

Abstract
Background
Cerebral malaria is a common complication of Plasmodium falciparum infection, and kills over a million people every year. People with cerebral malaria become unconscious, and often have protracted convulsions. It is unclear whether giving anticonvulsant drugs routinely to people with cerebral malaria will improve the outcome of treatment and prevent death.
Objectives
To evaluate the effect of routine anticonvulsant drugs in people with cerebral malaria.
Search methods
We searched the Cochrane Infectious Diseases Group Specialized Register (May 2004), CENTRAL (The Cochrane Library Issue 2, 2004), MEDLINE (1966 to May 2004), EMBASE (1988 to May 2004), LILACS (1982 to May 200), Science Citation Index (May 2004), African Index Medicus (1999), reference lists of articles, and research organizations. We also contacted the authors for additional information.
Selection criteria
Randomized and quasi‐randomized controlled trials of people with cerebral malaria. The trials compared anticonvulsant drugs started on admission to hospital with no anticonvulsant drug or placebo.
Data collection and analysis
Two reviewers independently extracted data from those trials eligible for inclusion. We assessed the risk of bias in the included trials by considering allocation sequence, concealment of allocation, blinding, and inclusion of all randomized participants. We used Review Manager (version 4.1) for the meta‐analysis and also explored possible sources of heterogeneity.
Main results
Three trials with a total of 573 participants met the inclusion criteria. These trials all compared phenobarbitone with placebo or no treatment. In the two trials with adequate allocation concealment, death was more common in the anticonvulsant group (Risk Ratio 2.0; 95% confidence interval 1.20 to 3.33; fixed effect model). In all three trials, phenobarbitone compared with placebo or no treatment was associated with fewer convulsions (Risk Ratio 0.30; 95% confidence interval 0.19 to 0.45; fixed effect model).
Authors' conclusions
Routine phenobarbitone in cerebral malaria is associated with fewer convulsions but possibly more deaths. Further trials with adequate design, more participants, and different doses of anticonvulsant drugs are needed.
23 April 2019
No update planned
Other
This is not a current question.
Plain language summary
Routine anticonvulsants for treating cerebral malaria
Plain language summary pending.
Background
There are an estimated 1.5 to 2.7 million deaths related to malaria each year (WHO 1997), and a large proportion of these deaths are preceded by cerebral malaria. About 10 to 30% of people with cerebral malaria die even when treated with effective antimalarial drugs (WHO 1990). Five to ten per cent of survivors have long term neurological disability, including epilepsy, paralysis, impaired hearing, cortical blindness, speech disorders, and learning disabilities (Warrell 1982, Brewster 1990, Bondi 1992, Meremikwu 1997).
People with cerebral malaria become unconscious (cannot be aroused even by pain) for several hours, and a high proportion have convulsions (Waller 1995, Waruiru 1996). The clinical manifestation of cerebral malaria is thought to occur because the majority of parasitized red blood cells occupy small blood vessels in the brain, which affects perfusion and metabolism in surrounding tissues (Pongparatn 1991, Pasvol 1995). Malaria reduces blood glucose levels in some individuals, and this can also cause convulsions and unconsciousness. Febrile convulsions can also occur in feverish children with malaria but, unlike in cerebral malaria, these are usually brief, and patients regain consciousness a few minutes after the convulsions (Molyneux 1995).
Convulsions due to cerebral malaria tend to be protracted, and can recur several times during the same illness. Observational studies have noted that cerebral malaria patients who have protracted or repeated convulsions have worse outcomes (Molyneux 1989, Jaffar 1997). Some researchers have also shown that people with cerebral malaria may have convulsions that are not obvious ('subtle convulsions'). These patients were shown to have abnormal electrical activity in the brain similar to those observed in convulsing patients, and may suffer from similar adverse outcomes (Molyneux 1995, Crawley 2001). Other research has shown that African children dying from cerebral malaria had deepening unconsciousness and convulsions, and raised intracranial pressure, probably resulting from brain oedema (Newton 1991, Newton 1994). It may be that convulsions in cerebral malaria contribute to more deaths by making brain anoxia (lack of oxygen) and oedema (swelling) worse, and raising the intracranial pressure.
The World Health Organization (WHO) recommends that people with cerebral malaria should be treated with antimalarial drug injections. Where necessary, they should also be treated for associated problems such as severe anaemia, using blood transfusion; low blood glucose, using glucose infusion; and convulsions, using anticonvulsant drugs (WHO 2000a).
