

Abstract
Background
Severe malaria results in over a million deaths every year, most of them in children aged under five years and living in sub‐Saharan Africa. This review examines whether treatment with artesunate, instead of the standard treatment quinine, would result in fewer deaths and better treatment outcomes.
Objectives
To compare artesunate with quinine for treating severe malaria.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register, CENTRAL (The Cochrane Library), MEDLINE, EMBASE, LILACS, ISI Web of Science, the metaRegister of Controlled trials (mRCT), conference proceedings, and reference lists of articles to November 2010.
Selection criteria
Randomized controlled trials comparing intravenous, intramuscular, or rectal artesunate with intravenous or intramuscular quinine for treating adults and children with severe malaria who are unable to take medication by mouth.
Data collection and analysis
Two authors independently assessed the eligibility and risk of bias of trials, and extracted and analysed data. The primary outcome was all‐cause death. Dichotomous outcomes were summarized using risk ratios (RR) and continuous outcomes by mean differences (MD). Where appropriate, we combined data in meta‐analyses.
Main results
Eight trials enrolling 1664 adults and 5765 children are included in this review.
Treatment with artesunate significantly reduced the risk of death both in adults (RR 0.61, 95% Confidence Interval (CI) 0.50 to 0.75; 1664 participants, five trials) and children (RR 0.76, 95% CI 0.65 to 0.90; 5765 participants, four trials)
In children, treatment with artesunate increased the incidence of neurological sequelae at the time of hospital discharge. The majority of these sequelae were transient and no significant difference between treatments was seen at later follow up.
Authors' conclusions
The evidence clearly supports the superiority of parenteral artesunate over quinine for the treatment of severe malaria in both adults and children and in different regions of the world.
11 April 2019
No update planned
Other
High certainty of evidence demonstrating artesunate is superior. All eligible published studies found in the last search (16 Nov, 2010) were included
Keywords: Adult; Child; Humans; Antimalarials; Antimalarials/administration & dosage; Antimalarials/therapeutic use; Artemisinins; Artemisinins/administration & dosage; Artemisinins/therapeutic use; Artesunate; Injections, Intramuscular; Injections, Intravenous; Malaria; Malaria/drug therapy; Malaria/mortality; Quinine; Quinine/administration & dosage; Quinine/therapeutic use; Randomized Controlled Trials as Topic
Plain language summary
Artesunate reduces death from severe malaria
Severe malaria occurs when infection with the malaria parasite is complicated by serious failure of the body's major organs, and results in over a million deaths every year. Sometimes severe malaria is associated with coma and is known as cerebral malaria. Following cerebral malaria a small proportion of children suffer with long‐term neurological disability.
This review of trials assessed the effectiveness of artesunate compared with the standard treatment quinine. Eight trials involving 1664 adults and 5765 children were identified, from study sites in Asia and Africa.
Treating adults in Asia with artesunate instead of quinine would prevent an extra 94 deaths for every 1000 patients treated. In trials involving children, the proportion of deaths was lower than in the trials involving adults. This lower risk of death results in a smaller benefit in children than in adults, but would still save an extra 26 lives for every 1000 children treated.
In the children who survived their illness, there were more neurological problems at the time of hospital discharge in those treated with artesunate than those treated with quinine. However, the majority of these neurological problems had resolved when they were reviewed 28 days later, and at this timepoint there was no difference between the two treatment groups.
Artesunate should be the drug of choice for adults and children with severe malaria worldwide.
Summary of findings
for the main comparison.
| Artesunate compared with quinine for treating severe malaria | ||||||
|
Patient or population: Children with severe malaria Settings: Malaria endemic areas Intervention: Artesunate Comparison: Quinine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Quinine | Artesunate | |||||
| Death | 109 per 1000 | 83 per 1000 (71 to 98) | RR 0.76 (0.65 to 0.9) | 5765 (4 studies1) | high2,3,4,5 | |
| Neurological sequelae at day 28 | 11 per 1000 | 14 per 1000 (8 to 22) | RR 1.23 (0.74 to 2.03) | 4857 (1 study6) | moderate7,8,9,10 | |
| Neurological sequelae at discharge | 28 per 1000 | 38 per 1000 (28 to 51) | RR 1.36 (1.01 to 1.83) | 5163 (3 studies11) | moderate2,3,4,12 | |
| Time to hospital discharge (days) | See comment | See comment | Not estimable | 113 (3 studies11) | moderate2,13,4,14 | |
| Hypoglycaemia episodes | 30 per 1000 | 19 per 1000 (13 to 26) | RR 0.62 (0.45 to 0.87) | 5765 (4 studies1) | high2,3,4,15 | |
| *The assumed risk was calculated by dividing the total number of events in the control group (across studies) by the total number of patients in the control group (across studies). This was numerically very similar to the median control group risk but is easier to link with the corresponding forest plot. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 One large multicentre trial (Dondorp 2010) and two small trials (Cao 1997, Eltahir 2010) have assessed artesunate vs quinine in children aged < 15 years. In addition one large multicentre study included a subgroup of children in this age group (Dondorp 2005) 2 No serious study limitations: All the trials adequately concealed allocation to be considered at low risk of bias. The trials were unblinded but this is unlikely to bias this objective outcome 3 No serious inconsistency: There was no statistical heterogeneity between the trials (I² = 0%). 4 No serious indirectness: Most of the data is from Dondorp 2010 which had centres in Mozambique, the Gambia, Ghana, Kenya, Tanzania, Nigeria, Uganda, Rwanda and the Democratic Republic of Congo, and used the established standard doses of artesunate and quinine (with loading dose). Of note the median age of children in this trial was 2.9 years in the quinine group and 2.8 in the artesunate group. 5 No serious imprecision: Both limits of the 95% CI of the pooled effect imply an appreciable clinical benefit with artesunate. The Number Needed To Treat to prevent one childhood death is 38. 6 Only one large multicentre trial (Dondorp 2010) reports this outcome. 7 Serious study limitations: 41/170 (24%) patients with neurological sequelae at discharge were not available for assessment at day 28. 8 No serious inconsistency: Not applicable as only one trial. 9 No serious indirectness: This trial (Dondorp 2010) had 11 centres throughout Africa and used the standard dosing of artesunate and quinine. The nature of the neurological sequelae is not described. 10 No serious Imprecision: The 95% CI around the absolute effect is narrow. The worst case scenario is a 1.2% increase in neurological sequelae at day 28 11 Three trials (Dondorp 2010, Dondorp 2005, and Cao 1997) report this outcome 12 Serious imprecision: The effect estimate is of a clinically important harm. However the 95% CI includes the possibility of no clinically important difference between the two interventions. 13 No serious inconsistency: None of the trials found evidence of an important difference between the two treatment groups 14 Serious imprecision: We were unable to pool the data as they were only reported as medians and range/intra quartile range. There is no evidence of a clinically important benefit with artesunate on this outcome. 15 No serious imprecision: The result is statistically significant in favour of artesunate. The current sample size is adequately powered to detect a 40% risk reduction with 80% power and 95% confidence.
2.
| Artesunate compared with quinine for treating severe malaria | ||||||
|
Patient or population: Adults with severe malaria Settings: Malaria endemic areas Intervention: Artesunate Comparison: Quinine | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Quinine | Artesunate | |||||
| Death | 241 per 1000 | 147 per 1000 (121 to 181) | RR 0.61 (0.5 to 0.75) | 1664 (5 studies1) | high2,3,4,5 | |
| Neurological sequelae at day 28 | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Neurological sequelae at discharge | 3 per 1000 | 9 per 1000 (2 to 44) | RR 2.97 (0.6 to 14.64) | 1259 (1 study6) | moderate7,8,9,10 | |
| Time to hospital discharge (days) | See comment | See comment | Not estimable | 113 (2 studies11) | moderate12,13,14,15 | |
| Hypoglycaemia episodes | 47 per 1000 | 17 per 1000 (9 to 32) | RR 0.36 (0.19 to 0.68) | 1372 (2 studies11) | high12,16,14,17 | |
| *The assumed risk was calculated by dividing the total number of events in the control group (across studies) by the total number of patients in the control group (across studies). This was numerically very similar to the median control group risk but is easier to link with the corresponding forest plot. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk Ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 One large multicentre trial (Dondorp 2005) and four smaller trials (Anh 1989, Anh 1995, Hien 1992, Newton 2003) have assessed artesunate vs quinine in adults 2 No serious study limitations: two of the smaller studies did not conceal allocation and none of the studies were blinded. However the majority of the data is from studies which did conceal allocation and the lack of blinding is unlikely to introduce bias for an objective outcome such as death. 3 No serious inconsistency: The point estimates of all five trials favoured artesunate. No significant statistical heterogeneity was detected (I² = 0%). 4 No serious indirectness: All five trials were from Asia, but from a variety of settings (Vietnam, Bangladesh, India, Myanmar, Indonesia and Thailand), and included age groups above 15/16 years. Of the four small trials; two did not give the loading dose of quinine, but there was no statistical heterogeneity between these two trials and the large multicentre trial which did give the loading dose. 5 No serious imprecision: Both limits of the 95% CI imply a clinically important benefit with artesunate. 6 Only one trial reported the incidence of neurological sequelae in adults (Dondorp 2005). This is unpublished data received from the authors. 7 No serious study limitations: This trial was unblinded but the nature of the sequelae makes an observer or reporting bias unlikely 8 No serious inconsistency: Not applicable as only one trial 9 No serious indirectness: This trial had sites in four different countries in Asia and used the established standard doses of artesunate and quinine (with loading dose). Of the 10 sequelae which occurred in this trial (the additional two were in children): Five were psychiatric sequelae, four were a persistent problem with balance, and two had a hemiparesis. 10 Serious imprecision: Neurological sequelae appear to be a rare event following severe malaria in adults. However the 95% CI includes the possibility of a clinically important harm with artesunate. 11 Two trials (Dondorp 2005 and Newton 2003) report this outcome. 12 No serious study limitations: Dondorp 2005 adequately concealed allocation to be considered at low risk of bias, Newton 2003 did not but is a much smaller trial. Neither trial was blinded 13 No serious inconsistency: Neither trial found a statistically significant difference in time to hospital discharge 14 No serious indirectness: This evidence is from multiple sites within Asia (Bangladesh, India, Myanmarand Indonesia) and both trials used standard drug doses. The data from Dondorp 2005 does include some children. 15 Serious imprecision: We were unable to pool data due to the way data were presented but there is no evidence of a benefit on this outcome with artesunate. 16 No serious inconsistency: There was no statistical heterogeneity (I² = 0%) 17 No serious imprecision: This result is statistically significant in favour of artesunate. The current sample size is adequately powered to detect a 75% risk reduction with 80% power and 95% confidence.
Background
Description of the condition
Malaria is a febrile illness caused by infection with the Plasmodium parasite, which is transmitted from person to person by mosquitos. Five species of plasmodium are known to cause disease in humans: P. falciparum, P. vivax, P. ovale, P. malariae, and P knowlesi (WHO 2010). P. falciparum is the most common malaria parasite worldwide, and is responsible for almost all of the severe disease and deaths (WHO 2000; WHO 2008).
Repeated exposure to malaria infection over 5 to 10 years can produce a naturally acquired immunity in humans, which is protective against the most severe forms of the disease (Doolan 2009). Consequently, in high transmission settings as seen in large parts of Africa, young children are most at risk prior to the acquisition of effective immunity, whereas in low transmission settings, or in travellers from non‐endemic areas, adults are often equally vulnerable to severe disease (WHO 2000).
Severe malaria is diagnosed on the basis of a positive blood slide or antigen test for malaria, plus the presence of clinical or laboratory markers which indicate vital organ dysfunction. These markers include impaired consciousness, coma, convulsions, respiratory distress, shock (systolic blood pressure < 70 mmHg in adults, < 50 mmHg in children), jaundice, haemoglobinuria, or severe acidosis or anaemia (WHO 2010). Cerebral malaria is a specific type of severe malaria characterised by an unrousable coma. Even with correct treatment cerebral malaria can have a mortality rate approaching 20%, and persistent neurological sequelae are seen in a small proportion of survivors (Jaffar 1997).
The standard treatment for severe malaria has been an intravenous infusion or intramuscular injection of quinine (WHO 2000). A loading dose of 20 mg/kg is recommended to reduce the time needed to reach effective concentrations in the blood, with subsequent dosing at 10 mg/kg at eight hourly intervals (White 1983b; van Der Torn 1996). A Cochrane Review found a significant reduction in fever clearance time and parasite clearance time with a loading dose compared with no loading dose but concluded that data were insufficient to demonstrate an impact on mortality (Lesi 2004).
