

Abstract
Background
Human African trypanosomiasis, or sleeping sickness, is a painful and protracted disease affecting people in the poorest parts of Africa and is fatal without treatment. Few drugs are currently available for second‐stage sleeping sickness, with considerable adverse events and variable efficacy.
Objectives
To evaluate the effectiveness and safety of drugs for treating second‐stage human African trypanosomiasis.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register (January 2013), CENTRAL (The Cochrane Library Issue 12 2012) , MEDLINE (1966 to January 2013), EMBASE (1974 to January 2013), LILACS (1982 to January 2013 ), BIOSIS (1926‐January 2013), mRCT (January 2013) and reference lists. We contacted researchers working in the field and organizations.
Selection criteria
Randomized and quasi‐randomized controlled trials including adults and children with second‐stage HAT, treated with anti‐trypanosomal drugs.
Data collection and analysis
Two authors (VL and AK) extracted data and assessed methodological quality; a third author (JS) acted as an arbitrator. Included trials only reported dichotomous outcomes, and we present these as risk ratio (RR) with 95% confidence intervals (CI).
Main results
Nine trials with 2577 participants, all with Trypansoma brucei gambiense HAT, were included. Seven trials tested currently available drugs: melarsoprol, eflornithine, nifurtimox, alone or in combination; one trial tested pentamidine, and one trial assessed the addition of prednisolone to melarsoprol. The frequency of death and number of adverse events were similar between patients treated with fixed 10‐day regimens of melarsoprol or 26‐days regimens. Melarsoprol monotherapy gave fewer relapses than pentamidine or nifurtimox, but resulted in more adverse events.
Later trials evaluate nifurtimox combined with eflornithine (NECT), showing this gives few relapses and is well tolerated. It also has practical advantages in reducing the frequency and number of eflornithine slow infusions to twice a day, thus easing the burden on health personnel and patients.
Authors' conclusions
Choice of therapy for second stage Gambiense HAT will continue to be determined by what is locally available, but eflornithine and NECT are likely to replace melarsoprol, with careful parasite resistance monitoring. We need research on reducing adverse effects of currently used drugs, testing different regimens, and experimental and clinical studies of new compounds, effective for both stages of the disease.
12 December 2018
Update pending
Authors currently updating
The update is due to be published in 2019.
Keywords: Animals; Humans; Trypanosoma brucei gambiense; Antiprotozoal Agents; Antiprotozoal Agents/adverse effects; Antiprotozoal Agents/therapeutic use; Drug Therapy, Combination; Drug Therapy, Combination/methods; Eflornithine; Eflornithine/therapeutic use; Melarsoprol; Melarsoprol/therapeutic use; Nifurtimox; Nifurtimox/therapeutic use; Pentamidine; Pentamidine/therapeutic use; Prednisolone; Prednisolone/therapeutic use; Randomized Controlled Trials as Topic; Recurrence; Trypanosomiasis, African; Trypanosomiasis, African/drug therapy
Plain language summary
Chemotherapy of second‐stage Human African trypanosomiasis
Human African trypanosomiasis (HAT), or sleeping sickness, is a painful and protracted disease transmitted through the bite of infected tsetse flies and it is found in rural parts of sub‐Saharan Africa. Sleeping sickness has two clinical phases but this review focuses only on treatment of the second‐stage, which is characterized by neurological changes and almost invariably fatal without treatment. There are only a few drugs currently available for second‐stage sleeping sickness, all with considerable adverse events and variable efficacy.
The review includes nine trials with 2577 participants. Each trial reported different comparisons of the drugs currently available to treat second stage HAT (melarsoprol, eflornithine, nifurtimox) so no meta‐analysis was possible.
Melarsoprol administration is intravenous and very painful, with many adverse reactions including a severe dysfunction of the brain, that can result in death. For this reason, trials were designed to evaluate shorter melarsoprol regimens. Giving melarsoprol for 10 days was found to be as effective as giving it for 26 days. Recently, nifurtimox and eflornithine combination therapy (NECT) was assessed. Few patients relapsed after NECT, which was generally well tolerated. It also has practical advantages: eflornithine has to be administered as a slow intravenous infusion thus requiring specialized health facilities and personnel, but nifurtimox is given orally. NECT uses less eflornithine doses and reduces the burden on health personnel and patients.
Considering that none of the current therapeutic options for HAT is optimal in terms of adverse events and ease of administration, it is essential that new anti‐trypanosomal compounds are developed and tested in experimental and clinical studies. In the meantime, local availability of the drugs and the status of health facilities and personnel will dictate choice of treatment. It is envisioned that melarsoprol, with its high level of adverse events, will be phased out in favour of eflornithine and NECT. The development of parasite resistance to the drugs needs to be carefully monitored. Future research should also focus on the reduction of the adverse effects of currently used drugs and better diagnostic tests.
Background
Human African trypanosomiasis (HAT), or sleeping sickness, is a disease caused by the protozoan parasite Trypanosoma brucei that is transmitted through the bite of infected tsetse flies. The disease occurs throughout sub‐Saharan Africa, wherever tsetse flies are found. In 2006, it was estimated that 50,000 to 70,000 individuals were infected (WHO 2006); between 2004 and 2008, the reported number of new cases was approximately 10,000 (WHO 2009). The eco‐distribution of tsetse flies is determined by the climate, presence of water, vegetation, and their requirement for blood meals (human or animals), but they are mostly found in rural and forested areas. Essential human activities such as farming, collecting water from natural sources, washing, and fishing, can increase contact between humans and tsetse flies and contribute to the spread of the disease (Pepin 2001). The incidence of HAT was reduced to very low levels by the end of the 1950s following large‐scale campaigns of active case detection and surveillance, and tsetse flies control campaigns (Cattand 2001). However, since the 1960s, the gradual breakdown of control programmes, aggravated by economic hardship, war, and civil strife in most endemic countries, resulted in an alarming resurgence of HAT, with epidemics in the Democratic Republic of Congo, Angola, Sudan, Uganda, and the Central African Republic, that resulted in the disruption of whole communities and with huge socioeconomic costs (Seed 2001). In the past few years the reported number of cases of sleeping sickness has again reduced due to increased control measures, although the exact number of cases is uncertain because of poor health services in most of the affected areas (Brun 2010).
Sleeping sickness is a painful and protracted disease which is almost invariably fatal without adequate treatment; treatment of infected individuals is crucial for reducing the trypanosome reservoir in humans and consequently for controlling the disease. The mostly rural distribution of the disease, civic unrest occurring in many regions affected, the financial and social constraints experienced by endemic countries, and the difficulties in diagnosing and effectively treating HAT, all contribute to make it one of the hardest diseases to control in sub‐Saharan Africa.
Two subspecies of Trypanosoma brucei can infect humans. T. b. gambiense causes a generally chronic form of sleeping sickness in West and Central Africa. T. b. rhodesiense, found in Eastern and Southern Africa, generally causes a more acute form of the disease. In both forms the disease is characterized by two clinical stages related to the propagation of the parasite in the infected host. In the first stage, when trypanosomes multiply in the haemolymphatic system, infected individuals experience intermittent episodes of fever and develop lymphadenopathy, and other non‐specific signs such as hepatosplenomegaly and skin rashes (Stich 2002). In the second stage of the disease, trypanosomes reach the central nervous system resulting in a chronic meningoencephalitis with headaches and extensive neurological changes, which result in severe sleep disturbances resembling narcolepsy, convulsions, semi‐coma, and death (Stich 2002).
Diagnosis and stage determination of HAT are problematic and cannot be based on clinical signs alone (Lejon 2005). The presence of parasites has to be demonstrated in body fluids, and, according to the World Health Organization (WHO), diagnosis of second‐stage HAT should be based on an examination of the cerebrospinal fluid (CSF) for trypanosomes, white blood cell (WBC) count of >5 WBC/μL in CSF, and increased total protein concentration (WHO 1998; WHO 2004). Patients with up to 5 WBC/μL in CSF are diagnosed with first‐stage HAT. There is some controversy about the correct staging of patients with 6 to 20 WBC/μL in CSF, as many patients in this 'grey zone' do not display typical symptoms of second‐stage HAT and can be cured with drugs that do not reach therapeutic levels in the brain (Lejon 2005). A WBC over 20/μL in CSF has been recommended by an expert panel as a cut‐off point for inclusion of patients in clinical trials for treatment of second‐stage HAT (WHO 2004).
Treatment for both stages of the disease is also complex. Treatment for the first stage relies on an early diagnosis, often missed due to the lack of specificity of the initial symptoms, and employs two drugs (pentamidine for Gambiense HAT and suramin for Rhodesiense disease) that have been used for more than 60 years and which can produce severe side effects. However, these drugs are still effective and in use (Brun 2010). In this review we will focus on the treatment of second‐stage HAT, which is problematic as drugs available are difficult to administer, and can cause severe adverse events and even death (Chappuis 2007).
Choice of drugs
Treatment of second‐stage HAT relies on melarsoprol, eflornithine, or nifurtimox − at present the only anti‐trypanosomal compounds that can reach therapeutic levels in the central nervous system. These drugs have been in use for many years, and their in vivo efficacy against HAT has been extrapolated after animal studies or, in the case of nifurtimox, after being used to treat American trypanosomiasis (Chagas disease). Also, the use of any of these drugs is complicated by multiple factors including the increasing incidence of therapeutic failures, painful administration, severe adverse reactions, availability, and high production costs.
Melarsoprol
Melarsoprol, a trivalent organic arsenical compound, has been the drug of choice for second‐stage HAT caused by either T. b. gambiense or T. b. rhodesiense since 1949. Melarsoprol is liposoluble and for this reason can cross the blood brain barrier (Nok 2003); however, being insoluble in water, it must be administered strictly intravenously after being dissolved in propylene glycol, which is highly irritating to tissues. As a result, the administration of melarsoprol is very painful (Nok 2003). The most appropriate regimen is not yet agreed upon and various regimens are currently in use. Melarsoprol causes a variety of adverse reactions, but the most serious is an encephalopathic syndrome. The incidence of this complication varies from 1.5% to 28% of all melarsoprol treatments, with a median associated fatality rate of 50% (Seixas 2005); because of this risk, treatment with melarsoprol requires hospitalization of the patient (Stich 2003). High rates of therapeutic failure have been observed in the Democratic Republic of Congo, Angola, and Sudan (Legros 2002).
Eflornithine
Eflornithine (difluoromethylornithine, DFMO), an irreversible inhibitor of ornithine decarboxylase, was developed as an anticancer drug in the 1970s, but it was found insufficiently active for this indication. Its activity against trypanosomes was demonstrated in laboratory animals in 1980. The first HAT patient was treated in 1981 (with oral eflornithine) and the intravenous formulation was registered in 1990. Eflornithine is effective against both stages of T. b. gambiense infection, but its effectiveness against T. b. rhodesiense is unreliable because of innate low sensitivity of this parasite. Eflornithine is difficult to administer as it requires four daily intravenous infusions for seven to 14 days (Burri 2003); recent trials have tested two daily eflornithine doses (Priotto 2009). Eflornithine can induce several adverse reactions such as anaemia, leucopenia, pancytopenia, gastroenteric symptoms, headaches, and sometimes seizures, but it is in general better tolerated than melarsoprol (Burri 2003). Therapeutic failures with eflornithine in the second stage of T. b. gambiense disease are uncommon, but relapse rates of 8.1% were reported (Balasegaram 2006) and need to be assessed.
Nifurtimox
Nifurtimox was originally developed and registered for use in American trypanosomiasis and only recently has been reserved for compassionate use in HAT when other treatments have failed. Nifurtimox has the advantage of being orally active. Both stages of infection with T. b. gambiense are susceptible to treatment with nifurtimox, but it has seldom been used as a first‐stage drug. Its efficacy against T. b. rhodesiense has not been assessed (Pepin 1994). Nifurtimox monotherapy has been used for patients who are refractory to melarsoprol, with high (75% to 85%) cure rates (Moens 1984, Van Nieuwenhove 1992). Adverse effects include dysfunctions of the central nervous system and of the gastrointestinal tract (Bouteille 2003).
Combination therapies and adjunctive treatments
Combining existing drugs may delay the onset of resistance and also help reduce dosages and adverse effects (Legros 2002). Combination therapies have been used on a compassionate basis in patients unresponsive to other drugs. Pre‐treatment with first‐stage drugs during second‐stage therapy with melarsoprol has frequently been used on an empirical basis to reduce melarsoprol toxicity. Combinations of nifurtimox with eflornithine, melarsoprol with nifurtimox, and nifurtimox and melarsoprol have been tested (Bouteille 2003); recent trials focusing on nifurtimox‐eflornithine combination therapy (NECT) are reported in this review (Priotto 2009).
Steroids have been used as an adjunctive treatment to melarsoprol to reduce the frequency of a drug‐induced encephalopathy. Some studies have shown a positive effect of prednisolone co‐administered with melarsoprol (Pepin 1989a), but the role of corticosteroids of different types and at different dosages needs to be properly assessed.
