

Abstract
Background
Malaria is the most common precipitating cause of crises in sickle cell disease in malaria‐endemic countries. Health professionals often recommend life‐long malaria chemoprophylaxis for people with sickle cell disease living in these areas. It is therefore important we have good evidence of benefit.
Objectives
To assess the effects of routine malaria chemoprophylaxis in people with sickle cell disease.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register (January 2006), Cochrane Cystic Fibrosis and Genetic Disorders Group Specialized Register (July 2006), CENTRAL (The Cochrane Library 2006, Issue 1), MEDLINE (1966 to January 2006), EMBASE (1974 to January 2006), LILACS (1982 to January 2006), and reference lists. We also contacted organizations and pharmaceutical companies.
Selection criteria
Randomized and quasi‐randomized controlled trials comparing chemoprophylaxis with any antimalarial drug given for a minimum of three months compared with a placebo or no intervention.
Data collection and analysis
Two authors independently applied the inclusion criteria, assessed the risk of bias in the trials, and extracted data. Dichotomous data were analysed using risk ratios (RR) and presented with 95% confidence intervals (CI).
Main results
Two trials with a total of 223 children with homozygous sickle cell disease met the inclusion criteria. A randomized controlled trial in Nigeria compared two different antimalarial drugs with a placebo, and reported that chemoprophylaxis reduced sickle cell crises (RR 0.17, 95% CI 0.04 to 0.83; 97 children), hospital admissions (RR 0.27, 95% CI 0.12 to 0.63; 97 participants), and blood transfusions (RR 0.16, 95% CI 0.05 to 0.56; 97 participants). A quasi‐randomized controlled trial of 126 children in Uganda compared an antimalarial drug plus antibiotics with no antimalarial plus placebo. Chemoprophylaxis reduced the number of episodes of malaria and dactylitis, and increased mean haemoglobin values in this trial.
Authors' conclusions
It is beneficial to give routine malaria chemoprophylaxis in sickle cell disease in areas where malaria is endemic.
4 November 2019
Update pending
New contributors needed
The CIDG Editors are looking for contributors to update and maintain this Cochrane Review. Contact the CIDG Managing Editor for further information.
Plain language summary
In areas where malaria is common, malaria drug prophylaxis benefits people with sickle cell disease
Sickle cell disease is a blood disorder, and means the part of the red blood cell that carries oxygen from the lungs to the body tissues (haemoglobin) is abnormal. It is a genetic disorder and occurs when people inherit abnormal genes from both parents. It is more common in people originating from tropical Africa, and the Caribbean, Mediterranean, Indian, and Middle Eastern regions. Some types of sickle cell disease cause more severe symptoms than others, but in general people with the disease may be tired and weak (due to anaemia), and have severe and recurrent pain in the bones (referred to as crises), have more infections, more problems with breathing, and more chance of having a stroke. Sickle cell disease contributes to the death of children in the first five years of life, and infants aged six to 12 months are particularly at risk. Malaria infection is known to trigger a sickle cell crisis. So preventing malaria in people with sickle cell disease may also help to reduce crises and all the problems that go along with it. Health professionals often recommend life‐long drugs to prevent malaria infections (chemoprophylaxis) for people with sickle cell disease living in these high risk areas for malaria. However, it is important to assess how effective this may be and what the adverse effects there might be from taking these drugs over a long period of time. This review of trials identified two small trials involving 223 people with homozygous sickle cell disease (commonly called sickle cell anaemia). These showed benefit in terms of reducing the number of sickle cell crises, blood transfusions, hospital admissions, and increasing the mean haemoglobin levels, but they did not collect data on potential adverse effects. More research is needed, therefore, to be sure of the benefits and to assess possible problems of drug resistance, and other potential long‐term adverse effects that may be associated with continuous treatment.
Background
Sickle cell disease is an inherited blood condition. It is more common in people with their origins in Equatorial Africa, and the Caribbean, Mediterranean, Indian, and Middle Eastern regions (Serjeant 1997; WHO 2000). It is caused by abnormalities in the production of haemoglobin, which is the part of the red blood cell that carries oxygen from the lungs to the body tissues. Sickle cell disease occurs when people inherit from both parents abnormal genes responsible for haemoglobin production.
There are two common genotypes:
Homozygous sickle cell disease (also known as sickle cell anaemia), where the sickle cell gene responsible for the production of abnormal haemoglobin S is inherited from both parents.
Sickle cell/haemoglobin C disease, where the person inherits one sickle cell gene from one parent, and another abnormal haemoglobin gene (haemoglobin C) from the other.