Adverse effects resulting from quinine therapy are common. Cinchonism (symptoms of quinine overdose) often occurs with conventional dose regimens. This usually mild and reversible symptom complex consists of tinnitus, deafness, dizziness, and vomiting, and may affect adherence (Alkadi 2007). Hypoglycaemia is a less common but more serious adverse effect (White 1983). Toxic levels of quinine can occur following rapid intravenous administration and can result in heart rhythm disturbances, blindness, coma, and even death (Alkadi 2007). In addition, there is limited evidence that the efficacy of quinine in severe malaria may be declining in some parts of South‐East Asia (Wongsrichanalai 2002).
Description of the intervention
Artesunate is one of a number of antimalarials derived from artemisinin, the active ingredient in a Chinese herbal remedy for fever, Artemesia annua. The artemisinin derivatives are now the recommended treatment for uncomplicated (less severe) malaria, when they are given orally in combination with a partner drug, as Artemisinin‐based Combination Therapy (ACT), to delay or prevent the development of artemisinin resistance (WHO 2010).
The artemisinin derivatives are generally regarded as safe in humans (Ribeiro 1998; Alkadi 2007; Nosten 2007). Animal studies using very high doses of artemisinins have demonstrated focal brain stem lesions particularly affecting the auditory pathways (Brewer 1994; Nontprasert 1998; Genovese 2000; Nontprasert 2000; Nontprasert 2002), but studies of brain stem function in humans, including audiometry, have failed to show any abnormality following repeated courses (Ribeiro 1998; Kissinger 2000). To date, only one nested case‐control study has demonstrated a significant audiometric hearing loss in factory workers treated with artemether‐lumefantrine for uncomplicated malaria compared with workers with no history of exposure to malaria infection or artemether‐lumefantrine (Toovey 2004). This result needs to be interpreted with caution due to a number of design limitations.
How the intervention might work
Deaths from severe malaria often occur during the first 24 to 48 hours following hospital admission. Consequently, to be effective antimalarial drugs need to achieve rapid therapeutic blood concentrations following administration.
Compared to quinine, the artemisinin derivatives have been shown to clear malaria parasites from the blood faster, and to have a broader spectrum of activity (ter Kuile 1993; Adjuik 2004). Importantly they are effective against young ring forms of the parasite before they sequester in the microcirculation of vital organs, a major pathophysiological step in the development of severe disease (ter Kuile 1993; WHO 2000).
Artesunate is the most studied artemisinin‐derivative for the treatment of severe malaria and may be given by intramuscular or intravenous injection. It has been shown to reliably reach peak concentrations within one hour of administration (Nealon 2002; Hien 2004).
Of the alternatives, artemether and arteether are available as oil‐based, intramuscular formulations. Artemether is prone to erratic and partial absorption (Karbwang 1997; Murphy 1997; Mithwani 2003), and arteether to low peak concentrations and slow absorption (Looareesuwan 2002; Li 2004). Systematic reviews of artemether and arteether compared to quinine have so far failed to show a reduction in mortality compared to quinine therapy, although the data are limited (AQMSG 2001; Afolabi 2004; Kyu 2009).
Why it is important to do this review
A Cochrane Review prepared in year 2000 assessed the effects of the artemisinin derivatives, including artesunate, for treating severe malaria (McIntosh 2000). This review has since been superseded by a series of Cochrane Reviews examining the different artemisinin derivatives.
This review was first published in 2006 and demonstrated the superiority of artesunate for treating adults in Asia, but found insufficient data to make firm conclusions in children. This update includes two additional, recently completed trials, focusing on artesunate versus quinine in African children.
Objectives
To compare artesunate with quinine for treating severe malaria.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials.
Types of participants
Adults and children with severe malaria who are unable to take medication by mouth.
Types of interventions
Intervention
Intravenous, intramuscular or rectal artesunate.
Control
Intravenous or intramuscular quinine.
Types of outcome measures
Primary outcomes
Death.
Secondary outcomes
Neurological sequelae.
Coma recovery time.
Time to hospital discharge.
Fever clearance time.
Parasite clearance time.
Adverse effects
Serious adverse effects resulting in discontinuation of treatment (eg biochemical abnormalities, cardiac effects).
Hypoglycaemia (symptomatic or asymptomatic).
Other adverse events, including tinnitus, hearing impairment, nausea, and vomiting.
Search methods for identification of studies
We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and ongoing).
Electronic searches
Databases
We searched the following databases using the search terms and strategy described in Table 3: Cochrane Infectious Diseases Group Specialized Register; Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library; MEDLINE; EMBASE; LILACS; and ISI Web of Science. We also searched the metaRegister of Controlled trials (mRCT) using artesunate and quinine as search terms.
1. Search methods: detailed search strategy.
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb | ISI Web of Science |
| 1 | malaria | malaria | malaria | malaria | malaria | malaria |
| 2 | quinine | quinine | quinine | quinine | quinine | quinine |
| 3 | artesunate | quinimax | quinimax | quinimax | artesunate | artesunate |
| 4 | artemisinin* | CINCHONA ALKALOIDS | CINCHONA ALKALOIDS | CINCHONA‐ALKALOID | artemisinin | arsumax |
| 5 | 3 or 4 | 2 or 3 or 4 | 2 or 3 or 4 | 2 or 3 or 4 | 3 or 4 | 3 or 4 |
| 6 | 1 and 2 and 5 | artesunate | artesunate | artesunate | 1 and 2 and 5 | 1 and 2 and 5 |
| 7 | — | arsumax | arsumax | arsumax | — | — |
| 8 | — | 6 or 7 | 6 or 7 | 6 or 7 | — | — |
| 9 | — | 1 and 5 and 8 | 1 and 5 and 8 | 1 and 5 and 8 | — | — |
| 10 | — | — | limit 9 to human | limit 9 to human | — | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Higgins 2008); upper case: MeSH or EMTREE heading; lower case: free text term.
Searching other resources
Conference proceedings
We searched the following conference proceedings for relevant abstracts: The 5th Multilateral Initiative on Malaria (MIM) Pan‐African Malaria Conference, 2 to 6 November 2009, Nairobi, Kenya; the 4th MIM Pan‐African Malaria Conference, 13 to 18 November 2005, Yaounde, Cameroon; the 4th European Congress on Tropical Medicine, 11 to 15 September 2005, Marseille, France; ACT NOW; the International Symposium on Malaria, 29 to 30 April 2004, Colombia, New York, USA; the 2nd International Malaria Research Conference, John Hopkins Malaria Research Institute, 25 to 26 March 2004, Maryland, USA; the 3rd MIM Pan‐African Conference, 18 to 22 November 2002, Arusha, Tanzania; and the 3rd European Congress on Tropical Medicine and International Health, 8 to 12 September 2002, Lisbon, Portugal.
Researchers, organizations, and pharmaceutical companies
We contacted individual researchers working in the field and the World Health Organization (WHO) for details of unpublished and ongoing trials.
Reference lists
We checked the reference lists of existing reviews and of all trials identified by the above methods.
Data collection and analysis
Selection of studies
All trials identified by the search strategy were screened by two authors working independently (Katharine Jones (KJ), Sarah Donegan (SD) or David Sinclair (DS)) and full reports of potentially relevant trials were obtained. Two authors independently applied the inclusion criteria to the full reports using an eligibility form and scrutinized publications to ensure each trial was included in the review only once. Trial authors were contacted for clarification if necessary. Disagreement was resolved by discussion with David Lalloo (DL).
Data extraction and management
Two authors independently extracted data using a data extraction form. For each outcome we aimed to extract the number of participants randomised and the number analysed in each treatment group. For dichotomous outcomes, we recorded the number of participants experiencing the event and the number assessed in each treatment group. For continuous outcomes, we extracted arithmetic means and standard deviations for each treatment group, together with the numbers assessed in each group. Where medians were used we also extracted the range or intra‐quartile range.
Assessment of risk of bias in included studies
DS and SD independently assessed the risk of bias for each trial using 'The Cochrane Collaboration's tool for assessing the risk of bias' (Higgins 2008). We followed the guidance to assess whether adequate steps had been taken to reduce the risk of bias across six domains: sequence generation; allocation concealment; blinding (of participants, personnel, and outcome assessors); incomplete outcome data; selective outcome reporting; and other sources of bias. We have categorized these judgments as 'yes' (low risk of bias), 'no' (high risk of bias), or 'unclear'. Where our judgement is unclear we attempted to contact the trial authors for clarification.
This information was used to guide the interpretation of the data that are presented.
Measures of treatment effect
Results were calculated using risk ratio (RR) for dichotomous data, and mean difference (MD) for continuous data. These effect estimates are presented with 95% confidence intervals (CI).
Dealing with missing data
If there was discrepancy between the number randomized and the number analysed, we calculated the percentage loss to follow up for each treatment group and reported this information.
Originally, we aimed to analyse data according to the intention‐to‐treat principle (all randomized participants should be analysed in the groups to which they were originally assigned). However, since for some trials it was unclear whether there was loss to follow up, we entered the number analysed into Review Manager 5 whenever these figures were available. By attempting to carry out a complete‐case analysis in this way, we have tried to avoid making assumptions about the outcomes of participants that were lost to follow up.
Assessment of heterogeneity
We looked for statistical heterogeneity by inspecting the forest plots for overlapping confidence intervals, applying the Chi2 test (P value < 0.10 considered statistically significant), and the I2 statistic (I2 value of 50% used to denote moderate levels of heterogeneity).
Assessment of reporting biases
We planned to construct funnel plots to look for evidence of publication bias, provided there were sufficient included trials to make this informative.
Data synthesis
We analysed the data using Review Manager 5, and where possible and appropriate we combined studies using a fixed‐effect model. If heterogeneity was detected but it was still considered clinically meaningful to combine studies, a random‐effects model was used. Medians and ranges are only reported in tables.
If arithmetic means were reported, normality of the data was checked by calculating the ratio of the mean over the standard deviation (Altman 1996). If this test suggested the data were skewed (ie if the ratio was less than two), we commented on this in the text but still combined the results in a meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We planned to investigate heterogeneity by conducting pre‐specified sub‐group analyses for the primary outcome. The potential sources of heterogeneity were allocation concealment, blinding, participant age (children versus adults), and drug regimen (loading dose versus no loading dose of quinine and use of any additional antimalarials).
Sensitivity analysis
Post hoc, we conducted a sensitivity analysis to investigate the robustness of the results to differences in trial design, by subgrouping the trials according to allocation concealment, participant age (children versus adults), type of severe malaria (cerebral versus non‐cerebral malaria), geographical region, drug regimen (loading dose versus no loading dose of quinine, and use of any additional antimalarials), route of administration (intravenous versus intramuscular route), and time since admission to hospital.
Results
Description of studies
Results of the search
The original search was conducted in November 2005 and identified 22 references of which four were duplicate trial reports. Six unique trials fulfilled the eligibility criteria and were included in the first version of this review.
An update search conducted in November 2010 identified a further six trials of which two were eligible and these are now included.
Included studies
The eight trials that met our inclusion criteria enrolled a total of 7429 participants (1664 adults and 5765 children).
Location
SIx trials were conducted in Asia; four took place in single centres in Vietnam (Anh 1989; Anh 1995; Cao 1997; Hien 1992), Newton 2003 had two centres in Thailand; and Dondorp 2005 had 11 centres throughout Bangladesh, Myanmar, India, and Indonesia. Of the two African studies; Eltahir 2010 was conducted at a single study site in Sudan, and Dondorp 2010 had 11 centres in nine African countries (Mozambique, The Gambia, Ghana, Kenya, Tanzania, Nigeria, Uganda, Rwanda, and the Democratic Republic of the Congo).
Source of funding
Four trials were funded by a medical research charity (Cao 1997; Dondorp 2005; Dondorp 2010; Newton 2003), one by the WHO (Anh 1995), one by a private sugar company (Eltahir 2010), and one received the study drugs from a pharmaceutical company (Anh 1989). Funding was not specified for Hien 1992.
Participants
Four trials enrolled only adults (Anh 1989; Anh 1995; Hien 1992; Newton 2003), of which three included only those with a diagnosis of cerebral malaria (Anh 1989, Anh 1995, Hien 1992). Three trials enrolled only children aged less than 15 years (Cao 1997; Dondorp 2010; Eltahir 2010), and one enrolled both adults and children (Dondorp 2005).
Dondorp 2005 and Dondorp 2010 used rapid diagnostic tests to confirm P. falciparum parasitaemia, and all the other trials used standard microscopy. Although standardized clinical definitions for severe malaria exist, the entry criteria were not consistent across trials.
Interventions
All trials compared artesunate with quinine, but the exact dosing and route of administration varied between trials.
Three trials (Dondorp 2005; Dondorp 2010; Eltahir 2010) administered both artesunate and quinine using the current recommended dosing schedules (artesunate: 2·4 mg/kg (intravenous or intramuscular) on admission, at 12 hours, at 24 hours, and then once daily until starting oral therapy, quinine: 20 mg/kg intravenous or intramuscular loading dose, then 10 mg/kg every 8 hours until starting oral therapy).
