 deficiency had no apparent effect on CD8+ T cell proliferation (Figures S4B and S4C). Also, there were minimal changes in the percentage of dead T cells in CD4+ and CD8+ T cell cultures (Figure S4D). Cumulatively, these findings indicate that NLRC3 acts intrinsically to attenuate the proliferation of CD4+ T cells but not CD8+ T cells and has a minimal effect on T cell death.
Figure 3. NLRC3 Negatively Regulates Cell Proliferation in a T-Cell-Intrinsic Manner.
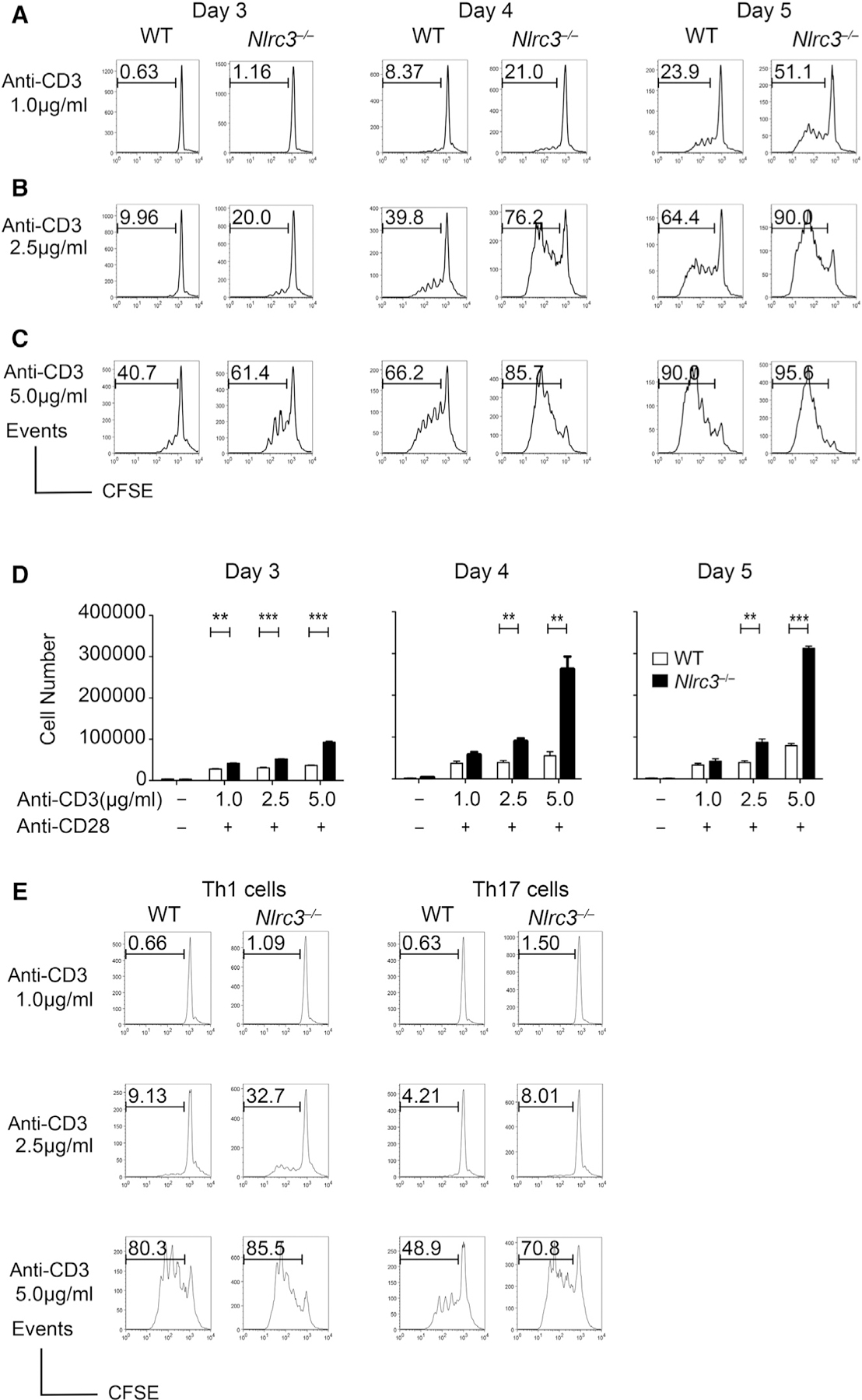
CD4+ T cells isolated from WT and Nlrc3−/− mice were stimulated with anti-CD3 (1, 2.5, and 5 μg/mL) and anti-CD28 (2 μg/mL) antibodies and incubated for 3–5 days.
(A) Histograms of CFSE-labeled CD4+ T cells after anti-CD3 (1.0 μg/mL) and anti-CD28 (2.0 μg/mL) treatment.
(B) Histograms of CFSE-labeled CD4+ T cells after anti-CD3 (2.5 μg/mL) and anti-CD28 (2.0 μg/mL) treatment.
(C) Histograms of CFSE-labeled CD4+ T cells after anti-CD3 (5.0 μg/mL) and anti-CD28 (2.0 μg/mL) treatment.
(D) Number of CD4+ T cells recovered from the cultures.
(E) CFSE-labeled CD4+ T cells isolated from WT and Nlrc3−/− mice were stimulated with anti-CD3 (1, 2.5, and 5 μg/mL) and anti-CD28 (2 μg/mL) antibodies under Th1 (IL-12) or Th17 (IL-6 and TGF-β) conditions. Histograms show CFSE fluorescence at day 3 of culture.
Results are representative of two or three experiments and represented as mean ± SEM. Statistical significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
Nlrc3–/– Mice Show Improved Protection against LCMV Infection
As an initial effort to evaluate the biologic consequence of Nlrc3−/− during an in vivo immune response, we infected Nlrc3−/− mice with LCMV-Armstrong, which causes an acute infection that is cleared by T cells within a week in immune-competent mice. After infection with LCMV-Armstrong, Nlrc3−/− mice showed significantly lower circulating virus at day 4 than did WT mice (Figure 4A), and both groups resolved the infection by day 8 (data not shown). The frequencies and the total number of LCMV-specific tetramer-binding DbGP33+ CD8+ T cells and cytokine-producing CD8+ T cells were unaffected by the loss of Nlrc3 at day 8 (Figures 4B and S5A). By contrast, the frequencies and the total number of LCMV-specific I-AbGP67+ CD4+ T cells were higher in Nlrc3−/− mice than in WT mice at day 8, and there was a modest but significant increase in the total number of cytokine-producing CD4+ T cells in Nlrc3−/− mice (Figure 4C). These data indicate that NLRC3 is dispensable for resolving an acute infection but limits peak CD4+ T cell responses.
Figure 4. Nlrc3–/– Mice Show Increased CD4+ T Cell Responses during LCMV Infection.
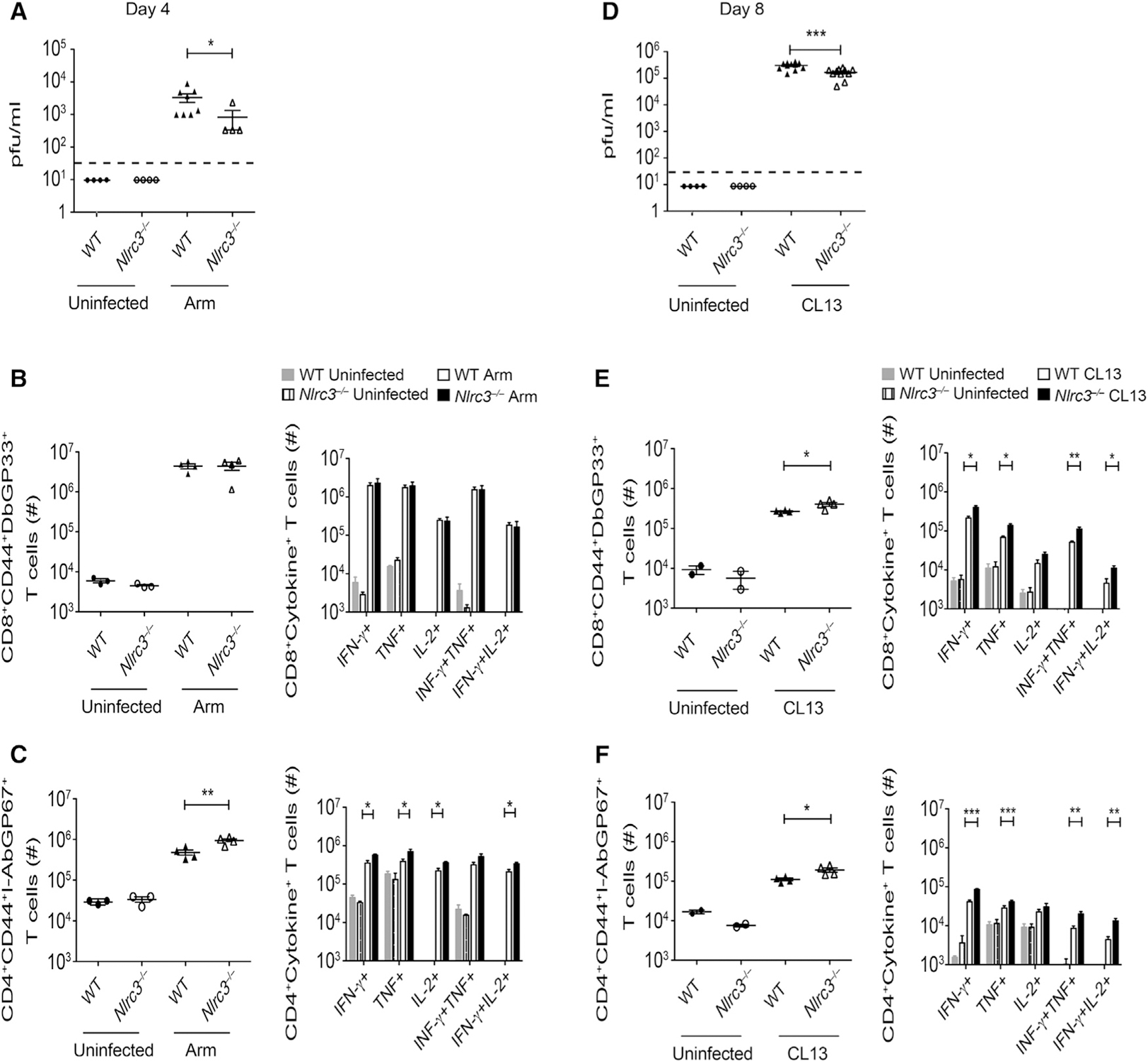
WT and Nlrc3−/− mice were given LCMV-Armstrong or LCMV-Clone 13 and analyzed for virus loads and T cell responses.