Two other genotypes are less common:
Sickle cell/beta + thalassaemia, where the person inherits one sickle cell gene from one parent, and another abnormal haemoglobin gene (thalassaemia) from the other. This is a mild form of the disease in which haemoglobin A is present.
Sickle cell/beta 0 thalassaemia, where the person inherits one sickle cell gene from one parent, and another abnormal haemoglobin gene (beta 0 thalassaemia) from the other. This is a severe form of the disease in which haemoglobin A is absent (Serjeant 1997; Ashley‐Koch 2000).
The frequency of the different genotypes found in sickle cell disease varies with populations. The highest frequencies of homozygous sickle cell disease in the world occur in sub‐Saharan Africa where 3% to 4% of the populations are affected (WHO 2000). In Jamaica, homozygous sickle cell disease is the most common (1 in 300), followed by sickle cell/haemoglobin C disease (1 in 500), with the thalassaemia variants being much less common (Serjeant 1997). The pattern in Nigeria is similar (Adekile 1999).
The sickle cell gene occurs commonly in areas of the world with intense malaria transmission. One of the reasons that the sickle cell gene has persisted is that it appears to be of benefit in 'carriers' of the gene. Carriers are people with one sickle cell gene and one normal haemoglobin gene. These people have some resistance to infection with Plasmodium falciparum malaria (Serjeant 1997; Adekile 1999; Ashley‐Koch 2000).
The illness seen in different genotypes varies with some causing more severe disease. Different forms of sickle cell disease may manifest the same problems and complications, some of which are potentially life threatening. Homozygous sickle cell disease and sickle cell/beta 0 thalassaemia tend to be more severe, whereas sickle cell/haemoglobin C disease and sickle cell/beta + thalassaemia are milder (Serjeant 1997).
The clinical manifestations of the disease are anaemia, recurring episodes (crises) of severe excruciating pain in the bones, an increased susceptibility to infections, the acute chest syndrome (a life threatening pneumonia‐like illness), and cerebrovascular accidents (strokes) (Serjeant 1997; Ashley‐Koch 2000).
Sickle cell disease appears to contribute to mortality especially in the first five years of life (Molineaux 1979a; Fleming 1989; Leikin 1989; Athale 1994; Platt 1994). Infants aged six to 12 months are particularly at risk (Rogers 1978; Serjeant 1997). In the USA, homozygous sickle cell disease reduces life expectancy to an average of 42 years for men and 48 years for women, even with specialized care and services (Platt 1994).
Malaria is the most common precipitating cause of crises in sickle cell disease in countries where malaria is endemic (Konotey‐Ahulu 1971a; Fleming 1989). Mortality and morbidity are increased in people with both sickle cell disease and malaria (Aluoch 1997; Ashley‐Koch 2000). One study found malaria parasites were the commonest infecting organism in people with homozygous sickle cell disease requiring hospitalizations in Nigeria (Maharajan 1983). Another study reported malaria infection as the precipitating cause in 133 of 848 consecutive admissions for crises in homozygous sickle cell disease in a hospital in Ghana (Konotey‐Ahulu 1971b).
An increased number of malaria attacks and deaths were observed in people with homozygous sickle cell disease who were not taking malaria chemoprophylaxis (Colbourne 1956; Adeloye 1971; Molineaux 1979a). Fewer deaths and crises were reported with malaria chemoprophylaxis, while growth, quality of life, as well as resistance to other infections, were thought to be improved (Konotey‐Ahulu 1971a; Fleming 1989). Therefore many authorities recommend life‐long malaria prophylaxis in people with homozygous sickle cell disease. Antimalarial drugs used for chemoprophylaxis include proguanil, pyrimethamine, and mefloquine (Okuonghae 1992; Ogala 1999; Nwokolo 2001).
Several factors have to be considered when starting life‐long malaria chemoprophylaxis. Poor adherence may occur as it is difficult to take drugs regularly. Adverse drug effects may develop, such as hair loss and mouth ulcers with proguanil, and neuropsychiatric reactions with mefloquine (WHO 2001). The development of natural immunity to malaria (particularly in children) could be impaired by chemoprophylaxis with the potential risk of severe malaria on stopping the treatment (Otoo 1988; Salako 2000). Drug resistance may develop increasing the cost of treating patients, since newer antimalarials are more expensive (WHO 1990). It is therefore important to assess the benefits and harms of this life‐long intervention carefully.
Objectives
To assess the effects of routine malaria chemoprophylaxis in people with sickle cell disease.
Methods
Criteria for considering studies for this review
Types of studies
Randomized and quasi‐randomized controlled trials.