Anh 1989, Anh 1995, and Hien 1992 gave 60 mg artesunate intravenously at admission, 4 hours, 24 hours and 48 hours. Cao 1997 gave 3 mg/kg intramuscular on admission then 2 mg/kg intramuscular at 12, 24, 48, and 72 hours, and Newton 2003 gave 2.4 mg/kg intravenously on admission, 1.2 mg/kg at 12 hours, and then 1.2 mg/kg every 24 hours until able to swallow. Two trials did not give the loading dose of quinine (Anh 1995; Hien 1992).
In addition six trials gave an additional oral antimalarial to at least one of the treatment arms, which was unmatched between the treatment arms (Anh 1989; Anh 1995; Cao 1997; Eltahir 2010; Hien 1992; Newton 2003). Two trials, Hien 1992 and Cao 1997, included an additional rectal artemisinin arm that was not pertinent to this review.
Supportive care
All eight trials reported measuring blood glucose on admission, but only five trials reported any subsequent active monitoring for hypoglycaemia. Newton 2003 tested all participants several times a day, Cao 1997 tested all participants with coma, prostration, jaundice or more than one complication every four hours for the first 24 hours and then every six hours, Anh 1989 tested all participants on days 1, 3, 7, and 14, and Eltahir 2010 tested all participants every six hours. Dondorp 2005 only measured blood glucose in those participants with clinical signs of hypoglycaemia.
Outcome measures (defined in Table 2)
All eight trials reported death as an outcome
Three trials reported neurological sequelae at discharge (Cao 1997; Dondorp 2005; Dondorp 2010). Six trials reported coma recovery time (Anh 1989; Hien 1992; Anh 1995; Cao 1997; Newton 2003; Eltahir 2010), and two trials reported time to eat, sit, and speak (Dondorp 2005; Dondorp 2010).
Five trials reported fever clearance time (Hien 1992; Anh 1995; Cao 1997; Newton 2003; Eltahir 2010). Reporting of parasite clearance time varied between trials and included parasite clearance times of 50%, 90%, 95%, and 100%, of which parasite clearance time of 50% was the most common (Anh 1989; Hien 1992; Anh 1995; Cao 1997; Newton 2003).
Four trials reported time to hospital discharge (Cao 1997; Newton 2003; Dondorp 2005; Dondorp 2010), and four trials reported adverse effects including hypoglycaemia (Cao 1997; Newton 2003; Dondorp 2005; Dondorp 2010).
Length of follow up
Cao 1997 specified that participants were asked to return for a follow up visit three weeks after discharge from hospital, and Dondorp 2010 followed those with neurological sequelae for 28 days. None of the other trials reported the length of follow‐up.
Excluded studies
Sixteen trials detected by the search specifications were excluded from the review (see 'Characteristics of excluded studies').
Risk of bias in included studies
See Figure 1 for a summary of the risk of bias assessments. Further details are presented in the 'Characteristics of included studies' tables.
1.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The generation of the allocation sequence was adequate in all eight trials and allocation concealment was adequate in six trials with only Anh 1989 and Newton 2003 using open randomization.
Blinding
In all eight trials, investigators were aware of treatment allocation. Participants were blind to the intervention in Hien 1992, and microscopists and data analysts were blind to the intervention in Dondorp 2005 and Dondorp 2010.
Incomplete outcome data
Newton 2003, Dondorp 2005 and Dondorp 2010 clearly state that no participants were lost to follow‐up. We were able to obtain individual patient data for one trial in which primary outcomes were available for all included participants (Cao 1997). For the remaining trials the number of participants randomized was used as the denominator in the analysis (Anh 1989; Hien 1992; Anh 1995; Eltahir 2010). As these were inpatient trials significant attrition is unlikely.
Selective reporting
No evidence of selective outcome reporting was detected.
Other potential sources of bias
No other sources of bias were identified.
Effects of interventions
Death
Treatment with artesunate significantly reduced the risk of death both in adults (RR 0.61, 95% CI 0.50 to 0.75; 1664 participants, five trials, Analysis 1.1, Figure 2) and children (RR 0.76, 95% CI 0.65 to 0.90; 5765 participants, four trials, Analysis 1.1, Figure 2). This reduction was consistent across all trials regardless of participant age or geographic region (I2 test for statistical heterogeneity = 0%, Analysis 1.1).
1.1. Analysis.
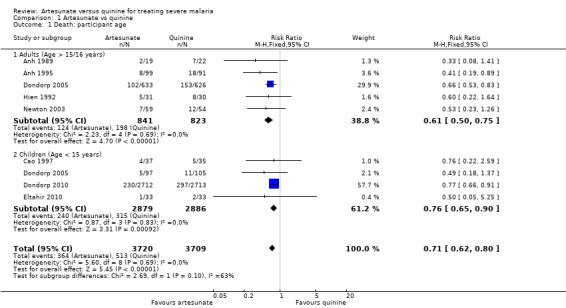
Comparison 1 Artesunate vs quinine, Outcome 1 Death: participant age.
2.
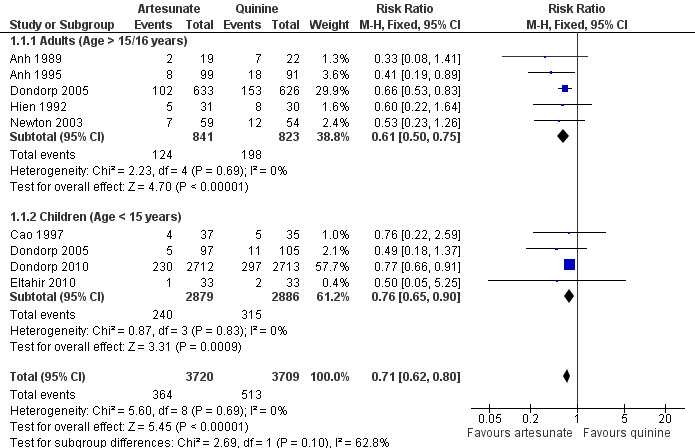
Forest plot of comparison: 1 Artesunate vs quinine, outcome: 1.1 Death: participant age [Relative effect].
Mortality was lower in the trials recruiting children and consequently the absolute reduction in mortality is lower than that seen in adults (absolute risk reduction: adults: 94 fewer deaths per 1,000 patients, 95% CI 60 fewer to 120 fewer; children: 26 fewer deaths per 1,000 patients, 95% CI 11 fewer to 38 fewer; see Table 2 and Table 1). This age effect is confounded by region, with the majority of the data in children coming from Africa (where the trial mortality was relatively low), and all the data in adults coming from Asia (where the trial mortality was higher).
Three trials report a subgroup analyses of deaths occurring within the first 48 hours following admission (Cao 1997; Dondorp 2005; Newton 2003) and one trial reports the number of deaths occurring within the first 24 hours (Dondorp 2010). Although the difference between groups did not reach statistical significance during these early time periods there were consistently fewer deaths in the groups treated with artesunate (6163 participants, four trials, Analysis 1.2).
1.2. Analysis.
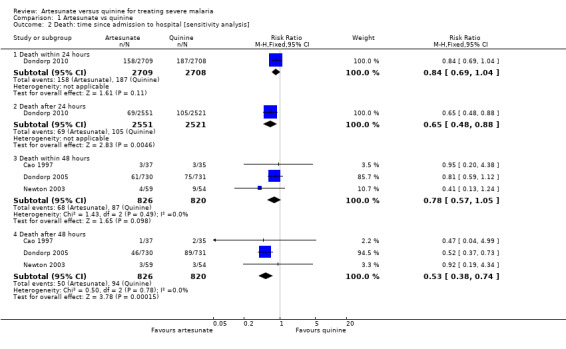
Comparison 1 Artesunate vs quinine, Outcome 2 Death: time since admission to hospital [sensitivity analysis].
In view of the significant variation in trial design we conducted sensitivity analyses excluding trials with inadequate allocation concealment, trials only included patients with cerebral malaria, and those with no loading dose of quinine, but these did not alter the significance of the result.
The two large multicentre trials (Dondorp 2005; Dondorp 2010) conducted multiple subgroup analyses according to the presence or absence of coma, anaemia, shock, acidosis, respiratory distress, or hyperparasitaemia at the time of admission. Mortality was consistently lower with artesunate in all of these subgroups but some were underpowered to show statistically significant differences.
Artesunate appears superior to quinine irrespective of intramuscular or intravenous administration (Analysis 1.3).
1.3. Analysis.
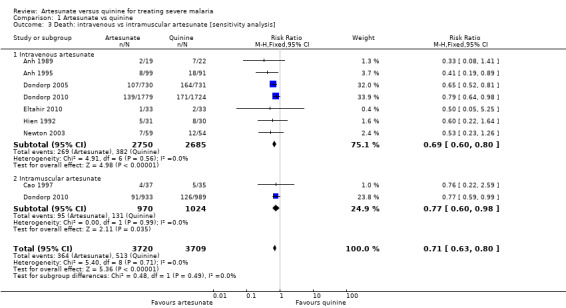
Comparison 1 Artesunate vs quinine, Outcome 3 Death: intravenous vs intramuscular artesunate [sensitivity analysis].
Neurological sequelae
At the time of hospital discharge, neurological sequelae were more common in those treated with artesunate than with quinine (RR 1.41, 95% CI 1.05 to 1.88; 6422 participants, three trials, Analysis 1.4, Figure 3). Of these three trials, only Dondorp 2005 included adults, and the incidence of neurological sequelae seems to be very low in this group (Analysis 1.4, Figure 3).
1.4. Analysis.
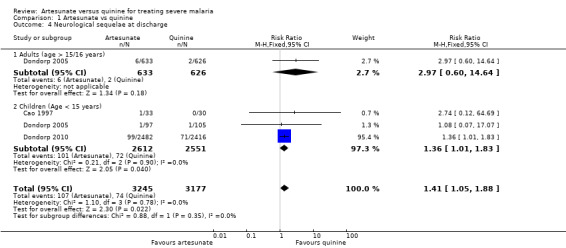
Comparison 1 Artesunate vs quinine, Outcome 4 Neurological sequelae at discharge.
3.
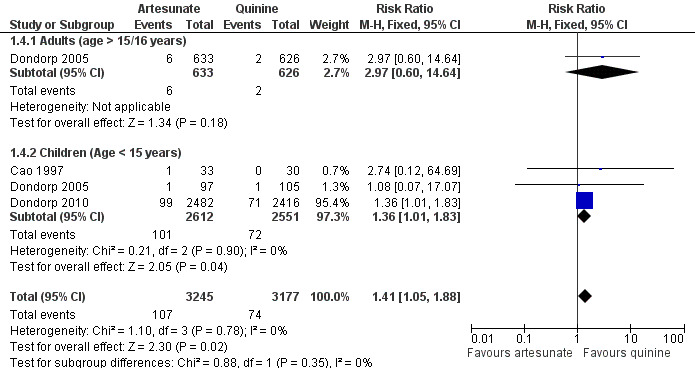
Forest plot of comparison: 1 Artesunate vs quinine, outcome: 1.9 Neurological sequelae at discharge.
One trial in children (Dondorp 2010) followed participants up until day 28 to see if these sequelae resolved. Of the 170 children with sequelae at the time of discharge, 129 (75.9%) were available for assessment on day 28, and 68 of these (52.7%) had fully recovered. At this time point the difference between groups was not statistically significant (RR 1.23, 95% CI 0.74 to 2.03; 4857 participants, one trial, Analysis 1.5).
1.5. Analysis.
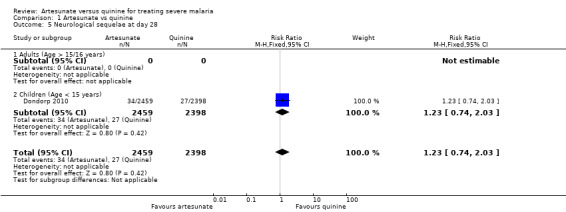
Comparison 1 Artesunate vs quinine, Outcome 5 Neurological sequelae at day 28.
Coma recovery time
Six trials report a measure of coma recovery time (Anh 1989; Anh 1995; Cao 1997; Eltahir 2010; Hien 1992; Newton 2003). The frequency of clinical monitoring to assess coma recovery varied between these trials and is likely to have influenced the result (see Table 4).
