

Abstract
The causes of essential hypertension remain an enigma. Interactions between genetic and external factors are generally recognized to act as aetiological mechanisms that trigger the pathogenesis of high blood pressure. However, the questions of what genes and factors are involved, and when and where such interactions occur, remain unresolved. Emerging evidence indicates that the hypertensive response to pressor stimuli, like many other physiological and behavioural adaptations, can become sensitized to particular stimuli. Studies in animal models show that, similarly to other response systems controlled by the brain, hypertensive response sensitization (HTRS) is mediated by neuroplasticity. The brain circuitry involved in HTRS controls the sympathetic nervous system. This Review outlines evidence supporting the phenomena of HTRS and describes the range of physiological and psychosocial stressors that can produce a sensitized hypertensive state. Also discussed are the cellular and molecular changes in the brain neural network controlling sympathetic tone involved in long-term storage of information relating to stressors, which could serve to maintain a sensitized state. Finally, this Review concludes with a discussion of why a sensitized hypertensive response might previously have been beneficial and increased biological fitness under some environmental conditions, and why today it has become a health-related liability.
Introduction
Environmental and physiological challenges encountered throughout one’s lifetime can induce long-lasting adaptations in physiological and behavioural systems. Many times the consequence of such experientially induced adaptations is increased biological fitness. However, in some cases, antecedent life events can act as strong aetiological factors that set the stage for subsequent challenges (or stressors) to trigger the onset of disease.
Among the important roles of the central nervous system (CNS) is the control of physiological and behavioural systems that maintain homeostasis as well as manage other complex functions, such as the integration of sensory and motor information and reproduction. Complex neural networks have been identified that control such functional systems. These networks are composed of many neuronal cell groups (nuclei) connected to one another by nerve pathways (tracts). Neural networks integrate and process incoming (afferent) information, which results in outgoing (efferent) signals that determine whether and to what extent particular behavioural, neural, immune or endocrine responses are elicited. Such networks are involved in both the short-term and long-term control of physiological and behavioural functions. Reflexes that are innate, hardwired and genetically determined mediate short-term responses to a particular stimulus. By contrast, long-term modifications can be introduced into neural networks to alter the control of functional systems. Neural networks also encode and store information about past events for later retrieval. This ‘memory’ property of adaptive neural control of effector systems involves neuroplasticity and enables new responses to be acquired or the magnitude of responses to a previously encountered stimulus to be adjusted in the face of new challenges or environmental changes.
One of the simplest forms of neurally mediated adaptation is response sensitization . The neural networks and cellular and molecular mechanisms involved in response sensitization have been studied in many functional systems, including pain, motivation, drug addiction, respiratory control, stress, salt appetite, and exercise. However, the hypertensive response to pressor stimuli has only recently1,2 been recognized as a mechanism that can become sensitized, and the neural mechanisms that mediate hypertensive response sensitization (HTRS) are, therefore, being actively investigated. Information from such studies provides a fresh understanding of what is likely to be an important causal mechanism of some forms of essential hypertension
In this Review, we describe insights into the mechanisms underlying HTRS derived from investigations into the conditions that induce this phenomenon. We also discuss how an induction-delay-expression (IND-DEL-EXP) experimental paradigm can be applied to investigate the neuroplasticity underlying HTRS and how activity-driven CNS neuroplasticity induced either in the perinatal period or in adulthood can maintain the propensity for increased sensitivity of the hypertensive response to pressor stimuli, perhaps even over the course of a lifetime. These topics will be introduced by discussing the nature of essential hypertension and the probable role of the sympathetic nervous system (SNS) in its pathogenesis. Also described are the types of stimuli and conditions that can activate and modify central neural networks, thereby changing the responsiveness of the SNS and altering the long-term regulation of blood pressure.
Physiological and psychosocial stress
For over 100 years, the SNS has been recognized as a key adaptive system that corrects factors that challenge homeostasis3–5. Actual dyshomeostasis or threats that might disrupt the stability of the internal environment activate the SNS. The latency of sympathetic activation (that is, the time interval between stimulus and response) under such circumstances is nearly instantaneous, and its effects are more rapid than those of other systems (such as endocrine, immune or behavioural responses) that are mobilized subsequently to restore dislocations from homeostatic norms.
The stimuli or conditions that increase sympathetic activity can vary over a range of intensities and produce graded degrees of regional sympathetic activation. For example, an external threat evokes strong behavioural (aggression or running away) and cardiovascular (a haemodynamic pattern characterized by increased cardiac output, increased skeletal muscle blood flow, and decreased flow to renal and mesenteric vascular beds) changes. This has been dubbed the ‘fight - flight’ response.
The term stress was used to describe both the sympathetic and the behavioural responses to noxious or threatening challenges3–5. Later research showed that behavioural and cardiovascular components of the fight - flight response could be elicited by stimulating the hypothalamus6,7, amygdala8, dorsal tegmentum and central grey9. In turn, these studies were followed by elucidation of the role of the hypothalamo–pituitary–adrenal axis in response to stressors, and the general adaptation syndrome10–12
A stressful stimulus (or stressor) can be characterized as physiological or psychosocial. Physiological stressors (also known as systemic, homeostatic or interoceptive stressors13–15) are challenges that result from an immediate disruption in homeostasis, such as hypoxia, hypovolaemia, extracellular hypertonicity or hypoglycaemia. By contrast, psychosocial stressors (also known as psychological, processive, neurogenic, mental or exteroceptive stressors13–15) do not immediately disrupt homeostasis but are perceived as posing a threat to an individual’s physical or psychological integrity. Examples of psychosocial stressors are restraint, conditioned fear and psychosocial defeat. Psychosocial stressors include both non-conditioned, prepotent stimuli, such as the sight, sound or odour of a predator, fear of heights or fear of snakes, and conditioned stimuli that, despite being initially neutral or innocuous, have become linked to aversive responses through associative learning (also termed classical or Pavlovian conditioning). Psychosocial stressors engage sensory receptors and their afferent pathways (related to sight, hearing, smell, touch and taste) and involve the limbic system (including the amygdala, hippocampus and medial prefrontal cortex) in information processing. Psychosocial stressors that elicit sustained or repeated SNS activation are hypothesized to be important causes of essential hypertension, discussed below16–19.
Essential hypertension
High blood pressure or hypertension is the leading risk factor for disability and death worldwide20,21. Hypertension increases the likelihood of many major disorders, including heart and kidney disease, stroke, dementia and retinopathy. In 2015 the number of adults with hypertension worldwide was estimated to be 1.13 billion22. A cross-sectional study of individuals aged 35–70 years from 17 countries estimated that 40.8% were hypertensive23. New guidelines24 announced in 2017 will further increase the number of individuals considered to have hypertension, as the new threshold values for systolic and diastolic blood pressure are lower than those used previously for diagnosis of hypertension25.
Patients in whom hypertension can be attributed to an underlying medical condition or medication are diagnosed as having secondary hypertension. However, these individuals account for 10% or less of the total hypertensive population. For the majority of hypertensive adults, the aetiology of the disease is unknown and they are consequently diagnosed as having primary (or essential) hypertension. Essential hypertension is recognized to be a multifactorial disorder involving multiple genes and environmental influences26,27. Almost certainly, several subtypes of essential hypertension will be identified, with different aetiologies and courses of development resulting from the interactions of multiple genetic and environmental factors.
The role of tissue perfusion factors
Increased systemic vascular resistance is the proximal cause and hallmark of established essential hypertension28,29. Early studies of the mechanisms responsible for cardiovascular homeostasis identified multiple interacting factors that control tissue perfusion, including chemical and neuromodulatory agents, and factors that control cardiac and vascular reactivity, blood volume, blood pressure and blood viscosity30. In recognition of the fact that blood pressure is a mechanism for controlling tissue perfusion, the mosaic theory of arterial hypertension proposed that these interacting factors were also likely to contribute to the pathogenesis of hypertension30,31. This theory led to the inevitable question of whether a fundamental fault in these systems causes essential hypertension32 — that is, whether a single cause of essential hypertension could be identified.
One approach to evaluate the relative contribution of various factors involved in the control of tissue perfusion and blood pressure regulation was to apply computer modeling with parallel animal experiments to test the relative contributions of various candidate factors33–35. These studies showed that normal kidneys have an infinite capacity to normalize blood pressure in response to changes in salt and water intake and excretion. By contrast, impairment in renal function reduces sodium and water excretion, which leads to increased extracellular volume and, in turn, to increases in blood volume and cardiac output. One route to increased systemic vascular resistance is thought to be mediated by the phenomenon of total body autoregulation, in which vasoconstriction is triggered by over-perfusion of tissues36.
The role of SNS overactivity
The role of the CNS in blood pressure regulation was initially thought to be limited to the baroreflexes and chemoreflexes. Such reflexes were considered to be involved only in short-term regulation of blood pressure; they were considered to have no role in the long-term control of blood pressure because these reflexes adapt to chronically high or low blood pressures37,38. Consequently, the role of the CNS in the regulation of blood pressure and in the pathogenesis of essential hypertension was largely disregarded.