Types of participants
Adults and children with sickle cell disease (proven by electrophoresis or DNA tests as appropriate) in a malaria‐endemic zone. This includes people with homozygous sickle cell disease, sickle‐cell/haemoglobin C disease, sickle cell/beta + thalassaemia, and sickle cell/beta 0 thalassaemia.
Types of interventions
Intervention
Chemoprophylaxis with any antimalarial drug given for a minimum of three months.
Control
Placebo or no intervention.
Types of outcome measures
Primary
Death.
Sickle cell painful crises (episodes of severe pain in the bones or abdomen).
Severe anaemia (haemoglobin concentration 5 g/dL or less) requiring blood transfusions.
Secondary
Malaria infection (diagnosed from blood film examinations).
Hospital admissions.
Haemoglobin concentrations in the absence of symptoms attributable to sickle cell disease or infection.
Adverse events that are fatal, life threatening, require hospitalization, or result in the discontinuation of treatment.
Search methods for identification of studies
We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and in progress).
Databases
We searched the following databases using the search terms and strategy described in Appendix 1: Cochrane Infectious Diseases Group Specialized Register (January 2006); Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library (2006, Issue 1); MEDLINE (1966 to January 2006); EMBASE (1974 to January 2006); LILACS (1982 to January 2006). We also searched Current Controlled Trials (www.controlled‐trials.com; January 2006) using malaria and sickle cell as search terms.
We also searched the haemoglobinopathies specialized register of controlled trials held at the editorial base of the Cochrane Cystic Fibrosis and Genetic Disorders Group (July 2006) using malaria and sickle cell as search terms.
Organizations and pharmaceutical companies
In 2002, we contacted the Georgia Comprehensive Sickle Cell Center (Atlanta, USA), the Sickle Cell Association of Ghana, the Jamaican Sickle Cell Unit of the Medical Research Council of Jamaica, and the pharmaceutical companies AstraZeneca UK Ltd (manufacturers of proguanil) and GlaxoSmithKline UK (manufacturers of pyrimethamine), for information about unpublished and ongoing trials.
Reference lists
We also checked the reference lists of all studies identified by the above methods.
Data collection and analysis
Selection of studies
We independently screened the results of the search strategy for potentially relevant trials and independently assessed them for inclusion in the review using a pre‐designed eligibility form based on the inclusion criteria.
Data extraction and management
We independently extracted data on the trial characteristics including the methods, participants, interventions, and outcomes. We resolved disagreements through discussion or by consulting an Editor of the Cochrane Infectious Diseases Group.
Assessment of risk of bias in included studies
Using a pre‐designed validity form, we independently assessed the risk of bias in the included trials using generation of the allocation sequence, allocation concealment, blinding, and the inclusion of all randomized participants. We assessed the methods used to generate the allocation sequence and conceal allocation to be adequate or inadequate according to Jüni 2001. We described who was blinded in each trial, and assessed the number of randomized participants included in the final analysis to be adequate if 90% or over.
Data synthesis
Where appropriate, we analysed data in Review Manager 5, using risk ratios (RR) and with 95% confidence intervals (CI) for dichotomous data. The included trials did not report continuous data and there were too few trials to assess heterogeneity; methods for these situations will be used should such data become available.
Results
Description of studies
We identified five potentially relevant trials. Two met the inclusion criteria (Warley 1965; Eke 2003; see 'Characteristics of included studies' for details) and three were excluded (see 'Characteristics of excluded studies').
Warley 1965 was conducted in Uganda in 1962 on 126 children with homozygous sickle cell disease who attended a clinic over a two‐year period. The trial was conducted in an area of stable malaria where transmission occurred all year round. The intervention group was given chloroquine tablets as malaria chemoprophylaxis every week. Children aged below three years received 100 mg and those above three years received 200 mg of chloroquine. The intervention group also received long‐acting benzathine penicillin injections of 1,200,000 units (all age groups) as antibiotic prophylaxis. The control group were given sterile water injections. The main outcome measures were episodes of malaria, dactylitis (swelling or tenderness of bones of the hands or feet), and haemoglobin levels.
Eke 2003 was conducted in Nigeria on 97 children with homozygous sickle cell disease who were attending clinic. The trial was held over a nine‐month period in an area of stable malaria endemicity. This included the rainy season, which is the period of maximum malaria transmission. Participants were aged one to 16 years and were treated with curative doses of antimalarial drugs on enrolment into the trial. They were then divided into three groups each to receive pyrimethamine (0.5 mg/kg/week) or proguanil (1.5 mg/kg/day) as malaria chemoprophylaxis, or placebo (vitamin C 1 mg/kg/day). The main outcome measures were episodes of malaria, bone pain crises (recurrent bilateral pattern of bone pains and including dactylitis), haemolytic crisis (presence of jaundice, anaemia, or splenomegaly in the absence of other precipitating cause), aplastic crisis (bone marrow aplasia from malarial infection), number of hospitalizations, number of blood transfusions, mean haemoglobin levels, and mean parasite density.