2. Definitions of outcome measures used in the review.
| Trial | Death | Neurological sequelae at discharge | Coma recovery time | Time to discharge | Fever clearance time | Parasite clearance time | Hypoglycaemia | Adverse effects |
| Anh 1989 | Death | Not reported | Mean value (h) reported but not defined | Not reported | Not reported | Mean value (h) reported but not defined. Parasite counts every 8 h until 2 consecutive slides were negative and then every 24 h | Not reported | Not reported |
| Anh 1995 | Death | Not reported | Mean value (h). Defined as time until consciousness regained. Glasgow Coma Scale measured every 12 h until regained consciousness | Not reported | Mean value (h) reported but not defined. Axillary temperature was recorded every 6 h until 4 consecutive temperatures were < 37.5 °C | Mean value (h) reported but not defined. Parasite count measured every 6 h until 3 consecutive blood smears were negative | Not reported | Not reported |
| Cao 1997 | Death | Number survived with neurological sequelae. Case definition for neurological sequelae: abnormal neurological signs and/or symptoms at time of discharge from hospital that were not present before onset of the episode of malaria as reported by the child's parents. All children had a full neurological examination on admission and at discharge from hospital (personal communication from author) | Median value (h) reported. Defined as time (h) for Blantyre Coma Score to become 5/5. Coma score assessed every 4 h (or more frequently if critically ill) for the first 24 h, and then every 6 h until discharge | Median value (d) reported | Median value (h) reported. Defined as time until temperature first dropped to 37.5 °C or below and remained below this level for at least 24 h. Axillary temperature measured every 4 h (or more frequently if critically ill) for the first 24 h, and then every 6 h until discharge | Median value (h) reported but not defined. Parasite count measured every 4 h (or more frequently if critically ill) for the first 24 h, and then every 6 h until discharge. Once 2 successive peripheral blood films had revealed no P. falciparum, no further blood film was made unless indicated clinically | Blood glucose < 2.2 mmol/L. Blood glucose measured every 4 h for first 24 h and then every 6 h until discharge from hospital if indicated (coma, prostration, jaundice, or > 1 complication ‐ personal communication from author) | Acute renal failure requiring dialysis, shock, convulsions, deterioration of coma score, gastrointestinal bleeding, anaemia requiring blood transfusion, chest infection, urinary tract infection, other infections, derangement of biochemical markers |
| Dondorp 2005 | Death | Neurological sequelae at discharge from hospital | Not reported | Median value (d) reported | Not reported | Not reported | Blood glucose < 2.2 mmol/L. Blood glucose checked in all patients on admission and then monitored on clinical indication | Not reported |
| Dondorp 2010 | Death | Severe neurological complications (initially assessed at discharge from hospital but the protocol was changed after 11% of patients had been enrolled, so that children who had not fully recovered at discharge were assessed 28 days after enrollment). | Not reported | Median value (d) reported | Not reported | Not reported | Not reported | Not reported |
| Eltahir 2010 | Death | Not reported | Mean value measured from administration of first antimalarial until the Glasgow coma score reached 15. Vital signs and coma scale were monitored every 15 mins for the first hour, then every 2 hours until 24 ours then every 6 hours. | Not reported | Mean value measured from administration of first antimalarial until the axillary temp first dropped below 37.5 and remained below for 24 hours | Mean value measured from administration of the first antimalarial until the first of two sequential negative blood films. Blood films were taken every 4 hours. | Blood glucose levels were measured every 6 hours. | Not reported |
| Hien 1992 | Death | Not reported | Mean, median, and mode values reported in hours. Defined as time to regain full consciousness (Glasgow Coma Scale of 15/15). Glasgow Coma Scale measured at 3‐h intervals until full recovery of consciousness, and at 6‐h intervals thereafter | Not reported | Mean value (h) reported. Defined as time (h) until "fever clearance". The axillary temperature was measured at 3‐h intervals until "fever clearance", and at 6‐h intervals thereafter | Mean value (h) reported but not defined. Parasite counts performed every 4 h for 12 h, then every 6 h until 3 consecutive films were negative | Not reported | None reported |
| Newton 2003 | Death | Not reported | Median value (h) reported. Defined as time to reach a Glasgow Coma Scale of 15 in those participants with a score < 11/15 on admission. Glasgow Coma Scale measured every 15 min for first h, at 2 h, and then every 2 h until 12 h, every 4 h from 12 to 24 h, and every 6 h from 24 h until the score reached 15 | Not reported | Median value (h) reported. Defined as time until the axillary temperature first dropped below 37.5 °C and remained below that level for 24 h. Axillary temperature measured every 15 min for the first h, at 2 h, and then every 2 h until 12 h, every 4 h from 12 to 24 h, and every 6 h from 24 h until fever cleared | Median value (h) reported. Defined as time to a 50% reduction in parasite density. Parasite counts were measured at 0, 1, 2, 4, 6, 8, 10, 12, 16, 20, and 24 h, and then every 6 h until 6 h after parasite clearance | Plasma glucose less than or equal to 2.2 mmol/L. Plasma glucose measured at 0, 4, 8, 12, 16, 20, and 24 h and then every 6 h | Seizures, bleeding and sepsis after admission, pulmonary oedema, oliguria, time in intensive care unit |
Three trials reported mean coma recovery time but the data from Eltahir 2010 were incompletely reported. There is no evidence of a difference between the groups, the data are skewed and the results inconsistent (231 participants, two trials, Analysis 1.6). Three trials reported median coma recovery time and again the results were inconsistent and no conclusions can be made (see Table 5).
1.6. Analysis.
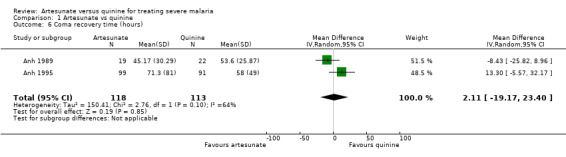
Comparison 1 Artesunate vs quinine, Outcome 6 Coma recovery time (hours).
3. Time‐to‐event data: medians, ranges, and modes.
| Outcome | Trial | Artesunate | Quinine | Comparative results reported in article |
| Coma recovery time (h): median (range), number | Hien 1992 | 35 (5 to 453); mode = 17; mean = 68.9 | 48 (7 to 144), mode = 43; mean = 58.1 | 'Not significantly different'. |
| Cao 1997 | 42 (4 to 228), n = 10 | 31 (4 to 66), n = 2 | ‐ | |
| Newton 2003 | 17 (1 to 125), n = 16 | 18 (1 to 188), n = 16 | 'P = 0.6' | |
| Eltahir 2010 | mean = 8.1 (SD not given), n = 4 | mean = 9.1, (SD not given), n = 5 | 'P=0.4' | |
| Time to localise pain (h): median (IQR), number | Dondorp 2010 | 12 (6 to 24), n = 698 | 12 (6 to 24), n = 726 | 'Hazard Ratio 0·87 (0·78–0·98), P = 0.0093' |
| Time to speak (h/days): median (IQR), number | Dondorp 2005 | 1 day (0.2 to 0.35), n = 730 | 1 day (0.2 to 0.21), n = 731 | 'P = 0.73' |
| Dondorp 2010 | 20 hrs (8 to 42), n = 664 | 18 hrs (11 to 36), n = 695 | 'Hazard Ratio 0·88 (0·79–0·99), P = 0.016' | |
| Time to hospital discharge (d): median (range/IQR), number | Cao 1997 | 8 (5 to 20), n = 33 | 8 (5 to 24), n = 29 | 'P = 0.99' |
| Dondorp 2005 | 5 (0 to 54), n = 623 | 5 (0 to 45), n = 567 | 'P = 0.20' | |
| Dondorp 2010 | 3 (IQR 2 to 5), n = 2478 | 3 (IQR 2 to 5) n = 2412 | 'P = 0.059' | |
| Fever clearance time (until first below 37.5 °C) (h): median (range), number | Cao 1997 | 4 (4 to 198), n = 35 | 8 (0 to 96), n = 35 | 'P = 0.17' |
| Newton 2003 | 11 (1 to 83), n = 42 | 13 (1 to 184), n = 42 | 'P = 0.2' | |
| Fever clearance time (until remains below 37.5 °C for 24 h) (h): median (range), number | Cao 1997 | 84 (4 to 198), n = 35 | 81 (0 to 246), n = 30 | 'P = 0.62' |
| Newton 2003 | 41 (3 to 138), n = 32 | 65 (12 to 383), n = 27 | 'P = 0.2' | |
| Time to parasite clearance of 50% (h): median (range), number | Cao 1997 | 5.7 (2.0 to 15.3), n = 35 | 13.2 (2.4 to 103.0), n = 32 | 'P < 0.0001' |
| Newton 2003 | 9.1 (0.3 to 37.2), n = 56 | 8.0 (0.2 to 46.0), n = 49 | 'P = 0.3' | |
| Time to parasite clearance of 90% (h): median (range), number | Cao 1997 | 12.0 (3.7 to 35.0), n=35 | 27.7 (7.5 to 107.0), n=32 | 'P < 0.0001' |
| Newton 2003 | 20.5 (2.8 to 50.11), n=54 | 24.7 (0.9 to 67.7), n=48 | 'P = 0.08' | |
| Time to parasite clearance of 100% (h): median (range), number | Cao 1997 | 36.0 (16.0 to126.0), n=34 | 84.0 (12.0 to 240.0), n=32 | 'P < 0.0001' |
In addition the two large multicentre trials (Dondorp 2005; Dondorp 2010) report median time to speak, and Dondorp 2010 reports median time to localise pain (see Table 5). Dondorp 2010 found the time to speak and localise pain to be slightly prolonged in those treated with artesunate but the clinical significance of this is unclear (Table 5).
Time to hospital discharge
Three trials reported this outcome as a median (Cao 1997; Dondorp 2005, Dondorp 2010). The results were consistent and showed no evidence of a difference between the groups (see Table 5). One trial reported this outcome as a mean (Newton 2003) with no evidence of a difference between the groups, but the data appeared skewed (113 participants, one trial, Analysis 1.7).
1.7. Analysis.
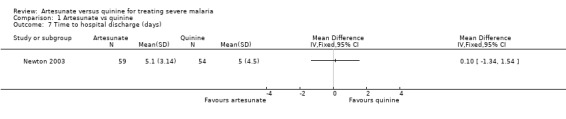
Comparison 1 Artesunate vs quinine, Outcome 7 Time to hospital discharge (days).
Fever clearance time
Three trials reported this outcome as a mean (Hien 1992; Anh 1995; Eltahir 2010). There is no evidence of a difference between the groups, although the data appeared to be skewed (317 participants, three trials, Analysis 1.8). Two trials reported this outcome as a median (Cao 1997; Newton 2003), and found no statistically significant difference between groups (see Table 5). The frequency with which fever was monitored differed between the trials (Table 4).
1.8. Analysis.
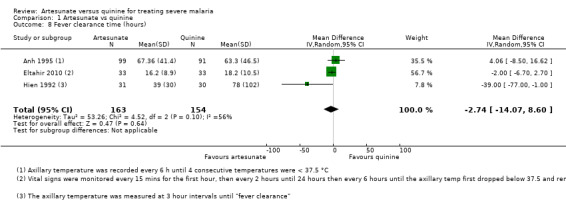
Comparison 1 Artesunate vs quinine, Outcome 8 Fever clearance time (hours).
Parasite clearance time (PCT)
Five trials report a measure of mean parasite clearance time.
Artesunate appears superior to quinine at reducing the mean 50% PCT (MD ‐8.14 hrs, 95% CI ‐11.55 to ‐4.73; 292 participants, three trials, Analysis 1.9), mean 90% PCT (MD ‐18.50 hrs, 95% CI ‐24.13 to ‐12.87; 61 patients; one trial, Analysis 1.9), mean 95% PCT (MD ‐10.69 hrs, 95% CI ‐20.27 to ‐1.10, 231 patients; two trials; Analysis 1.9), and mean 100% PCT (MD ‐9.77h 95% CI ‐18.11 to ‐1.44, 419 patients; four trials; Analysis 1.9).
1.9. Analysis.
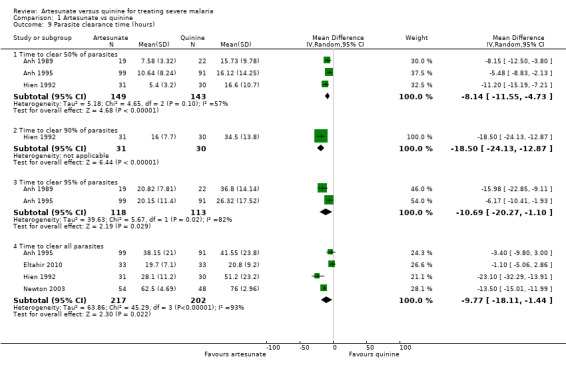
Comparison 1 Artesunate vs quinine, Outcome 9 Parasite clearance time (hours).
Two additional trials (Newton 2003; Cao 1997) reported median 50% and 90% PCT (see Table 5).
The frequency with which trials repeated malaria blood films differed between the trials (see Table 4).