In recent years, doctors have considered ways of further reducing the possible adverse consequences of convulsions in cerebral malaria, and have suggested that anticonvulsant drugs should be routinely given to all people with cerebral malaria whether or not they are convulsing at the time of admission (White 1995). This recommendation is based on the reasonable assumption that convulsions may themselves contribute to brain damage or increase the risk of death. Giving anticonvulsants to all cerebral malaria patients has the advantage of controlling unrecognized (as well as obvious) convulsions, and preventing further convulsions that cannot be reliably predicted. If, however, convulsions do not themselves damage the brain or make the prognosis worse, but are merely marker of a more severely affected brain, then preventing convulsions with routine anticonvulsant drugs may not improve the person's chance of surviving or escaping brain damage.
Anticonvulsant drugs are known to cause some adverse effects. For instance, phenobarbitone, phenytoin sodium, and diazepam may cause respiratory depression (Parfitt 1999). This may even lead to fatal respiratory failure in unconscious patients, especially where there are inadequate facilities for respiratory support. Although these three drugs are recommended for treating people who are convulsing (Rylance 1990, Winstanley 1992, Parfitt 1999, WHO 2000a), giving them routinely to all people with cerebral malaria could not be justified if they offer no additional benefits.
The uncertainty surrounding the routine use of anticonvulsant drugs in cerebral malaria has led us to prepare this systematic review, in which we consider whether all people with cerebral malaria should be given anticonvulsant drugs, whether or not they show visible signs of convulsing.
Objectives
To evaluate the effect of routine anticonvulsant drugs in people with cerebral malaria.
Methods
Criteria for considering studies for this review
Types of studies
Randomized and quasi‐randomized controlled trials.
Types of participants
People with cerebral malaria who fulfil all the following criteria:
coma defined as non‐localizing response to pain corresponding to either: (i) Blantyre coma score less than 4 or (ii) Glasgow coma score less than 11;
presence of asexual Plasmodium falciparum in peripheral blood;
normal cerebrospinal fluid.
Blantyre coma scale (Molyneux 1989) was modified from the widely used Glasgow coma scale (Teasdale 1974) to become more applicable to young children (WHO 2000a, WHO 2000b). The Blantyre coma scale has a range of 0 to 5 scores, while the Glasgow coma scale ranges from 3 to 14.
Types of interventions
Intervention: Anticonvulsant drugs started immediately after diagnosis and randomization.
Control: No routine anticonvulsant drugs, with or without placebo (anticonvulsants only given to treat convulsions according to clinical indications).
Types of outcome measures
Primary
Death within 6 months post‐randomization.
Proportion of people experiencing convulsions within 4 weeks of randomization.
Secondary
Proportion of people still in a coma by day 7 post‐randomization.
Deficits in cognitive function (measured with standard psychometric test).
Long term epilepsy (proportion still experiencing seizures by one year post‐randomization or taking antiepileptic drugs after recovery from cerebral malaria).
Proportion of people with a physical handicap at 12 months (measured with standard physical functioning scale)
Search methods for identification of studies
We attempted to identify all relevant studies regardless of language or publication status (published, unpublished, in press, and in progress).
We used the following topic search terms for all searching trial registers and databases: anticonvulsive agent; anticonvulsant; antiepileptic; seizure; seizure, epilepsy and convulsion; convulsion; malaria; brain malaria; and cerebral malaria. The full search strategy is available in Appendix 1.
We searched the Cochrane Infectious Diseases Group (CIDG) Specialized Register up to May 2004. Full details of the CIDG methods and the journals hand searched are published in The Cochrane Library in the section on Collaborative Review Groups.
We searched The Cochrane Central Register of Controlled Trials (CENTRAL), published in The Cochrane Library (Issue 2, 2004). This contains mainly reference information to randomized controlled trials and controlled clinical trials in health care.
We also searched the following electronic databases using search terms in combination with the search strategy for identifying trials developed by The Cochrane Collaboration and detailed in the Cochrane Reviewers' Handbook (Clarke 2000):MEDLINE (1966 to May 2004); EMBASE (1988 to May 2004); LILACS (1982 to May 2004); Science Citation Index (May 2004); and African Index Medicus (1999).
We contacted the Medical Research Council, The Gambia; Kenya Medical Research Institute; and the Wellcome Tropical Research Groups Thailand/Vietnam/Oxford for information on published, unpublished, or ongoing trials.
External referees checked the search strategy and helped to identify additional unpublished, ongoing, and planned trials. We also checked existing related reviews, and the citations of all the trials identified by the above methods. Where necessary, we contacted authors for additional information and clarification.