Risk of bias in included studies
See 'Characteristics of included studies' for details.
The allocation sequence was generated by alternate allocation in Warley 1965 and by block randomization (one in six) in Eke 2003. Allocation was therefore inadequately concealed in Warley 1965, while it was adequate in Eke 2003 as participants selected their intervention groups from sealed, opaque envelopes. Both trials were open: Warley 1965 was open because of the different routes of administration of the interventions, and the regimens were different in Eke 2003. Less than 30% of the participants were followed up for 12 months in Warley 1965 (inadequate), whereas Eke 2003 followed more than 90% of participants for nine months (adequate).
Effects of interventions
Chloroquine plus benzathine penicillin versus placebo (Warley 1965)
Chemoprophylaxis significantly reduced the number of episodes of dactylitis (swelling and tenderness of the bones of the hands or feet) 12 months after treatment (P < 0.1, trial authors' calculation); data were derived from the published average cumulative incidence of dactylitis (Table 3). Chemoprophylaxis also significantly reduced the number of episodes of malaria after 12 months of prophylaxis (RR 0.16, 95% CI 0.07 to 0.39; 157 children, Analysis 1.1) and increased the mean haemoglobin (P < 0.02, trial authors' calculation). Death, severe anaemia (requiring blood transfusions), hospital admissions, and adverse events were not reported.
1. Dactylitis crises 12 months after intervention, Warley 1965.
| Dactylitis crises | Chemoprophylaxis | Placebo |
| Monthly cumulated incidence (estimated from published graph) | 1.0 | 2.75 |
| Participants | 19 | 13 |
| Episodes: cumulated incidence x no. participants | 19 | 36 |
1.1. Analysis.
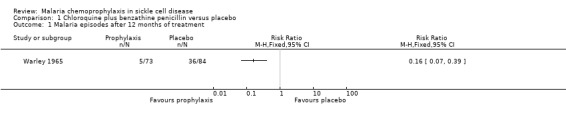
Comparison 1 Chloroquine plus benzathine penicillin versus placebo, Outcome 1 Malaria episodes after 12 months of treatment.
Proguanil or pyrimethamine versus placebo (Eke 2003)
Data for the antimalarial drugs were pooled in the analyses. Chemoprophylaxis did not reduce the number of deaths (97 children, Analysis 2.1), and the one death that was reported occurred in the placebo group and was attributed to viral hepatitis and presumed overwhelming septicaemia. Chemoprophylaxis did however have a positive impact on reducing the number of painful sickle cell crises (RR 0.17, 95% 0.04 to 0.83; 97 children, Analysis 2.2) and blood transfusions required from episodes of severe anaemia (RR 0.16, 95% 0.05 to 0.56; 97 children, Analysis 2.3).
2.1. Analysis.
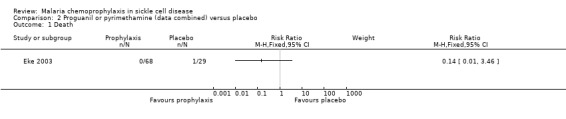
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 1 Death.
2.2. Analysis.
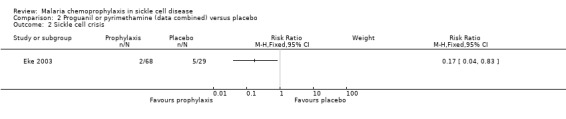
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 2 Sickle cell crisis.
2.3. Analysis.
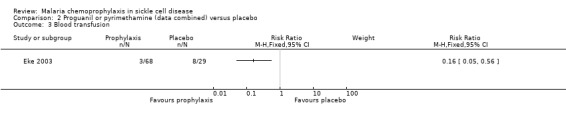
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 3 Blood transfusion.
For the secondary outcome measures, the number of hospital admissions was significantly reduced by chemoprophylaxis (RR 0.27, 95% CI 0.12 to 0.63; 97 participants, Analysis 2.5). There was no statistically significant difference in the number of malaria infections (97 children, Analysis 2.4), defined as body temperatures > 37.5 °C and malaria parasitaemia, or mean haemoglobin concentrations in the steady state (97 children, Analysis 2.6). No adverse events due to the drugs were reported.