However, overactivity of the SNS is now considered a major cause of essential hypertension39–42. Studies measuring sympathetic nerve traffic and norepinephrine spillover have provided direct evidence for sympathetic activation of the skeletal muscle, heart and kidney in the early and established stages of essential hypertension43,44. As essential hypertension progresses, this increased sympathetic drive contributes to end-organ damage45. Some patients with elevated SNS tone respond to treatment with sympatholytic drugs by a decrease in blood pressure. At least 50% of patients with primary hypertension are estimated to have this neurogenic form of essential hypertension46. Among the most important issues to be addressed for understanding the causes and course of essential hypertension is identification of the mechanisms responsible for inducing and initiating the early increase in SNS activity and for sustaining the potential for increased SNS drive. If the SNS does indeed play an important part in the pathogenesis of essential hypertension, the next questions that arise are what factors or mechanisms lead to this excess SNS activation, and how do they result in long-term increased systemic vascular resistance (FIG. 1).
Figure 1: The hypothetical role of neuroplasticity and hypertensive response sensitization in the aetiology and progression of essential hypertension.

Left∣ In the stimulus-naive state, exposure to stressors earlier in life activates the central neural network that controls sympathetic nervous system tone and raises blood pressure. Over time, repeated exposure to the stressors result in activity-driven neuroplasticity, which reprograms the central sympathetic neural network to increase sympathetic drive. Right ∣ In the trained state, stressors produce increased sympathetic drive, which results in increased vasoconstriction and/or increased blood volume and cardiac output. Collectively, these changes trigger an increase in vascular resistance33–35. If the increase in vascular resistance is sustained, vascular remodeling and chronic hypertension result.
The central sympathetic nervous system
The SNS and the parasympathetic nervous system are two branches of the autonomic nervous system (ANS). Both sympathetic and parasympathetic arms influence activity of the heart, and the sympathetic component acts on the vasculature. Portions of the ANS are located in both the periphery (peripheral ganglia and nerves) and CNS (brain and spinal cord). Insight into how the SNS contributes to the long-term control of blood pressure necessitates some familiarity with its central organization.
The CNS portion of the ANS is a neural network that receives and integrates inputs from somatic and visceral sensory systems. The consequence of this central processing is the generation of a pattern of autonomic and endocrine system responses that determine blood pressure on a moment-by-moment basis. The number of brain nuclei involved in the control of sympathetic tone and blood pressure is large, and their connections and functional interactions are complex47,48. It is likely that multiple sites within the neural network controlling SNS activity store information about prior SNS activations resulting from physiological and psychosocial stressors.
The neural systems implicated in sympathetic responses to physiological and psychosocial stressors share some common elements, but they also have some different components. Probably the best-characterized central circuitry controlling SNS tone is that related to systemic stressors affecting the control of blood pressure and body fluid homeostasis (reviewed elsewhere47–50). The brain receives information about blood pressure and extracellular fluid status through two different modes of body–brain communication: afferent neural input from the vasculature, and humoral input acting directly on target tissues in the brain. Mechanoreceptors in the venous and arterial vasculature sense changes in blood pressure and blood volume, causing a change in afferent nerve signaling to the nucleus tractus solitarius (NTS), which triggers corrective reflexes. Humoral signals, by contrast, act on two forebrain regions that lack a blood-brain barrier: the subfornical organ (SFO), and the organum vasculosum of the lamina terminalis (OVLT)51. The SFO contains receptors for angiotensin-II (ANG II) and the OVLT includes osmoreceptors that detect extracellular solute concentrations (mainly Na+). The SFO and OVLT, along with the median preoptic nucleus (MnPO), process information relevant to the status of blood volume, pressure and extracellular fluid osmolality52. (FIG. 2).
Figure 2: A midline schematic representing the circuitry of a portion of neural network controlling sympathetic tone and blood pressure regulation.

Activation of the neural pathways arising from rostral structures located along the lamina terminalis and running caudally to the spinal cord are normally activated by the physiological stressors of falls in blood volume and blood pressure. In components of the neural network, structures located along the lamina terminalis [i.e., subfornical organ (SFO), median preoptic nucleus (MnPO), organum vasculosum (OVLT)] and the hypothalamic paraventricular nucleus (PVN) have been implicated as structures where stressors produce neuroplasticity that mediates a sensitized hypertensive response.
The SFO is the primary forebrain target for ANG II and cells in the OVLT function as osmo- or Na+ -receptors. The MnPO, which lies inside the blood-brain barrier, receives input from both the SFO and OVLT and probably functions to process information about the status of intracellular and extracellular fluid compartments and blood pressure. The SFO, MnPO, and OVLT all provide input to the PVN. In turn, the PVN integrates this information with input from other sources (not shown) to influence preganglionic sympathetic neurons in the spinal cord [interomediolateral cell column (IML)] both directly and via the rostral ventrolateral medulla (RVLM). Represented are some additional areas implicated in cardiovascular control. These include the area postrema (AP), caudal ventrolateral medulla (CVLM), nucleus of the solitary tract (NTS) and parabrachial nucleus (PBN), which either directly or indirectly influence activity in the RVLM. Additional abbreviations: AC, anterior commissure; OC, optic chiasm. (Modified Figure based on figure in reference 116)
Hindbrain structures (the NTS and parabrachial nucleus (PBN) are connected to rostrally located structures in the hypothalamus, amygdala and along the lamina terminalis; reviewed elsewhere47–49,53). The hypothalamic paraventricular nucleus (PVN) is a key area in the hypothalamus that receives both ascending input from the hindbrain and descending input from the SFO, OVLT and MnPO. The PVN, therefore, functions as a key integrative node for processing information important for maintaining body fluid and cardiovascular homeostasis. Sympathetic premotor neurons projecting from the PVN innervate the intermediolateral cell column (IML) of the spinal cord, either directly or indirectly via the rostral ventrolateral medulla (RVLM). The IML contains the preganglionic cell bodies of sympathetic axons leaving the CNS, which act as the final common path for the SNS (FIG. 2).
In comparison to the neural pathways mediating responses to challenges associated with altered fluid balance and blood pressure, the network that controls SNS activity associated with psychosocial stressors is not very well defined. However, some major components have been identified, mainly by functional mapping 49,54–56. The amygdala is a key structure in mediating responses to psychosocial stressors. The short neural path from exteroceptive sensory receptors to the amygdala enables rapid activation of responses to environmental stressors that signal danger57. The amygdala also receives inputs from the medial prefrontal cortex and hippocampus, which are involved in memories of previously learned associations with conditioned stimuli54. In the diencephalon, the lateral hypothalamus /perifornical area55–58 and the dorsomedial hypothalamus have both been implicated in the cardiovascular components of the fight or flight response53,59,60. An output pathway projects from the dorsomedial hypothalamus to the rostral ventromedial medulla, and contains sympathetic premotor neurons that project to the IML49,53.
The renin–angiotensin–aldosterone system
The discovery of the enzyme renin and its substrate, angiotensinogen61, precipitated the elucidation of a major pressor system involving renin, angiotensinogen, angiotensin-I (ANG I), angiotensin-converting enzyme (ACE) and ANG II (reviewed elsewhere62). ANG II exerts its actions by binding to the type 1 and type 2 ANG II receptors (named AT1 and AT2, respectively). Because ANG II is a major determinant of circulating aldosterone levels, and ANG II and aldosterone have many common functions, this system is often referred to as the renin–angiotensin–aldosterone system (RAAS).
Several other elements of the RAAS have since been discovered. A homologue of ACE, ACE2, removes a carboxy-terminal phenylalanine from ANG II to produce a heptapeptide, ANG1–7, which binds to the Mas-related G-protein coupled receptor MRG (also known as MASR). ANG1–7, ACE2 and MASR are considered to be components of a counter-regulatory (that is, antihypertensive) arm of the RAAS62–64. ANG II itself could also have both hypertensive and antihypertensive actions, as the effects of ANG II binding to AT2 oppose those produced by its binding to AT162,63.
In the early 1970s, so-called brain renin or isorenin (a renin-like substance discovered in the brain, probably cathepsin D) was thought to be the first brain RAAS component65,66. It is now recognized that most components of the RAAS are synthesized in the CNS as well as at many sites in the body, including the vascular wall, heart, adipose tissue, haemopoietic bone marrow, lungs, and the gastrointestinal tract62,63. Although some controversy remains, work by many contributors has resulted in a general consensus that essentially all RAAS components are generated de novo within the mammalian CNS and that the weight of the evidence favours the existence of a brain RAAS67–71.
It is important to understand how the peripheral RAAS is related to the brain RAAS. Many early studies investigating communication between the peripheral and brain RAASs led to the proposal that circulating ANG II acts on the SFO in the mode of a circulating hormone to activate ANG II-containing efferent neurons that use ANG II as a neurotransmitter or neuromodulator 72–74. Substantial evidence continues to support this concept of humoral–neural coupling between the circulating and brain RAASs (reviewed elsewhere75). In the brain, ANG II is likely to function as an excitatory neuromodulator that acts in concert with excitatory neurotransmitters (such as glutamate) at synapses in the descending pathway between the SFO and IML to increase SNS activity75 (FIG. 2) and thereby induce activity-driven neuroplasticity.
Microglia and pro-inflammatory cytokines
Both neurons and brain glial cells participate in the orchestration of cardiovascular and metabolic functions76,77. The brain has its own innate immune system; microglia are macrophages that migrate into the brain early in development and become its resident immune cells. Both systemic and brain components of the immune system use cytokines for autocrine, paracrine and endocrine (extracellular) signaling involved in immunomodulation. Levels of pro-inflammatory cytokines, including tumour necrosis factor-α (TNF-α), interleukin (IL)-6 and IL-1β, are increased in cardiovascular disease states, and extensive evidence indicates that these cytokines contribute to SNS activation in heart failure and hypertension78,79.