(A) Viral load in serum at day 4 after LCMV-Armstrong infection (n = 4 for WT uninfected, n = 4 for Nlrc3−/− uninfected, n = 8 for WT with Armstrong infection, n = 4 for Nlrc3−/− with Armstrong infection). Data are from one experiment representative of two independent experiments.
(B) Total number of LCMV-specific DbGP33+ CD8+ cells per spleen (left) and GP33-specific cytokine-producing CD8+ cells (right) 8 days after LCMV-Armstrong infection (n = 3 for WT uninfected, n = 3 for Nlrc3−/− uninfected, n = 4 for WT with Armstrong infection, n = 4 for Nlrc3−/− with Armstrong infection). Data are from one experiment representative of two independent experiments.
(C) Total number of LCMV-specific I-AbGP67+ CD4+ T cells per spleen (left) and GP61–80-specific cytokine-producing CD4+ cells (right) 8 days after LCMV-Armstrong infection (n = 3 for WT uninfected, n = 3 for Nlrc3−/− uninfected, n = 4 for WT with Armstrong infection, n = 4 for Nlrc3−/− with Armstrong infection). Data are from one experiment representative of two independent experiments.
(D) Viral load in serum at day 8 after LCMV-Clone 13 infection (n = 4 for Nlrc3−/− uninfected, n = 11 for WT with Clone 13 infection, n = 11 for Nlrc3−/− with Clone 13 infection). Data are pooled from two independent experiments.
(E) Total number of LCMV-specific DbGP33–41+ CD8+ cells (left) and GP33-specific cytokine-producing CD8+ cells (right) 8 days after LCMV-Clone 13 infection (n = 2 for WT uninfected, n = 2 for Nlrc3−/− uninfected, n = 4 for WT with Clone 13 infection, n = 4 for Nlrc3−/− with Clone 13 infection). Data are from one experiment representative of three independent experiments.
(F) Total number of LCMV-specific I-AbGP66–77 CD4+ T cells (left) and GP61–80-specific cytokine-producing CD4+ cells (right) 8 days after LCMV-Clone 13 infection (n = 2 for WT uninfected, n = 2 for Nlrc3−/− uninfected, n = 4 for WT with Clone 13 infection, n = 4 for Nlrc3−/− with Clone 13 infection). Data are from one experiment representative of three independent experiments.
Each symbol represents one mouse. Data are shown as mean ± SEM. Statistical significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S5.
In contrast to LCMV-Armstrong, LCMV-Clone 13 replicates to a high titer and widely disseminates in WT mice and establishes a chronic infection. Immune control of LCMV-Clone 13 requires CD8+ T cells that are heavily dependent on the activity of CD4+ T cells and B cells. Nlrc3−/− mice had significantly lower amounts of virus in serum than did WT mice at day 8 (Figure 4D). There were significantly more virus-specific tetramer-positive CD8+ T cells in Nlrc3−/− mice than in WT mice at day 8, and there was an increase in CD8+ T cells that could make cytokines upon re-stimulation (Figures 4E and S5B). In addition, Nlrc3−/− mice displayed an increase in the frequency and number of LCMV-specific CD4+ T cells and cells capable of producing cytokines (Figures 4F), consistent with the notion that NLRC3 negatively regulates T cell responses during disseminated infection.
NLRC3 Functions within CD4+ T Cells to Inhibit T Cell Accumulation and Expression of Pro-inflammatory Cytokines
The data above suggest that NLRC3 negatively regulated the CD4+ T cell response to LCMV, but the effect was modest, and a concern is that NLRC3 has known roles in other cell types, thus masking its function in T cells. To elucidate the intrinsic role of NLRC3 in CD4+ T cells, we performed competitive adoptive-transfer experiments where LCMV-specific WT and Nlrc3−/− CD4+ T cells were co-transferred into WT recipients, and the two donor cell populations in the same hosts were directly compared after infection. Thus, we crossed Nlrc3−/− mice (CD45.2+) with LCMV-specific TCR transgenic SMARTA mice that express the congenic CD45.1 allele to generate Nlrc3−/− SMARTA mice (CD45.1+ CD45.2+). Equal numbers of WT SMARTA T cells (CD45.1+/+) and Nlrc3−/− SMARTA T cells (CD45.1+CD45.2+) were co-transferred to the same WT C57BL/6 mice (CD45.2+), which were then infected with LCMV-Clone 13 (Figure 5A). Donor WT SMARTA T cells were identified by CD45.1 expression, whereas Nlrc3−/− SMARTA T cells were identified by their co-expression of CD45.1 and CD45.2.
Figure 5. NLRC3 Inhibits CD4+ T Cell Expansion and Pro-inflammatory Cytokine Production during LCMV Infection In Vivo.

WT mice received equal numbers of WT or Nlrc3−/− SMARTA CD4+ T cells. The recipients were given LCMV-Clone 13, and donor cell response was analyzed at multiple times after infection.
(A) Schematic of dual-transfer experiment of wild type SMARTA cells (CD45.1+) and Nlrc3−/− SMARTA cells (CD45.1+CD45.2+) into B6 mice (CD45.2+) (left). Flow analysis of donor WT SMARTA cells (CD45.1+) and donor Nlrc3−/− SMARTA cells (CD45.1+CD45.2+) is also shown (right).
(B) Percentage of SMARTA cells in peripheral blood at days 8, 22, and 42 after LCMV-Clone 13 infection (day 8: n = 3 recipients for WT uninfected, n = 3 recipients for Nlrc3−/−, n = 10 recipients for WT with Clone 13 infection, n = 10 recipients for Nlrc3−/− with Clone 13 infection; days 22 and 42: n = 3 recipients for WT uninfected, n = 3 recipients for Nlrc3−/− uninfected, n = 8 recipients for WT with Clone 13 infection, n = 8 recipients for Nlrc3−/− with Clone 13 infection).
(C) Percentage and total number of splenic SMARTA cells and intracellular cytokine staining for IFN-γ, TNF, and IL-2 8 days after Clone 13 infection (n = 3 recipients for WT uninfected, n = 3 recipients for Nlrc3−/− uninfected, n = 5 recipients for WT with Clone 13 infection, n = 5 recipients for Nlrc3−/− with Clone 13 infection).
(D) Percentage and total number of splenic SMARTA cells and intracellular cytokine staining for IFN-γ, TNF, and IL-2 at 42 days of Clone 13 infection (n = 3 recipients for WT uninfected, n = 3 recipients for Nlrc3−/− uninfected, n = 8 recipients for WT with Clone 13 infection, n = 8 recipients for Nlrc3−/− with Clone 13 infection).
Each symbol represents one mouse. Data are from one experiment representative of two independent experiments and are shown as mean ± SEM. Statistical significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S5.
Nlrc3−/− SMARTA T cells expanded significantly more than WT SMARTA cells in the peripheral blood of the same recipient mice at both early and late time points during LCMV-Clone 13 infection (Figure 5B). Similarly, splenic Nlrc3−/− SMARTA T cells were more frequent and numerous than WT SMARTA T cells in the same mice at day 8 (Figure 5C, left two panels). The Nlrc3−/− splenic T cells continued to be more abundant at day 42 (Figure 5D, left two panels), whereas WT SMARTA T cell responses subsided in the same hosts. In addition to increased expansion, there were more Nlrc3−/− SMARTA T cells that could make cytokine at early and late time points (Figures 5C and 5D, right two panels). These findings suggest that CD4+ T cell expression of Nlrc3 limits cell expansion and maintenance over time during LCMV infection.