Adverse effects
No trial reported discontinuation of medication. With the exception of hypoglycaemia and tinnitus, all adverse effects reported could be attributable to malaria. Artesunate was associated with a statistically significant reduction in episodes of hypoglycaemia (RR 0.55, 95% 0.41 to 0.74; 7137 participants, 4 trials, Analysis 1.10, Figure 4).
1.10. Analysis.
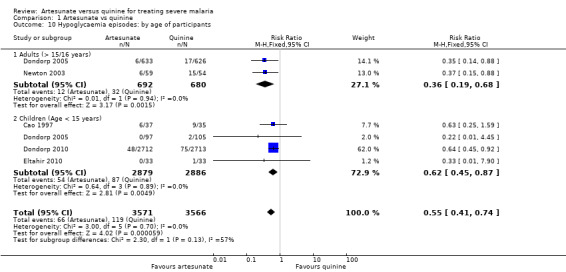
Comparison 1 Artesunate vs quinine, Outcome 10 Hypoglycaemia episodes: by age of participants.
4.
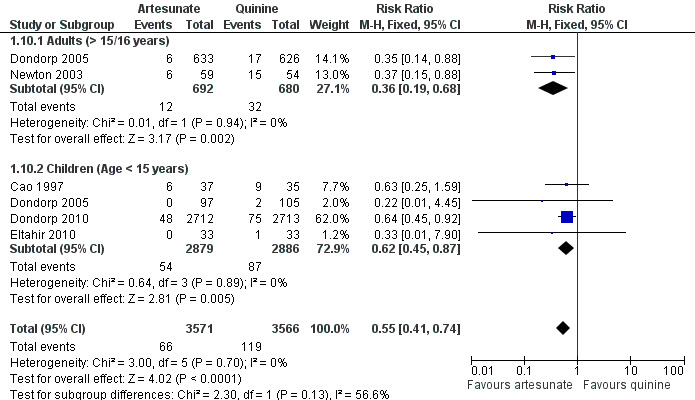
Forest plot of comparison: 1 Artesunate vs quinine, outcome: 1.15 Hypoglycaemia episodes: by age of participants.
Additional comments on adverse events taken from the original trial reports are given in Table 6.
4. Adverse event reporting.
| Study ID | Additional comments on adverse events |
| Anh 1989 | No comment on adverse events |
| Anh 1995 | No comment on adverse events |
| Cao 1997 | 'All 3 drug regimens were well tolerated, and no patient had to discontinue treatment because of adverse effects.' This study also conducted some cardiac monitoring on a non‐randomised subset of patients and does not report any significant differences between groups. |
| Dondorp 2005 | 'With the exception of hypoglycaemia there were no serious adverse effects that could be attributed to either treatment.' |
| Dondorp 2010 | 'We detected no severe adverse effects that could be attributed directly to drug toxicity. Although one patient treated with artesunate developed a mild urticarial rash, no severe type 1 hypersensitivity reactions were recorded.' |
| Eltahir 2010 | 'Following quinine infusion, 12 patients developed tinnitus and one hypoglycaemia. Abdominal pain and nausea were observed in three and four patients in artesunate and quinine groups, respectively.' |
| Hien 1992 | No comment on adverse events |
| Newton 2003 | 'Patients treated with quinine consistently developed cinchonism and had a significantly higher frequency of hypoglycemia. One patient had a probable adverse reaction to artesunate. This patient presented with parasitaemia of 31%, a plasma lactate level of 14.5 mmol/L, and a serum bilirubin level of 23 mg/dL and developed a widespread erythematous urticarial rash 17 h after treatment with intravenous artesunate was initiated.' |
Discussion
Summary of main results
Potential benefits of treating severe malaria with artesunate instead of quinine
Treating severe malaria with artesunate instead of quinine reduces the risk of death by 39% in adults (95% CI 25% to 50%), and 24% in children (95% CI 10% to 35%).
Artesunate also reduces episodes of hypoglycaemia during treatment by 45% (95% CI 26% to 59%).
Potential harms of treating severe malaria with artesunate instead of quinine
In adults neurological sequelae following treatment for severe malaria appears to be very low (< 1 %) and no difference has been shown between artesunate and quinine.
In children, treatment with artesunate appears to increase the incidence of neurological sequelae at the time of hospital discharge but the majority of these sequelae seem to resolve with time, and there is no evidence of a difference between the two treatments 28 days later.
Overall completeness and applicability of evidence
This review includes trials from multiple countries within Africa and Asia where malaria is most prevalent. Although there are no trials from South America it would be reasonable to generalise these findings to all geographic regions.
There is now adequate evidence to be confident of the results in both adults and children. However, it should be noted that these trials did not include a significant number of pregnant women.
Based on this trial data the absolute benefit in children appears lower than that seen in adults, primarily due to the lower mortality seen in children. This observation may be related to regional differences rather than participant age as the majority of child data is from Africa, and all the adult data is from Asia. Potential reasons for the lower risk of mortality seen in children in Dondorp 2010 include: inclusion of 'less severe' malaria, increased efficacy of quinine in Africa or true differences in the risk of death related to acquisition of partial immunity in Africa.
Quality of the evidence
Although several of the smaller trials suffer from methodological problems such as open randomization, unmatched additional oral antimalarials, or non‐standard dosing, these do not affect the overall quality of the evidence as the majority of the data is from large multicentre trials which do not suffer the same problems.
We consider the evidence for a reduction in mortality with artesunate to be high quality, and further research to establish this is unnecessary (see Table 1 and Table 2).
The increase in neurological sequelae is of a smaller magnitude than the reduction in deaths and seems to be temporary. The balance of benefits and harms is in favour of benefit with artesunate.
Economic commentary
To supplement the main systematic review of the effects on artesunate in the management of severe malaria, we sought to identify economic evaluations which compared the use of artesunate with the use of quinine. Systematic supplementary searches of the NHS Economic Evaluation Database (NHS EED) and the Health Economic Evaluations Database (HEED) identified four articles using the search terms outlined in Table 3. The articles were then screened for inclusion and two were discarded as they evaluated the use of rectal artesunate.
Lubell 2009 is a cost‐effectiveness analysis based on the large multicentre trial of artesunate in Asia (Dondorp 2005), and Lubell 2011 is based on the large multicentre trial from sub‐Saharan Africa (Dondorp 2010). Both studies take the service‐provider’s perspective, calculating the total costs to the health service, and no estimate was made of the economical benefit to the individual or society.
In both studies, the total cost of care using artesunate was marginally higher per patient than using quinine (in Asia: US$43.0 vs. US$32.4; in Africa: US$66.5 vs. US$63.5). However, given the large treatment effect of artesunate, the incremental cost per additional death averted was less than US$140 in both studies. For a brief summary of these two studies see Table 7.
5. Summary of cost‐effectiveness studies.
| Lubell 2009 | Lubell 2011 | |
| Location(s) | Asia | Sub‐Saharan Africa |
| Population | Mostly adults (86%) | Children |
| Type of analysis | Cost‐effectiveness | Cost‐effectiveness |
| Perspective taken | Provider | Provider |
| Main cost categories | Drug cost (but not administration costs) Inpatient care cost |
Drugs Fluids Laboratories Hotel |
| Currency | USD | USD |
| Price year | 2008 | 2009 |
| Sensitivity analysis? | Yes | Yes |
| Time horizon | Immediate | Immediate |
| Total cost per treatment episode artesunate vs. quinine | $43.0 vs. $32.4 | $66.5 vs. $63.5 |
| Number needed to treat | 13 | 41 |
| Incremental cost per death averted | $135.6 | $123 |
| Incremental cost per DALY averted | Not calculated | $3.8 |
It is important to highlight that neither of the identified economic evaluations were subjected to any formal critical appraisal and we do not attempt to draw any firm or general conclusions regarding the relative costs or efficiency of artesunate compared with quinine for the treatment of severe malaria. However, the available evidence indicates that, from an economic perspective, the use of artesunate for the treatment of severe malaria is a promising strategy when compared with quinine. Combined with the clinical data and the WHO treatment guidelines, this information may prove useful to those with responsibility for making local, regional, or national decisions regarding the management of individuals with severe malaria. However, end users of this review will need to assess the extent to which the economic evidence presented here may be applicable to their own setting.
Authors' conclusions
Implications for practice.
Intravenous artesunate should be the treatment of choice for adults and children presenting with severe malaria in any geographical region.
Implications for research.
Further research to examine the efficacy of artesunate versus quinine in children and adults is unnecessary. The safety of artesunate in pregnancy still needs to be determined, and trials are underway to examine the effects of artesunate when given repeatedly for multiple episodes of malaria. Toxicity from repeated dosing can not be ruled out on the basis of the evidence included in this review. Particular attention should be paid to the incidence of neurological sequelae.
What's new
| Date | Event | Description |
|---|---|---|
| 10 May 2012 | New search has been performed | updated with ecomonic evaluation |
| 10 May 2012 | New citation required but conclusions have not changed | Economic evaluation conducted by new author (R. Isba) and added to the review |
History
Protocol first published: Issue 2, 2006 Review first published: Issue 4, 2007
| Date | Event | Description |
|---|---|---|
| 12 April 2011 | Amended | Dates corrected (review assessed as up‐to‐date is 31 January 2011, not 2010 as previously stated). |
| 16 February 2011 | New citation required and conclusions have changed | Two new trials of artesunate versus quinine in African children have been added. |
| 28 November 2010 | New search has been performed | Major update. New Search November 2010. David Sinclair has joined the author team and become contact author, and Katharine Jones has stepped down from the author team. |
| 30 July 2009 | New search has been performed | New search conducted; no new trials for inclusion. Contact person changed. |
| 5 August 2008 | Amended | Converted to new review format with minor editing. |
Acknowledgements
Advice and support were provided by Prof Paul Garner, Manager of the Effective Health Care Research Programme Consortium (Effective Health Care RPC) at the Liverpool School of Tropical Medicine. This protocol was developed during a contract with the Effective Health Care RPC, which is funded by the UK Department for International Development (DFID). The authors would like to thank Dr Katharine Jones, first author on the original review, for her contributions to this point; Dr Jones stood down as an author at this update.
This document is an output from a project funded by the DFID for the benefit of developing countries. The views expressed are not necessarily those of DFID.
Data and analyses
Comparison 1. Artesunate vs quinine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death: participant age | 8 | 7429 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.62, 0.80] |
| 1.1 Adults (Age > 15/16 years) | 5 | 1664 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.50, 0.75] |
| 1.2 Children (Age < 15 years) | 4 | 5765 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.65, 0.90] |
| 2 Death: time since admission to hospital [sensitivity analysis] | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Death within 24 hours | 1 | 5417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.69, 1.04] |
| 2.2 Death after 24 hours | 1 | 5072 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.48, 0.88] |
| 2.3 Death within 48 hours | 3 | 1646 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.57, 1.05] |
| 2.4 Death after 48 hours | 3 | 1646 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.38, 0.74] |
| 3 Death: intravenous vs intramuscular artesunate [sensitivity analysis] | 8 | 7429 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.63, 0.80] |
| 3.1 Intravenous artesunate | 7 | 5435 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.60, 0.80] |
| 3.2 Intramuscular artesunate | 2 | 1994 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.60, 0.98] |
| 4 Neurological sequelae at discharge | 3 | 6422 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [1.05, 1.88] |
| 4.1 Adults (age > 15/16 years) | 1 | 1259 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.97 [0.60, 14.64] |
| 4.2 Children (Age < 15 years) | 3 | 5163 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.36 [1.01, 1.83] |
| 5 Neurological sequelae at day 28 | 1 | 4857 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.74, 2.03] |
| 5.1 Adults (Age > 15/16 years) | 0 | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Children (Age < 15 years) | 1 | 4857 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.74, 2.03] |
| 6 Coma recovery time (hours) | 2 | 231 | Mean Difference (IV, Random, 95% CI) | 2.11 [‐19.17, 23.40] |
| 7 Time to hospital discharge (days) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8 Fever clearance time (hours) | 3 | 317 | Mean Difference (IV, Random, 95% CI) | ‐2.74 [‐14.07, 8.60] |
| 9 Parasite clearance time (hours) | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Time to clear 50% of parasites | 3 | 292 | Mean Difference (IV, Random, 95% CI) | ‐8.14 [‐11.55, ‐4.73] |
| 9.2 Time to clear 90% of parasites | 1 | 61 | Mean Difference (IV, Random, 95% CI) | ‐18.5 [‐24.13, ‐12.87] |
| 9.3 Time to clear 95% of parasites | 2 | 231 | Mean Difference (IV, Random, 95% CI) | ‐10.69 [‐20.27, ‐1.10] |
| 9.4 Time to clear all parasites | 4 | 419 | Mean Difference (IV, Random, 95% CI) | ‐9.77 [‐18.11, ‐1.44] |
| 10 Hypoglycaemia episodes: by age of participants | 5 | 7137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.41, 0.74] |
| 10.1 Adults (> 15/16 years) | 2 | 1372 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.19, 0.68] |
| 10.2 Children (Age < 15 years) | 4 | 5765 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.45, 0.87] |
| 11 Hypoglycaemia episodes: by method of monitoring | 5 | 7137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.41, 0.74] |
| 11.1 Routine monitoring | 3 | 251 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.25, 0.85] |
| 11.2 Clinical monitoring | 1 | 1461 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.13, 0.79] |
| 11.3 Unclear | 1 | 5425 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.45, 0.92] |
1.11. Analysis.
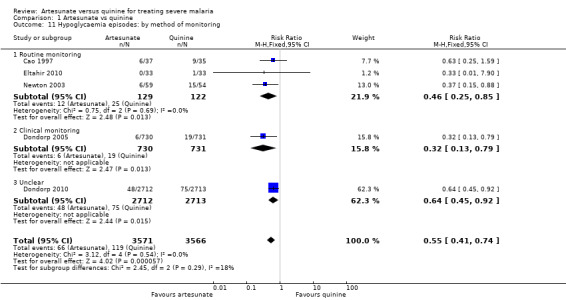
Comparison 1 Artesunate vs quinine, Outcome 11 Hypoglycaemia episodes: by method of monitoring.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Anh 1989.