Activated brain microglia release pro-inflammatory cytokines and upregulate their production of extracellular and intracellular reactive oxygen species80–82. Both spontaneously hypertensive rats and animals with ANG II-induced hypertension show microglial activation within the PVN, associated with increased levels of pro-inflammatory cytokines80–82 or targeted depletion of activated microglia within the PVN attenuates ANG II-induced hypertension and decreases both pro-inflammatory cytokine levels within the PVN and cardiac hypertrophy81,82. These results indicate that ANG II-induced microglial activation and the subsequent release of pro-inflammatory cytokines contribute to the development of hypertension. Of note, ANG II itself can activate microglia and stimulate the release of pro-inflammatory cytokines82,83.
Pro-inflammatory cytokines released in the periphery and in the brain as a consequence of RAAS activation can also upregulate AT1. Blood-borne pro-inflammatory cytokines can increase SNS activity and elicit a pressor response by acting on the SFO to increase brain RAAS activity and inflammation84–86. This finding suggests that the SFO-mediated sympathoexcitatory response to pro-inflammatory cytokines depends on having an ambient level of activity of both the brain RAAS and pro-inflammatory cytokines84–86.
Plasticity in neural networks
Late in the 19th century, the idea that the relationship between cells of the nervous system could be modified and that life experiences could produce such changes provided the first structural basis for memory (reviewed in 87). Half a century later, a functional hypothesis accounting for the storage of information involving the synapse was proposed88. According to this hypothesis, the close temporal contiguity of activation of a presynaptic neuron immediately followed by firing of a postsynaptic neuron strengthened the connection between the two cells.
Further support for this functional hypothesis was generated by experiments demonstrating that the strength of synapses could be modified by delivering high-frequency electrical stimulation to a pathway projecting onto hippocampal granule cells89. The stimulation induced facilitation of extracellular field potentials in the dentate gyrus of the hippocampus that lasted for a sustained duration, suggesting that the phenomenon was akin to memory. This enhanced response, known as long-term potentiation (LTP), can be induced at many other sites in the nervous system90,91, including the sympathetic ganglia92,93.
At the time LTP was discovered, investigators in the field of memory and learning classified learning as either associative or non-associative. Two forms of associative learning are classical or Pavlovian conditioning and instrumental conditioning (also known as operant conditioning). In both forms of associative learning, a neutral stimulus becomes associated with a response (or with another stimulus) as a result of the subject undergoing a conditioning procedure. Non-associative learning does not require a neutral stimulus; however, repeated presentations of a stimulus produce a change in the magnitude of an elicited response. A decrease in response amplitude after repeated stimulation is termed habituation, whereas an increase in response amplitude is termed sensitization. Habituation occurs when a stimulus provides little relevant information — particularly when it proves to be non-aversive and non-threatening. Response sensitization occurs when a stimulus is injurious or threatening, and activates defensive physiological and behavioural responses.
The study of response sensitization in mammals and lower species led to the discovery of many fundamental cellular and molecular mechanisms that are important for the formation and retention of memories94,95. Neural mechanisms responsible for sensitization have been investigated in many functional systems, including pain96–98, cravings for drugs of abuse99–102 or salt103,104, baroreceptor and chemoreceptor-reflexes105,106, reflex motor responses107, intermittent hypoxia108,109, respiration110,111, stress112,113 and exercise114,115. Progress in investigating the role and mechanisms of neuroplasticity in sensitization of many of these CNS-supervised functional systems has been rapid because the neural pathways controlling these responses have been defined. The neuroanatomy of the brain network controlling SNS activity and blood pressure has also been characterized. However, only recently have we recognized that HTRS exists and is mediated by maintained neuroplasticity1,116.
The evidence supporting the existence of a modifiable CNS-network-embedded controller of SNS activity and blood pressure is discussed in the next section. This CNS controller of sympathetic tone is adaptive and can alter long-term regulation of blood pressure as a result of prior experience117,118. In other words, an altered cardiovascular response is learned as a result of the presence of an antecedent stimulus. This sensitized capacity to increase SNS activity in response to physiological and psychosocial challenges provides a working hypothesis to explain how exposure to stressors earlier in life can, through vascular and renal mediators, lead to increased systemic vascular resistance and high blood pressure at a later time (FIG. 1).
Hypertensive response sensitization
IND-DEL-EXP experiments
Our research group has investigated sensitization of the hypertensive response using an IND-DEL-EXP experimental paradigm (BOX 1)1. In in an initial experiment a very low, non-pressor dose of ANG II was administered during the induction phase. After a 1-week delay period, animals exposed to the non-pressor dose of ANG II during the induction phase show a significantly enhanced hypertensive response during the expression phase, compared with control animals that received vehicle during the induction phase (BOX 1). A follow-up experiment demonstrated that the HTRS-inducing effects of ANG II were mediated by the CNS. Intracerebroventricular administration of very low (non-pressor) doses of ANG II during the induction phase had no sustained effect on blood pressure during the induction and delay phases yet still induced HTRS1 . Additional evidence implicating the CNS in HTRS was provided by experiments showing that intracerebroventricular administration of irbesartan (an AT1 antagonist) along with subcutaneous low-dose ANG II in the induction phase prevented HTRS1.
Box 1: Experiments showing hypertensive response sensitization.
In induction-delay-expression (IND-DEL-EXP) experiments, a stimulus is presented during an induction phase typically lasting 1–3 weeks. Subsequently, the individual is challenged with the same or another stimulus, and the magnitude of the elicited response is measured during the expression phase, which generally lasts 2 weeks. Depending upon the experiment, the interval between induction and expression is of variable duration (ranging between no delay and many weeks), and is referred to as the delay phase.
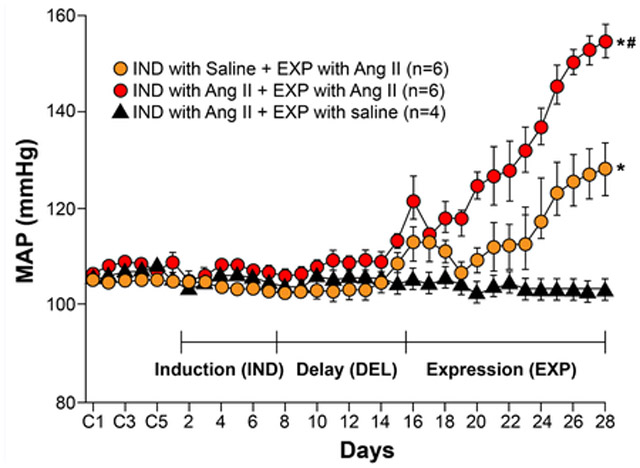
The above illustration shows typical findings from an IND-DEL-EXP experiment showing hypertensive response sensitization (HTRS)1. Three groups of adult male rats underwent implantation of telemetry probes to enable continuous recording of blood pressure and heart rate. After recovery from surgery, the animals received either saline or subcutaneous angiotensin II (ANG II) 10 ng/kg/min. The induction and delay phases each lasted 1 week. Blood pressure usually changes little during the induction phase; a transient increase in blood pressure can occasionally be seen in some models, but always returns to baseline before the start of the expression phase. The delay phase enabled the persistence of the effects of HTRS Induction to be assessed and any remaining exogenous ANG II to be metabolized. At the beginning of the expression phase, the rats received a further stimulus with either saline or subcutaneous ANG II 120 ng/kg/min. As this slow-pressor dose of ANG II does not immediately elicit hypertension, the initial rise in mean arterial blood pressure (MAP) seen in animals that received ANG II over the first 3–4 days of the expression phase probably represents the systemic vasoconstrictor action of ANG II. After 4 days the vasoconstrictor effect of ANG II is replaced by increased sympathetic drive, which continues to increase blood pressure241. *P <0.05 versus baseline, or versus induction with ANG II then expression with saline. #P <0.05 versus induction with saline then expression with ANG II. Figure redrawn and based on data presented in 1.
By employing the IND-DEL-EXP experimental design, it was possible to provide the first unambiguous demonstration that the hypertensive response can be sensitized1. The power of the IND-DEL-EXP method is that it disambiguates induction of a sensitized response from expression of sensitization. Most procedures used to induce hypertension conflate the time and processes involved in inducing sensitization with the period of expression of the hypertensive response. A retrospective view of the literature indicates that there are phenomena that in all likelihood are examples of HTRS but that have not been interpreted as involving the process of sensitization and of CNS neuroplasticity. Two good examples are the effects of the induction of hypertension by prolonged systemic administration of very low doses of ANG II and of the phenomenon of the rapid expression of hypertension following renal artery clipping-unclipping and re-clipping.
Large doses of ANG II administered systemically are well known to produce a rapid rise in blood pressure. However, in the mid-1960s, studies in the rabbit interestingly demonstrated that maintained infusions of very low doses of ANG II that initially produced no notable rise in blood pressure would after several hours or days produce frank hypertension119,120. This is in spite of the fact that circulating levels of ANG II were likely to reach study-state within a few minutes. This so-called slow-pressor effect of ANG II has been demonstrated in many species (including, dog121, rat122, mouse123, sheep124 and humans125). The capacity of the maintained administration of low doses of ANG II to progressively shift the dose-response curve upward and produce a greater rise in the blood pressure response was originally defined as “angiotensin auto-potentiation” 126,127. Now after recognizing that the hypertensive response can be sensitized, perhaps a more logical interpretation of the slow-pressor effect of ANG II and a mechanistic clarification of the concept of auto-potentiation is that as the low-slow pressor dose infusion of ANG II proceeds, a sensitized state is progressively being induced that reinforces and amplifies the ongoing rise in blood pressure.