Data collection and analysis
We independently applied the inclusion criteria to all identified trial reports. From those trials eligible for inclusion, we independently extracted data on the methods, types of participants, interventions, and outcomes. We compared the two sets of extracted data and discussed them to ensure accuracy and completeness. We planned to seek the opinion of an editor in the Cochrane Infectious Diseases Group if we disagreed on any points. Where necessary, we requested additional data from the trial authors. Only one author (Crawley 2000) responded, providing additional information on generation of allocation sequence, and some outcome measures.
We made comparisons between the groups receiving anticonvulsant drug(s) and the control groups. While our aim was to do intention‐to‐treat analysis, two of the trials (White 1988, Kochar 1997) failed to clearly specify how many people were lost to follow up. We therefore did not have enough data to apply intention‐to‐treat analysis. We assessed heterogeneity by visual examination of forest plot and Chi‐square test for heterogeneity. Where we decided to proceed with meta‐analysis despite heterogeneity, we used both a fixed and random effect model. We performed a sensitivity analysis of the trials with adequate allocation concealment (White 1988, Crawley 2000) with exclusion of the inadequately concealed trial (Kochar 1997). We calculated Risk Ratio with 95% confidence interval for all the outcomes since all were dichotomous data.
Results
Description of studies
Eligibility
Three trials met the inclusion criteria (White 1988, Kochar 1997, Crawley 2000). One trial published data in two separate reports (Kochar 1997).
Participants
A total of 573 participants were recruited into the 3 trials: 340 children in the Kenyan study (Crawley 2000), 185 adults in the Indian study (Kochar 1997), and 48 (mostly adults) in the Thai study (White 1988). More details are given in the 'Characteristics of included studies'.
Interventions
In all three trials the intervention group received a single dose of parenteral phenobarbitone. Doses varied from 3.5 mg/kg (maximum 200 g; White 1988) to 10 mg/kg (maximum 400 mg; Kochar 1997) and 20 mg/kg body weight (Crawley 2000).
The phenobarbitone was given intramuscularly to all the participants in two trials (White 1988, Kochar 1997). In the third, the first 23 (6.8%) participants received intravenous phenobarbitone while the rest received intramuscular phenobarbitone (Crawley 2000). Two trials compared phenobarbitone with placebo (Crawley 2000, White 1988), while the third trial had a no treatment control group (Kochar 1997).
Outcome measures
All three trials reported deaths and number of people experiencing convulsions within four weeks of hospitalization. Crawley 2000 reported patients who experienced three or more episodes of convulsions. Only one trial reported neurological problems, including visual impairment, physical handicap, and developmental delay (Crawley 2000). None provided data on long term epilepsy.
Risk of bias in included studies
Crawley 2000 used a sequentially numbered randomization register to generate the allocation sequence. The method used to generate allocation sequence was unclear in White 1988, and suboptimal in Kochar 1997, where the participants were allocated to receive either phenobarbitone or nothing on alternate days.
Allocation was concealed using sealed envelopes in White 1988 and by using a central code in Crawley 2000; the staff and participants were blinded in both. Allocation was not concealed in Kochar 1997 and the clinicians were not blind to the allocated group.
In Crawley 2000, 32 (9.4%) participants were either excluded after randomization (11) or lost to follow up (21). The authors of the other two trials did not state categorically that no participants were lost follow up. It is possible that some losses to follow up were not reported. The authors performed intention‐to‐treat analysis on their primary outcome of convulsion after randomization.
Effects of interventions
Death
We found statistically significant heterogeneity for this outcome of death within six months post‐randomization (Chi‐square = 9.91; p = 0.0071, Analysis 1.1). The overall fixed and random effects estimates of the Risk Ratio were 1.16 (95% confidence interval 0.85 to 1.58) and 1.30 (95% confidence interval 0.58 to 2.88) respectively. Both estimates indicate no statistically significant difference.
1.1. Analysis.
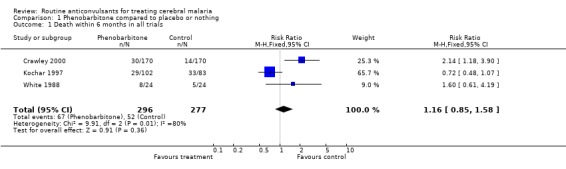
Comparison 1 Phenobarbitone compared to placebo or nothing, Outcome 1 Death within 6 months in all trials.