Future perspectives
No new drugs for treating late‐stage sleeping sickness were specifically developed from 1949 until very recently ‐ both nifurtimox and eflornithine were designed for other indications. The pharmaceutical industry has little economic incentive to research and develop new compounds for a disease with such a limited and unprofitable market. Around 2000, manufacturers even seriously considered abandoning the production of melarsoprol, eflornithine, suramin, and nifurtimox, and to considerably increase the price of pentamidine (Stich 2003). Fortunately, in May 2001, the pharmaceutical company Aventis (now Sanofi‐Aventis) agreed to guarantee the production of pentamidine, melarsoprol, and eflornithine for at least five years and to deliver these drugs free of charge to WHO. The agreement was renewed in 2006 and it is still ongoing.
However, recent research has resulted in the clinical development of two new compounds, fexinidazole and oxaborole. Fexinidazole (DNDi 2008) is a 5‐nitroimidazole and in experimental studies was found to be active against both T. b. gambiense and T. b. rhodesiense, to have a favourable safety profile and to be orally active. Phase II/III clinical trials of fexinidazole are going to take place in the Democratic Republic of the Congo, and the Central African Republic (DNDi 2012). Another oral drug candidate, an oxaborole‐6‐carboxamide (SCYX‐7158) is undergoing a Phase I clinical trial (DNDi 2012b).
As progress on the development of new drugs and their clinical applications is going to take time, the urgent short‐term advance to be made consists of preclinical investigations and of clinical trials to improve the effectiveness, safety, and ease of administration of monotherapy regimens using melarsoprol, eflornithine, or nifurtimox, and, most importantly, of regimens with combinations of these drugs. A multicentre NECT trial run by Medecines sans Frontieres, Epicentre, Drugs for Neglected Diseases Initiative (DNDi), and the Swiss Tropical institute, in collaboration with national Ministries of Health and HAT programs, has recently ended. On the basis of its results, a proposal for the inclusion of the combination of eflornithine and nifurtimox as treatment for second‐stage sleeping sickness in the WHO model list of essential medicines (EML) was submitted in November 2008 and approved in May 2009 (WHO 2010).
Our review aims to examine whether any of the current drugs, their combinations, or their combinations with adjunctive treatments at any particular dosage provides a definite advantage over other regimens for the treatment of second‐stage HAT, measured in terms of clinical outcomes and in relation to the severity of adverse effects.
Objectives
To evaluate the effectiveness and safety of drugs for treating second‐stage HAT.
Methods
Criteria for considering studies for this review
Types of studies
Randomized and quasi‐randomized controlled trials.
Types of participants
Adults and children with a primary diagnosis of second‐stage HAT, that is, having evidence of trypanosomal infection and a CSF analysis showing a WBC count of more than 5 cells/μL, with no upper limit, and/or the presence of trypanosomes.
Adults and children relapsing after treatment for second‐stage HAT.
Types of interventions
Intervention
Drugs for treating HAT, including melarsoprol, eflornithine, and nifurtimox. Drugs may be given alone, in combination (concomitantly or sequentially), or with an adjunctive treatment.
Control
Other drugs for treating HAT or different regimens of the intervention drugs (eg different dose, frequency, or route of administration).
Types of outcome measures
Primary
Death during treatment, up to one month after the last drug administration.
Overall mortality (for any reason, including HAT and treatment toxicity) up to one month after the last drug administration.
Relapse during follow up: trypanosomes detected in any body compartment (blood, lymph, or CSF) at any follow‐up examination (between one and 24 months after the last drug administration); or CSF leukocyte count > 50 WBC/μL CSF, or doubled from previous count, at any follow‐up examination; or CSF leukocyte count between 20 and 49 WBC/μL CSF together with symptoms strongly suggestive of relapse (worsened clinical condition since previous examination, with long lasting headache, mental and/or neurological disturbances, increased somnolence, recurrent fever, etc).
Secondary
Death likely to be due to HAT, up to one month after the last drug administration.
Relapse: trypanosomes detected in any body compartment (blood, lymph, or CSF) up to one month after the last drug administration.
Adverse events
Central nervous system adverse events: encephalopathy, seizures, confusion.
Bone marrow toxicity: anaemia, leucopenia, thrombocytopenia.
Gastrointestinal symptoms: diarrhoea, nausea and vomit.
Skin reactions
Infections
Cardiotoxicity.
Search methods for identification of studies
We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press, and in progress).
Databases
We searched the following databases using the search terms and strategy described in Appendix 1: Cochrane Infectious Diseases Group Specialized Register (January 2013); Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library 2012, Issue 12; MEDLINE (1966 to January 2013); EMBASE (1974 to January 2013) ; LILACS (1982 to January 2013); and BIOSIS (1926‐January 2013) . We also searched the metaRegister of Controlled Trials (mRCT, accessed 11 January 2013) using trypanosom* as the search term.
Conference proceedings
We searched the conference proceedings of The International Scientific Council for Trypanosomiasis Research and Control (ISCTRC) Conferences for relevant abstracts.
Researchers, organizations, and pharmaceutical companies
We attempted to locate unpublished and ongoing trials by contacting individual researchers working in the field; organizations including Médecins sans Frontières, Epicentre, Malteser, WHO, and TDR.
Data collection and analysis
Selection of studies
All trials identified through systematic literature searches were entered into a database that was screened independently by VL and JS for potentially relevant trials. VL retrieved the full articles of the potentially relevant trials. The three authors together applied the inclusion criteria on the potentially relevant trials using an eligibility form and prepared lists of included and excluded studies. We described the reasons for excluding studies in the 'Characteristics of excluded studies'.
Data extraction and management
VL and AK independently extracted data from the included studies using standardized data extraction forms. JS compared the two data extraction form and prepared a final version. VL entered the data into Review Manager 5. We extracted the number of participants randomized and analysed in each group. For each dichotomous outcome measured, we recorded the number of participants experiencing the event and the number analysed in each group.
Assessment of risk of bias in included studies
VL and AK independently assessed the methodological quality of the included trials using a standardized form. We assessed generation of randomization sequence, allocation concealment, blinding, and loss to follow up. We categorized the generation of allocation sequence and concealment to be adequate, inadequate, or unclear according to Jüni 2001. We assessed which party was blinded in each trial (study investigators, participants, or study assessors). We considered inclusion of 90% or more of the randomized participants in the analysis to be adequate, and less than 90% to be inadequate. Disagreements were resolved by discussion or by reassessment of the data extraction forms by JS. We also attempted to contact the trial authors for any information not specified or unclear.
Data synthesis
VL analysed data using Review Manager 5. Included trials only reported dichotomous outcomes. We did not perform a meta‐analysis. Results were presented in forest plots and tables and analysis were stratified by comparisons and by doses/regimens of the drugs.
Measure of effect
We presented outcomes for dichotomous data as risk ratio (RR) with 95% confidence intervals (CI).
Dealing with missing data
We analysed data extracted from the trials on an intention‐to‐treat basis when there were no missing data, or we used a complete‐case analysis, using the number of participants for whom outcomes were reported.
Results
Description of studies
Our first search in May 2010 identified 25 potentially eligible trials. Sixteen trials were excluded (see 'Characteristics of excluded studies') and nine met the inclusion criteria (see 'Characteristics of included studies').
A second search, for the updated version of this review in January 2013, identified no newly published RCTs but resulted in 2 ongoing trials of interest (DNDi 2012 and DNDi 2012b).
Trial location and participants
All included trials were conducted in one of the following countries: Democratic republic of Congo, Republic of Congo, Angola, Uganda, Cote d'Ivoire. Two multicenter trials (Pepin 2000 and .Priotto 2009 had sites in different countries. Eight trials were conducted in hospitals or specialized trypanosomiasis units, in one (Lejon 2003) the setting was unspecified. The oldest included trial is Pepin 1989a.
Four trials (Bisser 2007, Burri 2000, Pepin 2006, Priotto 2009) did not include young children but only adolescents and adults. One trial (Na‐Bangchang 2004) only included adults (over 18 years), the remaining four trials (Lejon 2003, Pepin 1989a, Pepin 2006, Priotto 2006) included both adults and young children.
Five trials (Bisser 2007, Burri 2000, Na‐Bangchang 2004, Pepin 2000, Priotto 2006) excluded pregnant patients. Except for Lejon 2003, Pepin 1989a and Priotto 2006, the remaining six trials excluded severely ill patients, defined either as in a "moribund condition", with "severe organ disease", "severe comorbidities" or in unarousable coma (Glasgow Coma score ≤ 8).
The total number of participants randomized in the included trials was 2577. All included patients suffered from T. b. gambiense HAT as we did not identify any RCT describing treatment for T. b. rhodesiense. Five trials (Bisser 2007, Pepin 2006, Priotto 2006, Priotto 2009) did not include participants that had a history of treatment for sleeping sickness (at any time or during the last 36 months), but three trials (Burri 2000, Lejon 2003, Pepin 1989a) did not mention this characteristic and Pepin 2000 included both new and relapsing cases.
Interventions
Seven trials tested the effectiveness of the currently available drugs to treat second stage HAT: melarsoprol, eflornithine, nifurtimox, used alone or in combination. Lejon 2003 tested the effectiveness of pentamidine, which is indicated to treat first stage HAT, in patients showing a CSF WBC count between 6 and 20 WBC/μL. Pepin 1989a tested melarsoprol with or without prednisolone as an adjunctive treatment to reduce the frequency of melarsoprol‐induced encephalopathy.
Two trials (Burri 2000 and Pepin 2006) compared different regimens of melarsoprol. Two trials (Na‐Bangchang 2004 and Pepin 2000) tested different regimens of eflornithine.
One trial (Bisser 2007) included comparisons between three monotherapies (two different regimens of melarsoprol, or nifurtimox alone) and a melarsoprol‐nifurtimox combination.
Priotto 2006 reports on comparisons between dual combinations of melarsoprol, eflornithine and nifurtimox; Priotto 2009 compares eflornithine monotherapy given for 14 days, with an eflornithine‐nifurtimox combination given for seven days.
Additional treatments
It is common practice to pre‐treat HAT patients against other diseases which are endemic in the same areas, such as malaria and helminthiasis. HAT patients are often treated with prednisolone to reduce the risk of melarsoprol‐induced encephalopathy, and in some of the included trials they received multivitamins, paracetamol or food rations (also given to their accompanying carers as hospitals and health centres in the endemic areas don't usually have enough resources to provide food). We have listed the details of the additional treatments for each trials in a separate table (Table 1).
1. Pre‐treatment and additional treatment of HAT patients.
| Trial | Pre‐treatment | Corticosteroids |
| Bisser 2007 |
|
Not systematically given, only for treatment of encephalopathy |
| Burri 2000 |
|
Prednisolone (1 mg/kg + decreasing doses) |
| Lejon 2003 | Not mentioned | Not mentioned |
| Na‐Bangchang 2004 |
|
Not mentioned |
| Pepin 1989 |
|
Not applicable |
| Pepin 2000 | Not mentioned | Not mentioned |
| Pepin 2006 |
|
Oral prednisolone (1 mg/kg) |
| Priotto 2006 |
|
Melarsoprol‐treated patients received oral prednisolone (1 mg/kg/day for 5 days + 0.5 mg/kg/day) until treatment completed |
| Priotto 2009 |
|
Not applicable |
Dosing and regimens
Melarsoprol monotherapy
Melarsoprol was always used intravenously; dosages and schedules tested varied between studies. A "standard regimen" (three series of 3.6 mg/kg/day for three days, with a seven day break between series for a total duration of 26 days) was used in Bisser 2007 and Pepin 2006. A shorter schedule of melarsoprol at 2.2 mg/kg/day for 10 days was used by Burri 2000, in comparison with a "standard Angolan schedule" over 26 days as follows: 1.2 mg/kg on day one, 2.4 mg/kg on day two, 3.6 mg/kg on day three and four, repeated on days 12‐15 and 23‐26. A 10 day schedule of melarsoprol at 2.16 mg/kg/day was used by Pepin 2006. Incremental melarsoprol was also used by Bisser 2007 at doses from 0.6 mg/kg to 1.8 mg/kg for the first three days, followed by the same dose of 1.8 mg/kg/day until day 10, and by Pepin 2006 where the doses started at 1.8 mg/kg up to 3.6 mg/kg for three series of three injections separated by seven days. Pepin 1989a used a 3.6 mg/kg dose of melarsoprol but the number of series and injections varied according to the white blood cell count of the patient ‐ this is known as a Neujean schedule.
Nifurtimox monotherapy
Nifurtimox was administered orally at 5 mg/kg every eight hours for 10 days in Bisser 2007.
Eflornithine monotherapy
Oral eflornithine was tested in Na‐Bangchang 2004 comparing doses of 400 mg/kg/day with 500 mg/kg/day. Pepin 2000 compared intravenous eflornithine (which is given a slow infusion) at 400 mg/kg/day for seven compared with 14 days in a trial including new and relapsing patients. Priotto 2009 tested eflornithine only given a slow infusion at 400 mg/kg/day for 14 days against the combination of nifurtimox‐eflornithine described below.