| Methods | Study design: An open label randomized controlled trial Study dates: Feb to Dec 1989 |
|
| Participants | Number: 41 enrolled Inclusion criteria: adults > 16 yr old with cerebral malaria (P. falciparum parasitaemia > 1000/mm3 and Glasgow Coma Scale of 14 or less not attributable to any cause other than malaria) Exclusions: not specified |
|
| Interventions | 1. Artesunate: 60 mg intravenous (IV) at 0, 4, 24, and 48 h
2. Quinine: 20 mg/kg IV loading dose over 4 h at 0 h then 10 mg/kg IV every 8 h until able to swallow then 10 mg/kg by mouth every 8 h until day 7 Additional antimalarials: none reported |
|
| Outcomes | 1. Death 2. Coma recovery time 3. Parasite clearance time of 50% 4. Parasite clearance time of 95% | |
| Notes | Location: Vietnamese hospital Transmission: not specified Funding: Roche Asian Research Foundation supplied artesunate (personal communication from author) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication with author: Random numbers table |
| Allocation concealment (selection bias) | High risk | Comment: Not done |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up occurred |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Anh 1995.
| Methods | Study design: An open label randomized controlled trial Study dates: Jul 1992 to May 1995 |
|
| Participants | Number: 190 enrolled Inclusion criteria: adults 15 to 65 yr with cerebral malaria (asexual P. falciparum parasitaemia and clinical signs of cerebral malaria alone or associated with other visceral complications) Exclusion criteria: associated P. vivax parasitaemia, pregnancy, and concomitant diseases such as diabetes mellitus, stroke, meningitis, head trauma, pulmonary tuberculosis, or AIDS |
|
| Interventions | 1. Artesunate: 60 mg intravenous (IV) at 0, 4, 24, and 48 h
2. Quinine: 10 mg/kg IV over 4 h at 0 h then 10 mg/kg IV every 8 h until able to swallow then quinine by mouth at similar doses every 8 h until day 7 Additional antimalarials: artesunate treatment arm given one dose of mefloquine by mouth 15 mg/kg at day 7; quinine none |
|
| Outcomes | 1. Death within 24 h
2. Death after 24 h
3. Coma recovery time
4. Fever clearance time
5. Parasite clearance time of 50%
6. Parasite clearance time of 95%
7. Parasite clearance time of 100% Not included in the review: 8. Time to sit 9. Time to take oral by self medication |
|
| Notes | Location: Vietnamese clinical research centre Transmission: not specified Funding: World Health Organization |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication with author: Central randomization |
| Allocation concealment (selection bias) | Low risk | Personal communication with author: Central randomization |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up are recorded |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Cao 1997.
| Methods | Study design: A 3‐arm open label randomized controlled trial Study dates: Aug 1992 to Mar 1995 |
|
| Participants | Number: 72 enrolled Inclusion criteria: children < 15 yr with severe malaria (asexual P. falciparum parasitaemia plus at least 1 of the following: coma (Blantyre Coma Scale less than or equal to 3), severe anaemia (capillary haematocrit < 15%) with parasitaemia (> 10,000/µL); hyperparasitaemia (> 10% parasitized red blood cells or parasitaemia > 500,000/µL); jaundice (obvious clinically or serum bilirubin > 48 µmol/L); hypoglycaemia (blood glucose < 2.2 mmol/L); spontaneous bleeding (eg gastrointestinal haemorrhage); shock (systolic blood pressure < 50 mmHg if aged < 6 yr, or < 70 mmHg if aged 6 to 14 yr); repeated generalized convulsions (3 or more in 24 h despite cooling); renal impairment (serum creatinine > 177 µmol/L, or urine output < 12 mL/kg/24 h that fails to improve despite rehydration) Exclusion criteria: severe diarrhoea, mixed infection with P. vivax, prior treatment with quinine > 60 mg/kg, artemisinin > 20 mg/kg, or artesunate > 2 mg/kg during the illness episode, or any antimalarial treatment continuing for > 48 h |
|
| Interventions | 1. Artesunate: 3 mg/kg intramuscular (IM) at 0 h then 2 mg/kg IM at 12, 24, 48, and 72 h
2. Quinine: 20 mg/kg intravenous (IV) loading dose over 4 h (omitted if pretreatment with quinine) then 10 mg/kg IV every 8 h up to day 7
3. [Not relevant to review: rectal artemisinin] Additional antimalarials given: artesunate treatment arm received one dose of mefloquine by mouth 15 mg/kg at 96 h; quinine treatment arm given one dose of sulfadoxine‐pyrimethamine 500 mg/25 mg on day 7 |
|
| Outcomes | 1. Death
2. Number survived with neurological sequelae
3. Fever clearance time (all patients, excluding superinfections)
4. Coma resolution
5. Parasite clearance time of 50%
6. Parasite clearance time of 90%
7. Parasite clearance time of 100%
8. Period in hospital
9. Hypoglycaemia
10. Adverse effects Not included in this review: 11. Number survived well 12. Time to death from admission 13. Number with acute renal failure 14. Shock 15. Convulsions 16. Deterioration of coma score 17. Gastrointestinal bleeding 18. Anaemia 19. Chest infection 20. Urinary tract infection 21. Other infection 22. Reticulocyte count at admission, on day 5, at discharge 23. Haematocrit at admission, on day 5, at discharge |
|
| Notes | Location: Vietnamese hospital Transmission: not specified Funding: Wellcome Trust of Great Britain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication with author: Computer generated |
| Allocation concealment (selection bias) | Low risk | Quote: 'Randomization slips were kept in sealed, consecutively numbered envelopes and opened only after a decision to include the patient in the study had been made' |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Personal communication from author: 100% analysed, no losses to follow‐up. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Dondorp 2005.
| Methods | Study design: An open label multi‐centre randomized controlled trial Study dates: Jun 2003 to May 2005 |
|
| Participants | Number: 1461 enrolled Inclusion criteria: adults and children > 2 yr with severe malaria (positive blood antigen stick test for P. falciparum and a diagnosis of severe malaria, according to the admitting physician) Exclusion criteria: convincing history of full treatment with quinine (40 mg/kg on the first day and 30 mg/kg on any subsequent day) or an artemisinin derivative for more than 24 h before admission, known allergy to 1 of the artemisinin derivatives or quinine |
|
| Interventions | 1. Artesunate: 2.4 mg/kg intravenous (IV) at 0, 12, and 24 h then 2.4 mg/kg IV every 24 h until able to swallow then 2 mg/kg by mouth until day 7
2. Quinine: 20 mg/kg IV loading dose then 10 mg/kg every 8 h until able to swallow then 10 mg/kg by mouth every 8 h until day 7 Additional antimalarials: both arms except in India and Bangladesh were given doxycycline (100 mg every 12 h for 7 d) once able to swallow |
|
| Outcomes | 1. In‐hospital death
2. Death within 48 h of entry
3. Death after 48 h of entry
4. In‐hospital death (blood‐smear positive)
5. Neurological sequelae
6. Time to discharge (median, intra quartile range, and range)
7. Hypoglycaemia after entry Not included in the review: 8. Combined outcome: in hospital death or neurological sequelae 9. Fetal death 10. Time to speak (median, intra quartile range, and range) 11. Time to eat (median, intra quartile range, and range) 12. Time to sit (median, intra quartile range, and range) 13. Convulsions after entry 14. Shock developing after entry 15. Blackwater fever developing after entry 16. Dialysis after entry 17. Vasopressor treatment after entry 18. Mechanical ventilation after entry |
|
| Notes | Location: hospitals in Bangladesh, Myanmar, India, and Indonesia Transmission: not specified Funding: Wellcome Trust grant |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'The two‐step randomisation was produced with a computer generated randomisation list' |
| Allocation concealment (selection bias) | Low risk | Quote: 'After informed consent was obtained, we signed and dated a numbered sealed envelope across the seal, then opened it to reveal a unique study number. This number did not indicate the treatment allocation, but referred to a separate sealed hardcover box, containing the study drug, case record form, and all disposables needed for drug administration and blood sampling' |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up are recorded. |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Dondorp 2010.
| Methods | Study design: An open label, multi‐centre randomized controlled trial Study dates: Oct 2005 to July 2010 |
|
| Participants | Number: enrolled Inclusion criteria: Age < 15 years (age criteria varied slightly between sites at the request of the respective ethics review boards), a positive rapid diagnostic test for P. falciparum, severe malaria (physicians opinion), written consent Exclusions: Prior full treatment with parenteral quinine, or an artemisinin derivative for more than 24 h. |
|
| Interventions | Each study site used either the intravenous or intramuscular route for both treatment arms 1. Artesunate (Guilin, China): 2·4 mg/kg on admission, at 12 h, at 24 h, and then once daily until starting oral therapy. 2. Quinine dihydrochloride (Indus Pharma, Pakistan): 20 mg salt per kg loading dose infused over 4 h (in 5–10 mL/kg of 5% dextrose), followed by a 10 mg salt per kg infusion over 2–8 h three times daily until starting oral therapy. (For intramuscular treatment the doses were the same as for intravenous treatment; quinine was diluted in normal saline to a concentration of 60 mg/mL, and injected into the anterior thigh. The loading dose was given as a split dose into each thigh). Once able to tolerate oral medication (but after a minimum of 24 h of parenteral treatment), all participants received oral artemether‐lumefantrine (Novartis, Switzerland) for 3 days with milk or fat) |
|
| Outcomes | 1. Death
2. Death or sequelae at 28 days
3. Malaria attributable mortality
4. Case fatality in HIV +ve children
5. Time to discharge
3. Neurological sequelae
4. Adverse events Not included in the review: 1. Development of coma 2. Convulsions developing after 6 hours 3. Severe anaemia after admission 4. Blackwater fever 5. Time to speak (median, intra quartile range) 6. Time to eat (median, intra quartile range) 7. Time to sit unsupported (median, intra quartile range) 8. Time to localise pain (median, intra quartile range) |
|
| Notes | Location: 11 centres in nine African countries (Mozambique, The Gambia, Ghana, Kenya,
Tanzania, Nigeria, Uganda, Rwanda, and Democratic Republic of the Congo). Transmission: variable Funding: The Wellcome Trust |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Randomisation was done by people unrelated to the study and provided to the study sites in blocks of 20'. |
| Allocation concealment (selection bias) | Low risk | Quote: 'Study numbers were kept inside opaque sealed paper envelopes. After full informed written consent was obtained, the next envelope, which contained a unique study box number, was opened by the study physician or nurse. Then the corresponding numbered sealed box was opened. This box contained the study drug, case record form (labelled with the unique study number), and all disposables needed for drug administration and blood sampling'. |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Quote: 'Although the trial was open label at each site, none of the investigators or triallists, apart from for the trial statistician (TEP), had access to the summaries of treatment allocations. When notes or case record forms were reviewed, all study drug details were removed to preserve masking' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants are included in the primary analysis. A secondary per‐protocol analysis including only those with proven malaria who received the full course of treatment excluded 149 (5.5%) from the artesunate arm and 161 (5.9%) from the quinine arm |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting. |
| Other bias | Low risk | One study site did study adults as well as children but these participants are not reported in the paper, or analysis. |
Eltahir 2010.
| Methods | Study design: An open label randomized controlled trial Study dates: Aug to Sep 2009 |
|
| Participants | Number: 66 enrolled Inclusion criteria: Children with slide‐confirmed, severe P. falciparum malaria, written informed consent Exclusions: None stated |
|
| Interventions | 1. Intravenous artesunate (Guilin; China): 2.4 mg/kg body weight given at 0, 12, and 24 h, and then daily.