Constricting renal arteries by various methods, including using small metal clips, produces hypertension over the course of several days. In rats with established two-kidney, one-clip renal hypertension (2K-1C), removal of the renal clip rapidly restores blood pressure to normal levels. However, re-clipping the renal artery a short time after its removal very quickly reestablishes the hypertensive state128,129. This reinstated hypertension occurs much more rapidly than the time it took to make the animals hypertensive with the original placement of the renal clips. Also shortly after the renal artery clips are removed or the clipped kidneys are ablated the pressor responses to exogenous renin or ANG II is significantly enhanced128–130. Notably, one unclipping-reclipping experiment implicated the CNS by demonstrating that after destroying the spinal cord there was no greater renin-induced pressor response in acutely unclipped 2K-1C rats129. Taken together, the clipping/unclipping/reclipping studies indicate that a control of cardiovascular function behaved as if it had a memory for a condition induced by renal clipping. Because the 2K-1C rat model of renal HT chronically increases circulating renin129, it is probable that an initial rise in ANG II acted through neuroplasticity to reprogram the CNS to trigger a sensitized HT response when the renal artery was re-clipped or exogenous renin-angiotensin was administered. Examination of the re-clipping and slow-pressor response phenomena in the context of recognition of HTRS invites investigations examining the nature of central neuroplasticity associated with the maintenance of the sensitized state.
Neuroplasticity accompanying hypertensive response sensitization
One of the advantages of using the IND-DEL-EXP protocol to investigate HTRS is that cellular and molecular changes in the CNS can be examined in control and experimental groups at the end of either the delay or expression phases Being able to collect tissue samples at the end of the delay phase is an especially important asset because the blood pressure of the experimental group is still normal and not different from that of the control group. This situation obviates any concern that results might be confounded by the differing levels of hypertension in the control and experimental groups, as is the case for samples collected at the end of the expression phase. Studies of mRNA and/or protein expression in the lamina terminalis and paraventricular nucleus at the end of the delay phase have proved to be particularly informative, because they indicate molecular changes that are sustained after induction. (FIG. 3)
Figure 3: Mechanisms involved in hypertensive response sensitization (HTRS) and neuroplasticity.

By studying changes after the induction of HTRS, important memory-related molecular changes have been identified that are maintained until the end of the delay period. Many neural systems involve ‘signature’ neurotransmitters or neuromodulatory mechanisms. For example, substance P and calcitonin gene-related peptide and their receptors are ubiquitous in pain signaling pathways97,98,242. As components of the brain renin–angiotensin–aldosterone system (RAAS) are upregulated after induction of hypertensive response sensitization (HTRS), RAAS components might reasonably be considered a signature of the pathways controlling sympathetic tone and blood pressure. Considerable evidence indicates that glutamate and glutamate receptors are critically involved in nearly all forms of neuroplasticity243,244, and most cells in the nervous system express at least one type of glutamate receptor245. This neurotransmitter depolarizes neurons by acting on ionotropic N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Most, if not all, cells in the network controlling sympathetic tone have both NMDA and AMPA receptors and receive glutamatergic input from other network neurons. Growth factors have multiple roles in neuroplasticity including altering membrane potentials, increasing protein synthesis, promoting cell viability, and morphological changes. Brain-derived neurotrophic factor (BDNF, probably the best-studied CNS growth factor) is associated with almost every aspect of neural and functional plasticity246 and acts through TrkB (also known as BDNF/NT3 growth factors receptor). Finally, long-term neuroplastic changes require the synthesis of new proteins, which requires activation of transcription factors, including c-Fos, FOSB and cAMP response element-binding protein (CREB). Red rectangles indicate signaling mechanisms implicated in the neuroplasticity underlying HTRS247. In future work, it will be important to determine how many structures in the neural network controlling sympathetic tone and blood pressure manifest detectable changes and how long such changes persist. Figure based on and modified from REF.116.
In every HTRS model studied to date using the IND-DEL-EXP paradigm, mRNAs related to components of the RAAS are upregulated at the end of the delay phase1,2,131–135. Evidence most consistent has been the observation of upregulation of AT1 mRNA. This finding is compatable with many other in vivo and in vitro studies showing upregulation of AT1 or increased binding of ANG II to AT1 after treatments that elevate levels of ANG II136–142, or after administration or mineralocorticoid agonist143–146. These observations are particularly important, because for most agonist-receptor interactions, increasing the concentration of a ligand will typically produce downregulation of its receptor. The functional significance of agonist-induced receptor upregulation in the RAAS is that it can function as a feed-forward mechanism that presumably would result in response amplification and contribute to HTRS.
Recognizing that very low doses of ANG II can sensitize the hypertensive response by reprogramming the CNS to make it more responsive to ANG II may help explain the long-standing therapeutic enigma observed in the treatment of many patients with essential hypertension. Specifically, HTRS and central neuroplasticity may help address the question of why RAAS antagonists are successful in lowering blood pressure in individuals with normal- or low- plasma renin activity147,148. In light of demonstrations of enhanced sensitization to ANG II and indications of neuroplasticity that includes upregulation of central AT1, it seems reasonable to speculate that in such patients prior exposure to challenges may have produced an increased the number of AT1 in the CNS to manifest HTRS. Consequently, the actions of RAAS antagonists to attenuate RAAS actions in the CNS would be more effective in reducing sympathetic tone and lowering blood pressure even in normal- or low- renin patients.
Sensitization is a form of non-associative memory. The study of the cellular and molecular basis of sensitization has significantly advanced knowledge about the nature of neuroplasticity underlying learning and memory94,149. The maintained molecular changes observed after HTRS induction in addition to those in the RAAS indicate that the neuroplasticity associated with HTRS are similar to those found in many other models of associative and non-associative learning and memory (FIG. 3). For example, in experiments that examined the effect of non-pressor doses of either ANG II or aldosterone given during induction of HTRS on growth factors in the lamina terminalis at the end of the delay phase, brain-derived neurotrophic factor mRNA and protein levels were increased, whereas vascular endothelial growth factor mRNA and protein levels remained unchanged136–142,150. Also, lamina terminalis tissues collected at the end of the delay phase showed no net increase in levels of the second messenger p38 mitogen-activated protein kinase (MAPK) nor transcription factor cyclic AMP-responsive element-binding protein (CREB). However, levels of the phosphorylated forms of each of these proteins increased significantly150.
Collectively, the characterization of several of sustained changes following the delay phase in components of the RAAS and other molecular indictors of synaptic plasticity is consistent with the idea that there is neuroplasticity in the neural network controling sympathetic tone and blood pressure (FIGS. 2 & 3). These changes are likely to represent an instantiation of memories of earlier life challenges that produced a neural state resulting in HTRS.
Cross-sensitization
Cross-sensitization describes the development of HTRS despite the stimulus used during the induction phase being different from that used during the expression phase. Aldosterone and ANG II interact cooperatively in the brain, indicating that both mechanisms must be intact for hypertension to occur in response to systemically administered ANG II or aldosterone151,152. Cross-sensitization was first confirmed by the results of IND-DEL-EXP studies, in which subcutaneous aldosterone given at a non-pressor dose during the induction phase led to HTRS when a slow-pressor dose of ANG II was administered at the beginning of the expression phase2. Evidence that the CNS is involved in cross-sensitization was provided by further experiments showing, first, that intracerebroventricular administration of aldosterone at a non-pressor dose during induction produced cross-sensitization when a slow-pressor dose of ANG II was delivered during the expression phase. Second, that intracerebroventricular administration of a mineralocorticoid receptor antagonist along with subcutaneous aldosterone during induction blocked HTRS2.
Many different stressors can act to cross-sensitize with ANG II to induce HTRS in IND-DEL-EXP experiments (TABLE 1). In the course of studying these stressors, our research group has also found many interventions that can block HTRS, some of which actually reverse the sensitized state (TABLE 2). The results of these studies provide insights into the nature of HTRS and into how it might be maintained by neuroplasticity (FIGS. 1 and 3). The Information that can be derived by varying the parameters applied during induction and expression phases and by changing the duration of the delay phase is illustrated by some examples of these studies, below.
Table 1.
Stimuli used to induce sensitization of the hypertensive response
| Stressor | Route of administration | Duration of challenge | Ref. |
|---|---|---|---|
| Systemic stressors | |||
| Non-pressor ANG II | Subcutaneous | 1 week | 1 |
| Non-pressor ANG II | Intracerebroventricular | 1 week | 1 |
| Non-pressor aldosterone | Subcutaneous | 1 week | 2 |
| Non-pressor aldosterone | Intracerebroventricular | 1 week | 2 |
| High-sodium diet | Food | 2 weeks | 116 |
| High-fat diet | Food | 3 weeks | 131 |
| Low-sodium diet | Food | 2 weeks | 116 |
| Hypotensive haemorrhage | NA | 3 times in 1 week | 248 |
| Lipopolysaccharide | Intraperitoneal | 3 times in 1 week | Unpublished |
| Leptin | Intracerebroventricular | 1 week | 134 |
| TNFα | Intracerebroventricular | 1 week | 131 |
| Processive stressor | |||
| Resident-intruder social defeat | External and/or psychosocial | 3 times in 1 week | 132, 133 |
| Perinatal challenge to offspring | |||
| Maternal gestational hypertension | ANG-elicited hypertension | 3 weeks of pregnancy | 191 |
| Maternal high-fat diet | Food | 3 weeks of pregnancy | 135 |
| Maternal high-fat diet | Food | 3 weeks of lactation | 135 |
| Maternal high-fat diet | Food | 3 weeks of pregnancy plus 3 weeks of lactation | 135 |
Expression of sensitization of the hypertensive response was tested by subcutaneous administration of angiotensin II 120 ng/kg/min. ANG, angiotensin; NA, not applicable; TNF, tumour necrosis factor-α.