Pentamidine monotherapy
Pentamidine was used in Lejon 2003 at a dose of 4 mg/kg for 10 days, given intramuscularly, for patients in the so‐called intermediate stage (ie with a CSF cell count between 6 and 20 cells/μL).
Combination therapies
1) Melarsoprol‐nifurtimox low‐dose combination (Bisser 2007) for 10 days: melarsoprol alone at 0.6 mg/kg intravenously on day one and at 1.2 mg/kg intravenously on day two, followed by eight days of oral nifurtimox at 7.5 mg/kg every twelve hours, combined with melarsoprol at 1.2 mg/kg intravenously/day.
2) Melarsoprol‐nifurtimox was used in Priotto 2006 at doses of 1.8 mg/kg/day intravenous melarsoprol for 10 days, and oral nifurtimox at 15 mg/kg/day every eight hours for 10 days.
3) Melarsoprol‐eflornithine (intravenous melarsoprol , eflornithine as slow infusion) was used in Priotto 2006: melarsoprol at 1.8 mg/kg/day for 10 days, eflornithine at 400 mg/kg/day, every six hours for seven days.
4) Melarsoprol + prednisolone (Pepin 1989a) added oral prednisolone at 1 mg/kg/day, given at intervals, to a Neujean schedule of melarsoprol.
5) Nifurtimox‐eflornithine was used in Priotto 2006 and in Priotto 2009 as oral nifurtimox at 15 mg/kg/day every eight hours for 10 days, added to eflornithine as a slow infusion at 400 mg/kg/day, every six hours for 7 days (Priotto 2006) or every 12 hours for seven days (Priotto 2009).
Outcome measures
The included trials measured different outcomes from our protocol: most trials did not differentiate between death due to the disease or due to treatment (encephalopathy caused by melarsoprol), and measured death related to treatment or within 30 days of ending it. Parasitological cure rates (trypanosomes in any body compartment) were measured usually within one day from the end of treatment.
Follow‐up was done at several time points, up to 24 months for all trials except Na‐Bangchang 2004 (12 months), Pepin 1989a (36 months), and Priotto 2009 (18 months). Outcomes measured at any follow‐up point included trypanosomes in any body compartment, an increase in WBC in CSF more than 50 cells/µL or a lower increase (20 to 49 cells/µL) together with symptoms typical of relapse. We grouped these outcomes as "relapse during follow‐up".
Adverse events
Adverse events represent one of the major measurable outcomes during sleeping sickness treatment, especially in trials which use melarsoprol which can cause severe encephalopathies.
All trials except Lejon 2003 reported in detail on several adverse events, (although different trials did not report exactly the same adverse events): central nervous system (CNS) alterations, gastrointestinal symptoms such as diarrhoea and abdominal pain, cardiac arrhythmia, leukopenia, neutropenia, infections, and skin reactions.
Risk of bias in included studies
We included nine randomized controlled trials. See 'Characteristics of included studies' for details, also Figure 1 and Figure 2.
1.
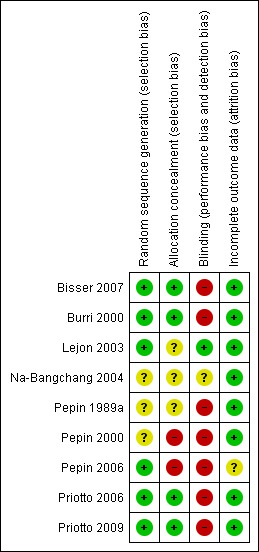
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
2.
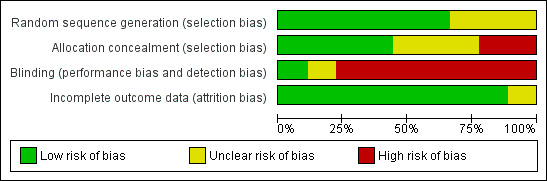
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
Generation of allocation sequence and concealment
Five trials reported block randomization. Four trials (Bisser 2007, Burri 2000,Priotto 2006 and Priotto 2009 ) reported computer‐generated randomization sequences, three trials did not describe how the participants were randomized, one reported that randomization was done in hospital without describing the methods used (Pepin 2000).
Four trials (Bisser 2007, Burri 2000, Priotto 2006 and Priotto 2009) reported using sealed, opaque envelopes to conceal allocation sequence. In two trials (Pepin 2000, Pepin 2006) allocation concealment was not attempted, the other trials did not report on it.
Blinding
Blinding of participants or clinical teams was not feasible in most of the included trials, because regimens and modes of administration of the drugs compared were too different to allow it. Only one trial mentioned that the assessors of clinical results were blinded (Burri 2000) and one trial (Lejon 2003) reported that the clinical team was blinded.
Inclusion of randomized participants
Five trials reported rates of follow‐up that were greater than 90% (Bisser 2007, Lejon 2003, Na‐Bangchang 2004, Priotto 2006, Priotto 2009). Two trials had follow‐up rates of less than 90 % – Burri 2000 at 88.4% and Pepin 2000 with 76.6%. For the remaining two trials it was not clear how many participants were lost to follow‐up, although from Pepin 1989a one could conclude that there was no loss to follow up. Numbers of losses to follow‐up are not mentioned in Pepin 2006 where, however, one of the three arms of the trial had to be stopped because of adverse events.
Effects of interventions
All adverse events described below are also listed in Table 2.
2. Adverse events.
| Comparison (Drug A vs Drug B) | Trial | n/Na | Adverse event | |
| Drug A | Drug B | |||
| Melarsoprol monotherapy | ||||
| Melarsoprol: graded (Angolan) vs fixed 10 days | Burri 2000 | 14/250 | 14/250 | Encephalopathy |
| 17/250 | 18/250 | Diarrhoea | ||
| 15/250 | 39/250 | Skin reactions | ||
| Melarsoprol: standard 3.6 mg vs graded 26 days | Pepin 2006 | 7/149 | 7/70 | Seizures |
| 10/149 | 3/70 | Confusion | ||
| 1/149 | 0/70 | Skin reactions | ||
| Melarsoprol: standard 3.6 mg vs incremental 10 days | Bisser 2007 | 4/69 | 5/70 | Encephalopathy |
| 7/69 | 5/70 | Diarrhoea | ||
| 14/69 | 11/70 | Nausea and vomiting | ||
| 19/69 | 13/70 | Infection (phlebitis) | ||
| Standard melarsoprol 3.6 mg vs fixed melarsoprol 10 days | Pepin 2006 | 10/149 | 6/170 | Confusion |
| 7/149 | 4/170 | Seizures | ||
| 1/149 | 6/170 | Skin reactions | ||
| Graded melarsoprol 26 days vs fixed melarsoprol 10 days | Pepin 2006 | 3/70 | 6/170 | Confusion |
| 7/70 | 4/170 | Seizures | ||
| 0/70 | 6/170 | Skin reactions | ||
| Eflornithine monotherapy | ||||
| Eflornithine 7 days vs 14 days | Pepin 2000 | 7/158 | 10/163 | Seizures |
| 13/158 | 26/163 | Diarrhoea | ||
| 7/158 | 13/163 | Nausea and vomiting | ||
| 5/158 | 24/163 | Infection | ||
| Oral eflornithine 500 vs 400 mg | Na‐Bangchang 2004 | 7/13 | 7/12 | Diarrhoea |
| 9/13 | 8/12 | Anaemia | ||
| 8/13 | 8/12 | Leukopenia | ||
| Comparisons between single drugs | ||||
| Standard melarsoprol 3.6 mg vs nifurtimox 14 days | Bisser 2007 | 4/69 | 1/70 | Encephalopathy |
| 7/69 | 10/70 | Diarrhoea | ||
| 14/69 | 17/70 | Nausea and vomiting | ||
| 19/69 | 0/70 | Infection (phlebitis) | ||
| Melarsoprol incremental 10 days vs nifurtimox 14 days | Bisser 2007 | 5/70 | 1/70 | Encephalopathy |
| 5/70 | 10/70 | Diarrhoea | ||
| 11/70 | 17/70 | Nausea and vomiting | ||
| 13/70 | 0/70 | Infection (phlebitis) | ||
| Standard (graded) melarsoprol vs pentamidine | Lejon 2003 | None recorded | None recorded | — |
| Combination therapies | ||||
| Melarsoprol vs melarsoprol+prednisolone | Pepin 1989a | 35/308 | 12/290 | Encephalopathy |
| 4/308 | 3/290 | Skin reactions | ||
| 8/308 | 8/290 | Infections | ||
| Standard melarsoprol 3.6 mg vs melarsoprol‐nifurtimox 10 days | Bisser 2007 | 4/69 | 2/69 | Encephalopathy |
| 7/69 | 7/69 | Diarrhoea | ||
| 14/69 | 12/69 | Nausea and vomiting | ||
| 19/69 | 6/69 | Infection (phlebitis) | ||
| Melarsoprol incremental 10 days vs melarsoprol‐nifurtimox | Bisser 2007 | 5/70 | 2/69 | Encephalopathy |
| 5/70 | 7/69 | Diarrhoea | ||
| 11/70 | 12/69 | Nausea and vomiting | ||
| 13/70 | 6/69 | infection (phlebitis) | ||
| Nifurtimox 14 days vs melarsoprol‐nifurtimox | Bisser 2007 | 1/70 | 2/69 | Encephalopathy |
| 10/70 | 7/69 | Diarrhoea | ||
| 17/70 | 12/69 | Nausea and vomiting | ||
| 0/70 | 6/69 | infection (phlebitis) | ||
| Eflornithine vs eflornithine‐nifurtimox | Priotto 2009 | 13/143 | 18/143 | Seizures |
| 41/143 | 9/143 | Diarrhoea | ||
| 29/143 | 69/143 | Nausea and vomiting | ||
| 1/143 | 2/143 | Anaemia | ||
| 10/143 | 2/143 | Neutropenia | ||
| 25/143 | 14/143 | Infection | ||
| 20/143 | 4/143 | Skin reactions | ||
| Eflornithine‐nifurtimox vs melarsoprol‐eflornithine | Priotto 2006 | 4/17 | 2/19 | Seizures |
| 4/17 | 8/19 | Diarrhoea | ||
| 1/17 | 4/19 | Nausea and vomiting | ||
| 0/17 | 0/19 | Skin reactions | ||
| 3/17 | 1/18 | Neutropenia | ||
| Eflornithine‐nifurtimox vs melarsoprol‐nifurtimox | Priotto 2006 | 4/17 | 4/18 | Seizures |
| 4/17 | 3/18 | Diarrhoea | ||
| 1/17 | 1/18 | Nausea and vomiting | ||
| 3/17 | 0/18 | Neutropenia | ||
| 0/17 | 1/18 | Skin reactions | ||
| Melarsoprol‐eflornithine vs melarsoprol‐nifurtimox | Priotto 2006 | 2/19 | 4/18 | Seizures |
| 8/19 | 3/18 | Diarrhoea | ||
| 4/19 | 1/18 | Nausea and vomiting | ||
| 1/18 | 0/18 | Neutropenia | ||
| 0/18 | 1/18 | Skin reactions | ||
an/N: number of participants with adverse event/total number of participants.
1. Melarsoprol monotherapy: dosages and regimens
1.1. Graded 26 days (Angolan schedule) versus fixed 10 days
Burri 2000 compared these regimens in two groups of 250 participants. There were no significant difference between the groups death during treatment (six in each group; Analysis 1.1), but the overall mortality was higher in the Angolan scheme: 12 versus 9; RR 1.33, 95% CI 0.57 to 3.11. The same number of participants in both groups (14/250) developed encephalopathy and there were no differences in the number of diarrhoea cases in the two groups of participants (17 and 18 respectively; Table 2). Participants treated for 10 days experienced a higher number of skin reactions (39/250 versus 15/250 for the 26 days schedule; Table 2). Number of relapses during follow up were higher in the Angolan scheme (5 versus 3) but not significantly different (RR1.67, 95% CI 0.40 to 6.90 Analysis 1.3)
1.1. Analysis.
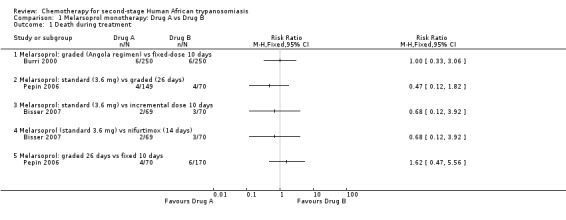
Comparison 1 Melarsoprol monotherapy: Drug A vs Drug B, Outcome 1 Death during treatment.
1.3. Analysis.
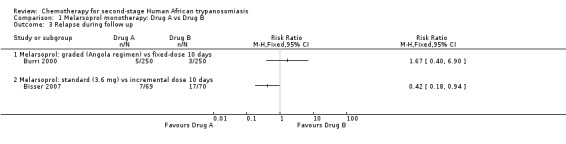
Comparison 1 Melarsoprol monotherapy: Drug A vs Drug B, Outcome 3 Relapse during follow up.