NLRC3 Reduces IFN-γ Production in Autoreactive CD4+ T Cells In Vitro
The above results indicate that NLRC3 attenuates prolonged T cell activation during a viral infection. We considered that NLRC3 might reduce T cell auto-reactivity in models of autoimmunity. To explore this issue, we bred Nlrc3−/− mice with the 2D2 transgenic strain, which expresses a TCR specific to the myelin antigen, MOG35–55 on I-Ab. CD4+ T cells from 2D2 mice can respond to MOG antigen upon challenge and induce inflammation in the spinal cord, leading to experimental autoimmune encephalomyelitis (EAE), a model for T-cell-mediated multiple sclerosis (MS). We first determined whether NLRC3 intrinsically controls MOG-specific T cell responses. Because NLRC3 reduces immune signaling in myeloid cells, we sought to determine the role of NLRC3 in T cells versus myeloid cells for T cell responses. Thus, Nlrc3−/− and WT CD4+ T cells were co-cultured with either Nlrc3−/− or WT dendritic cells (DCs) that were pulsed with MOG35–55. Significantly higher amounts of IFN-γ were detected in cultures with Nlrc3−/− 2D2 T cells than in those with Nlrc3+/+ 2D2 T cells irrespectively of the genotype of the DCs (Figure 6A). Similar findings were observed when CD4+ T cells were differentiated into Th1 cells by the addition of IL-12 and anti-IL-4 into the co-cultures (Figure 6B). Cultures containing Nlrc3−/− 2D2 T cells had higher concentrations of IFN-γ than those containing Nlrc3+/+ 2D2 T cells irrespectively of whether the DCs were Nlrc3+/+ or Nlrc3−/−. These data are consistent with the fact that NLRC3 acts within T cells to suppress IFN-γ expression.
Figure 6. Nlrc3–/– Mice Are More Susceptible to EAE.

(A) MOG35–55 coated DCs from WT and Nlrc3–/– mice were co-cultured with 2D2 CD4+ T cells from 2D2-Tg+Nlrc3+/+ or 2D2-Tg+Nlrc3–/– mice. The graph shows amounts of IFN-γ in the supernatant as measured by ELISA.
(B) ELISA analysis of IFN-γ in the supernatant at 36 hr co-culture with MOG35–55-stimulated DCs from WT and Nlrc3–/– mice and 2D2 CD4+ T cells from 2D2 Tg+Nlrc3+/+ or 2D2 Tg+Nlrc3–/– mice under Th1 condition.
(C) WT and Nlrc3–/– mice were immunized with MOG35–55 CFA and pertussis toxin for the induction of EAE. The graph shows the clinical score of EAE (n = 11 for WT mice, n = 10 for Nlrc3–/– mice). Data are pooled from two independent experiments.
(D) Total number of infiltrating IFN-γ+CD4+ T cells, IL-17+CD4+ T cells, IL-4+CD4+ T cells, and Foxp3+CD25+CD4+ T cells in the spinal cord 15 days after EAE (n = 9 for WT mice, n = 9 for Nlrc3–/– mice).
(E) CD4+ T cells were isolated from WT and Nlrc3–/– mice and adoptively transferred into Rag1–/– mice; 1 day later, the recipient mice were vaccinated for the induction of EAE. The graph shows the clinical score of EAE (n = 11 for Rag1–/– mice transferred with WT CD4+ T cells, n = 10 for Rag1–/– mice transferred with Nlrc3–/–CD4+ T cells). Data are pooled from two independent experiments.
(F) T cells were isolated from the draining lymph nodes of Rag1–/– recipients of WT or Nlrc3−/− CD4 T+ cells at day 18 after MOG vaccination and were re-stimulated with MOG35–55 antigen in a recall assay. Supernatants collected 48 hr later were analyzed by ELISA for IFN-γ and IL-17A (n = 3 for WT untreated [UT]), n = 3 for Nlrc3−/− UT, n = 4 for WT, n = 7 for Nlrc3−/−). Data are from one experiment representative of two independent experiments.
(G) The heatmap is based on RNA-seq data comparing MOG-reactive or -non-reactive CD4+ T cells from MS subjects and healthy control individuals.
(H) Mouse Th0 cells were untreated or treated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) and then analyzed by RNA-seq. The heatmap shows the relative amounts of RNA for WT and Nlrc3−/− CD4+ T cells.
Data are shown as mean ± SEM. Significance was determined by unpaired t test. *p < 0.05, **p < 0.01, ***p < 0.001.
Nlrc3–/– Mice Are More Susceptible to EAE
Next, we examined the in vivo role of NLRC3 in the EAE model to determine whether it plays a role in a CD4+ T-cell-driven disease model. Nlrc3−/− mice were more susceptible to EAE than WT mice and had significantly higher clinical scores in the effector phase (Figure 6C). We quantified the number of Th1, Th2, Th17, and regulatory T (Treg) cells infiltrating into the spinal cords of WT and Nlrc3−/− mice. Significantly higher numbers of IFN-γ+CD4+ T cells and IL-17+CD4+ cells were present in the spinal cords of Nlrc3−/− mice than in those of WT mice (Figure 6D), though there was no significant difference in the number of IL-4+CD4+ cells and FoxP3+CD25+CD4+ T cells.
To address whether NLRC3 plays a T-cell-intrinsic role in the EAE mouse model, we isolated CD4+ T cells from either unimmunized WT or Nlrc3−/− mice and transferred them into separate Rag1–/– recipients (Nlrc3+/+); we immunized the recipients with MOG35–55 to induce EAE. Rag1–/– recipients receiving Nlrc3−/− CD4+ T cells developed significantly worse EAE than those receiving WT CD4+ T cells (Figure 6E). At day 18, after MOG35–55 stimulation and before the onset of clinical symptoms, T cells were isolated from the draining lymph nodes of the recipients and re-stimulated in vitro with MOG35–55 peptide. Nlrc3−/− CD4+ T cells expressed more IFN-γ and IL-17A than WT CD4+ T cells (Figure 6F). Collectively, these findings indicate that NLRC3 acts in a cell-intrinsic fashion to suppress CD4+ T cells in the EAE model.
To more broadly identify the genes that might be altered by NLRC3, we analyzed a public database of MS patients (Figure 6G). We examined RNA sequencing (RNA-seq) data from the NCBI Gene Expression Omnibus (GEO) (Cao et al., 2015) to identify gene expression patterns relevant to human NLRC3 expression. Gene expression was compared in MOG-non-reactive versus MOG-reactive CD4+ T cells isolated from healthy control individuals and MS patients. CD4+ T cells from healthy control individuals expressed high amounts of NLRC3, whereas expression was reduced in CD4+ T cells from subjects with MS, especially in MOG-specific CD4+ T cells where IFNG, IL17A, IL17F, AHR, IL2, and TNF were elevated. These indicate a negative correlation between NLRC3 and Th1 and Th17 cell signatures. To establish a causal relationship between Nlrc3 and T cell response genes, we performed RNA-seq analysis on Th0 WT and Nlrc3−/− CD4+ T cells stimulated with or without anti-CD3 and anti-CD28 (Figure 6H). The heatmap showed that Nlrc3−/− resulted in the elevation of a number of genes associated with Th1, Th17, and T cell activation, including Tnf, Ifng, Tbx21, Il17a, Il17f, and Nfatc1. This is consistent with the human data. Nlrc3−/− T cells also showed elevated Foxp3 and Il4. It will be of interest in the future to perform similar analyses under Treg- or Th2-cell-inducing conditions.
NLRC3 Suppresses Immune Signaling in CD4+ T Cells
We next explored the signaling pathway(s) that NLRC3 uses to limit CD4+ T cell activation and proliferation. Purified Nlrc3−/− CD4+ but not CD8+ T cells stimulated with anti-CD3 and anti-CD28 showed enhanced phosphorylation of p65 and STAT1, a marker for IFN-γ signaling (Figure 7A). However, phosphorylation of ZAP70, a tyrosine kinase adaptor that lies downstream of CD3-zeta and is important for proximal TCR signaling, was not altered by NLRC3 (Figures 7B and S6A). Additionally, NLRC3 did not affect the phosphorylation of PLC-g1, another molecule that is recruited to the TCR signaling complex (Figure S6B). WT DCs induced the nuclear translocation of NFAT-1 from the cytoplasm of WT CD4+ T cells, and Nlrc3−/− CD4+ T cells showed a similar degree of NFAT-1 translocation (Figure S6C). T-bet and Eomes transcription factors (Szabo et al., 2002) are important for IFN-γ transcription in T cells; however, T-bet and Eomes were similarly phosphorylated in Nlrc3−/− and WT CD4+ T cells after DC-induced stimulation (Figure S6D). However, Nlrc3−/− T cells showed more phosphorylated STAT1 than WT CD4+ T cells (Figure S6D). The inability to detect these differences by western blot, in contrast to RNA-seq, is most likely due to the sensitivity limit of western blots. Because NLRC3 inhibits mTOR pathways in colon epithelial cells (Karki et al., 2016), we considered that NLRC3 might negatively regulate signaling along the mTOR pathway in CD4+ T cells. Anti-CD3- and anti-CD28-mediated stimulation of Nlrc3−/− CD4+ T cells led to enhanced phosphorylation of 4E-BP1, which lies downstream of mTOR, but there was no detectable difference in mTOR or AKT phosphorylation (Figures 7C and S6E). Recently, several kinases have been shown to phosphorylate 4E-BP1 independently of mTOR, and NLRC3 could cause changes in 4E-BP1 in the absence of mTOR activation (Qin et al., 2016).