2.5. Analysis.
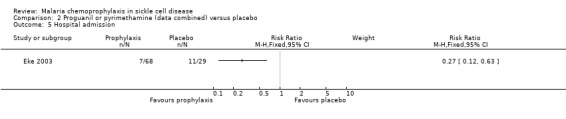
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 5 Hospital admission.
2.4. Analysis.
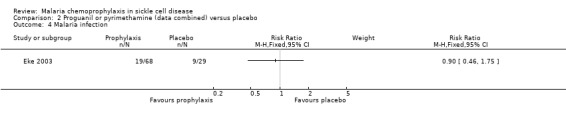
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 4 Malaria infection.
2.6. Analysis.
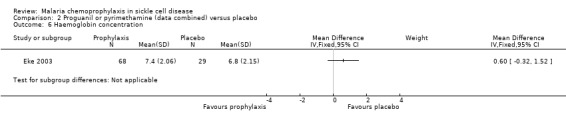
Comparison 2 Proguanil or pyrimethamine (data combined) versus placebo, Outcome 6 Haemoglobin concentration.
Discussion
We identified two trials that compared malaria chemoprophylaxis with no intervention or placebo in children with homozygous sickle cell disease. Eke 2003 had antimalarial chemoprophylaxis as the sole intervention, while Warley 1965 had antimalarial chemoprophylaxis intervention combined with antibiotics. Both trials showed improved clinical outcomes.
Warley 1965 reported improved outcomes of fewer episodes of malaria, dactylitis, and higher mean haemoglobin levels with chemoprophylaxis. It could be argued that this was the result of the combined effect of the antimalarial and antibiotic prophylaxis. Either bacterial infections or malaria can precipitate a crisis in sickle cell disease, although the reduction in malaria infections was most probably due to the antimalarial intervention. Eke 2003 showed reduced episodes of bone pain crises, hospital admissions, and blood transfusions that were solely due to the effects of the antimalarial prophylaxis. It had been previously reported that malaria chemoprophylaxis in sickle cell disease reduces morbidity from malaria infections and is thought to improve resistance to other infections as well (Konotey‐Ahulu 1971a; Fleming 1989). Eke 2003 supports this finding as malaria chemoprophylaxis not only improved clinical outcomes and decreased hospitalization rates from malaria infections, but from other causes as well.
The trials did not address the important issues of drug resistance to antimalarial chemotherapy, which may explain the varying results obtained from the chemoprophylaxis group or the potential for impairment of development of natural immunity to malaria.
In areas where malaria is endemic, chemoprophylaxis is often prescribed to people with homozygous sickle cell disease. We have found evidence to support this practice. We found no evidence to confirm or refute the benefits of malaria chemoprophylaxis in other types of sickle cell disease, that is, sickle‐cell/haemoglobin C disease, sickle cell/beta + thalassaemia, and sickle cell/beta 0 thalassaemia.
Authors' conclusions
Implications for practice.
Malaria chemoprophylaxis is widely used in homozygous sickle cell disease (commonly called sickle cell anaemia) because of the associated morbidity and mortality. Existing policies recommending prophylaxis have been based on case reports, observational studies, and consensus. The trials included in this review support these policies in people with homozygous sickle cell disease. There is inadequate evidence to support or refute giving routine antimalarial chemoprophylaxis in other forms of sickle cell disease in areas where malaria is endemic.
Implications for research.
Randomized controlled trials are required to evaluate the use of malaria chemoprophylaxis in sickle cell disease. In homozygous sickle cell disease there is such a strong clinical relationship between sickle cell crises, anaemia, and malaria that it would be difficult to justify another placebo‐controlled trial. As those with homozygous sickle cell disease are often placed on chemoprophylaxis in endemic areas, a comparison of the different antimalarial regimens, such as proguanil versus pyrimethamine or proguanil versus mefloquine, could be made. In the other forms of the disease, antimalarial drugs, such as proguanil, pyrimethamine, and mefloquine, should be given as prophylaxis with placebo or other proven methods of preventing malaria (such as insecticide‐treated bed nets) as the control intervention. Trials evaluating specific methods to ensure adherence to long‐term drug therapy, development of drug resistance, and adverse events with the drugs should also be carried out.