1.2. Standard (3.6 mg) versus graded 26 days
In Pepin 2006 a standard dose of melarsoprol was compared to a graded dose, both given for 26 days. The group of participants receiving the standard dose had a lower risk of death during treatment (RR 0.47, 95% CI 0.12 to 1.82; Analysis 1.1) and a lower number of seizures (7/149 compared to 7/70 for graded melarsoprol). As the clinical team was not blinded, enrolment in the graded melarsoprol arm was stopped early.
1.3. Standard (3.6 mg) versus incremental 10 days
Bisser 2007 found higher overall mortality (9/70) in the group that received incremental melarsoprol for 10 days than in the group of patients treated with standard melarsoprol (5/69, Analysis 1.2). The risk of relapse during follow‐up was lower in the standard melarsoprol group (RR 0.42, 95% CI 0.18 to 0.94; Analysis 1.3). Numbers of participants suffering encephalopathy, diarrhoea, nausea and vomiting were similar in the two groups but the longer standards schedule resulted in more phlebitis 19/69 versus 13/70 (see Table 2).
1.2. Analysis.
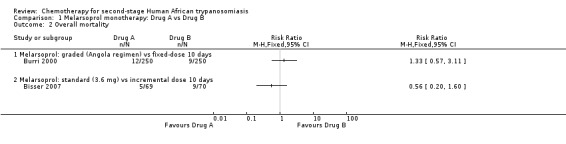
Comparison 1 Melarsoprol monotherapy: Drug A vs Drug B, Outcome 2 Overall mortality.
1.4. Standard melarsoprol (3.6 mg) versus fixed 10 days
Pepin 2006 evaluated the effectiveness of a standard dose of melarsoprol given over 26 days compared with a shorter regimen of 10 consecutive daily injections, finding that the frequency of death during treatment did not vary significantly (4/149 for the standard dose versus 6/170 for the 10 days regimen, Analysis 1.1). The number of neurological adverse events (seizures, confusion) was also similar in the groups (see Table 2) but the group receiving a shorter schedule had more skin reactions (6/170 versus only 1/149 in the standard melarsoprol group).
1.5. Graded 26 days versus fixed 10 days
Pepin 2006 reported that the frequency of death during treatment was not significantly different between groups receiving the two treatments (Analysis 1.1). Seizures were more frequent in the participants receiving graded melarsoprol (7/70) than in those receiving the fixed 10‐day schedule (4/170). Skin reactions were experienced with the shorter schedule (6/170).
2. Eflornithine monotherapy: dosages and regimens
Different regimens for eflornithine monotherapy were tested in two trials.
Pepin 2000 compared the same dose of 400 mg/kg/day, given as a slow intravenous infusion every six hours, for seven or 14 days, in groups of patients recruited from four different sites. Participants treated for seven days had lower risk of death during treatment (RR 0.21, 95% CI 0.02 to 1.75) than those on the 14‐day schedule (Analysis 2.1), and fewer of them suffered gastrointestinal symptoms (Table 2), but the shorter schedule resulted in more relapses during follow up (28/158 against 14/163; RR 2.06, 95% CI 1.13 to 3.77 Analysis 2.3).
2.1. Analysis.
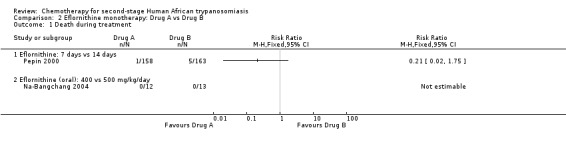
Comparison 2 Eflornithine monotherapy: Drug A vs Drug B, Outcome 1 Death during treatment.
2.3. Analysis.
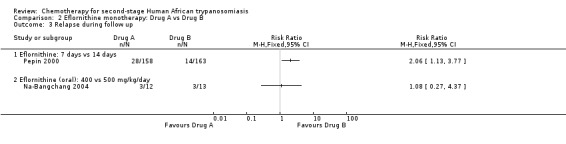
Comparison 2 Eflornithine monotherapy: Drug A vs Drug B, Outcome 3 Relapse during follow up.
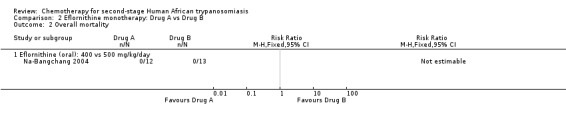
Na‐Bangchang 2004 used eflornithine orally, as this may constitute a more convenient way of administration than the slow intravenous infusion, and compared two doses of 500 and 400 mg/kg/day in two small groups of participants (13 and 12).There were no deaths in the two groups, and rates of relapse (Analysis 2.3) and adverse events (diarrhoea, anaemia, leukopenia) were similar.
3. Comparisons between single drugs (monotherapies)
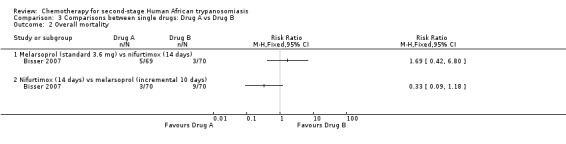
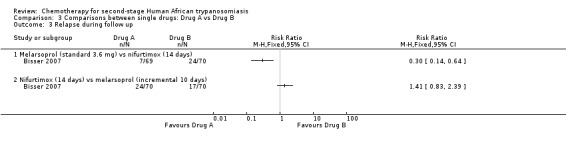
3.1. Melarsoprol (standard 3.6 mg) versus nifurtimox (14 days)
This comparison was tested by Bisser 2007. The frequency of death during treatment or overall mortality was not significantly different in the two groups (Analysis 3.1 and Analysis 3.2), but patients receiving melarsoprol had fewer relapses during follow up (RR 0.30, 95% CI 0.14 to 0.64; Analysis 3.3). Encephalopathy was more frequent in the melarsoprol group (4/69 participants) than in the nifurtimox group (1/70), and experienced a high number of infections (phlebitis; 19/69), but participants receiving nifurtimox had more gastrointestinal symptoms such as nausea and vomiting.
3.1. Analysis.
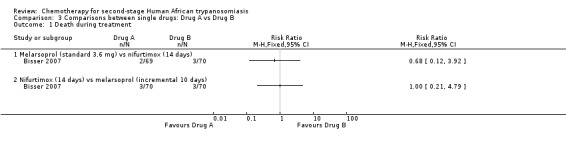
Comparison 3 Comparisons between single drugs: Drug A vs Drug B, Outcome 1 Death during treatment.
3.2. Analysis.
Comparison 3 Comparisons between single drugs: Drug A vs Drug B, Outcome 2 Overall mortality.
3.3. Analysis.
Comparison 3 Comparisons between single drugs: Drug A vs Drug B, Outcome 3 Relapse during follow up.
3.2. Melarsoprol (incremental 10 days) versus nifurtimox (14 days)
This comparison was tested in Bisser 2007 in two groups of 70 participants each. There was no difference in the number of deaths during treatment (three in each group), but the overall mortality was much lower in the nifurtimox group – three versus nine (RR 0.33, 95% CI 0.09 to 1.18; Analysis 3.2), while the number of relapses was higher in participants who has received nifurtimox (24/70) than in those treated with melarsoprol (17/70; Analysis 3.3). Participants in the melarsoprol group had a high number of phlebitis (13/70; Table 2).
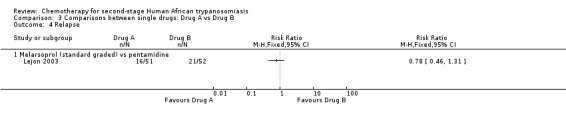
3.3. Melarsoprol (standard (graded)) versus pentamidine
Lejon 2003 compared melarsoprol with pentamidine, which is more commonly used for first stage HAT, in participants with 20 or fewer cells in CSF. The only reported outcome is relapse, which was more frequent in participants treated with pentamidine (Analysis 3.4). No adverse events were recorded.
3.4. Analysis.
Comparison 3 Comparisons between single drugs: Drug A vs Drug B, Outcome 4 Relapse.
4. Combination therapies
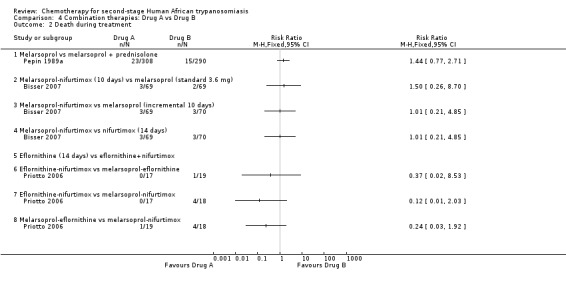
4.1. Melarsoprol versus melarsoprol + prednisolone
In Pepin 1989a, a Neujean schedule of melarsoprol was compared to the same schedule with added oral prednisolone. The differences in the number of deaths and relapses in the two groups were not statistically significant (Analysis 4.2 and Analysis 4.4), but participants who received melarsoprol only had a much higher number of encephalopathies (35/308) compared with those who also received prednisolone (12/290; Table 2).
4.2. Analysis.
Comparison 4 Combination therapies: Drug A vs Drug B, Outcome 2 Death during treatment.
4.4. Analysis.
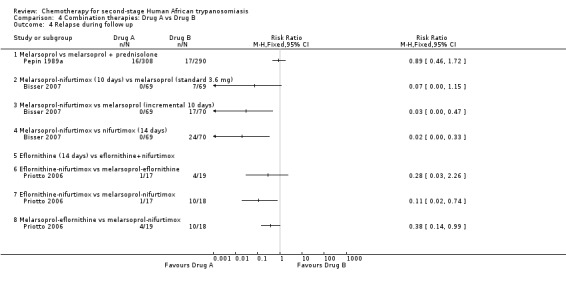
Comparison 4 Combination therapies: Drug A vs Drug B, Outcome 4 Relapse during follow up.
4.2. Melarsoprol (standard 3.6 mg) versus melarsoprol‐nifurtimox (10 days)
There was no statistically significant difference in the number of deaths between the two groups of 69 participants each, but participants treated with melarsoprol‐nifurtimox had no relapses compared with seven occurring in the melarsoprol group (RR 0.07, 95% CI 0.00 to 1.15; Analysis 4.4) lower numbers of encephalopathy cases (2/69 versus 4/69 for standard melarsoprol), and lower numbers of phlebitis (6/69 versus 19/69; Table 2).
4.3. Melarsoprol (incremental 10 days) versus melarsoprol‐nifurtimox
Bisser 2007 reported no relapses in the group of participants who received the combination of melarsoprol and nifurtimox (RR 0.03, 95% CI 0.00 to 0.47; Analysis 4.4); differences in the number of deaths during treatment were not significant (Analysis 4.2). Melarsoprol‐nifurtimox also reduced the number of encephalopathies (2/69 versus 5/70), and of phlebitis (6/69 versus 13/70; Table 2) but the two groups had similar numbers of gastrointestinal symptoms.
4.4. Nifurtimox (14 days) versus melarsoprol‐nifurtimox
Melarsoprol‐nifurtimox was compared to nifurtimox monotherapy in Bisser 2007: there were no relapses in the 69 participants receiving the drug combination (RR 0.02, 95% CI 0.00 to 0.33; Analysis 4.4). Overall mortality was higher in the melarsoprol‐nifurtimox group of participants (6/69 versus 3/70 deaths; Analysis 4.3), who also reported two cases of encephalopathy against only one case in the nifurtimox group, and six cases of phlebitis (Table 2). Participants treated with nifurtimox alone had slightly more nausea and vomiting (Table 2).
4.3. Analysis.
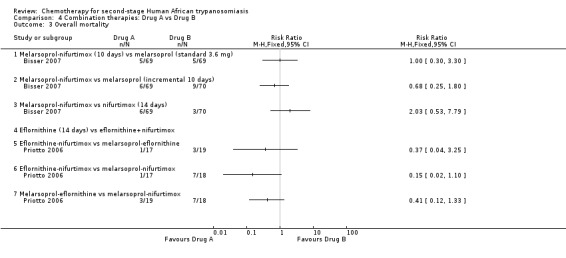
Comparison 4 Combination therapies: Drug A vs Drug B, Outcome 3 Overall mortality.
4.5. Eflornithine versus eflornithine‐nifurtimox
Priotto 2009 compared eflornithine monotherapy given every six hours for 14 days, with a eflornithine‐nifurtimox combination (eflornithine given every 12 hrs for seven days + oral nifurtimox for 10 days). The results (Analysis 4.2; Analysis 4.3; Analysis 4.4) show that although number of deaths were similar, there were more relapses during follow‐up in the eflornithine group. Participants treated with eflornithine‐nifurtimox had more seizures (18/143) than those receiving eflornithine only (13/143), and also experienced more nausea and vomiting (69/143 versus 29/143), but they did not experience diarrhoea and also had fewer cases of neutropenia (2/143 versus 10/143). Eflornithine caused more infections and more skin reactions when used alone than when combined with nifurtimox (Table 2).