The scope for investigating sources of heterogeneity is limited because only three trials were included. We carried out a sensitivity analysis of the two studies with adequate allocation concealment (Crawley 2000, White 1988) and found no detectable statistically significant heterogeneity (Chi‐square 0.26; p = 0.61, Analysis 1.2). The overall fixed and random effect estimates of the Risk Ratio of death of 2.00 (95% confidence interval 1.20 to 3.33) and 1.98 (95% confidence interval 1.19 to 3.28) respectively, showed that in the adequately concealed trials, the risk of death was increased if phenobarbitone was used.
1.2. Analysis.
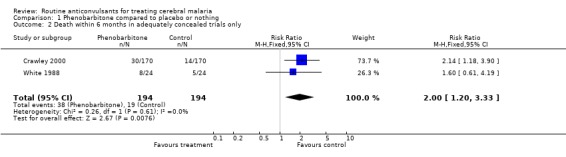
Comparison 1 Phenobarbitone compared to placebo or nothing, Outcome 2 Death within 6 months in adequately concealed trials only.
Convulsions
We found no heterogeneity for this outcome of proportion of people experiencing convulsions within four weeks of randomization. The pooled estimate shows that participants treated with phenobarbitone were statistically significantly less likely to have convulsions within four weeks of going to hospital (Risk Ratio 0.30; 95% confidence interval 0.19 to 0.45; fixed effect model; Analysis 1.3). Crawley 2000 also reported that the placebo group was also more likely to have prolonged convulsions (23/170) than the phenobarbitone group (9/170). The other trials did not provide such detailed information on the pattern of convulsions.
1.3. Analysis.
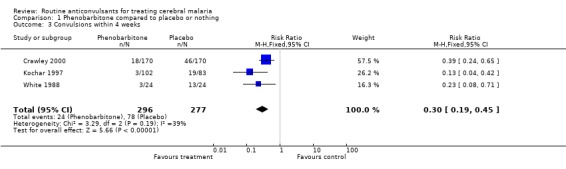
Comparison 1 Phenobarbitone compared to placebo or nothing, Outcome 3 Convulsions within 4 weeks.
Other outcomes
None of the trials reported a standard assessment of physical handicap at 12 months follow up. However one trial (Crawley 2000), reported that the risk of neurological problems (described by authors as "permanent sequelae") up to the third month of follow up was not statistically significantly different between the phenobarbitone (9/170) and the placebo (15/170) groups (Risk Ratio 0.60; 95% confidence interval 0.27 to 1.33, Analysis 1.4).
1.4. Analysis.
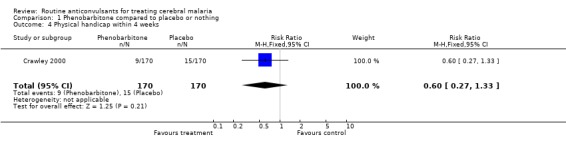
Comparison 1 Phenobarbitone compared to placebo or nothing, Outcome 4 Physical handicap within 4 weeks.
None of the trials provided data on coma by day 7, deficits in cognitive function, or long term epilepsy. Additional information supplied by Crawley 2000 confirmed that none of the people in that trial was still in coma by day 7.
Discussion
Of the three trials included in this review, two used adequate methods of allocation concealment and were double blinded (White 1988, Crawley 2000). The third trial used a quasi method of randomization, had no allocation concealment, and was not blinded (Kochar 1997). Although no losses to follow up were described in two trials (Kochar 1997, White 1988), there were no categorical statements that all the randomized participants were followed up; this left us uncertain as to whether the analyses are truly intention‐to‐treat. Crawley 2000 reported the loss of 5% of the randomized participants to follow up. The sample sizes of the treatment (n = 102) and control (n = 83) groups in Kochar 1997 differ remarkably, suggestive of possible bias in allocation or follow up of participants. In summary, two of the three included trials were of good methodological quality (White 1988, Crawley 2000), while the third was of poor methodological quality and prone to greater bias (Kochar 1997).
We found statistically significant heterogeneity for our primary outcome of death within six months post‐randomization. This is an indication that the data sets from the respective trials differ so much that combination of the data sets in meta‐analysis would give misleading results. We tried to explore the source of this heterogeneity, but found ourselves limited because only three trials were eligible for inclusion in this review. The differences in the methodological quality of trials could explain this heterogeneity, since when we analysed the two trials of good methodological quality, we found no statistically significant heterogeneity. However, given the low power of the test for heterogeneity and the small size of the trials, this does not necessarily mean that heterogeneity is absent. Pooled estimates suggest that mortality will be increased if people are treated with phenobarbitone compared to placebo. This is statistically significant for the subgroup of the two trials of good methodological quality and suggests that the risk of death doubles, with a number needed to harm of approximately 10.