Figure 7. NLRC3 Suppresses Immune Signaling in CD4+ T Cells In Vitro and In Vivo.

(A and B) Western blot analyses of purified splenic CD4+ T cells and CD8+ T cells stimulated by anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies.
(C) Western blot analyses of purified splenic CD4+ T cells stimulated by anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies.
(D) Western blot analyses of spinal cords from CD4+ T-cell-transferred Rag1–/– mice 18 days after induction of EAE. Each lane represents a spinal cord from a different mouse.
(E) Densitometric analysis of (D).
(F) WT and Nlrc3−/− CD4+ T cells stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) antibodies, and then endogenous TRAF6 immune-precipitation was performed. The western blot shows K63-linked ubiquitination (K63-Ub) of TRAF6.
(G) CD4+ T cells from spleens of WT or Nlrc3−/− mice were stimulated or not with anti-CD3 and anti-CD28 antibodies for 24 hr and then assayed for ECA) or OCR. Cells were measured over time and exposed (at the indicated time points) to glucose, oligomycin, and 2-DG for ECAR measurement or to oligomycin, FCCP, and rotenone for OCR assessment. ECARs and OCRs were recorded three times per condition. The basal ECAR, glycolysis (ECAR after glucose addition), glycolytic capacity (maximal ECAR after subtracting the ECAR following 2-DG exposure), and glycolytic reserve (difference between oligomycin-induced maximal ECAR and glucose-induced glycolytic flux) were calculated.
(H) Cells were cultured as in (G) except that basal OCR, ATP-linked mitochondrial respiration (difference between basal respiration and OCR after the addition of oligomycin), maximal respiration (FCCP-induced maximal OCR minus the portion of nonmitochondrial OCR due to rotenone treatment), and spare respiratory capacity (difference between maximal OCR and basal respiration) were measured.
(I) CD4+ T cells from WT or Nlrc3−/− mice were stimulated with anti-CD3 and anti-CD28 antibodies (Stim) together with BAY11–7082 inhibitor (1 mM) or DMSO for 24 hr and then subjected to ECAR analysis.
(J) ECAR analysis of CD4+ T cells from WT or Nlrc3−/− mice that were stimulated for 24 hr with anti-CD3 and anti-CD28 antibodies (Stim) in the presence of QNZ inhibitor (1 mM) or DMSO.
Data are from one experiment representative of two or three independent experiments. Data are shown as mean ± SEM. Significance was determined by Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figures S6 and S7.
The data above suggest that NLRC3 attenuates NF-κB and 4E-BP1 activation but not TCR proximal signaling. To assess whether NLRC3 negatively regulates NF-κB signaling in vivo, we transferred CD4+ T cells from WT or Nlrc3−/− mice into Rag1–/– mice that were then given MOG35–55 peptide. T cells were isolated from spinal cords 18 days after MOG35–55 vaccination and analyzed by western blot (Figures 7D and 7E). Nlrc3−/− CD4+ T cells showed increased phosphorylation of p65, whereas mTOR, Akt, and S6K, the last of which resides downstream of mTOR, were not altered. 4E-BP1 was below the limits of detection for these in vivo samples (data not shown). These findings suggest that NLRC3 regulates canonical NF-κB signaling in CD4+ T cells.
NLRC3 interacts with TRAF6 in peritoneal macrophages to attenuate K63-linked ubiquitination of TRAF6, a post-translational modification that activates TRAF6 in innate cells (Schneider et al., 2012). To determine whether NLRC3 regulates this step to control NF-κB signaling in T cells, we performed a co-immunoprecipitation assay to evaluate K63-linked ubiquitination of TRAF6 in CD4+ T cells isolated from WT and Nlrc3−/− mice. We detected faster K63-linked ubiquitination of TRAF6 in Nlrc3−/− CD4+ T cells than in WT CD4+ T cells at the 5 min time point (Figure 7F). In addition, TRAF6 and p-p65 were more abundant in Nlrc3−/− CD4+ T cells than in WT CD4+ T cells (Figure 7F). These findings suggest that NLRC3 negatively regulates K63-linked ubiquitination of TRAF6 and reduces downstream NF-κB signaling.
NLRC3 Diminishes Metabolic Pathways to Attenuate T Cell Activation in a NF-κB-Dependent Manner
T cell activation and differentiation are associated with metabolic rewiring (Pearce et al., 2013), such that aerobic glycolysis is a metabolic hallmark of activated T cells and Th1 cell differentiation (Peng et al., 2016). Because our results indicate that NLRC3 negatively regulates IFN-γ production and proliferation in activated CD4+ T cells in vitro and in vivo, we investigated whether the absence of NLRC3 would alter T cell metabolism. Using the Seahorse Extracellular Flux XF24 Analyzer, we estimated the bioenergetics profile of naive and activated CD4+ T cells by measuring extracellular acidification rates (ECARs) and oxygen consumption rates (OCRs), indicators of glycolytic activity and mitochondrial oxidative phosphorylation (OXPHOS), respectively (TeSlaa and Teitell, 2014; van der Windt et al., 2016). Glycolytic and mitochondrial function of WT and Nlrc3−/− CD4+ T cells were determined 24 hr after stimulation with anti-CD3 and anti-CD28 (basal rate) and after the addition of compounds that modulate ECARs or OCRs (Figures 7G–7J).
WT and Nlrc3−/− CD4+ T cells were in a low metabolic state without stimulation, whereas TCR signaling promptly increased the glycolytic flux, especially in Nlrc3−/− CD4+ T cells (Figure 7G). Although the glycolytic responses were comparable between WT and Nlrc3−/− CD4+ cells when exposed to glucose, Nlrc3−/− T cells reached a significantly higher maximum ECAR (glycolytic capacity) after the addition of the ATP synthase inhibitor oligomycin, which shuts down OXPHOS to force cells to use glycolysis as the main energy source (Figure 7G). Untreated WT or Nlrc3−/− CD4+ T cells exhibited lower mitochondrial metabolic profiles than their activated counterparts. T cell activation increased the mitochondria respiration rate in both genotypes, but the increase in Nlrc3−/− CD4+ T cells was significantly higher than that observed in WT CD4+ T cells. The absence of Nlrc3 in activated CD4+ T cells also resulted in higher usage of OXPHOS for driving ATP production (ATP-linked respiration) and maximized OXPHOS activity after cell exposure to FCCP (carbonylcyanide-4-trifluoro-methoxyphenylhydrazone), which uncouples oxygen consumption from ATP production and raises OCR to a maximal value, as seen for the maximum ECAR measurement (Figure 7H). The higher glycolytic capacity, glycolytic reserve, and maximal mitochondrial respiration in active Nlrc3−/− CD4+ T cells indicate a greater ability to operate at maximal metabolic capacity and cope with sudden ‘‘high energy demand.’’ This capability most likely fuels the energy necessary for the enhanced proliferation and cytokine production in Nlrc3−/− CD4+ T cells.
We next assessed whether the higher glycolytic response in Nlrc3−/− CD4+ T cells is dependent on the increased NF-κB signaling, as observed earlier. Simultaneous treatment of CD4+ T cells with anti-CD3 and anti-CD28 antibodies and the NF-κB inhibitor BAY11–7082, which targets IκBα phosphorylation, abolished the induction of a glycolytic response by TCR signaling activation and resulted in similar ECARs between activated WT and Nlrc3−/− CD4+ T cells (Figures 7I and S7A). To exclude off-target and toxic effects of BAY11–7082, we evaluated a different NF-κB inhibitor, QNZ, which is a quinazoline derivative (Tobe et al., 2003). QNZ also eliminated the difference between WT and Nlrc3−/− CD4+ T cells (Figures 7J and S7B). Treatment of unstimulated or anti-CD3- and anti-CD28-stimulated T cells with BAY11–7082 did not result in changes in cell death (Figure S7C) but did result in reduced NF-κB activation (Figure S7D). These results suggest that NLRC3 controls CD4+ T cell metabolism by limiting maximal glycolytic and mitochondrial metabolism and by attenuating TCR-induced glycolytic flux in an NF-κB-dependent manner. In total, our findings indicate that NLRC3 attenuates CD4+ T cell responses by targeting pathways downstream of TCR signaling (TRAF6 ubiquitination and NF-κB activation) and by interfering with T cell metabolism. Thus, NLRC3 is an innate immune receptor that guards against excessive T cell proliferation and cytokine expression and demonstrates crossover regulatory functions in adaptive immunity.