4.6. Eflornithine‐nifurtimox versus melarsoprol‐eflornithine
Priotto 2006 evaluated this comparison in two relatively small groups of participants (17 and 19 respectively). There were significantly fewer deaths in the group treated with eflornithine‐nifurtimox (RR 0.37, 95% CI 0.02 to 8.53 for deaths during treatment; (Analysis 4.2); RR 0.37, 95% CI 0.04 to 3.25 for overall mortality, (Analysis 4.3). The risk of relapse during follow up was significantly smaller for participants treated with eflornithine‐nifurtimox (RR 0.28, 95% CI 0.03 to 2.26,( Analysis 4.4). The eflornithine‐nifurtimox group reported more seizures (4/17) and more participants developed neutropenia (3/17) than in the melarsoprol‐eflornithine group; however the latter had more gastrointestinal symptoms (see Table 2).
4.7. Eflornithine‐nifurtimox versus melarsoprol‐nifurtimox
There was a significant lower risk of death during treatment (RR 0.12, 95% CI 0.01 to 2.03, Analysis 4.2), overall mortality (RR 0.15, 95% CI 0.02 to 1.10, Analysis 4.3), and relapse during follow up (RR 0.11, 95% CI 0.02 to 0.74, Analysis 4.4) in the group of participants treated with eflornithine‐nifurtimox. The two groups reported similar number of adverse events (seizures, gastrointestinal symptoms; Table 2), but more eflornithine‐nifurtimox participants developed neutropenia (3/17 versus 0/18).
4.8. Melarsoprol‐eflornithine versus melarsoprol‐nifurtimox
There were fewer deaths during treatment (RR 0.24, 95% CI 0.03 to 1.92, Analysis 4.2), less overall mortality (RR 0.41, 95% CI 1.12 to 1.33, Analysis 4.3), and fewer relapses (RR 0.38, 95% CI 0.14 to 0.99, Analysis 4.4) in the group that received melarsoprol‐eflornithine. Twice as many participants in the melarsoprol‐nifurtimox group had seizures (4/18) but fewer developed diarrhoea, nausea, and vomiting than those treated with melarsoprol‐eflornithine (Table 2). Only one participant in the melarsoprol‐eflornithine group suffered neutropenia.
Discussion
This review's objectives were to assess the effectiveness and safety of drugs used for treating second‐stage human African trypanosomiasis. All the trials we identified refer to treatment for HAT caused by T. b. gambiense; we did not find any RCT reporting on treatment for HAT caused by T.b. rhodesiense. Trials on treatment of T. b. rhodesiense would have been analysed and discussed separately from the T. b. gambiense trials and will be included, if available, in future updates of the review.
Trials of treatment for sleeping sickness encounter logistic, organizational and clinical difficulties that have to be taken into consideration when assessing trial design and methodological quality. The number of drugs available is very limited, routes of administration are painful or difficult to secure under field conditions, and toxicity high. Also, the quantity of drugs available may be a limiting factor under field circumstances, and clinical trials may have to be interrupted because of local political instability. Drug regimens were mainly empirically developed and scarce pharmacokinetic data are available. Pharmaceutical companies have little commercial interest in developing new drugs for HAT. For all these reasons, efforts have been focused on optimizing and minimizing drug regimens and reducing adverse events. We grouped the treatments tested in the included randomized controlled trials as drug monotherapies (melarsoprol regimens, eflornithine regimens), comparisons between monotherapies and in more recent trials, comparisons of several drug combinations. No meta‐analysis was possible as no two trials compared the same treatment.
Some aspects of the methodological quality of most trials were not optimal, but this was related to the characteristics of the treatment under investigations. The different routes of administration and regimes under comparison would not have allowed blinding of participants and medical personnel in any of the trials. Allocation concealment and randomization methods however were adequate and well described in most trials. Furthermore, the priority in the past was mainly to treat the enormous existing number of patients in order to be able to control the disease; we did not include studies which were not randomized, but many of these excluded publications reported on important medical observations. This applies in particular to T. b. rhodesiense second‐stage disease in which no randomized controlled trials were found. Rhodesiense HAT tends to occur in self‐limited epidemic outbreaks or as isolated cases, and the reported cases represent less than 10% of all HAT cases (Simarro 2008).
1) Monotherapies ‐ melarsoprol and eflornithine
Melarsoprol can induce a life‐threatening encephalopathy in a large percentage of treated patients (Seixas 2005) and trials assessing melarsoprol have been aimed at minimizing doses of the drug and length of treatment while maintaining anti‐trypanosome activity.
Fixed 10‐day regimens were found to be as effective as those of 26 days and resulted in similar levels of adverse events (Burri 2000 and Pepin 2006). They offer however significant practical advantages (fewer painful injections, less drug used and shorter treatment duration). Slightly different graded 26‐day melarsoprol regimens were used in Burri 2000 and Pepin 2006, and an incremental 10‐day regimen was tested in Bisser 2007. Although Burri 2000 did not report differences in outcomes or adverse events between the graded schedule and the 10‐day schedule, the other two trials showed that graded or incremental melarsoprol resulted in higher death rates, higher number of seizures (Pepin 2006) and more relapses (Bisser 2007), suggesting that incremental melarsoprol schedules should be abandoned (Pepin 2006).
A large multinational non‐randomized clinical study (Schmid 2005) also confirmed the effectiveness (non‐inferiority) of the shorter 10‐day melarsoprol schedule, in comparison with the standard 26 days of treatment, with regard to cure rates and adverse events. The applicability of this abridged 10‐day melarsoprol schedule to Rhodesiense HAT patients has recently been tested in a utilization study in two trial centres in Uganda an Tanzania (IMPAMEL IIII), showing similar levels of adverse events with historical controls. The potential implementation of this abridged melarsoprol schedule to second stage Rhodesiense HAT patients is currently being evaluated, but the trial was non‐randomized and outside the inclusion criteria of this review.
Pepin 1989a showed that the addition of prednisolone to melarsoprol reduced the number of encephalopathy cases and associated mortality. Prednisolone and other corticosteroids had been used as an adjunctive treatment to melarsoprol for many years but this was the first randomized trial to test it and indicate its effectiveness. This trial is however of insufficient methodological quality (Figure 1). Prednisolone and prednisone are currently still in use in patients receiving melarsoprol as no suitable alternative encephalopathic syndrome preventive treatment has been identified, but their effectiveness remains unclear.
Eflornithine is effective against T. b. gambiense and induces less severe adverse events than melarsoprol, but it has to be administered as a slow intravenous infusion every six hours and this is difficult under field conditions. Pepin 2000 tested a 7‐day against a 14‐day regimen and showed that treating for seven days resulted in fewer deaths and fewer adverse events in patients relapsing from a previous episode of sleeping sickness, making it a suitable alternative in this kind of patient. The 7‐day regimen was however less effective than the 14‐day schedule especially for new cases. Na‐Bangchang 2004 tested oral eflornithine at two slightly different dosages (500 and 400 mg/kg/day). There were no deaths in this trial, and similar levels of adverse events between the two patient groups, but oral eflornithine seemed not to reach adequate levels in plasma and CSF, and further development of this administration route was abandoned.
3) Comparisons between single drugs
Bisser 2007 tested two melarsoprol regimens, a standard 26‐day regimen and an incremental 10‐day regimen, against nifurtimox. The two melarsoprol regimens were more effective at preventing relapses than nifurtimox alone, but induced more cases of encephalopathies, and overall mortality was highest with incremental melarsoprol.
Melarsoprol also gave fewer relapses than pentamidine (Lejon 2003) but no other outcomes or adverse events were reported in this trial. The results of this trial indicate that the use of pentamidine (commonly used for first stage HAT) in patients in the so‐called intermediate stage is hazardous and that better markers of disease stage are needed to allow its safe use in this clinical situation.
4) Combination therapies
Since 2006, two‐drugs combinations between any of the drugs used in second‐stage HAT (melarsoprol, eflornithine and nifurtimox) have been tested in randomized controlled trials.
Bisser 2007 tested a combination of melarsoprol and nifurtimox, given for 10 days, against standard or incremental melarsoprol regimens, and against nifurtimox alone, in an equivalence trial. Melasorprol‐nifurtimox was more effective than monotherapies at reducing the number of relapses but adverse events were comparable between groups and encephalopathies were reported in all regimens which included melarsoprol.
A trial testing comparing melarsoprol‐nifurtimox with melarsoprol‐eflornithine and nifurtimox‐eflornithine had to be interrupted because of the high number of deaths, due to reactive encephalopathy, reported in the melarsoprol‐nifurtimox group (Priotto 2006). The same trial showed that patients receiving nifurtimox‐eflornithine had a lower risk of relapse and fewer deaths than those receiving drug combinations with melarsoprol. Following this initial observation a much larger multi‐site trial to compare eflornithine with nifurtimox‐eflornithine (thus completely removing melarsoprol) was planned, implemented in selected HAT treatment centres and recently completed (Priotto 2009). The combination of nifurtimox and eflornithine (NECT) was shown to give fewer relapses and was generally well tolerated. A satisfactory safety profile of NECT was confirmed in a non‐randomized study in the Democratic Republic of Congo, which included children and pregnant women (Schmid 2012), and in a recent cohort study (Alirol 2013) which also included children . A major advantage of NECT is the reduction in the frequency and number of eflornithine slow infusions to twice a day, thus reducing the burden on health personnel and patients alike. Priotto 2009 was designed as a non‐inferiority trial and its clinical results confirmed the non‐inferiority of NECT to eflornithine alone; other considerations are the practical advantages of using NECT in terms of drug quantities, personnel time and logistic costs. Furthermore, the combined use of two drugs should decrease the emergence of resistance.
Future perspectives
NECT was approved by the Expert Committee on the Selection and Use of Essential Medicines at its 17th meeting on 30 April 2009 and was included in the WHO Essential List of Medicines for the treatment of human African trypanosomiasis (WHO 2010). Treating second‐stage HAT patients without the need for melarsoprol has the clear advantage of removing the risk of encephalopathic syndromes and other severe adverse events. NECT has this and other significant advantages in comparison with other therapies, but it still requires two drugs, used for several days, involving specialized health personnel. Further refinements and reductions of this combination need to be tested, and additional field studies as well as the establishment of an appropriate dose regimen for NECT in children are planned (DNDi 2008).
The development of new (and easier to use) drugs would represent a big step forward for the management of second‐stage HAT. A promising recent initiative is the announcement by DNDi that fexinidazole is entering clinical development for HAT and that an agreement was signed with Sanofi‐Aventis for its further development. Fexinidazole is a 5‐nitroimidazole and in experimental studies was found to be active against both T. b. gambiense and T. b. rhodesiense. Phase II studies of oral fexinidazole in humans are ongoing (DNDi 2012). Another new product, Oxaborole SCYX‐7158, also for oral administration, is undergoing Phase I studies (DNDi 2012b).
After several decades of scarce attention, the past few years have seen a new impetus in the fight against HAT, due in good part to an efficient co‐ordination and collaboration between different agencies, researchers, and national trypanosomiasis programmes, the diminution of social upheavals, capacity building activities and the free provision of diagnostic and reagents and medicines. The situation has improved even in the few years since the protocol for this Cochrane review was first published (2006): the total number of HAT cases decreased 68% between 1995 and 2006 (Simarro 2008). Clinical trials of high methodological quality have been completed since then (despite no significant reduction in the logistic challenges to be faced by trialists). So the practical implications of these latest trials go beyond their clinical results to also include a framework for planning and executing trials in resource‐poor settings.
There are signs that the use of melarsoprol will decline. An analysis of pooled data from 11,668 patients from different countries showed that its effectiveness was lower than eflornithine (Balasegaram 2009). In the absence of a direct randomized comparison between melarsoprol, eflornithine and NECT, this is a relevant finding due to the number of patients treated from several different locations and a comprehensive analysis of the outcomes. Also, although the choice of HAT therapy is often dictated by local conditions of availability, active public‐private partnerships have allowed Gambiense HAT endemic countries to increase the use of eflornithine and NECT, resulting in a decrease in the percentage of melarsoprol treated patients from 86% in 2004 to 51% in 2008 (WHO 2009). Parasite resistance is less likely to develop with a combination such as NECT, but a system of monitoring will be needed to monitor the effectiveness of drug regimens over time.
It is imperative that studies on the reduction of the adverse effects of currently used drugs, testing different regimens, and experimental and clinical studies on the development of new anti‐trypanosomal compounds, effective for both stages of the disease, also continue taking place.
Authors' conclusions
Implications for practice.
Choice of therapy for second stage Gambiense HAT in the next few years will continue to be dictated by local conditions of availability and logistic difficulties, but it is envisioned that melarsoprol, with its high level of adverse events, will be phased out in favour of eflornithine and NECT. Parasite resistance to the drugs as well as their effectiveness need to be carefully monitored in large cohort studies.
Implications for research.
It is essential that future research focus on the reduction of the adverse effects of currently used drugs, tests on different regimens, and experimental and clinical studies on the development of new anti‐trypanosomal compounds, effective for both stages of the disease. Development of new diagnostic tools, both to improve disease confirmation and to precisely determine the stage of the disease, and to avoid the need for lumbar punctures performed under unsafe conditions, is also necessary.