What's new
| Date | Event | Description |
|---|---|---|
| 21 July 2008 | Amended | Converted to new review format with minor editing. |
History
Protocol first published: Issue 1, 2002 Review first published: Issue 3, 2003
| Date | Event | Description |
|---|---|---|
| 17 August 2006 | New citation required and conclusions have changed | 2006, Issue 4 (substantive update): The conclusions of the review have changed due to inclusion of information obtained from the new trials that have been carried out since it was first published. We have subjected the results of the included trials to meta‐analysis. The search for trials has been updated. |
| 11 August 2006 | New search has been performed | 2006, Issue 4 (substantive update): The conclusions of the review have changed due to inclusion of information obtained from the new trials that have been carried out since it was first published. We have subjected the results of the included trials to meta‐analysis. The search for trials has been updated. |
| 8 May 2003 | Amended | 2003, Issue 3 (first review version): The order of the outcome measures has changed from those in the protocol. This is because our primary interest is how the clinical manifestations of the disease are affected by malaria chemoprophylaxis. Sickle cell painful crises and severe anaemia have been moved from secondary to primary outcome measures; and episodes of malaria has been moved to secondary outcome measures where it has been renamed malaria infection. We have also clarified in the secondary outcome measures that the steady state in which haemoglobin concentrations are measured is that in which symptoms attributable to sickle cell disease or infection are absent. |
Acknowledgements
The protocol for this review was developed during the Mentorship Programme organized by the Cochrane Infectious Diseases Group, May to June 2001. The UK Department for International Development (DFID) supports the editorial base of the Cochrane Infectious Diseases Group through a project funded by the DFID for the benefit of developing countries. The views expressed are not necessarily those of DFID.
Appendices
Appendix 1. Search methods: search strategies for database
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb |
| 1 | malaria | malaria | malaria | malaria | malaria |
| 2 | sickle cell disease | sickle cell disease | sickle cell disease | sickle cell disease | sickle cell disease |
| 3 | sickle cell anaemia | ANEMIA, SICKLE CELL | ANEMIA, SICKLE CELL | SICKLE‐CELL‐ANEMIA | sickle cell anaemia |
| 4 | 2 or 3 | haemoglobin SC | haemoglobin SC | HEMOGLOBINOPATHY | 2 or 3 |
| 5 | 1 and 4 | haemoglobin SS | haemoglobin SS | haemoglobin SC | 1 and 4 |
| 6 | — | haemoglobin SD | haemoglobin SD | haemoglobin SS | — |
| 7 | — | thalassaemia | thalassaemia | haemoglobin SD | — |
| 8 | — | drepanocyt* | drepanocyt* | thalassaemia | — |
| 9 | — | meniscocytosis | meniscocytosis | drepanocyt$ | — |
| 10 | — | 2‐9/or | 2‐9/or | 2‐9/or | — |
| 11 | — | 1 and 10 | 1 and 10 | 1 and 10 | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Higgins 2005); upper case: MeSH or EMTREE heading; lower case: free text term.
Data and analyses
Comparison 1. Chloroquine plus benzathine penicillin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Malaria episodes after 12 months of treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 2. Proguanil or pyrimethamine (data combined) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Sickle cell crisis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Blood transfusion | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Malaria infection | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Hospital admission | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6 Haemoglobin concentration | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Eke 2003.
| Methods | Randomized controlled trial Generation of allocation sequence: block randomization (1 in 6); equal allocation Allocation concealment: participants selected their intervention groups from sealed opaque envelopes Blinding: none Inclusion of all randomized participants in analysis: > 90% participants followed up for 9 months (32/35 proguanil group, 29/30 placebo group, and 100% pyrimethamine group) |
|
| Participants | 97 children with homozygous sickle cell disease (sickle cell anaemia genotype SS) | |
| Interventions | 1. Pyrimethamine (0.5 mg/kg/week) 2. Proguanil (1.5 mg/kg/day) 3. Placebo (vitamin C; 1 mg/kg/day for 9 months) | |
| Outcomes | 1. Deaths 2. Malaria infections 3. Bone pain crises 4. Haemolytic crises 5. Aplastic crisis 6. Number of hospitalizations 7. Number of blood transfusions 8. Mean haemoglobin levels 9. Mean parasite density 10. Splenomegaly | |
| Notes | Location: University of Port Harcourt Teaching Hospital, Nigeria | |
Warley 1965.