DISCUSSION
Immune regulatory pathways are critical for preventing over-zealous and detrimental inflammation and autoimmunity. A prominent example is the adverse reaction associated with the use of immune checkpoint inhibitors in cancer treatment (Friedman et al., 2016), which can result in severe autoimmunity. Although most studies of T cell checkpoint proteins have focused on molecules long known to be prominent in T cells (Sharpe and Pauken, 2018), the discovery of alternative immune checkpoints is an area of active pursuit because of their relevance to cancer immunotherapy, sepsis control, and auto-immunity. In this study, we have described an unappreciated role for an innate immune receptor member during T cell responses to infection and T-cell-mediated diseases. NLRC3 exhibited an intrinsic role in T cells, and its deficiency in CD4+ T cells resulted in greater activation, proliferation, and inflammatory cytokine production. The relevance of this finding is evident in both viral infection and autoimmunity. Ablation of Nlrc3 led to enhanced antiviral cytokine production and improved control of LCMV. In parallel, Nlrc3−/− mice exhibited hyper-inflammatory T cell responses in the EAE model and displayed more severe paralysis than wild-type mice. Gene profiling also indicated that NLRC3 affects Th1 and Th17 effector cells, an observation found in both the EAE model in mice and human MOG-specific CD4+ T cells from MS patients.
Mechanistically, NLRC3 reduced NF-κB activation by attenuating K63-linked ubiquitination of TRAF6 in T cells after TCR stimulation. This is consistent with a previous study showing that NLRC3 impedes K63 ubiquitination of TRAF6 to attenuate NF-κB activation in peritoneal macrophages (Schneider et al., 2012). The effect of NLRC3 on NF-κB activation was also observed in vivo after adoptive transfer of CD4+ T cells. NF-κB activation regulates glycolysis after T cell stimulation, whereas the glycolytic pathway promotes optimal proliferation and inflammatory cytokine production in Th1 cells (Peng et al., 2016). However, the activation of CD4+ T cells, but not CD8+ T cells, is also accompanied by an increase in oxidative metabolism (Cao et al., 2014). The dual reduction of glycolysis and oxidative metabolism by NLRC3 could explain the suppressive effect of NLRC3 on CD4+ T cells but not on CD8+ cells, although this remains to be explored. The negative regulatory function of NLRC3 is consistent with its reduced expression in activated CD4+ T cells, suggesting that activation of these cells requires the lowering of Nlrc3 expression to permit proliferation and inflammatory cytokine induction. In total, these findings define NLRC3 as a checkpoint protein of CD4+ T lymphocyte activation.
Although the roles of PRRs among innate immune cells are well described, their functions in T cells are less studied. Among TLRs, TLR2 is the most widely expressed in T cells (Imanishi et al., 2007), and its role in T cell proliferation and IFN-γ production is documented (Reynolds and Dong, 2013). TLR2 promotes Th17 cell proliferation and cytokine production, and the loss of TLR2 in CD4+ T cells dramatically ameliorates EAE (Reynolds et al., 2010). TLR4 signaling also directly regulates CD4+ T cells function (González-Navajas et al., 2010). Finally, the TLR adaptor MyD88 augments PI-3 kinase signaling in T cells (Gelman et al., 2006). The expression of most NLRs in T cells is low to nil. However, Nlrp12 / mice develop an atypical EAE disease via enhanced IL-4 production in T cells (Lukens et al., 2015), and another EAE study reached a different conclusion to suggest that NLRP12 mitigates inflammatory myeloid cells (Gharagozloo et al., 2015). Similar to NLRP12, NLRP3 can bind and regulate the Il4 promoter (Bruchard et al., 2015). No study has shown a role for NLR in IFN-γ expression in T cells.
TCR signaling results in the activation of membrane proximal events followed by downstream signals NF-κB, NFAT and AP-1, which lead to IFN-γ production by T cells. NLRC3 did not cause detectable changes in proximal TCR signals but rather reduced downstream immune signaling, which is consistent with its role in the cytosol. Our findings are consistent with the notion that NLRC3 acts in a T-cell-intrinsic fashion to limit CD4+ T cell responses. For example, Nlrc3−/− CD4+ T cells induced heightened EAE symptoms in Rag1–/– mice. Additionally, Nlrc3−/− SMARTA TCR transgenic CD4+ T cells responded more vigorously than Nlrc3+/+ SMARTA cells in the same LCMV-infected mice, consistent with the fact that NLRC3 acts within CD4+ T cells to limit early CD4+ T cell responses during infection. Over the course of chronic LCMV infection, Nlrc3−/− T cells sustained cytokine responses at times when WT T cell responses had subsided, suggesting that NLRC3 might be involved in resolving immune responses during protracted infection. It is also notable that the inhibitory role of NLRC3 on T cells is moderate, suggesting that it acts as a rheostat rather than an on-off switch. Hence, NLRC3 might be attractive as a target of checkpoint blockade because its inhibition could improve T cell responses without precipitating overt auto-immunity.
In summary, we have identified NLRC3 as a negative CD4+ T cell regulator affecting both Th1 and Th17 cells in a T-cell-intrinsic fashion to limit cellular activation, proliferation, and downstream IFN-γ and TNF expression through interference with NF-κB signaling. Nlrc3−/− mice showed exacerbated T-cell-mediated autoimmunity and prolonged CD4+ T cell responses to virus infection, suggesting that NLRC3 acts as an intrinsic attenuator of T cell activity. NLRC3-targeted therapies could be attractive as treatment strategies for alleviating T-cell-mediated inflammatory disease or for resolving chronic viral infection. Consistent with our findings, a recent paper has reported that NLRC3 attenuates Th1 and Th17 responses during M. tuberculosis infection, indicating that NLRC3 also lowers T cell response against bacteria (Hu et al., 2018). Together, these results indicate that NLRC3 serves as a general mitigator of CD4+ T cell activation in response to viruses, bacteria, and auto-antigens.
STAR*METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TruStain fcX (anti-mouse CD16/32) | Biolegend | Cat#101320, RRID: AB_1574975 |
| Anti-mouse CD3, APC, clone 17A2 | Biolegend | Cat#100236, RRID: AB_2561456 |
| Anti-mouse CD3, FITC, clone 17A2 | Biolegend | Cat#100204, RRID: AB_312661 |
| Anti-mouse CD4, APC, clone RM4–5 | Biolegend | Cat#100516, RRID: AB_312719 |
| Anti-mouse CD4, PE, clone RM4–5 | Biolegend | Cat#100511, RRID: AB_312714 |
| Anti-mouse CD4, PerCP, clone RM4–5 | Biolegend | Cat#100538, RRID: AB_893325 |
| Anti-mouse CD4, FITC, clone RM4–5 | Biolegend | Cat#100510, RRID: AB_312713 |
| Anti-mouse CD8, APC, clone 53–6.7 | Biolegend | Cat#100712, RRID: AB_312751 |
| Anti-mouse CD8, PE, clone 53–6.7 | Biolegend | Cat#100707, RRID: AB_312746 |
| Anti-mouse CD8, FITC, clone 53–6.7 | Biolegend | Cat#100706, RRID: AB_312745 |
| Anti-mouse CD25, PE, clone 3C7 | Biolegend | Cat#101904, RRID: AB_312847 |
| Anti-mouse CD44, FITC, clone IM7 | Biolegend | Cat#103006, RRID: AB_312957 |
| Anti-mouse CD45.1, PE, clone A20 | Biolegend | Cat#110708, RRID: AB_313497 |
| Anti-mouse CD45.1, FITC, clone A20 | Biolegend | Cat#110706, RRID: AB_313495 |
| Anti-mouse CD45.2, APC, clone 104 | Biolegend | Cat#109814, RRID: AB_389211 |
| Anti-mouse CD45.2, PE, clone 104 | Biolegend | Cat#109808, RRID: AB_313445 |
| Anti-mouse CD45RB, APC, clone C363–16A |
Biolegend | Cat#103320, RRID: AB_2565229 |
| Anti-mouse CD62L, APC, clone MEL-14 | Biolegend | Cat#104412, RRID: AB_313099 |
| Anti-mouse CD69, FITC, clone H1.2F3 | Biolegend | Cat#104506, RRID: AB_313109 |
| Anti-mouse B220, PE, clone B6.220 | Biolegend | Cat#352604RRID: AB_10933245 |
| Anti-mouse NK1.1, APC, clone PK136 | Biolegend | Cat#108710, RRID: AB_313397 |