Table 2.
Treatments that block or reverse sensitization of the hypertensive response.
| Blocking treatment | Route of administration | Sensitizing challenge | Ref. |
|---|---|---|---|
| Mineralocorticoid antagonist | Intracerebroventricular | Blocks induction with subcutaneous aldosterone | 2 |
| Leptin inhibitor | Intracerebroventricular | Blocks induction with high-fat diet feeding | 134 |
| Minocycline | Intracerebroventricular | Blocks induction with high-fat diet, intracerebroventricular ANG II and intracerebroventricular leptin | 131,134 |
| Pentoxifylline | Intracerebroventricular | Blocks induction with high-fat diet, intracerebroventricular ANG II and intracerebroventricular leptin | 131,134 |
| AT1 antagonist | Intracerebroventricular | Blocks induction with high-fat diet, subcutaneous ANG II and intracerebroventricular leptin | 1,131,134 |
| Renal denervation | NA | Reverses sensitization induced by maternal gestational hypertension | 191 |
| Spontaneous exercise | NA | Delays expression of sensitization by maternal gestational hypertension | Unpublished |
| Oestrogen | Intracerebroventricular | Blocks induction with subcutaneous ANG II in male rats and ovariectomized female rats | 192 |
| Raloxifene | Intracerebroventricular | Blocks induction with subcutaneous ANG II in male rats and ovariectomized female rats | Unpublished |
| Valproate | Subcutaneous | Blocks induction with subcutaneous ANG II | Unpublished |
| Sodium butyrate | Intraperitoneal | Blocks induction with subcutaneous ANG II | Unpublished |
| Dizocilpine (MK-801) | Subcutaneous | Blocks induction with subcutaneous ANG II | 247 |
| AP-5 | Intracerebroventricular | Blocks induction with subcutaneous ANG II | 247 |
| ANG1–7 | Intracerebroventricular | Blocks induction with subcutaneous ANG II | 192 |
ANG, angiotensin; AP-5, (2R)-amino-5-phosphonovaleric acid, an N-methyl-D-aspartate receptor antagonist; AT1, type-1 angiotensin II receptor; NA, not applicable.
High dietary fat intake and the role of central leptin and inflammation.
The current obesity epidemic makes it clear that increased adiposity is a major risk factor for hypertension. Obesity and consumption of a high fat diet are both widely accepted to produce a chronic state of low-grade inflammation in the CNS, which is characterized by increased activation of microglia and astrocytes, and increased CNS expression of genes encoding pro-inflammatory cytokines (TNF-α, IL-6 and IL-1β)153. Increased CNS levels of pro-inflammatory cytokines also contribute to elevated SNS activity in obesity154.
Growing evidence from human and animal studies indicates that activation of the RAAS is involved in the pathogenesis of both obesity and obesity-related hypertension155. Increased ANG II levels during diet-induced obesity (DIO) activate NADPH oxidase via AT1, leading to increased production of reactive oxygen species and further increased transcription of genes encoding pro-inflammatory cytokines155. Thus, the RAAS and pro-inflammatory cytokines mutually reinforce each other’s actions in the periphery and in the CNS to generate sympathoexcitation and hypertension156,157.
Results from clinical trials suggest that in addition to its anti-hypertensive effects, pharmacological inhibition of the RAAS also protects against the development of DIO and type 2 diabetes mellitus158. Obese Zucker rats show increased sensitivity to the blood-pressure-elevating effect of ANG II, and RAAS blockade lowers blood pressure in obese rats more than it does in lean rats, despite the obese animals having lower plasma renin activity than their lean counterparts159. In rabbits, 3 weeks of a high-fat diet led to increases in blood pressure, heart rate and renal sympathetic nerve activity, and this model of obesity-induced hypertension is known to be of neurogenic origin160–162. Interestingly, although animals with DIO placed on a normal diet were able to restore their body weight, insulin levels and leptin sensitivity to normal, their blood pressure, renal sympathetic nerve activity and levels of central RAAS components and pro-inflammatory cytokines remained high163. These results indicate that obesity itself might induce hypothalamic inflammation and sensitization of the brain to circulating sympathoexcitatory factors (such as those from the RAAS) and that these factors might drive hypertension.
In light of the relationship between obesity and essential hypertension and the need to understand how adiposity results in high blood pressure, our group conducted IND-DEL-EXP experiments to test whether a high-fat diet could produce HTRS. Rats were placed on a high-fat diet for 3 weeks (the induction phase). At the end of this period, body weight, white adipose tissue mass and plasma leptin levels were all increased. Tissue samples from the lamina terminalis showed increased levels of mRNAs encoding components of the brain RAAS (namely, renin, ACE and AT1), the pro-inflammatory cytokines TNF-α and IL-6, and a marker of microglial activation (CD11b)131. In parallel experiments, animals on the high-fat diet for three weeks were then returned to a normal diet and immediately challenged during an expression phase with a slow-pressor dose of ANG II. This evoked HTRS in the animals that had been on the high-fat diet. Taken together, these results suggest that a high-fat-diet-induced upregulation of the RAAS and pro-inflammatory cytokines in the CNS, which participated in the induction of HTRS. The capacity of the RAAS and pro-inflammatory cytokines to mutually upregulate their expression is an important factor in central feed-forward pathways that induce HTRS.
In obese humans and in animal models of DIO, increased SNS activity and high blood pressure are both associated with increased levels of leptin and activation of the leptin–melanocyte stimulating hormone-α–melanocortin receptor 4 pathway164,165. Brain structures that mediate the central actions of leptin include the arcuate nucleus (ARC), PVN, SFO and hindbrain. Microinjection of leptin into the ARC or PVN resulted in increased SNS activity166,167, whereas deletion of the leptin receptor (LEPR) in the ARC or SFO attenuated this leptin-elicited increase in SNS activity166,168. Leptin-deficient or LEPR-deficient mice demonstrate attenuated microglial activation and reduced levels of inflammatory mediators169, findings that are consistent with leptin having a pro-inflammatory function. Furthermore, a high-fat diet and obesity both activate microglia and astrocytes and increase levels of pro-inflammatory cytokines in the SFO and PVN170. These effects are ANG II-dependent, as some responses are reversed by deletion of AT1 in the PVN170. SNS responses to leptin are also dependent on central AT1, as intracerebroventricular infusion of losartan attenuated SNS activity in response to leptin171. Additional evidence indicates that obesity-associated activation of the hypothalamic inflammatory pathway mediates the CNS functions of leptin172. These findings suggest that the interactions and synergism between leptin, the RAAS and pro-inflammatory cytokines are responsible for the increase in SNS activity that occurs early in the onset of obesity.
Obesity, a high-fat diet, leptin, pro-inflammatory cytokines and microglial interactions all have a role in HTRS. In IND-DEL-EXP experiments, central administration of leptin during the induction phase mimicked the HTRS-inducing effects of a high-fat diet, whereas intracerebroventriclar administration of a leptin receptor antagonist prevented the induction of HTRS by 3 weeks of a high-fat diet134. Intracerebroventricular TNF-α was as effective in inducing HTRS as intracerebroventricular ANG II given at a non-pressor dose. TNF-α given during the induction phase resulted in increased levels of mRNAs encoding components of the RAAS and increased levels of markers of inflammation and microglial activation in lamina terminalis structures131. Central inhibition of AT1, TNF-α synthesis or microglial activation all blocked the induction of HTRS by either leptin or high fat diet. These inhibitors also blocked the upregulation of RAAS activity and the rise in indicators of inflammation in the lamina terminalis131,134.
Taken together, the results of these studies provide insight into the role of metabolic factors and the actions of the brain RAAS and the brain innate immune system that mediate the production of HTRS. Accordingly, we hypothesize that eating a high-fat diet increases the effects of leptin on and in the brain, which activates the brain RAAS and upregulates inflammatory mechanisms that result in reprogramming the control of SNS activity and HTRS (FIG. 4). The consequence of these changes is that HTRS increases the likelihood that frank hypertension will develop if these challenges are sustained or when additional hypertensinogenic stressors are encountered later in life.
Figure 4: How a high-fat diet and obesity might induce hypertensive response sensitization.

High-fat diet and obesity both increase circulating levels of both angiotensin II (ANG II) and leptin. Circulating ANG II and leptin act on the subfornical organ (SFO) and/or the hypothalamic arcuate nucleus (ARC), both of which have projections to the hypothalamic paraventricular nucleus (PVN). Actions of ANG II and leptin in the SFO, ARC, and/or PVN activate the brain renin–angiotensin–aldosterone system and inflammatory mechanisms that result in inducing sensitization by increasing synaptic efficiency in the SFO and/or PVN.
Gestational hypertension and maternal high dietary fat intake.
Maternal health during pregnancy is strongly associated with the cardiovascular health of her adult offspring. Many studies have demonstrated that the offspring of mothers with gestational hypertension have increased blood pressure in childhood and adolescence, and that these children are at an increased risk of developing hypertension in adulthood173–178. This risk to the offspring is graded and greatest in those whose mothers had the most severe hypertensive signs, such as early onset hypertension or pre-eclampsia177,179. Animal experiments also demonstrate that many kinds of prenatal insult, including uteroplacental insufficiency, protein restriction, chronic secondary hypertension and glucocorticoid treatment during pregnancy, lead to hypertension in the offspring180.