Another potential source of heterogeneity is the dose of phenobarbitone. Crawley 2000 used the highest dose (20 mg/kg) and has the highest mortality rate. This is biologically plausible, as higher doses of phenobarbitone are more sedating and can cause depression of the respiratory centre. However, there was no clear trend among the three included trials that indicated mortality increases as the dose of phenobarbitone increases. Crawley 2000 also gave other anticonvulsants to participants who convulsed for longer than five minutes. Such participants first received intravenous diazepam (0.3 mg/kg/dose; maximum of two doses), and subsequently intramuscular paraldehyde (0.2 ml/kg), followed by phenytoin infusion (20 mg/kg) if convulsions persisted. A commentary on this study has raised a question on the possible effect of the additional doses of diazepam on the participants (Molyneux 2000). Kochar 1997 also gave participants additional diazepam and phenytoin to control convulsions while White 1988 used diazepam alone. It would therefore be difficult to attribute the observed heterogeneity to these additional anticonvulsant drugs alone.
The age of trial participants may have contributed to the heterogeneity. Crawley 2000 included young children while the other two trials included mainly adults. It is possible that younger children are more susceptible to the sedative effects of phenobarbitone and hence to respiratory depression and death. Extrapolating this observation to adult populations may be misleading given that the trial with the highest number of adults (Kochar 1997) was of poor methodological quality. Because only three trials were eligible for inclusion in this review, and because we only had access to aggregate level data, we were unable to explore the sources of heterogeneity further.
We found no heterogeneity for convulsions within four weeks of follow up, our second primary outcome. Phenobarbitone was associated with statistically significantly fewer convulsions during follow up. This effect is however outweighed by the possibility of a higher risk of mortality.
Crawley 2000 provided data on physical handicap; there was no statistically significant difference between the treatment and placebo groups. We found no data on the other outcomes that we had planned to assess in this review namely, coma by day 7; deficits in cognitive function; and long term epilepsy.
Authors' conclusions
Implications for practice.
Giving phenobarbitone routinely to people with cerebral malaria reduces the number of convulsions, but may increase the risk of death. Phenobarbitone should not be given routinely to people with cerebral malaria until there is more evidence on how it can be used without causing harm.
Implications for research.
Anticonvulsant drugs have potential for benefit. Larger clinical trials using lower doses of phenobarbitone or other anticonvulsant drugs, such as phenytoin, are needed to determine whether or not routine anticonvulsant drugs are beneficial in treating cerebral malaria. It is also necessary to consider the cost implications of any choice of anticonvulsant drugs used in these trials because many people in countries where falciparum malaria is endemic may be unable to afford expensive anticonvulsant drugs. In this sense, phenobarbitone has the advantage of being relatively cheap.
What's new
| Date | Event | Description |
|---|---|---|
| 9 November 2008 | Amended | Converted to new review format with minor editing. |
History
Protocol first published: Issue 2, 2000 Review first published: Issue 2, 2002
| Date | Event | Description |
|---|---|---|
| 21 May 2004 | New search has been performed | New studies sought but none found. |
| 26 May 2003 | Amended | Contact details modified. |
Acknowledgements
We gratefully acknowledge the very helpful contributions of Professor Malcom Molyneux, Professor Kevin Marsh, and Dr Jane Crawley. The reviewers have received technical assistance from both the Cochrane Infectious Diseases Group and the Cochrane Epilepsy Group.