| Methods | Quasi‐randomized controlled trial Generation of allocation sequence: alternate allocation Allocation concealment: none Blinding: none Inclusion of all randomized participants in analysis: < 30% of participants followed up for 12 months (13/60 intervention group and 19/66 control group) |
|
| Participants | 126 children with homozygous sickle cell disease (sickle cell anaemia genotype SS) | |
| Interventions | 1. Chloroquine tablet (100 mg < 3 years; 200 mg > 3 years) plus benzathine penicillin injection (1,200,000 units) 2. No tablets plus sterile water injection | |
| Outcomes | 1. Parasitaemia 2. Dactylitis 3. Mean haemoglobin 4. Fall in haemoglobin | |
| Notes | Location: Mulago Hospital, Uganda Date: August 1962 to May 1964 |
|
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Hendrickse 1966 | Compared different antimalarial regimens in people with sickle cell anaemia; did not compare the interventions to placebo or to no intervention |
| Molineaux 1979b | Longitudinal research project on epidemiology of malaria conducted in 1970 in Garki, Northern Nigeria |
| Terlouw 2004 | Study participants had the sickle cell trait (haemoglobin genotype AS) and not sickle cell disease |
Differences between protocol and review
2003, Issue 3 (first review version): The order of the outcome measures has changed from those in the protocol. This is because our primary interest is how the clinical manifestations of the disease are affected by malaria chemoprophylaxis. Sickle cell painful crises and severe anaemia have been moved from secondary to primary outcome measures; and episodes of malaria has been moved to secondary outcome measures where it has been renamed malaria infection. We have also clarified in the secondary outcome measures that the steady state in which haemoglobin concentrations are measured is that in which symptoms attributable to sickle cell disease or infection are absent.
Contributions of authors
Oluseyi Oniyangi wrote the protocol, extracted and analysed data, and drafted the review. Aika Omari helped write the protocol, extracted and analysed data, and helped draft the review. Both authors have been involved in updating the review.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
External sources
Department for International Development, UK.
Declarations of interest
None known.
Unchanged
References
References to studies included in this review
Eke 2003 {published data only}
- Eke FU, Anochie I. Effects of pyrimethamine versus proguanil in malaria chemoprophylaxis in children with sickle cell disease: a randomized, placebo‐controlled, open‐label study. Current Therapeutic Research 2003;64(8):616‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Warley 1965 {published data only}
- Lewthwaite CJ. A trial of chemoprophylaxis in sickle‐cell anaemia. East African Medical Journal 1962;39(5):196‐9. [PubMed] [Google Scholar]
- Warley MA, Hamilton PJ, Marsden PD, Brown RE, Merselis JG, Wilks N. Chemoprophylaxis of homozygous sicklers with antimalarials and long‐acting penicillin. British Medical Journal 1965;2(5453):86‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Hendrickse 1966 {published data only}
- Hendrickse RG, Barnes PM. Sickle cell anaemia: report of a therapeutic trial. West African Medical Journal 1966;15(2):55‐64. [PubMed] [Google Scholar]
Molineaux 1979b {published data only}
- Molineaux L, Gramiccia G. The Garki Project: research on the epidemiology and control of malaria in the Sudan savanna of West Africa. Geneva: World Health Organization, 1980. [Google Scholar]
Terlouw 2004 {published data only}
- Terlouw DJ, Desai MR, Wannemuehler KA, Kariuki SK, Pfeiffer CM, Kager PA, et al. Relation between the response to iron supplementation and sickle cell haemoglobin phenotype in preschool children in western Kenya. American Journal of Clinical Nutrition 2004;79(3):466‐72. [DOI] [PubMed] [Google Scholar]
Additional references
Adekile 1999
- Adekile AD. Haemoglobinopathies. In: Azubuike JC, Nkangineme KEO editor(s). Paediatrics and child health in a tropical region. Owerri, Nigeria: African Educational Services, 1999:195‐206. [ISBN. 978 ‐ 2411 ‐ 76 ‐ 0] [Google Scholar]
Adeloye 1971
- Adeloye A, Luzzatto L, Edington GM. Severe malarial infection in a patient with sickle‐cell anaemia. British Medical Journal 1971;2(759):445‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aluoch 1997
- Aluoch JR. Higher resistance to Plasmodium falciparum infection in patients with homozygous sickle cell disease in western Kenya. Tropical Medicine & International Health 1997;2(6):568‐71. [DOI] [PubMed] [Google Scholar]
Ashley‐Koch 2000
- Ashley‐Koch A, Young Q, Olney RS. Sickle haemoglobin (HbS) allele and sickle cell disease: a HuGE review. American Journal of Epidemiology 2000;151(9):839‐45. [DOI] [PubMed] [Google Scholar]
Athale 1994
- Athale UH, Chintu C. Clinical analysis of mortality in hospitalized Zambian children with sickle cell anaemia. East African Medical Journal 1994;71(6):388‐91. [PubMed] [Google Scholar]
Colbourne 1956
- Colbourne MJ, Edington GM. Sickling and malaria in the Gold Coast. British Medical Journal 1956;i(4970):784‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fleming 1989
- Fleming AF. The presentation, management and prevention of crisis in sickle cell disease in Africa. Blood Reviews 1989;3(1):18‐28. [DOI] [PubMed] [Google Scholar]
Higgins 2005
- Higgins J, Green S, editors. Highly sensitive search strategies for identifying reports of randomized controlled trials in MEDLINE. Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [updated May 2005]; Appendix 5b. www.cochrane.org/resources/handbook/hbook.htm (accessed 1 March 2006).