What's new
| Date | Event | Description |
|---|---|---|
| 18 June 2013 | New citation required but conclusions have not changed | Updated search. No new included studies. |
| 25 February 2013 | New search has been performed | Updated the search for trials, added two ongoing trials, changed search dates in the review, added a few 'Additional References', slightly modified the Discussion. |
Acknowledgements
This document is an output from a project funded by the UK Department for International Development (DFID) for the benefit of low‐ and middle‐income countries. The views expressed are not necessarily those of DFID.
Appendices
Appendix 1. Search methods: detailed search strategies
| Search set | CIDG SRa | CENTRAL | MEDLINEb | EMBASEb | LILACSb | BIOSIS |
| 1 | Human African trypanosom* | Human African trypanosom* | Human African trypanosom* | Human African trypanosom$ | Human African trypanosom* | Human African trypanosom* |
| 2 | HAT | HAT | HAT | HAT | HAT | HAT |
| 3 | sleeping sickness | sleeping sickness | sleeping sickness | sleeping sickness | sleeping sickness | sleeping sickness |
| 4 | 1 or 2 or 3 | 1 or 2 or 3 | 1 or 2 or 3 | 1 or 2 or 3 | 1 or 2 or 3 | 1 or 2 or 3 |
| 5 | melarsoprol | melarsoprol | melarsoprol | melarsoprol | melarsoprol | melarsoprol |
| 6 | eflornithine | eflornithine | eflornithine | eflornithine | eflornithine | eflornithine |
| 7 | DFMO | DFMO | DFMO | DFMO | DFMO | DFMO |
| 8 | nifurtimox | nifurtimox | nifurtimox | nifurtimox | nifurtimox | nifurtimox |
| 9 | 5 or 6 or 7 or 8 | 5 or 6 or 7 or 8 | 5 or 6 or 7 or 8 | 5 or 6 or 7 or 8 | 5 or 6 or 7 or 8 | 5 or 6 or 7 or 8 |
| 10 | 4 and 9 | 4 and 9 | TRYPANOSOMIASIS, AFRICAN/DRUG THERAPY | 4 and 9 | 4 and 9 | 4 and 9 |
| 11 | — | — | 9 or 10 | limit 10 to human | limit 10 to human | limit 10 to human |
| 12 | — | — | limit 11 to human | — | — | — |
aCochrane Infectious Diseases Group Specialized Register. bSearch terms used in combination with the search strategy for retrieving trials developed by The Cochrane Collaboration (Lefebvre 2011); upper case: MeSH or EMTREE heading; lower case: free‐text term.
Data and analyses
Comparison 1. Melarsoprol monotherapy: Drug A vs Drug B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death during treatment | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Melarsoprol: graded (Angola regimen) vs fixed‐dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Melarsoprol: standard (3.6 mg) vs graded (26 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Melarsoprol: standard (3.6 mg) vs incremental dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Melarsoprol (standard 3.6 mg) vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Melarsoprol: graded 26 days vs fixed 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Overall mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Melarsoprol: graded (Angola regimen) vs fixed‐dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Melarsoprol: standard (3.6 mg) vs incremental dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Relapse during follow up | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Melarsoprol: graded (Angola regimen) vs fixed‐dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Melarsoprol: standard (3.6 mg) vs incremental dose 10 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Eflornithine monotherapy: Drug A vs Drug B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death during treatment | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Eflornithine: 7 days vs 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Eflornithine (oral): 400 vs 500 mg/kg/day | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Overall mortality | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Eflornithine (oral): 400 vs 500 mg/kg/day | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Relapse during follow up | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Eflornithine: 7 days vs 14 days | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Eflornithine (oral): 400 vs 500 mg/kg/day | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
2.2. Analysis.
Comparison 2 Eflornithine monotherapy: Drug A vs Drug B, Outcome 2 Overall mortality.
Comparison 3. Comparisons between single drugs: Drug A vs Drug B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death during treatment | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Melarsoprol (standard 3.6 mg) vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Nifurtimox (14 days) vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Overall mortality | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Melarsoprol (standard 3.6 mg) vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Nifurtimox (14 days) vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Relapse during follow up | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Melarsoprol (standard 3.6 mg) vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Nifurtimox (14 days) vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Relapse | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Melarsoprol (standard graded) vs pentamidine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 4. Combination therapies: Drug A vs Drug B.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death due to HAT | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Eflornithine‐nifurtimox vs melarsoprol‐eflornithine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Eflornithine‐nifurtimox vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Melarsoprol‐eflornithine vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Death during treatment | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Melarsoprol vs melarsoprol + prednisolone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Melarsoprol‐nifurtimox (10 days) vs melarsoprol (standard 3.6 mg) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Melarsoprol‐nifurtimox vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 Melarsoprol‐nifurtimox vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 Eflornithine (14 days) vs eflornithine+nifurtimox | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 Eflornithine‐nifurtimox vs melarsoprol‐eflornithine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.7 Eflornithine‐nifurtimox vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.8 Melarsoprol‐eflornithine vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Overall mortality | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Melarsoprol‐nifurtimox (10 days) vs melarsoprol (standard 3.6 mg) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Melarsoprol‐nifurtimox vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Melarsoprol‐nifurtimox vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Eflornithine (14 days) vs eflornithine+nifurtimox | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Eflornithine‐nifurtimox vs melarsoprol‐eflornithine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Eflornithine‐nifurtimox vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Melarsoprol‐eflornithine vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Relapse during follow up | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Melarsoprol vs melarsoprol + prednisolone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Melarsoprol‐nifurtimox (10 days) vs melarsoprol (standard 3.6 mg) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Melarsoprol‐nifurtimox vs melarsoprol (incremental 10 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Melarsoprol‐nifurtimox vs nifurtimox (14 days) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Eflornithine (14 days) vs eflornithine+nifurtimox | 0 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 Eflornithine‐nifurtimox vs melarsoprol‐eflornithine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Eflornithine‐nifurtimox vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.8 Melarsoprol‐eflornithine vs melarsoprol‐nifurtimox | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
4.1. Analysis.
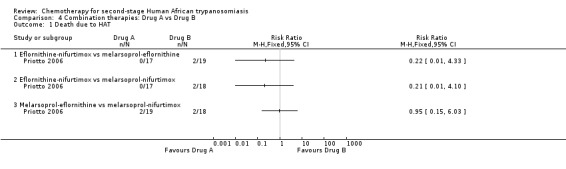
Comparison 4 Combination therapies: Drug A vs Drug B, Outcome 1 Death due to HAT.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bisser 2007.
| Methods | Open randomized equivalence trial Generation of allocation sequence: "Block randomisation was prepared by the statistician prior to the start of the study. A randomisation list was generated by means of the statistical analysis system S.A.S 8.0 (SAS Institute, Cary, USA) " ‐ personal communication by the author. and block randomization Allocation concealment: "closed envelopes containing the treatment type and randomisation number were prepared." personal communication by the author Blinding: not feasible Inclusion of all randomized participants: ITT profile included, 93.8% participants completed treatment Enrollment February‐August 1998, follow up ends in 2000 |
|
| Participants | Number randomized: 278 Inclusion criteria: living in the study area; age > 15 years; parasitologically confirmed second stage T. b. gambiense infection; no history of treatment for sleeping sickness Exclusion criteria: pregnancy; Glasgow coma score < 8; active tuberculosis; potential central nervous system (CNS) infection; severe organ disease Diagnosis and follow‐up methods: routine parasitological methods + cerebrospinal fluid (CSF) cell count > 20 cells/μL |
|
| Interventions | 1. Standard melarsoprol: 3.6 mg/kg/day for 3 days; 3 series with 7‐day intervals; intravenous 2. Incremental melarsoprol: 10‐day incremental melarsoprol (0.6 mg/kg day 1, 1.2 mg/kg day 2, and 1.8 mg/kg at days 3 to 10; intravenous 3. Nifurtimox monotherapy: 5 mg/kg every 8 h for 14 days; oral 4. Melarsoprol‐nifurtimox: consecutive 10‐day melarsoprol‐nifurtimox low‐dose combination; (2 days of melarsoprol alone at 0.6 mg/kg on day 1, 1.2 mg/kg on day 2, and from days 3 to 10: melarsoprol at 1.2 mg/kg + nifurtimox 7.5 mg/kg every 8 h); melarsoprol given intravenously; nifurtimox given orally All participants pretreated with chloroquine for 3 days and with mebendazole Prednisolone was given for treatment of encephalopathy |
|
| Outcomes | 1. Relapse 2. Death after treatment 3. Cure at ≥24 months 4. Adverse events | |
| Notes | Location: Equator Province, Democratic Republic of Congo Setting: hospital Financial support: Belgian Ministry of Foreign Affairs, Directorate General for Development Cooperation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block computer‐generated randomisation was prepared before the study |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used. |
| Blinding (performance bias and detection bias) All outcomes | High risk | Regimens and modes of administration were too different to allow blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT profile included, 93.8% participants completed treatment. |
Burri 2000.
| Methods | Randomized controlled equivalence trial Generation of allocation sequence: randomization in blocks of 10 Allocation concealment: opaque sealed envelopes Blinding: not feasible Inclusion of all randomized participants: ITT profile included, 88.4 % participants completed treatment Duration: not mentioned |
|
| Participants | Number randomized: 500 Inclusion criteria: age > 14 years; confirmed second‐stage T. b. gambiense infection; trypanosomes in CSF or > 5 WBC/μL in CSF. Exclusion criteria: Glasgow coma score < 8; pregnancy; active tuberculosis Diagnosis and follow‐up methods: cerebrospinal fluid (CSF) examination, double centrifugation |
|
| Interventions | 1. Melarsoprol: 1.2 mg/kg on day 1, 2.4 mg/kg on day 2, 3.6 mg/kg on day 3, and 3.6 mg/kg on day 4; 3 series repeated at 7‐day intervals; administered intravenously 2. Melarsoprol: 2.2 mg/kg/day per 10 days; administered intravenously All participants were pretreated with chloroquine, mebendazole, multivitamins, and paracetamol Prednisolone was given to all participants at 1 mg/kg followed by decreasing doses |
|
| Outcomes | 1. Cure rates at 24 h after treatment 2. Death 3. Relapse 4. Adverse events | |
| Notes | Location: Kwanza Norte Province, Angola Setting: Trypanosomiasis Units Funding: Swiss Agency for Development and Cooperation, partly World Health Organization: Division of Control of Tropical Diseases. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation in blocks of ten was done during pretreatment |
| Allocation concealment (selection bias) | Low risk | Non transparent, sealed envelopes were used |
| Blinding (performance bias and detection bias) All outcomes | High risk | Stated: "A masked trial design was not possible" because of substantial differences between the two treatment schedules |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis included; 88.4% participants analysed (not adequate) |
Lejon 2003.
| Methods | Open randomized equivalence trial Generation of allocation sequence: block randomization Allocation concealment: not specified Blinding: Field team was blinded "for blocking procedures" Inclusion of all randomized participants: ITT or per protocol profile not included, but participants completed treatment Duration: not mentioned |
|
| Participants | Number randomized: 103 Inclusion criteria: age 2 to 59 years, presence of T. b. gambiense in blood, lymph nodes or CSF, > 5 WBC/μL in CSF, ≤20 WBC/μL in CSF Exclusion criteria: previous treatment for trypanosomiasis Diagnosis and follow‐up method: double centrifugation |
|
| Interventions | 1. Pentamidine 4 mg/kg for 10 days 2. Melarsoprol (3 series of 3 injections at increasing doses) |
|
| Outcomes | Relapse | |
| Notes | Location: Arua District, Uganda Setting: not specified Source of funding: Médecins Sans Frontières |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 'Block randomisation with uniform allocation..' |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned |
| Blinding (performance bias and detection bias) All outcomes | Low risk | The team on the field was blinded for blocking procedures' |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | no mention of ITT or per protocol analysis, however 98 out of 103 participants completed treatment and were analysed |
Na‐Bangchang 2004.
| Methods | Randomized controlled trial Generation of allocation sequence: method not mentioned Allocation concealment: not mentioned Blinding: not mentioned Inclusion of all randomized participants: ITT or per protocol profile not included, but all 25 participants completed treatment Duration: 2000 to 2002 |
|
| Participants | Number randomized: 25 Inclusion criteria: age 18 to 69 years; bodyweight 43 to 63 kg; parasitologically confirmed second‐stage T. b. gambiense infection Exclusion criteria: pregnancy; lactating women; Glasgow coma scale < 8; chronic medical condition or critically ill Diagnosis and follow‐up methods: Miniature anion exchange centrifugation technique and double centrifuge for detection of trypanosomes, cerebrospinal fluid (CSF) WBC |
|
| Interventions | 1. Eflornithine, oral, 400 mg/kg/day (100 mg/kg every 6 h) for 14 days 2. Eflornithine, oral, 500 mg/kg/day (125 mg/kg every 6 h) for 14 days All participants pretreated with chloroquine and albendazole |
|
| Outcomes | 1. Cure rates
2. Death
3. Adverse events Pharmacokinetics analysis |
|
| Notes | Location: Daloa, Cote d'Ivoire Setting: research centre Source of funding: UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were randomly allocated but method not specified |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Not mentioned in report |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data from all 25 patients were analysed |
Pepin 1989a.