2. Intravenous quinine (Shanghai; China): 20 mg/kg loading dose infused over 4 h then 10 mg/kg infused over 2–4 h three times a day. Once oral therapy was tolerated participants received artesunate+ sulfadoxine‐pyrimethamine or quinine tablets to complete treatment. |
|
| Outcomes | 1. Death
2. Coma recovery time
3. Parasite clearance time
4. Fever clearance time
5. Adverse events Not included in the review: |
|
| Notes | Location: Sudan Transmission: unstable Funding: Sudanese Sugar Company and Assalaya Sugar Factory, Sudan |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Individuals were randomised (by computer‐generated numbers)' |
| Allocation concealment (selection bias) | Low risk | Quote: 'computer generated numbers were sealed in individual envelopes and securely stored' |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Hien 1992.
| Methods | Study design: A 3‐arm open label randomized controlled trial Study dates: 1989 to 1990 |
|
| Participants | Number: 61 enrolled Inclusion criteria: cerebral malaria (P. falciparum parasitaemia with clinical signs of malaria and a Glasgow Coma Scale < 10) Exclusion criteria: not specified |
|
| Interventions | 1. Artesunate: 60 mg intravenous (IV) at 0, 4, 24, and 48 h
2. Quinine: 500 mg IV over 4 h then 500 mg IV every 8 h until able to swallow then 500 mg by mouth every 8 h until day 14
3. [Not relevant to review: rectal artemisinin] Additional antimalarials: artesunate treatment arm given one dose of mefloquine (by mouth 500 mg) once able to swallow |
|
| Outcomes | 1. Fever clearance time
2. Parasite clearance time of 50% 3. Parasite clearance time of 90% 4. Parasite clearance time of 100% 5. Time to regain full consciousness 6. Death Not included in the review: |
|
| Notes | Location: intensive care unit in Vietnam Transmission: not specified Funding: artesunate was provided by Professor Li Guo Qiao |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication from author: We did randomization by hand using tables of randomization from a statistic book |
| Allocation concealment (selection bias) | Low risk | Personal communication from author: All treatment codes were sealed in opaque envelopes which were only opened when patients had been recruited into trial based on inclusion criteria |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses to follow‐up are recorded |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
Newton 2003.
| Methods | Study design: An open label randomized controlled trial Study dates: May to July 1994 and 1995 to 2001 |
|
| Participants | Number: 113 enrolled, 100 analysed Inclusion criteria: adults aged 15 yr or above with severe malaria (single‐species P. falciparum parasitaemia > 0.1% plus at least 1 of following: Glasgow Coma Scale < 11; haematocrit < 20% with asexual parasitaemia > 100,000/µL; total serum bilirubin > 50 µmol/L with asexual parasitaemia > 100,000/µL; serum creatinine > 264 mol/µL with urine output < 400 mL/24 h; systolic blood pressure < 80 mmHg with cool extremities; asexual parasitaemia > 10%; plasma lactate level > 4 mmol/L; plasma glucose level < 2.2 mmol/L; plasma venous bicarbonate level < 15 mmol/L Exclusion criteria: pregnancy, contraindications to study drugs, artesunate, mefloquine, or significant quinine (> 2 g) intake in the previous 24 h |
|
| Interventions | 1. Artesunate: 2.4 mg/kg intravenous (IV) at 0 h then 1.2 mg/kg at 12 h then 1.2 mg/kg every 24 h until able to swallow then 12 mg/kg by mouth every 24 h over 7 days
2. Quinine: 20 mg/kg IV over 4 h loading dose then 10 mg/kg IV every 8 until able to swallow then 10 mg/kg by mouth until day 7 Additional antimalarials: once able to swallow some participants in both arms were given additional antimalarials, but the drug given varied during the trial; AS: no additional antimalarial (n = 22), mefloquine 15 mg/kg (n = 1), mefloquine 25 mg/kg in 2 doses (n = 22), doxycycline 100 mg every 12 h for 7 d (n = 14); quinine: no additional antimalarial (n = 20), tetracycline 250 mg every 12 h for 7 d (n = 19), doxycycline 100 mg every 12 h for 7 days (n = 15) |
|
| Outcomes | 1. Fever clearance time
2. Parasite clearance time of 50% 3. Parasite clearance time of 90% 4. Parasite clearance time of 100% 5. Time to regain full consciousness 6. Death 6. Hypoglycaemia 7. Adverse effects Not included in the review: |
|
| Notes | Location: 2 hospitals in Thailand Transmission: seasonal low intensity Funding: Wellcome Trust of Great Britain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication with author: Random codes were created in Excel using the 'Randbetween' command. |
| Allocation concealment (selection bias) | High risk | Quote: 'The randomization was open' |
| Blinding (performance bias and detection bias) Objective outcomes: Death | Low risk | Comment: An open‐label trial is unlikely to bias an objective outcome like death |
| Blinding (performance bias and detection bias) Subjective outcomes: Others | High risk | Comment: An open label trial. No attempt was made to blind participants, providers or outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13 patients were excluded after randomization for not meeting the criteria for severe malaria 5 (%) in the artesunate arm and 8 (%) in the quinine arm |
| Selective reporting (reporting bias) | Low risk | No evidence of selective reporting |
| Other bias | Low risk | No other bias identified |
n: number of participants.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aguwa 2010 | Artemether versus quinine |
| Awad 2003 | Not a randomized controlled trial |
| Barnes 2004 | Not severe malaria |
| Bounyasong 2001 | Not severe malaria |
| Haroon 2005 | A quasi‐randomized controlled trial in which the first patient was allocated a treatment at random and then future patients were allocated their treatment using an alternating pattern |
| Krudsood 2003 | Not a randomized controlled trial |
| Li 1990 | Not severe malaria |
| McGready 2001a | Not severe malaria |
| McGready 2001b | Not a randomized controlled trial |
| Mohanty 2004 | Not a randomized controlled trial (quasi‐randomized) |
| Newton 2001 | Treatment comparison is artesunate versus artesunate and quinine |
| Osanuga 2009 | Artemether versus quinine |
| Phu 2010 | Artesunate versus artemether |
| Pukrittayakamee 2004 | Not severe malaria |
| Win 1992 | Not a randomized controlled trial |
| Zhao 2001 | Not severe malaria |
Differences between protocol and review
2007, Issue 4 (first review version): We removed the requirement for all participants to fulfil the WHO definition for severe malaria (Gilles 2000) in view of the large number of participants this would have excluded from the review as the largest included trial used a clinical case definition.
We changed the intervention from "parenteral artesunate" to "intravenous, intramuscular, or rectal artesunate" to clarify that trials using artesunate suppositories would be included in the review.
We subgrouped "neurological sequelae" into "neurological sequelae at discharge" and "neurological sequelae at day 28".
We added a number of sensitivity analyses post‐hoc after noting significant variation in study design across trials.
We presented data for hypoglycaemia in a forest plot rather than a table as stated in the protocol to reflect the clinical importance of this outcome.
Contributions of authors
Katharine Jones and Sarah Donegan assessed the eligibility and methodological quality of trials, extracted and analysed data, and completed the first published version of the review. David Sinclair replaced Katharine Jones for the 2011 update of this review. David Lalloo contributed to the design and writing of the review. Rachel Isba designed and conducted the economic commentary.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
University of Liverpool, UK.
External sources
Department for International Development (DFID), UK.
Declarations of interest
Dr David Lalloo was part of the data and safety monitoring committee for the two Dondorp trials. This committee is independent, does not run or gain anything from the trial, and has a main role of protecting participants.
Unchanged
References
References to studies included in this review
Anh 1989 {unpublished data only}
- Anh TK. Efficacy of qinghaosu in Plasmodium falciparum malaria. World Health Organization report [M20/181/38].
- Anh TK, Kim NV, Bich NN, Huong N.ng, Phuong N.V, et al. Randomized comparative study of artesunate intravenously and quinine in loading dose IV on severe and complicated malaria. Unpublished report.
Anh 1995 {published data only}
- Anh TK. Standard dose quinine & IV artesunate in treatment of severe and complicated falciparum malaria [WHO/Geneva/TDCRC/CRH]. Malaria Symposium & Workshop, Vungtau 1992.
- Anh TK, Binh TQ, Kim NV, et al. Comparative study of intravenous artesunate followed by oral mefloquine versus intravenous quinine in the treatment of severe and complicated malaria in Viet Nam. Unpublished report 1995.
- Anh TK, Binh TQ, Kim NV, et al. Comparative study of intravenous artesunate followed by oral mefloquine versus intravenous quinine in the treatment of severe and complicated malaria in Vietnam. Symposium on Tropical Medicine, Sanya, China. Institute Guangzhou University of TCM, 1995; Vol. 12:8‐9.
Cao 1997 {published data only}
- Cao XT, Bethell DB, Pham TP, Ta TT, Tran TN, Nguyen TT, et al. Comparison of artemisinin suppositories, intramuscular artesunate and intravenous quinine for the treatment of severe childhood malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 1997;91(3):335‐42. [DOI] [PubMed] [Google Scholar]
Dondorp 2005 {published data only}
- Dondorp A, Nosten F, Stepniewska K, Day N, White N. Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet 2005;366(9487):717‐25. [DOI] [PubMed] [Google Scholar]
Dondorp 2010 {published data only}
- Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open‐label, randomised trial. Lancet 2010;376(9753):1647‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eltahir 2010 {published data only}
- Eltahir HG, Omer AA, Mohamed AA, Adam I. Comparison of artesunate and quinine in the treatment of Sudanese children with severe Plasmodium falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2010;104(10):684‐6. [DOI] [PubMed] [Google Scholar]
Hien 1992 {published data only}
- Hien TT, Arnold K, Vinh H, Cuong BM, Phu NH, Chau TT, et al. Comparison of artemisinin suppositories with intravenous artesunate and intravenous quinine in the treatment of cerebral malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 1992;86(6):582‐3. [DOI] [PubMed] [Google Scholar]
- Vinh H, Arnold K, Cuong B, Phu N, Chau T, Hao N. Treatment of cerebral malaria comparing artemisinin suppositories with intravenous artesunate and intravenous quinine. XIIIth International Congress for Tropical Medicine and Malaria; Pattaya, Thailand. 1992:9. [DOI] [PubMed]
Newton 2003 {published data only}
- Newton PN, Angus BJ, Chierakul W, Dondorp A, Ruangveerayuth R, Silamut K, et al. Randomized comparison of artesunate and quinine in the treatment of severe falciparum malaria. Clinical Infectious Diseases 2003;37(1):7‐16. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aguwa 2010 {published data only}
- Aguwa CN, Ukwe CV, Adibe MO. A comparative study of quinine and artemether in the treatment of severe malaria in Nigerian children [Tropical Journal of Pharmaceutical Research]. 2010 9;1:11‐17. [Google Scholar]
Awad 2003 {published data only}
- Awad MI, Alkadru AM, Behrens RH, Baraka OZ, Eltayeb IB. Descriptive study on the efficacy and safety of artesunate suppository in combination with other antimalarials in the treatment of severe malaria in Sudan. American Journal of Tropical Medicine and Hygiene 2003;68(2):153‐8. [PubMed] [Google Scholar]
Barnes 2004 {published data only}
- Barnes KI, Mwenechanya J, Tembo M, McIlleron H, Folb PI, Ribeiro I, et al. Efficacy of rectal artesunate compared with parenteral quinine in initial treatment of moderately severe malaria in African children and adults: a randomised study. Lancet 2004;363(9421):1598‐605. [DOI] [PubMed] [Google Scholar]
Bounyasong 2001 {published data only}
- Bounyasong S. Randomized trial of artesunate and mefloquine in comparison with quinine sulfate to treat P. falciparum malaria pregnant women. Journal of Medical Association of Thailand 2001;84(9):1288‐99. [PubMed] [Google Scholar]
Haroon 2005 {published data only}
- Haroon N, Amichandwala K, Solu M. Comparative efficacy of quinine and artesunate in the treatment of severe malaria: A randomized controlled trial. JK Science 2005;7(1):32‐5. [Google Scholar]
Krudsood 2003 {published data only}
- Krudsood S, Wilairatana P, Vannaphan S, Treeprasertsuk S, Silachamroon U, Phomrattanaprapin W, et al. Clinical experience with intravenous quinine, intramuscular artemether and intravenous artesunate for the treatment of severe malaria in Thailand. Southeast Asian Journal of Tropical Medicine and Public Health 2003;34(1):54‐61. [PMC free article] [PubMed] [Google Scholar]
Li 1990 {published data only}
- Li G, Guo X, Fu L. A randomised comparative study of artesunate versus quinine dihydrochloride in the treatment of falciparum malaria. Clinical Trials on Qinghaosu and its Derivatives; Guangzhou, China. 1990; Vol. 1:50‐8.