Appendices
Appendix 1. Search methods: detailed search strategies
| MEDLINE (OVID) | EMBASE (OVID) |
| [1] anticonvulsants/ [2] anticonvulsant$.tw. [3] antiepileptic$.tw. [4] seizures/ [5] seizure$.tw. [6] convulsions/ [7] convulsion$.tw. [8] 1 or 2 or 3 or 4 or 5 or 6 or 7 [9] exp malaria/ [10] malaria, cerebral/ [11] malaria.tw. [12] 9 or 10 or 11 [13] 8 and 12 [14] randomized controlled trials/ [15] randomized‐controlled‐trial.pt. [16] controlled‐clinical‐trial.pt. [17] random allocation/ [18] double‐blind method/ [19] single‐blind method/ [20] 14 or 15 or 16 or 17 or 18 or 19 [21] exp clinical trials/ [22] clinical‐trial.pt. [23] (clin$ adj trial$).ti,ab. [24] ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ti,ab. [25] placebos/ [26] placebo$.ti,ab. [27] random$.ti,ab. [28] 21 or 22 or 23 or 24 or 25 or 26 or 27 [29] research design/ [30] comparative study/ [31] exp evaluation studies/ [32] follow‐up studies/ [33] prospective studies/ [34] (control$ or prospective$ or volunteer$).ti,ab. [35] 30 or 31 or 32 or 33 or 34 [36] 20 or 28 or 29 or 35 [37]13 and 36 [38] limit 37 to human | [1] anticonvulsive agent/ [2] anticonvulsant$.tw. [3] antiepileptic.tw. [4] seizure/ [5] "seizure, epilepsy and convulsion"/ [6] convulsion/ [7] seizure$.tw. [8] convulsion$.tw. [9] 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 [10] exp malaria/ [11] brain malaria/ [12] malaria.tw. [13] 10 or 11 or 12 [14] 9 and 13 [15] randomized controlled trial/ [16] controlled study/ [17] case control study/ [18] randomization/ [19] double blind procedure/ [20] single blind procedure/ [21] (control$ adj trial$).ti,ab. [22] 15 or 16 or 17 or 18 or 19 or 20 or 21 [23] exp clinical trials/ [24] exp multicenter study/ [25] (clin$ adj trial$).ti,ab. [26] (clin$ adj stud$).ti,ab. [27] ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ti,ab. [28] placebo/ [29] placebo$.ti,ab. [30] random$.ti,ab. [31] 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 [32] methodology/ [33] comparative study/ [34] evaluation/ [35] follow up/ [36] prospective study/ [37] (control$ or prospectiv$ or volunteer$).ti,ab. [38] 33 or 34 or 35 or 36 or 37 [39] 22 or 31 or 32 or 38 [40]14 and 39 [41] limit 40 to human |
Data and analyses
Comparison 1. Phenobarbitone compared to placebo or nothing.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Death within 6 months in all trials | 3 | 573 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.85, 1.58] |
| 2 Death within 6 months in adequately concealed trials only | 2 | 388 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [1.20, 3.33] |
| 3 Convulsions within 4 weeks | 3 | 573 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.19, 0.45] |
| 4 Physical handicap within 4 weeks | 1 | 340 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.27, 1.33] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Crawley 2000.
| Methods | Randomized Placebo controlled Parallel trial |
|
| Participants | Number screened: 440 Number included: 170 placebo; 170 intervention; 184 male; 156 female Inclusion criteria: age between 9 months and 3 years; unrousable coma (non‐localizing or Blantyre score ≤ 3); falciparum parasitaemia Exclusion criteria: afebrile epilepsy; significant neurodevelopmental problem; prior treatment with phenobarbitone or phenytoin in present illness; lumbar puncture revealed meningitis |
|
| Interventions | Intervention: intramuscular phenobarbitone; single dose 20 mg/kg = 0.1 ml/kg Control (placebo): 90% propylene glycol (vehicle of parenteral phenobarbitone) in similar ampoule (0.1 ml/kg) |
|
| Outcomes | 1. Death 2. Seizure 3. Coma recovery time 4. Neurological sequelae at discharge and at 3 months | |
| Notes | Study location: Kilifi, Kenya Losses to follow up: 17 Intention‐to‐treat analysis only for primary outcome |
|
Kochar 1997.
| Methods | Block randomization based on days of admission Inadequate concealment of allocation, no blinding Losses to follow up/post‐randomization exclusion not stated |
|
| Participants | Age: 14 to 74 years Intervention group: 102 Control group: 83 Inclusion criteria: Plasmodium falciparum in peripheral blood smear; coma for at least half an hour (Glasgow coma score ≥ 6); no generalized seizure in previous 2 hours; exclusion of other causes of fever with altered consciousness Exclusion criteria: diabetes; space‐occupying lesion; psychiatric illness; alcoholism; head injury; chronic renal failure |
|
| Interventions | Intervention: injection phenobarbitone 10 mg/kg body weight (not more than 400 mg) Control: nothing given |
|
| Outcomes | 1. Death 2. Seizure | |
| Notes | Study location: malaria epidemics in Bikaner, Rajastan, India | |
White 1988.