Increased renal sympathetic nerve activity and over activity of the RAAS and pro-inflammatory cytokines in the periphery have been implicated as causal mechanisms in prenatal programming of hypertension. In human studies, the adolescent sons of mothers who had high blood pressure during pregnancy had increased aldosterone levels, a trend towards increased circulating renin activity, and increased SNS activity before and during isometric exercise181,182. In animal models, renal denervation or chronic blockade of both the RAAS and inflammation reduced hypertension (via restoration of renal and arterial function) in the offspring of dams that had received different types of insult during pregnancy183–187. The RAAS has a key role not only in the generation of increased basal blood pressure but also in the development of enhanced renal sympathetic and pressor responses to physical stress observed in male offspring of pregnant dams exposed to protein restriction188,189.
Besides exploring the roles of the peripheral RAAS and sympathetic nerve activity in the development of hypertension in prenatally programmed hypertensive animals, a few studies have implicated the brain RAAS in hypertension associated with antenatal nutrient deprivation. Intracerebroventricular injection of an ACE inhibitor or an AT1 antagonist significantly reduced the blood pressure of animals previously subjected to maternal protein restriction in utero. Expression of AT1 in the SFO and the OVLT was increased in these animals compared with controls190.
The time between prenatal interventions and the development of hypertension in adult animals is long compared to the delay periods used in our IND-DEL EXP studies of HTRS. Consequently, these studies have provided a good way to investigate the duration of HTRS. Prenatal induction of HTRS was accomplished by subjecting pregnant dams to ANG II-elicited gestational hypertension. The adult male offspring of these dams had normal resting blood pressures but showed HTRS when challenged with a slow-pressor dose of ANG II during a 2-week expression phase, which began when the rats were 10 weeks old191. Protein and/or mRNA expression of components of the RAAS, markers of microglial activation and brain levels of pro-inflammatory cytokines (which indicate activation of the brain’s innate immune system) were also increased in these offspring191. These experiments demonstrate that reprogramming of the mechanisms controlling SNS activity and blood pressure during the prenatal period produces long-lasting phenotypic changes in the CNS as well as sensitized responsiveness to a hypertensinogenic challenge during adulthood.
Various interventions during the delay period (that is, between weaning of these rats at 3 weeks of age and pressor challenge at 10 weeks of age) can abrogate HTRS. For example, in one experimental group, the ACE inhibitor captopril was administered in the drinking water beginning from weaning until the rats were 8 weeks old. Another experimental group underwent renal denervation at 8 weeks of age. Both interventions blocked HTRS in 10-week-old male offspring of dams with gestational hypertension. Consistent with these findings, the upregulation of mRNAs encoding components of the brain RAAS and indices of CNS inflammation also were normalized in captopril-treated animals191.
As noted above, many human and animal studies have found that female subjects in comparison to male offspring are frequently protected from the effects of negative earlier life experiences producing hypertension later in life. We have also seen that females are protected against the induction of HTRS192,193. Unfortunately, an in-depth discussion of the nature of the female-related protection against HTRS and the role of estrogen and other anti-hypertensive factors prominent in females is precluded by space limitations placed on the present review.
In humans as well as in experimental animals, maternal obesity and maternal high dietary fat intake have adverse consequences for health of her offspring by altering their responses to environmental challenges and predisposing them to cardiovascular and metabolic disease194–197. For example, the offspring of rabbits fed a high-fat diet have elevated renal sympathetic nerve activity and pressor responses to central leptin, increased sympathetic responses to ghrelin and an exaggerated sympathetic response to acute air-jet stress198. These observations suggest that the elevations in blood pressure and renal sympathetic nerve activity are attributable to changes in central pathways that regulate SNS activity198. Also, a maternal high-fat diet has been associated with hypothalamic inflammation, impaired baroreflex sensitivity and reduced heart rate variability in the offspring, implicating abnormalities in autonomic control199–201. The effects of maternal obesity and a maternal high-fat diet on HTRS, and the influence of a maternal high-fat diet on the autonomic function of her offspring have been studied in female rats fed a high-fat diet beginning 8 weeks before conception and continuing throughout either pregnancy and lactation, just during pregnancy, or just during lactation135. The 10-week-old adult offspring of these rats, which had initially normal blood pressure, showed a blunted cardiac baroreflex and an elevated autonomic responsiveness of blood pressure and heart rate to an acute challenge with either intracerebroventricular ANG II or TNF-α135. These offspring also showed upregulated mRNA expression of RAAS components and increases in levels of NADPH oxidase and pro-inflammatory cytokines in the lamina terminalis and PVN in response to being challenged with a slow-pressor dose of ANG II during the expression phase135.
Taken together, these results indicate that a maternal high-fat diet during either pregnancy or lactation is sufficient for early-life reprogramming of the central neural network controlling SNS activity and blood pressure, thereby producing HTRS and predisposing the offspring to an increased risk of hypertension in adulthood.
The perinatal period has long been recognized as a critical time during which many interventions are particularly effective in reprogramming many physiological systems and particularly the nervous system202,203. One of the consequences the presentation of challenges to offspring via the mother is that it can induce a sensitized state that is maintained and that can be expressed as hypertension when exposed to stressors much later in life. Perinatal challenges presented to the offspring represent a predisposition for expressing hypertension that are likely to last a lifetime.
Psychosocial stressors.
Social defeat is an example of a psychosocial stressor that can induce HTRS when administered in a resident-intruder experimental paradigm. These experiments involve introducing a smaller adult male rat, the intruder, into the cage of a larger male, the resident, who is well established in his living quarters. During such pairings, the two animals reliably take on different behavioural roles. The resident immediately assumes the role of the dominant animal and the intruder becomes submissive and displays psychosocial defeat. In addition to behavioural submission, the defeated intruder manifests cardiovascular changes typical of reactions to stressors204.
This paradigm is considered to be an excellent model of post-traumatic stress disorder (PTSD), a psychiatric illness characterized by persistent emotional and mental stress following traumatic events205,206. Patients with PTSD are at increased risk of developing hypertension and cardiovascular disease207,208. Both patients with PTSD and animal models of PTSD are characterized by increased SNS activity, decreased cardiac vagal control and baroreflex dysfunction207–212. Enhanced sympathetic activity and blunted baroreceptor reflex sensitivity is also associated with elevated levels of pro-inflammatory cytokines and activation of the peripheral and brain RAASs213,214.
Three resident-intruder pairings (made every other day during a 1-week induction period) followed by a 3-day delay period reliably produced HTRS in the intruder. In addition, lamina terminalis tissue collected at the end of the delay period showed increased mRNA expression of AT1, IL-1β, IL-6 and TNF-α132. Pretreatment with captopril or administration of a TNF-α antagonist during the induction period abrogated the HTRS produced by resident-intruder pairings, and also blocked the increased expression of RAAS components and proinflammatory markers in the brain133.
The results of these resident-intruder studies provide insight into the mechanisms by which psychosocial stress, such as that associated with PTSD, induces HTRS. Information derived from such studies will be useful in generating new therapeutic strategies for patients with mental health and cardiovascular disease comorbidities.
Salt-sensitive hypertension.
High dietary sodium chloride (table salt) intake is widely viewed to be a contributor to the pathogenesis of hypertension215–217. The effect of high salt intake on blood pressure varies between individuals in both humans and nonhuman species, as environmental and genetic factors both contribute to salt sensitivity215,218,219. Rigorously defined protocols have been developed to diagnose salt sensitivity215,220, and it is estimated that in the United States, about 51% of patients with hypertension and about 26% of normotensive individuals are salt-sensitive221.
Growing evidence indicates that the brain is involved in the control of both sympathetic tone and salt sensitivity221–225. Accordingly, the effects of systemic ANG II or aldosterone administration were studied in IND-DEL-EXP experiments to see if these hypertensinogenic challenges would produce salt sensitivity150. Rats were implanted with probes to enable continuous recording of blood pressure and heart rate. After recovery from surgery, they received 1 week of systemic treatment with either vehicle or non-pressor doses of either ANG II or aldosterone (the induction phase). After a 1-week delay period, the rats were given 2% saline as their sole drinking fluid226,227. Blood pressure increased significantly from baseline in all animals; however, the increase was significantly greater in the groups that had previously received ANG II or aldosterone. Blood pressure in all three groups fell to pre-exposure levels when saline was replaced with plain water. Additional studies found that salt sensitivity can also be produced by intracerebroventricular administration of non-pressor doses of either ANG II or aldosterone during a 1-week induction period.
The finding that salt sensitivity can be experimentally induced is important for two reasons. First, these experiments show that HTRS can be demonstrated during the expression phase by using a treatment (high salt intake) other than a slow-pressor dose of ANG II. Second, they demonstrate that the expression of salt sensitivity does not require a genetic predisposition or the administration of another pressor agent in conjunction with high salt intake. The CNS can be programmed to display HTRS when excessive amounts of sodium are subsequently consumed.
The functional relevance of adaptation
Substantial evidence from HTRS studies implicates the brain RAAS in reprogramming of the central neural network that controls SNS activity and determines long-term blood pressure regulation. The observation that stressors induce a sustained upregulated expression of components of the RAAS is of particular importance, as this feed-forward mechanism is critical for the memory-related processes that mediate HTRS. However, the brain RAAS is not the only system that includes mechanisms for long-term information storage.