| Anti-mouse IFN-γ, PE, clone XMG1.2 | Biolegend | Cat#505808, RRID: AB_315402 |
| Anti-mouse IFN-γ, FITC, clone XMG1.2 | Biolegend | Cat#505806, RRID: AB_315400 |
| Anti-mouse TNF, APC, clone MP6-XT22 | Biolegend | Cat#506308, RRID: AB_315429 |
| Anti-mouse TNF, PE, clone MP6-XT22 | Biolegend | Cat#506306, RRID: AB_315427 |
| Anti-mouse IL-2, APC, clone JES6–5H4 | Biolegend | Cat#503810, RRID: AB_315304 |
| Anti-mouse IL-2, FITC, clone JES6–5H4 | Biolegend | Cat#503806, RRID: AB_315300 |
| Anti-mouse T-bet, APC, clone 4B10 | Biolegend | Cat#644813, RRID: AB_10896913 |
| Anti-mouse Stat1 (Y701), Alexa Fluor 647, clone: 4a | BD Bioscience | Cat#562070 RRID: AB_10896129 |
| Anti-actin, HRP, clone: C4 | Santa Cruze Technology | Cat#47778 RRID: AB_2714189 |
| Goat anti-rabbit, HRP, polyclonal | Jackson ImmunoResearch labs | Cat#111–035-144, RRID: AB_2307391 |
| Goat anti-mouse, HRP, polyclonal | Jackson ImmunoResearch labs | Cat#115–035-146, RRID: AB_2307392 |
| Anti-Histone H3 | Millpore sigma | Cat#07–690 RRID: AB_417398 |
| Anti-NF-κB p65, clone: D14E12 | Cell Signaling Technology | Cat#8242 RRID: AB_10859369 |
| Anti-pp65 (S536), clone: 93H1 | Cell Signaling Technology | Cat#3033 RRID: AB_331284 |
| Anti-STAT1 | Cell Signaling Technology | Cat#9172 RRID: AB_10693929 |
| Anti-pSTAT1 (Tyr701), clone: 58D6 | Cell Signaling Technology | Cat#9167 RRID: AB_561284 |
| Anti-ZAP70, clone: D1C10E | Cell Signaling Technology | Cat#3165 RRID: AB_2218656 |
| Anti-pZAP70 (Tyr319)/Syk(Tyr352), clone: 65E4 | Cell Signaling Technology | Cat#2717 RRID: AB_2218658 |
| Anti-PLC-g1 | Cell Signaling Technology | Cat#2822 RRID: AB_10691689 |
| Anti-pPLC-g1 (Tyr783), clone: D6M9S | Cell Signaling Technology | Cat#14008 |
| Anti-NFAT, clone: D43B1 | Cell Signaling Technology | Cat#5861 RRID: AB_10835147 |
| Anti-mTOR | Cell Signaling Technology | Cat#2972 RRID: 330978 |
| Anti-pmTOR (Ser2448), clone: D9C2 | Cell Signaling Technology | Cat#5536 RRID: AB_10691552 |
| Anti-AKT | Cell Signaling Technology | Cat#9272 RRID: AB_329827 |
| Anti-pAKT (Thr308), clone: 244F9 | Cell Signaling Technology | Cat#4056 RRID: AB_331163 |
| Anti-pAKT (Ser473), 587F1 | Cell Signaling Technology | Cat#4051 RRID: AB_331158 |
| Anti-4E-BP1, clone: 53H11 | Cell Signaling Technology | Cat#9644 RRID: AB_2097841 |
| Anti-p4E-BP1 (Thr37/46), clone: 236B4 | Cell Signaling Technology | Cat#2855 RRID: AB_560835 |
| Anti-S6K, clone 49D7 | Cell Signaling Technology | Cat#2708 RRID: AB_390722 |
| Anti-pS6K (Thr389) | Cell Signaling Technology | Cat#9205 RRID: AB_330944 |
| Anti-cMyc, clone: D84C12 | Cell Signaling Technology | Cat#5605 RRID: AB_1903938 |
| Anti-pc-Myc (Ser629. Clone: E1J4K | Cell Signaling Technology | Cat#13748 RRID: AB_2687518 |
| Anti-JNK | Cell Signaling Technology | Cat#9252 RRID: AB_2250373 |
| Anti-pJNK (Thr183/Tyr185) | Cell Signaling Technology | Cat#9251 RRID: AB_331659 |
| Anti-ERK½, clone: 3A7 | Cell Signaling Technology | Cat#9107 RRID: AB_2235073 |
| Anti-pERK1/1 (Thr202/Tyr204), clone: D13.14.4E | Cell Signaling Technology | Cat#4370 RRID: AB_2315112 |
| Anti-TRAF6, Clone: D-10 | Santa Cruz Biotechnology | Cat# sc-8409 RRID: AB_628391 |
| Anti-K63 Ub, Clone: D7A11 | Cell Signaling Technology | Cat# 5621 |
| Bacterial and Virus Strains | ||
| LCMV (Armstrong) | Whitmire laboratory | N/A, generated in house |
| LCMV (Clone 13) | Whitmire laboratory | N/A, generated in house |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MOG35–55 | GenemeSynthesis,Inc | Cat#SP-51716–1 |
| Pertussis Toxin | LIST Biological Laboratories | Cat#180 |
| Killed M.tuberculosis H37 Ra | ThermoFish Scientific | Cat#DF3114–33-8 |
| Freund’s Adjuvant, complete | Sigma-Aldrich | Cat#F5381 |
| Brefeldin A Solution (1,000X) | Biolegend | Cat#420601 |
| Fixation Buffer | Biolegend | Cat#420801 |
| Intracellular Permeabilization Wash Buffer | Biolegend | Cat#421002 |
| Biotinylated DbGP33–41 monomer | NIH Tetramer core | N/A |
| APC-conjugated I-AbGP67 tetramer | NIH Tetramer core | N/A |
| CellTrace™ CFSE Cell Prokiferation Kit | ThermoFisher Scientific | Cat#C34554 |
| Ghost Dyes™ Reb 780 | TONBO bioscience | Cat#13–0865 |
| CD3e Monoclonal Antibody | Invitrogen | Cat#16–0031-85 |
| CD28 Monoclonal Antibody | Invitrogen | Cat#16–0281-85 |
| Phorbol 12-myristate 13-acetate (PMA) | Abcam | Cat#ab120297 |
| Ionomycin calcium salt | Sigma-Aldrich | Cat#10634 |
| BAY11–7082 | Selleck chem | Cat#S2913 |
| QNZ | Selleck chem | Cat#S4902 |
| Glucose solution | GIBCO | Cat#A2494001 |
| L-Glutamine | GIBCO | Cat#25030081 |
| Oligomycin A | Sigma-Aldrich | Cat#75351 |
| Rotenone | Sigma-Aldrich | Cat#R8875 |
| Carbonyl cyanide 4-(trifluoromethoxy) Phenylhydrazone (FCCP) | Sigma-Aldrich | Cat#C2920 |
| 2-Deoxy-D-glucose (2-DG) | Sigma-Aldrich | Cat#D8375 |
| Seahorse XF Calibrant Solution | Agilent | Cat#100840–000 |
| Seahorse XF Base Medium | Agilent | Cat#103335–100 |
| RPMI | GIBCO | Cat#12–167F |
| FBS | GIBCO | Cat#26140–079 |
| ACK lysing buffer | GIBCO | Cat#A1049201 |
| Recombinant Murine GM-CSF | Peprotech | Cat#315–03 |
| Percoll | GE Healthcare Percoll Centrifugation Media | Cat#17089102 |
| RIPA Buffer With Triton X-100 | Boston Bioproducts | Cat#BP-116TX |
| PhosStop | Roche | Cat#4906845001 |
| Complete Ultra Tablets | Roche | Cat#5892791001 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74106 |
| Mouse IFN-γ ELISA MAX™ Standard | Biolegend | Cat#430803 |
| Mouse TNF-a ELISA MAX™ Standard | Biolegend | Cat#430903 |
| Mouse IL-2 ELISA MAX™ Standard | Biolegend | Cat#431003 |
| Mouse IL-17 ELISA MAX™ Standard | Biolegend | Cat#432503 |
| EasySep™ Mouse Naive CD4+ T cell isolation Kit | STEMCELL | Cat#19765 |
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Cat#130–049-201 |
| CD4+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat#130–104-454 |
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat#130–104-075 |
| CD11c Microbeads UltraPure, mouse | Miltenyi Biotec | Cat#130–108-338 |
| Experimental Models: Cell lines | ||
| Vero-E6 | Michael Buchmeier | The Scripps Research Institute |
| BHK-21 | American Type Culture Collection | Cat#CCL-10 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat#000664 |
| Mouse: B6.Nlrc3–/– | Ting laboratory | N/A (Schneider et al., 2012) |
| Mouse: C57BL/6-Tg(Tcra2D2, Tcrb2D2) 1 Kuch/J | Jackson Laboratory | Cat#006912 |
| Mouse: B6.129S7-Rag1tmMomIJ | Jackson Laboratory | Cat#0002216 |
| Mouse: B6.Cg-Ptprca Pepcb Tg(TcrLCMV) 1Aox/PpmJ (SMARTA/CD45.1) | Whitmire laboratory | N/A |
| Oligonucleotides | ||
| PCR primer: Nlrc3 | Thermofisher | Assay ID: Mm00615968 Cat#4331182 |
| PCR primer: 18 s rRNA | Thermofisher | Cat#433760F |
| Software and Algorithms | ||
| Flo-Jo Software (version 9.8.3) | FlowJo | https://www.flowjo.com/ |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com |
| ImageJ bundle with Java1.80_101 | ImageJ | https://imagej.nih.gov |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jenny Ting (jenny_ting@med.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Nlrc3−/− mice have been previously described (Schneider et al., 2012, Zhang et al., 2014). C57BL/6J (WT), 2D2 TCR transgenic mice and Rag1–/– mice were purchased from The Jackson Laboratory. SMARTA TCR transgenic mice were provided by Dr. Jason K. Whitmire at the University of North Carolina. Nlrc3−/− mice were crossed with SMARTA TCR transgenic mice to generate SMARTA Tg+Nlrc3−/− mice and with 2D2 TCR transgenic mice to generate 2D2 Tg+Nlrc3–/– mice. Mice were bred and maintained in specific pathogen-free conditions in the animal facilities at UNC-Chapel Hill. All mice used for experiments were between 8 to 12 weeks of age and female mice were used in EAE experiments. All mouse studies were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and Institutional Animal Care and Use Committee guidelines of the University of North Carolina Chapel Hill.