| Methods | Randomized Placebo controlled parallel trial Treatment concealed in sealed envelopes Losses to follow up/post‐randomization exclusion not stated |
|
| Participants | Number screened not given Number included: 24 intervention; 24 placebo; 40 male; 8 female Inclusion criteria: cerebral malaria (unrousable coma in falciparum malaria) Exclusion criteria: other causes of coma |
|
| Interventions | Intervention: intramuscular phenobarbitone (Gardenal Sodium: May & Baker); single dose of 200mg (1 ml ampoule) for participants > 60 kg weight; 3.5 mg/kg (or 0.0175 ml/kg) for < 60 kg weight Placebo: saline in identical ampoule (same dose in 'ml' as intervention) |
|
| Outcomes | 1. Death 2. Seizure 3. Coma recovery time | |
| Notes | Setting: Chantaburi and Kanchanaburi, Thailand | |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Kuile 1992 | Some participants (9/21) did not have cerebral malaria |
| Winstanley 1992 | Participants not randomized |
Contributions of authors
Martin Meremikwu raised the research question and wrote the first draft of the protocol. Subsequently both reviewers (Martin Meremikwu and Tony Marson) contributed to the preparation of the protocol, literature search, selection of trials, data extraction, analysis, and writing the text of the review.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK.
Department for International Development, UK.
External sources
Department for International Development, UK.
University of Calabar, Nigeria.
Declarations of interest
We certify that we have no affiliations with or involvement in any organization or entity with a direct financial interest in the subject matter of the review (eg, employment, consultancy, stock ownership, honoraria, expert testimony).
Unchanged
References
References to studies included in this review
Crawley 2000 {published data only}
- Crawley J, Waruiru C, Mithwani S, Mwangi I, Watkins W, Ouma D, et al. Effect of phenobarbital on seizure frequency and mortality in childhood cerebral malaria: a randomised, controlled intervention study. Lancet 2000;355:701‐6. [DOI] [PubMed] [Google Scholar]
Kochar 1997 {published data only}
- Kochar DK, Kumawat BL, Bajya HN, Chauhan S, Kochar SK, Agarwal RP. Prophylactic role of single dose phenobarbitone in preventing convulsions in cerebral malaria. Journal of Association of Physicians in India (JAPI) 1997;45(2):123‐4. [Google Scholar]
- Kochar DK, Shubhakaran, Kumewat BL, Kochar SK. Seizures in cerebral malaria (letter). Quarterly Journal of Medicine 1997;91:605‐7. [DOI] [PubMed] [Google Scholar]
White 1988 {published data only}
- White NJ, Looareesuwan S, Phillips RE, Chanthavanich P, Warrell DA. Single dose phenobarbitone prevents convulsions in cerebral malaria. Lancet 1988;2(8602):64‐6. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Kuile 1992 {published data only}
- Kuile F, Nosten F, Chongsuphajaisidhi T, Holloway P, Maelankirri L, White NJ. Absorption of intramuscular phenobarbitone in children with severe falciparum malaria. European Journal of Clinical Pharmacology 1992;42(1):107‐10. [DOI] [PubMed] [Google Scholar]
Winstanley 1992 {published data only}
- Winstanley PA, Newton CRJC, Pasvol G, Kirkham FJ, Mberu E, Peshu N, et al. Prophylactic phenobarbitone in young children with severe falciparum malaria: pharmokinetics and clinical effects. British Journal of Clinical Pharmacology 1992;33:149‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Bondi 1992
- Bondi FS. The incidence and outcome of neurological abnormalities in childhood cerebral malaria: a long‐term follow‐up of 62 survivors. Transactions of the Royal Society of Tropical Medicine and Hygiene 1992;86:17‐9. [DOI] [PubMed] [Google Scholar]
Brewster 1990
- Brewster DR, Kwiatkowski D, White NJ. Neurological sequelae of cerebral malaria in children. Lancet 1990;336:1039‐43. [DOI] [PubMed] [Google Scholar]
Clarke 2000
- Clarke M, Oxman AD, editors. Optimal search strategy. Cochrane Reviewers' Handbook 4.1 [updated June 2000]; Appendix 5c. In: The Cochrane Library [database on disk and CD‐ROM]. The Cochrane Collaboration. Oxford: Update Software; 2001, Issue 2.