The acquired or adaptive immune system employs T and B memory cells to quickly and specifically recognize an antigen the body has previously encountered, which enables the initiation of a corresponding immune response. However, several components of the innate immune system228, including brain microglia229, also have memory capacities, and this newly recognized function of the innate immune system in the brain brings neurological and immune memory mechanisms in close proximity to one another. The emerging picture of mechanisms involved in HTRS is that microglia and pro-inflammatory cytokines are as likely as the brain RAAS to have important roles in inducing and maintaining HTRS (FIG. 4). Essentially, these mechanisms predict future events, a theme that is explored further below.
Phenotypic plasticity
The concept of phenotypic plasticity is related to the likelihood that a given genotype will produce different phenotypes under varied environmental conditions230. In many physiological and behavioural systems, response sensitization is advantageous for defending an organism’s integrity against the consequences of recurring challenges. For example, immune memory enables a more rapid, amplified response to be generated against a previously encountered pathogen. Rather than strict adherence to information carried in the genome, phenotypic plasticity involves mechanisms that alter genetic expression and neuroplasticity. Under some — but not necessarily all — conditions, the consequence of phenotypic plasticity is increased biological fitness.
Selection pressure and mismatch
Many species spent much of their evolutionary history under environmental conditions that posed a constant threat of circulatory collapse217. Acute extracellular fluid losses — from injury-induced blood loss, from pathogen-induced emesis and/or diarrhoea, and water and sodium loss via perspiration were (and still are) frequent threats to survival in hunter-gatherer populations in the hot African savanna. Furthermore, under natural conditions, full recovery from severe hypovolaemia requires the behavioural competence necessary for acquiring adequate amounts of water and sodium, which are both necessary for restoring extracellular volume and for maintaining blood pressure. Access to these commodities can be severely limited, especially during a prolonged dry season. For an individual living under such circumstances, survival of a hypovolaemic challenge might require adaptations that favour a phenotype that maintains blood pressure and staves off uncompensated hypovolaemic shock. Therefore, the capacity to enhance a sympathetically driven hypertensive response, mediated by neuroplasticity and CNS reprogramming, would confer biological fitness in an ecological niche where hypovolaemic shock and circulatory collapse had a high probability of occurrence. However, under different circumstances, such an environmentally triggered phenotypic adaptation might contribute to the pathogenesis of hypertension.
Essential hypertension and related cardiovascular diseases are pathologies of old age. At the time in human evolutionary history when selection for a phenotype that conferred protection against hypovolaemia and hypotension presumably occurred, the life span of most individuals was probably 30–40-years231, and morbidity and mortality from cardiovascular disorders would be limited. Moreover, as individuals would not develop hypertension and its consequences until after their prime reproductive years, the selection pressure for mechanisms that promoted survival in the face of circulatory shock would be greater than that exerted by the manifestations of cardiovascular disease in aged individuals.
Ambient conditions, at least in western societies, have changed dramatically since this selection for what was once an adaptive trait. This fact is particularly relevant in considering the functions of the RAAS, a system whose major components have been present since the divergence of bony fishes (about 400 million years ago)232. The interest in the RAAS for countless researchers and clinicians has been its role in body fluid homeostasis and blood pressure control. By contrast, the role of the RAAS as a major mediator of the stress response has received relatively little attention. Stressors increase activity in both the systemic and brain RAASs (reviewed elsewhere233). The RAAS is readily activated not only by challenges that produce hypovolaemia and blood pressure dyshomeostasis, but also by a wide range of stressors, including academic examinations234, restraint235, immobilization236, avoidance conditioning237, swimming238, pain238, hypoglycaemia239 and chronic mild stress240. Cumulative activation of the systemic and/or brain RAAS might induce a progressively greater and greater sensitized state of the CNS, and when sufficiently intense or sustained hypertensinogenic challenges are present (or when counter-regulatory or protective mechanisms begin to fail, as they do in ageing individuals), frank essential hypertension ensues. Thus, if plasticity-promoting phenotypic change was indeed incorporated into the ancestral human genome to protect against hypovolaemic shock, this trait may now have become a liability.
Conclusions
In summary, the recognition that a wide range of physiological and psychosocial challenges to actual or perceived potential homeostatic disruption can lead to HTRS provides a new paradigm for considering the causes and course of essential hypertension. Experiencing repeated challenges or stressors over the course of a lifetime can leave their mark by introducing neuroplastic changes in the brain network controlling sympathetic tone and blood pressure. By employing the IND-DEL-EXP experimental model, we have been able to begin to characterize the nature and range of stressors that induce HTRS and to investigate the sites of cellular and molecular neuroplasticity that relate the encoding of earlier events to the ongoing control of sympathetic tone and blood pressure.
One might be discouraged by realizing that stressor-induced insults are maintained as memories in the CNS and that the probability of expressing frank hypertension increases as time passes (TABLE 1). However, a basis for optimism remains, as pharmacological, surgical or behavioural interventions might be able to reverse the mediators and the expression of HTRS (TABLE 2).
The number of studies using the IND-DEL-EXP paradigm that link changes in the neural control of the circulation to mechanisms involving sensitization of the hypertensive response is as of yet small and the work is in its infancy. The recent demonstration of the phenomenon of HTRS and the identification of some accompanying indicators of CNS neuroplasticity allowing a sensitized state to be maintained over extended durations raises many new questions that need to be answered. Some examples of such new issues are presented in TABLE 3. Addressing many of these questions will provide deeper insights into the causes, treatment and prevention of essential hypertension.
Table 3:
Important Questions to be Addressed for a Better Understanding of the Role and Mechanisms of Neuroplasticity in Hypertensive Response Sensitization
|
Key points.
The aetiology of essential hypertension is still unknown.
Emerging evidence has shown that the hypertensive response can undergo sensitization.
Hypertensive response sensitization (HTRS) involves neuroplasticity induced by a wide range of physiological and behavioural challenges (stressors) occurring throughout life.
The cellular and molecular changes that mediate HTRS are located and maintained in the central neural network that controls sympathetic nervous system activity.
The neuroplasticity of the sympathetic nervous system provides adaptive blood pressure control, such that an increased hypertensive response (to physiological or psychosocial stressors) is learned and subsequently remembered.
Recognition of HTRS and the centrally mediated mechanisms driving the sensitized state provides a new paradigm for understanding essential hypertension and developing new strategies for its prevention and treatment.
Acknowledgements
The authors thank M. Dennis of the Department of Psychological and Brain Sciences at the University of Iowa for help in preparing the manuscript. The authors’ work described in this Review was supported by US National Institute of Health (NIH) grants HL14388 (AKJ), MH080241 (AKJ), HL 98207 (AKJ &BX), HL73986 (AKJ), HL84027 (AKJ), and HL139575 (AKJ)
GLOSSARY
- Response sensitization
Sensitization is operationally defined and occurs when repeated administration of a stimulus results in an increase in the magnitude of a response.
- General adaptation syndrome
A term describing the three predictable stages of behavioural and physiological responses to stressors. The ‘alarm reaction’ stage provides a burst of energy to deal with the onset of a stressor. In the ‘resistance’ stage, the body attempts to overcome or adapt to the stressor. Maintenance of the resistance stage is hypothesized to lead to ‘exhaustion’, associated with depletion of bodily resources, morbidity and mortality.
- Stressor
A threatening or noxious stimulus that produces a stress response and is associated with state defined as stress (i.e., an inferred state or hypothetical construct).
- Classical or Pavlovian conditioning
A learning paradigm first developed by the physiologist Ivan Pavlov. A biologically potent stimulus (such as food or an electric shock) is paired with a previously neutral stimulus (such as a tone or light). Pairing produces an association between the two stimuli, such that the neutral stimulus comes to elicit a response similar to that originally produced by a prepotent stimulus.
- Limbic system
An extensive set of phylogenetically old, interconnected brain structures located in the rostral part of the nervous system (forebrain). The limbic system was originally identified as a functional system related to emotion. Today, limbic structures are implicated in the control of many physiological, behavioural and cognitive functions.
- Lamina terminalis
The single layer of ependymal cells that form the rostral wall of the third cerebral ventricle. Four structures — the subfornical organ, median preoptic nucleus, the organum vasculosum of the lamina terminalis and the anterior commissure — lie immediately rostral to the lamina terminalis and are often, albeit technically erroneously, commonly referred to as the lamina terminalis.
- Neuromodulator
A substance released by neurons that acts to increase or decrease the actions of neurotransmitters. Neuromodulators affect large numbers of neurons by acting in a diffuse paracrine fashion, in contrast to the tight coupling between neurons using synaptic neurotransmitters to communicate.
- Cytokines
Proteins that are important in autocrine, paracrine, and endocrine signaling, particularly in the immune system. Pro-inflammatory cytokines promote inflammation whereas anti-inflammatory cytokines reduce inflammation. Adipokines are cytokines secreted by adipose tissue.
- Long-term potentiation
The strengthening of synapses that results from increased neural activity. Long-term potentiation facilitates synaptic transmission between adjacent neurons.
- Operant conditioning
Also known as instrumental conditioning. A type of learning in which a response is modified by positive or negative reinforcement: that is, by association with the presentation of either a reward (such as food) or a punishment (such as electric shock).