Virus
Mice received 2 3 105 plaque-forming units (PFU) of the LCMV Armstrong strain by intraperitoneal injection or 2 3 106 PFU of the LCMV Clone13 strain by intravenous tail-vein injection. Viral stocks of LCMV were generated from infected BHK-21 cells. Viral titer in serum, liver, lung, and kidney were quantified by plaque assay on Vero cell monolayers.
EAE
EAE was induced in age-matched WT and Nlrc3−/− female mice by subcutaneous injection of 250 mg MOG35–55 peptide per mouse emulsified in complete Freund’s adjuvant (CFA). The dosage of total heat-killed Mycobacterium tuberculosis was 200 mg per mouse. On days 0 and 2, mice received intraperitoneal injection of 200 ng pertussis toxin. Symptoms of EAE were monitored daily using a standard clinical score ranging from 0 to 5 as follows: 0, asymptomatic; 1, tail paralyzed; 2, hind limb paresis; 3, both hind limbs paralyzed; 4, forelimbs paralyzed; and 5, moribund, as previously described (Stromnes and Goverman, 2006).
METHOD DETAILS
In vitro CD4+ T cell activation assay
Spleen cells were isolated from WT and Nlrc3−/− mice. CD4+ T cells were negatively selected using mouse CD4+ T Cell Isolation Kit or EasySep™. Mouse Naive CD4+ T cell Isolation Kit. Cells were cultured in 96 round-bottom plates with plate-bound anti-CD3 (1 to 5 μg/mL) and soluble anti-CD28 (2 μg/mL) antibodies or soluble PMA (50 ng/mL) and Ionomycin (1 μg/mL) in complete RPMI1640 media with 10% fetal bovine serum, 1% penicillin, 100 μg/mL streptomycin, 4 mM L-glutamine, 55 mM 2-Mercaptoethanol, 1 mM sodium pyruvate, and nonessential amino acids.
CD4+ T cell adoptive transfer
Spleens were removed from naive WT and Nlrc3−/− mice, and CD4+ T cells were positively selected from the splenocytes population using L3T4 MicroBeads. CD4+ T cells (2.5 3 106 per mouse) were injected via tail vein injection into Rag1–/– female mice. One day later, the recipient mice were subjected to EAE induction.
Competitive T cell transfers
Splenocytes were isolated from WT (CD45.1+) and Nlrc3−/− (CD45.1+CD45.2+) SMARTA TCR transgenic mice. The frequency of CD4+ T cells was determined by flow cytometry. 1×104 WT SMARTA CD4+ T cells and 1×104 Nlrc3−/− SMARTA CD4+ T cells were mixed together and co-transferred into WT C57BL/6J (CD45.2+) recipient mice via tail vein injection. The cells were allowed to engraft for 1 to 2 days before the recipient mice were infected with LCMV-Clone 13.
MOG35–55 recall assay
Rag1–/– female mice received CD4+ T cells (2.5 3 106 per mouse) intravenously and, one day later, MOG35–55 peptide to induce to EAE. Eighteen days after EAE induction, draining lymph nodes were removed. A total of 106 cells were re-stimulated with 100 μg/mL MOG35–55 in complete RPMI1640 media for 48 hr. Culture supernatants were analyzed for IFN-γ and IL-17A by ELISA.
T cell proliferation assay
CD4+ T cells were isolated from WT and Nlrc3−/− mice by negative MACS sorting techniques. Purified CD4+ T cells were labeled with 5.0 mM CFSE and incubated at room temperature for 5 m. Labeled CD4+ T cells were then stimulated with anti-CD3 (1.0 or 2.5 or 5.0 μg/mL) and anti-CD28 (2 μg/mL) for 72, 96, or 120 hr in complete RPMI1640 media. Proliferation was determined by measuring the dilution of CFSE via flow cytometery.
DC purification and DC-2D2 T cell co-culture
Bone marrow cells isolated from from WT or Nlrc3−/− mice were cultured in the presence of granulocyte and macrophage colony-stimulating factor (GM-CSF). After 6 days of incubation, DCs were positively isolated using CD11c MicroBeads. CD4+ T cells were positively selected from splenocytes of unimmunized 2D2 Tg+Nlrc3+/+ or 2D2 Tg+Nlrc3−/− mice using the CD4 (L3T4) MicroBeads or FACS sorting as CD4+ Va3.2+ CD25 CD44lo CD62Lhi. A total of 2.5 3 104 DCs from either WT or Nlrc3−/− mice were pulsed for 2 hr with 50 μg/mL MOG35–55 and then co-cultured with 2D2 Tg+Nlrc3+/+ or 2D2 Tg+Nlrc3−/− T cells at a ratio of 1:10 in 96 round-bottom plate in a complete RPMI1640 media. The presence of IFN-γ and IL-17A in the supernatant was analyzed by ELISA at multiple times.
In vitro Th1 and Th17 differentiation assays
DC and 2D2 T cell co-cultures were performed in the presence of cytokines and neutralizing antibodies as follows. Th1 conditions: IL-12 (10 ng/mL) and/or anti-IL-4 (10 μg/mL). Th17 conditions: IL-6 (20 ng/mL), TGF- (2 ng/mL) and/or IL-23 (20 ng/mL), anti-IL-4 (10 μg/mL) and anti-IFN-γ (10 μg/mL). ELISA was used to measure IFN-γ and IL-17A in the supernatants.
Flow cytometry staining
Single cell suspensions of spleen cells were surface stained ex vivo with fluorescent antibodies to T cell markers. To identify LCMV-specific T cells, cell suspensions were exposed to APC-conjugated tetramers (DpGP33 and I-AbGP67). The intracellular cytokine staining assay was performed by re-stimulating splenocytes with LCMV peptides in the presence of brefeldin for 5 hr. Cells were surface stained for CD8, CD4 and activation markers. The cells were fixed and permeabilized for intracellular staining using the Transcription Factor Buffer Set, BD Cytofix/Cytoperm Fixation/Permeabilization Kit and Perm Buffer III (BD). Intracellular cytokines or transcription factor were measured using fluorescent antibodies and quantified using CyAn (Beckman-Coulter) or FACSCalibur (BD) cytometers. The results were analyzed using FlowJo software (Tree Star).
ELISAs
The concentrations of IFN-γ, IL-17A, IL-2, and TNF in cell culture supernatants was determined by ELISA according to the manufac-turer’s instructions.
Quantitative Real-time PCR
Total RNA was harvested from CD4+ T cells using RNeasy Mini Kit. cDNA was generated by reverse transcription using M-MLV (Invitrogen) and oligo-dT according to the manufacturer’s instructions. Q-PCR was performed for murine Nlrc3 using the TaqMan® Gene Expression Assay.
RNA-seq analyses
The Human RNaseq data analyses were obtained from the NCBI GEO database, with accession number GSE66763 (Cao et al. 2015). Raw sequence data were downloaded and analyzed by using SRAdownload, HISAT2 (Ver. 2.0.5.2), and Cufflink (Ver. 2.2.1.0) tools from the usegalaxy.org. Heatmaps of gene expression were generated using GENE-E (Broad Institute). Mouse RNA-seq data were obtained using CD4+ T cells from WT and Nlrc3−/− mouse treated with or without anti-CD3 (5 μg/mL) and anti-CD28 (2 μg/mL) for 24 hr, followed by total RNA isolation. Each condition was prepared in quadruplicate for each RNA-seq experiment. Beijing Genomics Institute (BGI) performed library preparation using Agilent 2100 Bio analyzer (Agilent RNA 6000 Nano Kit) and conducted sequencing on a BGISEQ-500 platform. Single-end FASTQ sequences were aligned to the mouse genome (GRCm38/mm10) using HISAT /Bowtie2. Differential gene expression analysis was performed using DESeq2 package with raw gene counts output from Rsubread. Counts for Nlrc3 were based on number of RNA sequences in the region of the deleted region of Nlrc3 in Nlrc3−/− mouse: from final 103 bp of exon 2 to first 35 bp of exon4 of Nlrc3 (Schneider et al., 2012). A heatmap of mouse RNA-seq data was generated as described above for human data.
Ubiquitination assay
For in vitro ubiquitination assay, 2 3 106 primary mouse CD4+ T cells were stimulated by anti-CD3 (5 μg/mL) and anti-CD28 (1 μg/mL) and then lysed 0, 5, and 10 min later in 100 ml 2% SDS isotonic buffer (50 mM Tris, pH 7.8, 150 mM NaCl, 1% (vol/vol) Nonidet-P40, 2% SDS and 10% (vol/vol) glycerol). Lysates were boiled for 15 min for removal of any noncovalent interactions, and then were centrifuged for 10 min at 16,000 g, and then supernatants were transferred to a new tube. Lysates were diluted 10-fold with 1% Triton X-100 buffer (50 mM Tris, pH 7.8, 150 mM NaCl, 1% (vol/vol) Nonidet-P40 and 10% (vol/vol) glycerol) and substrate molecules were immunoprecipitated with anti-TRAF6 and protein G Sepharose beads. TRAF6 and K63 ubiquitination of TRAF6 was detected by western blot with anti-TRAF6 and anti-K63 Ub respectively.