| Methods | Prospective randomized trial Generation of allocation sequence: not described Allocation concealment: not specified Blinding: not done Inclusion of all randomized participants: ITT or per protocol profile not included, but 598 participants out of 620 enrolled, completed treatment (96.4%) Study conducted between March 1984 and October 1988 |
|
| Participants | Number randomized: 620 Inclusion criteria: parasitologically confirmed cases of T. b. gambiense Diagnosis and follow up methods: standard parasitological investigations and white cell count (WCC) |
|
| Interventions | 1. Melarsoprol: 3.6 mg/kg; 2 series of 3 injections if WCC < 20, 3 series of 3 injections if WCC = 20 to 99, or 3 series of 4 injections if WCC ≥100; 1‐week interval between first and second series, and 2‐week interval between second and third series 2. Melarsoprol + prednisolone: melarsoprol same as group 1; oral prednisolone as a single daily dose of 1 mg/kg up to a maximum of 40 mg started on the day before first dose of melarsoprol; given throughout first series, first interval, and second series of melarsoprol; discontinued over 3 days after second series, resumed on the day before third series, and discontinued over 3 days after the end of the third series Pretreatment: mebendazole, chloroquine, and suramin 24 h before first melarsoprol dose |
|
| Outcomes | 1. Relapse 2. Death 3. Encephalopathy and other adverse events | |
| Notes | Location: Nioki, Zaire (Democratic Republic of Congo) Setting: hospital Source of funding: Canadian International Development Agency |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomization was not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 96.4% participants completed treatment |
Pepin 2000.
| Methods | Multicentre randomized controlled trial Generation of allocation sequence: not specified Allocation concealment: "There was no concealment of allocation as far as I can remember", personal communication from the author Blinding: not feasible Inclusion of all randomized participants: 84% patients followed up to 1 year Enrolment started July 1993, continued until February 1996, follow up completed in April 1998 |
|
| Participants | Number randomized: 321 Inclusion criteria: parasitologically confirmed new patients and relapsing patients; likely to complete follow up; age 3 to 77 years Exclusion criteria (in new cases only): pregnancy; moribund patients Diagnosis and follow‐up methods: standard parasitological investigation and cerebrospinal fluid (CSF) count |
|
| Interventions | 1. Eflornithine: 400 mg/kg/day (100 mg/kg every 6 h), intravenous, for 7 days 2. Eflornithine: 400 mg/kg/day (100 mg/kg every 6 h), intravenous, for 14 days Pretreatment or additional prednisolone: not mentioned |
|
| Outcomes | 1. Treatment failure 2. Death 3. Adverse events | |
| Notes | Locations: Cote d'Ivoire, Congo, Democratic Republic of Congo, Uganda Setting: 3 hospitals and one trypanosomiasis unit Source of funding: UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | 'Randomisation was carried out in each hospital' Methods not stated |
| Allocation concealment (selection bias) | High risk | Not mentioned or described in trial report but the author was contacted and stated it was not done |
| Blinding (performance bias and detection bias) All outcomes | High risk | The authors state that blinding was not feasible |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 84% patients were followed up to 1 year (65% up to 2 years). |
Pepin 2006.
| Methods | Randomized controlled trial Generation of allocation sequence: block randomization, method not specified Allocation concealment: not done ( personal communication from the author) Blinding: not feasible Inclusion of all randomized participants: unclear how many participants completed treatment Enrolment started in April 1996 and was stopped in December 1998 for Arm C, enrolment in the other two arms continued until December 2001. Follow up data were accumulated until January 2004 |
|
| Participants | Number randomized: 389 inclusion criteria: age > 13 years; parasitologically confirmed new cases of T. b. gambiense trypanosomiasis; > 5 WBC/μL in CSF Exclusion criteria: past history of treatment for trypanosomiasis; "moribund condition"; resident outside area Diagnosis and follow‐up methods: standard parasitological investigation and cerebrospinal fluid (CSF) count |
|
| Interventions | 1. Melarsoprol: intravenous; 3.6 mg/kg/day; 3 series of 3 injections per day x 3 days, repeat after 7‐day interval; total duration 26 days 2. Melarsoprol: intravenous; 2.16 mg/kg/day once a day for 10 days 3. Melarsoprol: intravenous; graded dosing; 1.8/2.16/2.52/2.88/3.24/3.6 mg/kg/day; 3 series of 3 injections per day x 3 days, repeat after 7 days interval; total duration of 26 days Pretreatment: all participants treated with pentamidine, chloroquine, and thiabendazole All patients received oral prednisolone at 1 mg/kg |
|
| Outcomes | 1. Cure rates 2. Relapse 3. Adverse events | |
| Notes | Location: Nioki, Democratic Republic of Congo Setting: hospital Source of funding: not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 'Block randomisation (blocks of ten)' |
| Allocation concealment (selection bias) | High risk | Not mentioned in article but author stated it was not done |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not feasible |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | not mentioned in report |
Priotto 2006.
| Methods | Randomized, open‐label, active control, parallel clinical trial Generation of allocation sequence: randomization list in blocks of 18 was electronically generated Allocation concealment: randomization list and block size were blinded from the field team. Sealed and numbered opaque envelopes were used Blinding: not feasible due to the different administration modes of the drugs inclusion of all randomized participants: ITT and per protocol analysis done, (>90%of all participants randomized into the trial were included in the analysis) Enrolment started in march 2001, suspended in November 2001 for ethical reasons |
|
| Participants | Number randomized: 54 Inclusion criteria: confirmed second‐stage T. b. gambiense infection with trypanosomes detected in CSF, or trypanosomes detected in blood or lymph nodes with > 5 WBC/μL in CSF Exclusion criteria: pregnancy; residency outside area; body weight under 10 kg; history of treatment for trypanosomiasis in the previous 2 years Diagnosis and follow‐up methods: double centrifugation of cerebrospinal fluid (CSF); quantitative buffy coat (QBC) technique in blood |
|
| Interventions | 1. Melarsoprol‐nifurtimox: melarsoprol given intravenously, 1.8 mg/kg/day for 10 days; nifurtimox given orally, 15 mg/kg/day, every 8 h for 10 days 2. Melarsoprol‐eflornithine: melarsoprol given intravenously, 1.8 mg/kg/day for 10 days; eflornithine given intravenously, 400 mg/kg/day, every 6 h for 7 days 3. Nifurtimox‐eflornithine: respective doses as in groups 1 and 2 All participants were pretreated with albendazole, those with positive malaria diagnosis were treated with Fansidar (sulfadoxine and pyrimethamine), and those positive for microfilariae were treated with ivermectin Melarsoprol‐treated participants received oral prednisolone (1 mg/kg/day for 5 days + 0.5 mg/kg/day) until treatment complete |
|
| Outcomes | 1. Cure rates 2. Adverse events | |
| Notes | Location: Arua District, Uganda Setting: trypanosomiasis centre Source of funding: MSF and Embassy of France in Uganda |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Electronic block randomisation |
| Allocation concealment (selection bias) | Low risk | Sealed, numbered opaque envelopes |
| Blinding (performance bias and detection bias) All outcomes | High risk | Not feasible ‐ explicitly stated as such |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT and Per protocol analysis done, adequate (>90% participants analysed). |
Priotto 2009.
| Methods | Multi‐center, randomized, open label, non‐inferiority trial Generation of allocation sequence:randomization list in blocks of 10 was electronically generated at the study headquarters Allocation concealment: randomization list was concealed from the field team Blinding: not feasible as different modes of administration inclusion of all randomized participants: ITT and per protocol analysis done, (> 90% of all participants randomized into the trial were included in the analysis) Enrolment started August 2003 and completed June 2008 |
|
| Participants | Number randomized: 287 Inclusion criteria: over 15 yrs Exclusion criteria: severe comorbidities, haemoglobin <5 g/dL, inability to complete 18 months follow up Diagnosis and follow‐up method: double centrifugation of CSF, QBC technique in blood, latex IgM in CSF |
|
| Interventions | 1) Eflornithine, IV (slow infusion), 400 mg/kg/day (100 mg kg every 6 hrs) for 14 days 2) Eflornithine + nifurtimox; N = oral; E = IV (slow infusion); E = 400 mg/kg/day, every 12 hrs for 7 days; N = 15 mg/kg, every 8 hrs for 10 days. Malaria + patients were treated with artemether‐lumefantrine |
|
| Outcomes | Cure rates, death, relapse, adverse events. | |
| Notes | Locations: Nkayi (Republic of Congo); Isangi, Dipumba, Katanda (Democratic Republic of Congo). Source of funding: MSF, DNDi |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | randomized through block randomization in blocks of ten |
| Allocation concealment (selection bias) | Low risk | Adequate ‐ numbered non‐transparent envelopes were used |
| Blinding (performance bias and detection bias) All outcomes | High risk | a blinded design was not acceptable because of differences in administration |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | trial profile included and analysis per protocol, ITT and safety analysis done, 94.1% participants included |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| APTED 1957 | Not randomized |
| Balasegaram 2006 | Retrospective analysis |
| Burri 1995 | Not a comparative study |
| Butler 1957 | Not a comparative study |
| Ceccaldi 1953 | Not a comparative study |
| Chappuis 2005 | Not a prospective trial |
| Eozenou 1989 | Not a comparative study |
| Janssens 1977 | Not randomized |
| Khonde 1997 | Not a comparative study |
| Moens 1984 | Not a comparative study |
| Mpia 2002 | Not a comparative study |
| Ogada 1973 | Retrospective analysis |
| Pepin 1985 | Retrospective analysis |
| Pepin 1989 | Not a comparative study |
| Schmid 2005 | Not a comparative study |
| Van Nieuwenhove 1985 | Not a comparative study |
Characteristics of ongoing studies [ordered by study ID]
DNDi 2012.
| Trial name or title | Pivotal Study of Fexinidazole for Human African Trypanosomiasis in Stage 2 |
| Methods | Allocation: Randomized, Endpoint Classification: Safety/Efficacy Study, Intervention Model: Parallel Assignment, Masking: Single Blind |
| Participants | 15 years old or more, male or female, with Karnofsky index>50, with parasitologically confirmed late‐stage African trypanosomiasis infection with T. b. gambiense in the blood and/or lymph and/or CSF, or WBC >20/µL detected in the CSF to document stage 2 infection. |
| Interventions | Fexinidazole Compared to Nifurtimox‐Eflornithine Combination Therapy |
| Outcomes | success or failure at 18 months follow‐up visit |
| Starting date | September 2012 |
| Contact information | Antoine Tarral (DNDi), Victor Kande, HAT National Control Program in DRC. |
| Notes |
DNDi 2012b.
| Trial name or title | Human African Trypanosomiasis: First in Man Clinical Trial of a New Medicinal Product, the SCYX‐7158 |
| Methods | Allocation: Randomized, Endpoint Classification: Safety Study, Masking: Double Blind (Subject, Caregiver, Investigator, Outcomes Assessor) |
| Participants | Male healthy volunteers 18 to 45 years of age, of sub‐Saharan African origins. |
| Interventions | SCYX‐7158 compared to placebo |
| Outcomes | Safety, Tolerability, Pharmacokinetics and Pharmacodynamics |
| Starting date | February 2012 |
| Contact information | Antoine Tarral (DNDi), Lionel Hovsepian (SGS Aster, Paris). |
| Notes |
Differences between protocol and review
The title was changed to "Chemotherapy for second‐stage Human African trypanosomiasis".
A. Kennedy joined the review team after the protocol was published.
We deviated from the protocol as follows: updated and modified the Background section; reduced the number and slightly modified the outcome measures; modified the list of adverse events to include skin reactions and infections.
As the trials included different treatments, we did not perform meta‐analysis, subgroup analysis and investigation of heterogeneity, or sensitivity analysis.
Contributions of authors
V Lutje developed and drafted the protocol; J Seixas provided comments and reviewed it. All three authors contributed to data extraction, A Kennedy assessed risk of bias, V Lutje run the literature searches, entered data into Review Manager 5 and prepared the first draft of the review, all three authors contributed to revisions and to the final version.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
External sources
Department for International Development (DFID), UK.
Declarations of interest
None known.