Crawley 2001
- Crawley J, Smith S, Muthinji P, Marsh K, Kirkham F. Electroencephalographic and clinical features of cerebral malaria. Archives of Diseases in Childhood 2001;84(3):247‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaffar 1997
- Jaffar S, Hensbroeke MB, Palmer A, Schneider G, Greenwood B. Predictors of a fatal outcome following childhood cerebral malaria. American Journal of Tropical Medicine and Hygiene 1997;57:20‐4. [DOI] [PubMed] [Google Scholar]
Meremikwu 1997
- Meremikwu MM, Asindi AA, Ezedinachi ENU. The pattern of neurological sequelae of childhood cerebral malaria among survivors in Calabar, Nigeria. Central African Journal of Medicine 1997;43:231‐4. [PubMed] [Google Scholar]
Molyneux 1989
- Molyneux ME, Taylor TE, Wirima JJ, Borgstein A. Clinical features and prognostic indicators in paediatric cerebral malaria: a study of 131 comatose Malawian children. Quarterly Journal of Medicine 1989;71:441‐59. [PubMed] [Google Scholar]
Molyneux 1995
- Molyneux ME. The clinical manifestations and diagnosis of malaria. Bailliere's Clinical Infectious Diseases 1995;2:271‐292. [Google Scholar]
Molyneux 2000
- Molyneux ME. Impact of malaria on the brain and its prevention. Lancet 2000;355:671‐672. [DOI] [PubMed] [Google Scholar]
Newton 1991
- Newton CRJC, Kirkham FJ, Winstanley PA, Pasvol G, Peshu N, Warrell DA, et al. Intracranial pressure in African children with cerebral malaria. Lancet 1991;337:573‐6. [DOI] [PubMed] [Google Scholar]
Newton 1994
- Newton CRJC, Peshu N, Kendall B, Kirkham FJ, Sowunmi A, Waruiru C, et al. Brain swelling and ischaemia in Kenyans with cerebral malaria. Archives of Diseases in Childhood 1994;70:281‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parfitt 1999
- Parfitt K. Antiepileptics. In: Parfitt K editor(s). Martindale: The complete drug reference. 2nd Edition. London: Pharmaceutical Press, 1999:335‐66. [Google Scholar]
Pasvol 1995
- Pasvol G, Clough B, Carlsson J, Snounou G. The pathogenesis of falciparum malaria. Bailliere's Clinical Infectious Diseases 1995;2(2):249‐70. [Google Scholar]
Pongparatn 1991
- Pongparatn E, Riganti M, Punpoowong B, Aikawa M. Microvascular sequestration of parasitised erythrocytes in human falciparum malaria: a pathological study. American Journal of Tropical Medicine and Hygiene 1991;44:168‐75. [DOI] [PubMed] [Google Scholar]
Rylance 1990
- Rylance GW. Treatment of epilepsy and febrile convulsions in children. Lancet 1990;336:488‐91. [DOI] [PubMed] [Google Scholar]
Teasdale 1974
- Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;ii:81‐84. [DOI] [PubMed] [Google Scholar]
Waller 1995
- Marsh K, Forster D, Waruiru C, Mwangi I, Winstanley M, Marsh V, et al. Clinical features and outcome of severe malaria in Gambian children. Clinical Infectious Diseases 1995;21:577‐87. [DOI] [PubMed] [Google Scholar]
Warrell 1982
- Warrell DA, Looareesuwan S, Warrell MJ, Kasemsarn P, Intaraprasert R, Bunnag D, et al. Dexamethasone proves deleterious in cerebral malaria. A double‐blind trial in 100 comatose patients. New England Journal of Medicine 1982;306:313‐9. [DOI] [PubMed] [Google Scholar]
Waruiru 1996
- Waruiru CM, Newton CRJ, Forster D, New L, Winstanley P, Mwangi I, et al. Epileptic seizures and malaria in Kenyan children. Transactions of the Royal Society of Tropical Medicine and Hygiene 1996;90:152‐5. [DOI] [PubMed] [Google Scholar]
White 1995
- White NJ. Controversies in the management of severe falciparum malaria. Bailliere's Clinical Infectious Diseases 1995;2(2):309‐30. [Google Scholar]
WHO 1990
- World Health Organization. Severe and complicated malaria. World Health Organization Malaria Action Programme. Transactions of the Royal Society of Tropical Medicine and Hygiene 1990;84 Suppl 2:1‐65. [PubMed] [Google Scholar]
WHO 1997
- World Health Organization. Malaria situation in 1994. Weekly Epidemiological Record 1997;72:269‐76. [Google Scholar]
WHO 2000a
- World Health Organization. Management of severe malaria. 2nd Edition. Geneva: World Health Organization, 2000. [Google Scholar]
WHO 2000b
- World Health Organization. Severe falciparum malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 2000;94(suppl 1):1‐90. [PubMed] [Google Scholar]