Western Blot
Spinal cords were homogenized in RIPA lysis buffer supplemented with complete proteinase inhibitor cocktail and PhoSTOP phosphatase inhibitors. Primary cells were lysed in RIPA buffer. Protein lysates were cleared of insoluble material through centrifugation, and the resulting protein lysates were subjected to SDS-PAGE. Proteins were wet transferred to 0.2 mm nitrocellulose membranes (BioRad Laboratories), which were blocked using 5% non-fat milk in 1% TBS-T buffer for 1 hr at room temperature. The membranes were incubated overnight using the following primary antibodies: anti-actin HRP, anti-Histone H3, anti-p65, anti-pp65, anti-STAT1, anti-pSTAT1, anti-ZAP70, anti-pZAP70, anti-PLC-g1, anti-pPLC-g1, anti-NFAT1, anti-mTOR, anti-pmTOR, anti-AKT, anti-pAkt, anti-Akt, anti-4EBP1, anti-p4EBP1, anti-S6K, anti-pS6K, anti-c-Myc, anti-pc-Myc, anti-JNK, anti-pJNK, anti-ERK½ or anti-pERK½. All primary antibodies were used at a 1:1,000 dilution in 5% non-fat milk. Membranes were washed in TBS-T and incubated with the following appropriate secondary antibodies: goat anti-rabbit-HRP or goat anti-mouse HRP. All secondary antibodies were used at a 1:40,000 dilution in 5% non-fat milk. Protein bands were visualized following exposure of the membranes to ECL substrate solution (ThermoFisher) and quantified by densitometryc analysis using ImageJ software.
Metabolism Assays
CD4+ T cells were isolated from WT and Nlrc3−/− mice and stimulated with anti-CD3 and anti-CD28 antibodies with or without the NF-κB inhibitor (BAY 11–7082 or QNZ) for 24 hr. The cells were then plated in glucose-free media with 100 mM sodium pyruvate and 2mM glutamine for ECAR measurements and with 100 mM sodium pyruvate and 2.5 M glucose for OCR measurements for 1 hr in absence of CO2 at 37 C prior to the start of the metabolism assay. ECAR and OCR rates were assessed at basal conditions and after the addition of glucose (10 mM), oligomycin (1mM), and 2-Deoxyglucose (20 mM) for ECAR, and after the addition of oligomycin (1mM), FCCP (1 mM), and rotenone (1mM) for OCR. Compounds were added at the indicated time points and the assay was performed using a Seahorse Extracellular XF24 analyzer. Three measurements were recorded for basal metabolic rates and following each injection
QUANTIFICATION AND STATISTICAL ANALYSIS
P values less than 0.05 were considered statistically significant for all datasets. Statistical significance between two groups was assessed by unpaired t tests. Statistical analyses and graphing of data were performed using GraphPad Prism® Software.
Supplementary Material
Highlights.
Nlrc3 expression in primary T cells is reduced by TCR signaling
NLRC3 restrains anti-viral and autoreactive T cell responses and cytokine expression
NLRC3 limits immune signaling and metabolic pathways in CD4+ T cells
ACKNOWLEDGMENTS
We thank the Flow Cytometry Core Facility at University of North Carolina at Chapel Hill for providing core and technical support. This work was supported by NIH grants AI029564, CA156330, DK094779, and CA10068 (the last to the National Multiple Sclerosis Society) to J.P.-Y.T. and U19AI109965 to J.P.-Y.T. and J.K.W. T.U. was supported by the Japanese vaccine research fellowship (Japan Foundation for Pediatric Research). We thank Dr. Shumpei Yokota and Dr.Shuichi Ito for editing the manuscript and Dr. Vijay Kuchroo for generously providing the 2D2 transgenic strain.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.immuni.2018.10.008.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allen IC (2014). Non-inflammasome forming NLRs in inflammation and tumorigenesis. Front. Immunol 5, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, Moore CB, Schneider M, Lei Y, Davis BK, Scull MA, Gris D, Roney KE, Zimmermann AG, Bowzard JB, et al. (2011). NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 34, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RM, Davis BK, Uronis JM, Herfarth HH, et al. (2012). NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J, Lamkanfi M, and Kanneganti TD (2012). NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015). Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol 33, 257–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, Boidot R, Humblin E, Hamman A, Chalmin F, Berger H, et al. (2015). The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol 16, 859–870. [DOI] [PubMed] [Google Scholar]
- Bruey JM, Bruey-Sedano N, Newman R, Chandler S, Stehlik C, and Reed JC (2004). PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J. Biol. Chem 279, 51897–51907. [DOI] [PubMed] [Google Scholar]
- Cao Y, Rathmell JC, and Macintyre AN (2014). Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE 9, e104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, and Hafler DA (2015). Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med 7, 287ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti BJ, Davis BK, Zhang J, O’connor W Jr., Williams KL, and Ting JP-Y (2005). CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J. Biol. Chem 280, 18375–18385. [DOI] [PubMed] [Google Scholar]
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, and Wang RF (2010). NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141, 483–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, and Wang RF (2012). NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat. Immunol 13, 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman CF, Proverbs-Singh TA, and Postow MA (2016). Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol 2, 1346–1353. [DOI] [PubMed] [Google Scholar]
- Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, and Turka LA (2006). The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 25, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharagozloo M, Mahvelati TM, Imbeault E, Gris P, Zerif E, Bobbala D, Ilangumaran S, Amrani A, and Gris D (2015). The nod-like receptor, Nlrp12, plays an anti-inflammatory role in experimental autoimmune encephalomyelitis. J. Neuroinflammation 12, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, and Raz E (2010). TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J. Clin. Invest 120, 570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, and Ting JP (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Du X, Huang Y, Fu Y, Yang Y, Zhan X, He W, Wen Q, Zhou X, Zhou C, et al. (2018). NLRC3 negatively regulates CD4+ T cells and impacts protective immunity during Mycobacterium tuberculosis infection. PLoS Pathog 14, e1007266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Hara H, Suzuki S, Suzuki N, Akira S, and Saito T (2007). Cutting edge: TLR2 directly triggers Th1 effector functions. J. Immunol 178, 6715–6719. [DOI] [PubMed] [Google Scholar]
- Karki R, Man SM, Malireddi RK, Kesavardhana S, Zhu Q, Burton AR, Sharma BR, Qi X, Pelletier S, Vogel P, et al. (2016). NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 540, 583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechtenberg BC, Mace PD, and Riedl SJ (2014). Structural mechanisms in NLR inflammasome signaling. Curr. Opin. Struct. Biol 29, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Zhao Z, Yang Z, Meng Q, Tan P, Xie W, Qin Y, Wang RF, and Cui J (2016). USP38 inhibits type I interferon signaling by editing TBK1 ubiquitination through NLRP4 signalosome. Mol. Cell 64, 267–281. [DOI] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schrӧder GF, Fitzgerald KA, Wu H, and Egelman EH (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens JR, Gurung P, Shaw PJ, Barr MJ, Zaki MH, Brown SA, Vogel P, Chi H, and Kanneganti TD (2015). The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity 42, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, and Jones RG (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, and Li MO (2016). Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Jiang B, and Zhang Y (2016). 4E-BP1, a multifactor regulated multi-functional protein. Cell Cycle 15, 781–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JM, and Dong C (2013). Toll-like receptor regulation of effector T lymphocyte function. Trends Immunol 34, 511–519. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, and Dong C (2010). Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity 32, 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, Davis BK, Allen IC, Holl EK, Ye Z, et al. (2012). The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat. Immunol 13, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AH, and Pauken KE (2018). The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol 18, 153–167. [DOI] [PubMed] [Google Scholar]
- Stromnes IM, and Goverman JM (2006). Active induction of experimental allergic encephalomyelitis. Nat. Protoc 1, 1810–1819. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, and Glimcher LH (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338–342. [DOI] [PubMed] [Google Scholar]
- TeSlaa T, and Teitell MA (2014). Techniques to monitor glycolysis. Methods Enzymol 542, 91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe M, Isobe Y, Tomizawa H, Nagasaki T, Takahashi H, Fukazawa T, and Hayashi H (2003). Discovery of quinazolines as a novel structural class of potent inhibitors of NF-kappa B activation. Bioorg. Med. Chem 11, 383–391. [DOI] [PubMed] [Google Scholar]
- van der Windt GJ, Chang CH, and Pearce EL (2016). Measuring Bioenergetics in T Cells Using a Seahorse Extracellular Flux Analyzer. Curr. Protoc. Immunol 113, 1–14, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ, and Wang RF (2011). NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 34, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M, and Kanneganti TD (2011). The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, et al. (2014). NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 40, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.