Unchanged
References
References to studies included in this review
Bisser 2007 {published data only}
- Bisser S, N'Siesi FX, Lejon V, Preux PM, Nieuwenhove S, Miaka Mia Bilenge C, et al. Equivalence trial of melarsoprol and nifurtimox monotherapy and combination therapy for the treatment of second‐stage Trypanosoma brucei gambiense sleeping sickness. Journal of Infectious Diseases 2007; Vol. 195, issue 3:322‐9. [DOI] [PubMed]
Burri 2000 {published data only}
- Burri C, Nkunku S, Merolle A, Smith T, Blum J, Brun R. Efficacy of new, concise schedule for melarsoprol in treatment of sleeping sickness caused by Trypanosoma brucei gambiense: a randomised trial. Lancet 2000; Vol. 355, issue 9213:1419‐25. [DOI] [PubMed]
- Schmid C, Nkunku S, Merolle A, Vounatsou P, Burri C. Efficacy of 10‐day melarsoprol schedule 2 years after treatment for late‐stage gambiense sleeping sickness. Lancet 2004;364(9436):789‐90. [DOI] [PubMed] [Google Scholar]
Lejon 2003 {published data only}
- Legros D, Erphas O, Priotto G, Hutin Y, Mbulamberi DB, Gastellu Etchegorry M, et al. [Essai clinique randomise ouvert comparant le melarsoprol a la pentamidine pour le traitment des patients souffrant de trypanosomiase a Trypanosoma brucei gambiense en stade 2 precoce en Ouganda]. Medecine Tropicale 2001;61(3):278. [Google Scholar]
- Lejon V, Legros D, Savignoni A, Etchegorry MG, Mbulamberi D, Buscher P. Neuro‐inflammatory risk factors for treatment failure in "early second stage" sleeping sickness patients treated with pentamidine. Journal of Neuroimmunology 2003; Vol. 144, issue 1‐2:132‐8. [DOI] [PubMed]
Na‐Bangchang 2004 {published data only}
Pepin 1989a {published data only}
Pepin 2000 {published data only}
- Pepin J, Khonde N, Maiso F, Doua F, Jaffar S, Ngampo S, et al. Short‐course eflornithine in Gambian trypanosomiasis: a multicentre randomized controlled trial. Bulletin of the World Health Organization 2000; Vol. 78, issue 11:1284‐95. [PMC free article] [PubMed]
Pepin 2006 {published data only}
Priotto 2006 {published data only}
- Priotto G, Fogg C, Balasegaram M, Erphas O, Louga A, Checchi F, et al. Three Drug Combinations for Late‐Stage Trypanosoma brucei gambiense Sleeping Sickness: A Randomized Clinical Trial in Uganda. PLoS Clinical Trials 2006; Vol. 1, issue 8:e39. [DOI] [PMC free article] [PubMed]
Priotto 2009 {published data only}
- Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, et al. Nifurtimox‐eflornithine combination therapy for second‐stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non‐inferiority trial. Lancet 2009;374(9683):56‐64. [DOI] [PubMed] [Google Scholar]
- Priotto G, Kasparian S, Ngouama D, Ghorashian S, Arnold U, Ghabri S, et al. Nifurtimox‐eflornithine combination therapy for second‐stage Trypanosoma brucei gambiense sleeping sickness: a randomized clinical trial in Congo. Clinical infectious diseases 2007;45(11):1435‐42. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
APTED 1957 {published data only}
Balasegaram 2006 {published data only}
- Balasegaram M, Harris S, Checchi F, Ghorashian S, Hamel C, Karunakara U. Melarsoprol versus eflornithine for treating late‐stage Gambian trypanosomiasis in the Republic of the Congo. Bulletin of the World Health Organization 2006; Vol. 84, issue 10:783‐91. [DOI] [PMC free article] [PubMed]
Burri 1995 {published data only}
- Burri C, Blum J, Brun R. Alternative application of melarsoprol for treatment of T. B. gambiense sleeping sickness. Preliminary results. Annales de la Société Belge de Médecine Tropicale 1995; Vol. 75, issue 1:65‐71. [PubMed]
Butler 1957 {published data only}
Ceccaldi 1953 {published data only}
- Ceccaldi J. [New contribution on arsobal (R.P. 3854) therapy of trypanosomiasis; its effectiveness in former trypanosomiasis patients, with meningoencephalitis, resistant to trypanocides customarily used.]. Bulletin de la Societe de Pathologie Exotique et de ses Filiales 1953; Vol. 46, issue 1:95‐121. [PubMed]
Chappuis 2005 {published data only}
Eozenou 1989 {published data only}
- Eozenou P, Jannin J, Ngampo S, Carme B, Tell GP, Schechter PJ. [A trial treatment with eflornithine of trypanosomiasis caused by Trypanosoma brucei gambiense in the Peoples Republic of the Congo]. Medecine Tropicale (Marseilles) 1989; Vol. 49, issue 2:149‐54. [PubMed]
Janssens 1977 {published data only}
- Janssens PG, Muynck A. Clinical trials with "nifurtimox" in African trypanosomiasis. Annales de la Société Belge de Médecine Tropicale 1977; Vol. 57, issue 4‐5:475‐80. [PubMed]
Khonde 1997 {published data only}
Moens 1984 {published data only}
- Moens F, Wilde M, Ngato K. [Clinical trial of nifurtimox in human African trypanosomiasis]. Annales de la Société Belge de Médecine Tropicale 1984; Vol. 64, issue 1:37‐43. [PubMed]
Mpia 2002 {published data only}
Ogada 1973 {published data only}
Pepin 1985 {published data only}
- Pepin J, Tetreault L, Gervais C. [The use of oral corticosteroids in the treatment of human African trypanosomiasis: a retrospective survey in Nioki, Zaire]. Annales de la Société Belge de Médecine Tropicale 1985; Vol. 65, issue 1:17‐29. [PubMed]
Pepin 1989 {published data only}
- Pepin J, Milord F, Mpia B, Meurice F, Ethier L, DeGroof D, et al. An open clinical trial of nifurtimox for arseno‐resistant Trypanosoma brucei gambiense sleeping sickness in central Zaire. Transactions of the Royal Society of Tropical Medicine and Hygiene 1989; Vol. 83, issue 4:514‐7. [DOI] [PubMed]
Schmid 2005 {published data only}
- Schmid C, Richer M, Bilenge CM, Josenando T, Chappuis F, Manthelot CR, et al. Effectiveness of a 10‐day melarsoprol schedule for the treatment of late‐stage human African trypanosomiasis: confirmation from a multinational study (IMPAMEL II). Journal of Infectious Diseases 2005; Vol. 191, issue 11:1922‐31. [DOI] [PubMed]
Van Nieuwenhove 1985 {published data only}
- Nieuwenhove S, Schechter PJ, Declercq J, Bone G, Burke J, Sjoerdsma A. Treatment of gambiense sleeping sickness in the Sudan with oral DFMO (DL‐alpha‐difluoromethylornithine), an inhibitor of ornithine decarboxylase; first field trial. Transactions of the Royal Society of Tropical Medicine and Hygiene 1985; Vol. 79, issue 5:692‐8. [DOI] [PubMed]
References to ongoing studies
DNDi 2012 {published and unpublished data}
- Pivotal Study of Fexinidazole for Human African Trypanosomiasis in Stage 2. Ongoing study September 2012.
DNDi 2012b {published and unpublished data}
- Human African Trypanosomiasis: First in Man Clinical Trial of a New Medicinal Product, the SCYX‐7158. Ongoing study February 2012.
Additional references
Alirol 2013
- Alirol E, Schrumpf D, Amici Heradi J, Riedel A, Patoul C, Quere M, et al. Nifurtimox‐eflornithine combination therapy for second‐stage gambiense human African trypanosomiasis: medecins sans frontieres experience in the democratic republic of the congo. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2013;56(2):195‐203. [PUBMED: 23074318] [DOI] [PubMed] [Google Scholar]
Balasegaram 2009
- Balasegaram M, Young H, Chappuis F, Priotto G, Raguenaud ME, Checchi F. Effectiveness of melarsoprol and eflornithine as first‐line regimens for gambiense sleeping sickness in nine Medecins Sans Frontieres programmes. Transactions of the Royal Society of Tropical Medicine and Hygiene 2009;103(3):280‐90. [DOI] [PubMed] [Google Scholar]
Bouteille 2003
- Bouteille B, Oukem O, Bisser S, Dumas M. Treatment perspectives for human African trypanosomiasis. Fundamental and Clinical Pharmacology 2003;17(2):171‐81. [DOI] [PubMed] [Google Scholar]
Brun 2010
- Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet 2010;375(9709):148‐59. [DOI] [PubMed] [Google Scholar]
Burri 2003
- Burri C, Brun R. Eflornithine for the treatment of human African trypanosomiasis. Parasitology Research 2003;90 Suppl 1:49‐52. [DOI] [PubMed] [Google Scholar]
Cattand 2001
- Cattand P, Jannin J, Lucas P. Sleeping sickness surveillance: an essential step toward elimination. Tropical Medicine and International Health 2001;6(5):348‐61. [DOI] [PubMed] [Google Scholar]
Chappuis 2007
- Chappuis F. Melarsoprol‐free drug combinations for second‐stage Gambian sleeping sickness: the way to go. Clinical Infectious Diseases 2007;45(11):1443‐5. [PUBMED: 17990226] [DOI] [PubMed] [Google Scholar]
DNDi 2008
- DNDi. Nifurtimox‐Eflornithine Combination Therapy: An Improved Treatment for Sleeping Sickness. DNDi Newsletter November 2008; Vol. http://www.dndi.org/newsletters/17/10_1.htm.
IMPAMEL III
- IMPAMEL III. http://www.swisstph.ch/services/medicines‐research/pharmaceutical‐medicine/experience/impamel‐iii.html.
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Available from www.cochrane‐handbook.org: The Cochrane Collaboration, 2011. [Google Scholar]
Legros 2002
- Legros D, Ollivier G, Gastellu‐Etchegorry M, Paquet C, Burri C, Jannin J, et al. Treatment of human African trypanosomiasis‐present situation and needs for research and development. Lancet Infectious Diseases 2002;2(7):437‐40. [DOI] [PubMed] [Google Scholar]
Lejon 2005
- Lejon V, Buscher P. Review Article: cerebrospinal fluid in human African trypanosomiasis: a key to diagnosis, therapeutic decision and post‐treatment follow‐up. Tropical Medicine and International Health 2005;10(5):395‐403. [DOI] [PubMed] [Google Scholar]
Nok 2003
- Nok AJ. Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human African trypanosomiasis. Parasitology Research 2003;90(1):71‐9. [DOI] [PubMed] [Google Scholar]
Pepin 1994
- Pepin J, Milord F. The treatment of human African trypanosomiasis. Advances in Parasitology 1994;33:1‐47. [DOI] [PubMed] [Google Scholar]
Pepin 2001
- Pepin J, Meda HA. The epidemiology and control of human African trypanosomiasis. Advances in Parasitology 2001;49:71‐132. [DOI] [PubMed] [Google Scholar]
Review Manager 5 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2008.
Schmid 2012
- Schmid C, Kuemmerle A, Blum J, Ghabri S, Kande V, Mutombo W, et al. In‐Hospital Safety in Field Conditions of Nifurtimox Eflornithine Combination Therapy (NECT) for T. b. gambiense Sleeping Sickness. PLoS neglected tropical diseases 2012;6(11):e1920. [PUBMED: 23209861] [DOI] [PMC free article] [PubMed] [Google Scholar]
Seed 2001
- Seed JR, Boykin DW. Chemotherapy of African trypanosomiasis. In: Black SJ, Seed JR editor(s). The African trypanosomes. Series: World Class Parasites, Vol. 1. Boston: Kluwer Academic Publishers, 2001:65‐78. [Google Scholar]
Seixas 2005
- Seixas J. Investigations on the encephalopathic syndrome during melarsoprol treatment of Human African Trypanosomiasis [PhD thesis]. Lisbon: Instituto de Higiene e Medicina Tropical, 2005. [Google Scholar]
Simarro 2008
- Simarro PP, Jannin J, Cattand P. Eliminating human African trypanosomiasis: where do we stand and what comes next?. PLoS Medicine 2008;5(2):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stich 2002
- Stich A, Abel PM, Krishna S. Human African trypanosomiasis. BMJ 2002;325(7357):203‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stich 2003
- Stich A, Barret MP, Krishna S. Waking up to sleeping sickness. Trends in Parasitology 2003;19(5):195‐7. [DOI] [PubMed] [Google Scholar]
Van Nieuwenhove 1992
- Nieuwenhove S. Advances in sleeping sickness therapy. Annales de la Société Belge de Médecine Tropicale 1992;72 Suppl 1:39‐51. [PubMed] [Google Scholar]
WHO 1998
- Control and surveillance of African trypanosomiasis. Report of a WHO Expert Committee. World Health Organization Technical Report Series 1998; Vol. 881, issue I‐VI:1‐114. [PubMed]
WHO 2004
- World Health Organization. Recommendations of the Informal consultation on Issue for Clinical Product development for human African Trypanosomiasis. http://www.who.int/neglected_diseases/mediacentre/Final%20Clinical_products_for_trypanosomiasis%20November%2007.pdf 2004.
WHO 2006
- WHO. Human African trypanosomiasis (sleeping sickness): epidemiological update. Releve epidemiologique hebdomadaire / Section d'hygiene du Secretariat de la Societe des Nations = Weekly epidemiological record / Health Section of the Secretariat of the League of Nations 2006;81(8):71‐80. [PubMed] [Google Scholar]
WHO 2009
- WHO Secretariat. Progress Report [Control of African trypanosomiasis]. (http://who.int/gb/ebwha/pdf_files/EB126/B126_38‐en.pdf).
WHO 2010
- WHO. WHO model list of essential medicines, 16th list (updated). March 2010; Vol. http://www.who.int/medicines/publications/essentialmedicines/Updated_sixteenth_adult_list_en.pdf.