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Konotey‐Ahulu 1971a
- Konotey‐Ahulu FID. Malaria and sickle‐cell disease. British Medical Journal 1971;2(763):710‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Konotey‐Ahulu 1971b
- Konotey‐Ahulu FID, Serjeant G, White JM. Treatment and prevention of sickle cell crisis. Lancet 1971;2(7736):1255‐6. [PubMed] [Google Scholar]
Leikin 1989
- Leikin SL, Gallagher D, Kinney TR, Sloan D, Klug P, Rida W. Mortality in children and adolescents with sickle cell disease. Pediatrics 1989;84(3):500‐8. [PubMed] [Google Scholar]
Maharajan 1983
- Maharajan R, Fleming AF, Egler L. Pattern of infections among patients with sickle cell anaemia requiring hospital admission. Nigerian Journal of Paediatrics 1983;10:13‐7. [Google Scholar]
Molineaux 1979a
- Molineaux L, Fleming AF, Cornille‐Brogger R, Kagan I, Storey J. Abnormal haemoglobins in the Sudan savanna of Nigeria. III. Malaria, immunoglobulins and antimalarial antibodies in sickle cell disease. Annals of Tropical Medicine and Parasitology 1979;73(4):301‐10. [DOI] [PubMed] [Google Scholar]
Nwokolo 2001
- Nwokolo C, Wamebe C, Akinyanju O, Raji AA, Audu BS, Emodi IJ, et al. Mefloquine versus proguanil in short‐term malaria chemoprophylaxis in sickle cell anaemia. Clinical Drug Investigation 2001;21(8):537‐44. [Google Scholar]
Ogala 1999
- Ogala WN. Malaria. In: Azubuike JC, Nkanginieme KEO editor(s). Paediatrics and child health in a tropical region. African Educational Services, 1999:427‐37. [ISBN. 978 ‐ 2411 ‐ 76 ‐ 0] [Google Scholar]
Okuonghae 1992
- Okuonghae HO, Nwankwo MU, Offor E. Malarial parasitaemia in febrile children with sickle cell anaemia. Journal of Tropical Pediatrics 1992;38(2):83‐5. [DOI] [PubMed] [Google Scholar]
Otoo 1988
- Otoo LN, Snow RW, Menon A, Byass P, Greenwood BM. Immunity to malaria in young Gambian children after a two‐year period of chemoprophylaxis. Transactions of the Royal Society of Tropical Medicine and Hygiene 1988;82(1):59‐65. [DOI] [PubMed] [Google Scholar]
Platt 1994
- Platt OS, Brambila DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. New England Journal of Medicine 1994;330(23):1639‐44. [DOI] [PubMed] [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Rogers 1978
- Rogers DW, Clarke JM, Cupidore L, Ramlal AM, Sparke BR, Serjeant GR. Early deaths in Jamaican children with sickle cell disease. British Medical Journal 1978;1(6126):1515‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Salako 2000
- Salako L. Malaria in Nigeria. Proceedings of the 31st Annual General and Scientific Meeting of the Paediatric Association of Nigeria; 2000 January 28; Abuja. Abuja: Paediatric Association of Nigeria, 2000.
Serjeant 1997
- Serjeant GR. Sickle‐cell disease. Lancet 1997;350(9079):725‐30. [DOI] [PubMed] [Google Scholar]
WHO 1990
- WHO Scientific Group on the Chemotherapy of Malaria. Practical chemotherapy of malaria: report of a WHO scientific group [meeting held in Geneva from 5 to 12 June 1989]. Technical Report Series 805. Geneva: World Health Organization, 1990. [PubMed] [Google Scholar]
WHO 2000
- World Health Organization. Updated estimates of the frequency of the haemoglobin disorders in each country. www.who.ch/programmes/ncd/hgm/haemogl.htm (accessed May 2001).
WHO 2001
- Global Parnership to Roll Back Malaria. The use of antimalarial drugs: report of a WHO informal consultation, 13‐17 November 2000 [WHO/CDS/RBM/2001.33]. Geneva: World Health Organization, 2001. [Google Scholar]