McGready 2001a {published data only}
- McGready R, Cho T, Samuel, Villegas L, Brockman A, Vugt M, et al. Randomized comparison of quinine‐clindamycin versus artesunate in the treatment of falciparum malaria in pregnancy. Transactions of the Royal Society of Tropical Medicine and Hygiene 2001;95(6):651‐6. [DOI] [PubMed] [Google Scholar]
McGready 2001b {published data only}
- McGready R, Cho T, Keo NK, Thwai KL, Villegas L, Looareesuwan S, et al. Artemisinin antimalarials in pregnancy: a prospective treatment study of 539 episodes of multidrug‐resistant Plasmodium falciparum. Clinical Infectious Diseases 2001;33(12):2009‐16. [DOI] [PubMed] [Google Scholar]
Mohanty 2004 {published data only}
- Mohanty AK, Rath BK, Mohanty R, Samal AK, Mishra K. Randomized control trial of quinine and artesunate in complicated malaria. Indian Journal of Pediatrics 2004;71(4):291‐5. [DOI] [PubMed] [Google Scholar]
Newton 2001 {published data only}
- Newton PN, Chierakul W, Ruangveerayuth R, Silamut K, Teerapong P, Krudsood S, et al. A comparison of artesunate alone with combined artesunate and quinine in the parenteral treatment of acute falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2001;95(5):519‐23. [DOI] [PubMed] [Google Scholar]
Osanuga 2009 {published data only}
- Osonuga OA, Osonuga IO. Parasitaemia changes in the course of treatment of severe malaria patients with artemether and quinine (A preliminary study). Macedonian Journal of Medical Sciences 2009;2(4):319‐323. [Google Scholar]
Phu 2010 {published data only}
- Phu NH, Tuan PQ, Day N, Mai NT, Chau TT, Chuong LV, et al. Randomized controlled trial of artesunate or artemether in Vietnamese adults with severe falciparum malaria. Malaria 2010;9(97):doi: 10.1186/1475‐2875‐9‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pukrittayakamee 2004 {published data only}
- Pukrittayakamee S, Chotivanich K, Chantra A, Clemens R, Looareesuwan S, White NJ. Activities of artesunate and primaquine against asexual‐ and sexual‐stage parasites in falciparum malaria. Antimicrobial Agents and Chemotherapy 2004;48(4):1329‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Win 1992 {published data only}
- Win K, Than M, Thwe Y. Comparison of combinations of parenteral artemisinin derivatives plus oral mefloquine with intravenous quinine plus oral tetracycline for treating cerebral malaria. Bulletin of the World Health Organization 1992;70(6):777‐82. [PMC free article] [PubMed] [Google Scholar]
Zhao 2001 {published data only}
- Zhao J. Artesunate for 321 patients with falciparum malaria in the Republic of Mali. Chinese Journal of New Drugs and Clinical Remedies 2001;20(4):275‐7. [Google Scholar]
Additional references
Adjuik 2004
- Adjuik M, Babiker A, Garner P, Olliaro P, Taylor W, White N, International Artemisinin Study Group. Artesunate combinations for treatment of malaria: meta‐analysis. Lancet 2004;363(9402):9‐17. [DOI] [PubMed] [Google Scholar]
Afolabi 2004
- Afolabi BB, Okoromah CAN. Intramuscular arteether for treating severe malaria. Cochrane Database of Systematic Reviews 2004, Issue 4. Art. No.: CD004391. [DOI: 10.1002/14651858.CD004391.Pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Alkadi 2007
- Alkadi HO. Antimalarial Drug Toxicity: A review. Chemotherapy 2007;53:385‐391. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313(7066):1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
AQMSG 2001
- The Artemether‐Quinine Meta‐analysis Study Group. A meta‐analysis using individual patient data of trials comparing artemether with quinine in the treatment of severe falciparum malaria. Transactions of the Royal Society of Tropical Medicine 2001;95(6):637‐50. [DOI] [PubMed] [Google Scholar]
Brewer 1994
- Brewer T, Grate SJ, Peggins JO, Weina PJ, Petras JM, Levine BS, et al. Fatal neurotoxicity of arteether and artemether. American Journal of Tropical Medicine and Hygiene 1994;51(3):251‐9. [DOI] [PubMed] [Google Scholar]
Doolan 2009
- Doolan DL, Dobano C, Baird JK. Acquired immunity to malaria. Clinical Microbiology Reviews 2009;22(1):13‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Genovese 2000
- Genovese RF, Newman DB, Brewer TG. Behavioural and neural toxicity of the artemisinin antimalarial, arteether, but not artesunate and artelinate in rats. Pharmacology, Biochemistry, and Behaviour 2000;67(1):37‐44. [DOI] [PubMed] [Google Scholar]
Gilles 2000
- Gilles HM. Management of severe malaria: a practical handbook. 2nd Edition. Geneva: World Health Organization, 2000. [Google Scholar]
Hien 2004
- Hien TT, Davis TM, Chuong LV, Ilett KF, Sinh DX, Phu NH, et al. Comparative pharmacokinetics of intramuscular artesunate and artemether in patients with severe falciparum malaria. Antimicrobial Agents and Chemotherapy 2004;48(11):4234‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors), Cochrane Handbook of Systematic Reviews of Intervention. Version 5.0.0 (updated February 2008). The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org. John Wiley & Sons Ltd.
Jaffar 1997
- Jaffar S, Hensbroek M, Palmer A, Schneider G, Greenwood B. Predictors of a fatal outcome following childhood cerebral malaria. American Journal of Tropical Medicine and Hygiene 1997;57(1):20‐4. [DOI] [PubMed] [Google Scholar]
Karbwang 1997
- Karbwang J, Na‐Bangchang K, Congpuong K, Molunto P, Thanavibul A. Pharmacokinetics and bioavailability of oral and intramuscular artemether. European Journal of Clinical Pharmacology 1997;52:307‐10. [DOI] [PubMed] [Google Scholar]
Kissinger 2000
- Kissinger E, Hien T, Hung N, Nam N, Tuyen N, Dinh B, et al. Clinical and neurophysiological study of the effects of multiple doses of artemisinin on brain‐stem function in Vietnamese patients. American Journal of Tropical Medicine and Hygiene 2000;63(1‐2):48‐55. [DOI] [PubMed] [Google Scholar]
Kyu 2009
- Kyu HH, Fernandez E. Artemisinin derivatives versus quinine for cerebral malaria inAfrican children: a systematic review. Bulletin of the World Health Organization 2009;87:896‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lesi 2004
- Lesi A, Meremikwu M. High first dose quinine regimen for treating severe malaria. Cochrane Database of Systematic Reviews 2004, Issue 3. [DOI: 10.1002/14651858.CD003341.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2004
- Li Q, Lugt CB, Looareesuwan S, Krudsood S, Wilairatana P, Vannaphan S, et al. Pharmacokinetic investigation of the therapeutic potential of artemotil (ß‐arteether) in Thai patients with severe Plasmodium falciparum malaria. Am J Trop Med Hyg 2004;71(6):723‐731. [PubMed] [Google Scholar]
Looareesuwan 2002
- Looareesuwan S, Oosterhuis B, Schilizzi BM, Sollie FAE, Wilairatana P, Krudsood S, et al. Dose‐finding and efficacy study for i.m. artemotil (beta‐arteether)and comparison with i.m. artemether in acute uncomplicated P. falciparum malaria. British Journal of Clinical Pharmacology 2002;53(5):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lubell 2009
- Lubell Y, Yeung S, Dondorp AM, Day NP, Nosten F, Tjitra E, et al. Cost‐effectiveness of artesunate for the treatment of severe malaria. Trop Med Int Health 2009;14(3):332‐7. [DOI] [PubMed] [Google Scholar]
Lubell 2011
- Lubell Y, Riewpaiboon A, Dondorp AM, Seidlein L, Mokuolu OA, Nansumba M, et al. Cost‐effectiveness of parenteral artesunate for treating children with severe malaria in sub‐Saharan Africa.. Bulletin of the World Health Organization 2011;89(7):504‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
McIntosh 2000
- McIntosh HM, Olliaro P. Artemisinin derivatives for treating severe malaria. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD000527] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mithwani 2003
- Mithwani S, Aarons L, Kokwaro GO, Majid O, Muchohi S, Edwards G, et al. Population pharmacokinetics of artemether and dihydroartemisinin following single intramuscular dosing of artemether in African children with severe falciparum malaria. British Journal of Clinical Pharmacology 2003;57(2):146‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Murphy 1997
- Murphy SA, Mberu E, Muhia D, English M, Crawley J, Waruiru C, et al. The disposition of intramuscular artemether in children with cerebral malaria; a preliminary study. Transactions of the Royal Society of Tropical Medicine and Hygiene 1997;91(3):331‐4. [DOI] [PubMed] [Google Scholar]
Nealon 2002
- Nealon C, Dzeing A, Müller‐Römer U, Planche T, Sinou V, Kombila M, et al. Intramuscular bioavailability and clinical efficacy of artesunate in gabonese children with severe malaria. Antimicrobial Agents and Chemotherapy 2002;46(12):3933‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nontprasert 1998
- Nontprasert N, Nosten‐Bertrand M, Pukrittayakamee S, Vanijanonta S, Angus BJ, White N. Assessment of the neurotoxicity of parenteral artemisinin derivatives in mice. American Journal of Tropical Medicine and Hygiene 1998;59(4):519‐22. [DOI] [PubMed] [Google Scholar]
Nontprasert 2000
- Nontprasert A, Pukrittayakamee S, Nosten‐Bertrand M, Vanijanonta S, White NJ. Studies of the neurotoxicity of oral artemisinin derivatives in mice. American Journal of Tropical Medicine and Hygiene 2000;62(3):409‐12. [DOI] [PubMed] [Google Scholar]
Nontprasert 2002
- Nontprasert A, Pukrittayakamee S, Prakongpan S, Supanaranond W, Looareesuwan S, White NJ. Assessment of neurotoxicity of oral dihydroartemisinin in mice. Transactions of the Royal Society of Tropical Medicine and Hygiene 2002;96(1):99‐101. [DOI] [PubMed] [Google Scholar]
Nosten 2007
- Nosten F, White NJ. Artemisinin‐based combination treatment of falciparum malaria. American Journal of Tropical Medicine and Hygiene 2007;77 (6 Suppl):181‐92. [PubMed] [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Ribeiro 1998
- Ribeiro IR, Olliaro P. Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Medecine Tropicale 1998;58 Suppl 3:50‐3. [PubMed] [Google Scholar]
ter Kuile 1993
- ter Kuile F, White NJ, Holloway P, Pasvol G, Krishna S. Plasmodium falciparum: in vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Experimental Parasitology 1993;76(1):85‐95. [DOI] [PubMed] [Google Scholar]
Toovey 2004
- Toovey S, Jamieson A. Audiometric changes associated with the treatment of uncomplicated falciparum malaria with co‐artemether. Transactions of the Royal Society of Tropical Medicine and Hygiene 2004;98(5):261‐7. [DOI] [PubMed] [Google Scholar]
van Der Torn 1996
- Torn M, Thuma PE, Mabeza GF, Biemba G, Moyo VM, McLaren CE, et al. Loading dose of quinine in African children with cerebral malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 1998;92(3):325‐31. [DOI] [PubMed] [Google Scholar]
White 1983
- White NJ, Warrell DA, Chanthavanich P, Looareesuwan S, Warrell MJ, Krishna S, et al. Severe hypoglycemia and hyperinsulinemia in falciparum malaria. New England Journal of Medicine 1983;309(2):61‐6. [DOI] [PubMed] [Google Scholar]
White 1983b
- White NJ, Looareesawan S, Warrell DA, Warrell MJ, Chanthavanich P, Bunnag D, et al. Quinine Loading Dose in Severe Malaria. American Journal of Tropical Medicine and Hygiene 1983;32:1‐5. [DOI] [PubMed] [Google Scholar]
WHO 2000
- WHO. Severe and complicated malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2000;94(suppl 1):1‐90. [PubMed] [Google Scholar]
WHO 2008
- WHO Global Malaria Programme. World Malaria Report: 2008. World Health Organization, 2008. [Google Scholar]
WHO 2010
- World Health Organization. Roll Back Malaria Dept. Guidelines for the treatment of malaria; Second edition. Geneva: World Health Organization, 2010. [Google Scholar]
Wongsrichanalai 2002
- Wongsrichanalai C, Pickard AL, Wernsdorfer WH, Meshnick SR. Epidemiology of drug‐resistant malaria. Lancet Infectious Diseases 2002;2(4):209‐18. [DOI] [PubMed] [Google Scholar]