Footnotes
Competing interests
The authors declare no competing interests.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Further information
AMYPAD: http://amypad.eu
US National Institute on Aging–Alzheimer’s Association: https://alz.org/aaic/nia-aa.asp
Poster
Poster title: www.urlofposter.com
References
- 1.Xue B, Zhang Z, Johnson RF & Johnson AK Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension 59, 459–466, doi: 10.1161/hypertensionaha.111.185116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue B, Zhang Z, Roncari CF, Guo F & Johnson AK Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension 60, 1023–1030, doi: 10.1161/hypertensionaha.112.196576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon WB Wisdom of the Body. (WW Norton & Co, 1932). [Google Scholar]
- 4.Cannon WB The interrelations of emotions as suggested by recent physiological researches. Amer J Psychol 25, 256–282 (1914). [Google Scholar]
- 5.Cannon WB Bodily changes in pain, hunger, fear, and rage. (Appleton-Century-Crofts, 1929). [Google Scholar]
- 6.Hess WR & Brugger M Das subkortikale Zentrum der affektiven Abewehrreaktion. Helv Physiol Acta 1, 33–52 (1943). [Google Scholar]
- 7.Ranson SW Some Functions of the Hypothalamus: Harvey Lecture, December 17, 1936. Bull. N. Y. Acad. Med 13, 241–271 (1937). [PMC free article] [PubMed] [Google Scholar]
- 8.Hilton SM & Zbrozyna AW Amygdaloid region for defence reactions and its efferent pathway to the brain stem. J. Physiol. 165, 160–173 (1963). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abrahams VC, Hilton SM & Zbrozyna A Active muscle vasodilatation produced by stimulation of the brain stem: its significance in the defence reaction. J. Physiol 154, 491–513 (1960). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selye H Stress and the general adaptation syndrome. Br. Med. J 1, 1383–1392 (1950). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selye H The physiology and pathology of exposure to stress, a treatise based on the concepts of the general-adaptation syndrome and the diseases of adaptation. (ACTA, Inc. Medical Publishers, 1950). [Google Scholar]
- 12.Szabo S, Tache Y & Somogyi A The legacy of Hans Selye and the origins of stress research: a retrospective 75 years after his landmark brief “letter” to the editor# of nature. Stress 15, 472–478, doi: 10.3109/10253890.2012.710919 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Herman JP & Cullinan WE Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 20, 78–84 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Pacak K & Palkovits M Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr. Rev. 22, 502–548, doi: 10.1210/edrv.22.4.0436 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Sawchenko PE, Li HY & Ericsson A Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog. Brain Res. 122, 61–78 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Esler M et al. Chronic mental stress is a cause of essential hypertension: presence of biological markers of stress. Clin. Exp. Pharmacol. Physiol 35, 498–502, doi: 10.1111/j.1440-1681.2008.04904.x (2008). [DOI] [PubMed] [Google Scholar]
- 17.Folkow B Physiological aspects of primary hypertension. Physiol. Rev 62, 347–504 (1982). [DOI] [PubMed] [Google Scholar]
- 18.Folkow B Psychosocial and central nervous influences in primary hypertension. Circulation 76, I10–19 (1987). [PubMed] [Google Scholar]
- 19.Folkow B Mental “stress” and hypertension. Evidence from animal and experimental studies. Integr. Physiol. Behav. Sci 26, 305–308 (1991). [DOI] [PubMed] [Google Scholar]
- 20.Forouzanfar MH et al. Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990–2015. JAMA 317, 165–182, doi: 10.1001/jama.2016.19043 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Lim SS et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2224–2260, doi: 10.1016/s0140-6736(12)61766-8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet 389, 37–55, doi: 10.1016/s0140-6736(16)31919-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chow CK et al. Prevalence, awareness, treatment, and control of hypertension in rural and urban communities in high-, middle-, and low-income countries. JAMA 310, 959–968 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Whelton PK et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71, 1269–1324, doi: 10.1161/hyp.0000000000000066 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Chobanian AV et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 42, 1206–1252, doi: 10.1161/01.HYP.0000107251.49515.c2 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Carretero OA & Oparil S Essential hypertension : part II: treatment. Circulation 101, 446–453 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Cowley AW Jr. et al. Report of the National Heart, Lung, and Blood Institute Working Group on epigenetics and hypertension. Hypertension 59, 899–905, doi: 10.1161/hypertensionaha.111.190116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayet J & Hughes A Cardiac and vascular pathophysiology in hypertension. Heart 89, 1104–1109 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulvany MJ Small artery remodelling in hypertension. Basic Clin. Pharmacol. Toxicol 110, 49–55, doi: 10.1111/j.1742-7843.2011.00758.x (2012). [DOI] [PubMed] [Google Scholar]
- 30.Page IH Pathogenesis of arterial hypertension. J. Am. Med. Assoc 140, 451–458 (1949). [DOI] [PubMed] [Google Scholar]
- 31.Page IH The mosaic theory of arterial hypertension--its interpretation. Perspect. Biol. Med 10, 325–333 (1967). [DOI] [PubMed] [Google Scholar]
- 32.Sambhi MP Fundamental Fault in Hypertension. (M Nijhoff Kluwer, 1984). [Google Scholar]
- 33.Guyton AC, Coleman TG & Granger HJ Circulation: overall regulation. Annu. Rev. Physiol 34, 13–46, doi: 10.1146/annurev.ph.34.030172.000305 (1972). [DOI] [PubMed] [Google Scholar]
- 34.Guyton AC, Coleman TG, Young DB, Lohmeier TE & DeClue JW Salt balance and long-term blood pressure control. Annu. Rev. Med 31, 15–27, doi: 10.1146/annurev.me.31.020180.000311 (1980). [DOI] [PubMed] [Google Scholar]
- 35.Guyton AC et al. Integration and control of circulatory function. Int. Rev. Physiol 9, 341–385 (1976). [PubMed] [Google Scholar]
- 36.Guyton AC The relationship of cardiac output and arterial pressure control. Circulation 64, 1079–1088 (1981). [DOI] [PubMed] [Google Scholar]
- 37.Cowley AW Jr. & Guyton AC Baroreceptor reflex effects on transient and steady-state hemodynamics of salt-loading hypertension in dogs. Circ. Res 36, 536–546 (1975). [DOI] [PubMed] [Google Scholar]
- 38.Liard JF, Cowley AW Jr., McCaa RE, McCaa CS & Guyton AC Renin, aldosterone, body fluid volumes, and the baroreceptor reflex in the development and reversal of Goldblatt hypertension in conscious dogs. Circ. Res 34, 549–560 (1974). [DOI] [PubMed] [Google Scholar]
- 39.Folkow B Sympathetic nervous control of blood pressure. Role in primary hypertension. Am. J. Hypertens 2, 103s–111s (1989). [DOI] [PubMed] [Google Scholar]
- 40.Grassi G & Ram VS Evidence for a critical role of the sympathetic nervous system in hypertension. J. Am. Soc. Hypertens 10, 457–466, doi: 10.1016/j.jash.2016.02.015 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Julius S & Majahalme S The changing face of sympathetic overactivity in hypertension. Ann. Med 32, 365–370 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Mancia G & Grassi G The autonomic nervous system and hypertension. Circ. Res 114, 1804–1814 (2014). [DOI] [PubMed] [Google Scholar]
- 43.DiBona GF Sympathetic nervous system and hypertension. Hypertension 61, 556–560, doi: 10.1161/hypertensionaha.111.00633 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Grassi G, Mark A & Esler M The sympathetic nervous system alterations in human hypertension. Circ. Res 116, 976–990, doi: 10.1161/circresaha.116.303604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mancia G, Grassi G, Giannattasio C & Seravalle G Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 34, 724–728 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Esler M, Lambert E & Schlaich M Point: Chronic activation of the sympathetic nervous system is the dominant contributor to systemic hypertension. J Appl Physiol (1985) 109, 1996–1998; discussion 2016, doi: 10.1152/japplphysiol.00182.2010 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Dampney RA Functional organization of central pathways regulating the cardiovascular system. Physiol. Rev 74, 323–364 (1994). [DOI] [PubMed] [Google Scholar]
- 48.Guyenet PG The sympathetic control of blood pressure. Nat. Rev. Neurosci 7, 335–346, doi: 10.1038/nrn1902 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Dampney RA Central neural control of the cardiovascular system: current perspectives. Adv. Physiol. Educ 40, 283–296, doi: 10.1152/advan.00027.2016 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Spyer KM Annual review prize lecture. Central nervous mechanisms contributing to cardiovascular control. J. Physiol. 474, 1–19 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Johnson AK & Gross PM Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 7, 678–686 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Johnson AK & Thunhorst RL The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front. Neuroendocrinol. 18, 292–353 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Dampney RA Central mechanisms regulating coordinated cardiovascular and respiratory function during stress and arousal. Am. J. Physiol. Regul. Integr. Comp. Physiol 309, R429–443, doi: 10.1152/ajpregu.00051.2015 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Phelps EA & LeDoux JE Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron 48, 175–187, doi: 10.1016/j.neuron.2005.09.025 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Smith OA, Astley CA, DeVito JL, Stein JM & Walsh KE Functional analysis of hypothalamic control of the cardiovascular responses accompanying emotional behavior. Fed. Proc 39, 2487–2494 (1980). [PubMed] [Google Scholar]
- 56.Smith OA, DeVito JL & Astley CA Neurons controlling cardiovascular responses to emotion are located in lateral hypothalamus-perifornical region. Am. J. Physiol 259, R943–954, doi: 10.1152/ajpregu.1990.259.5.R943 (1990). [DOI] [PubMed] [Google Scholar]
- 57.Iwata J, LeDoux JE & Reis DJ Destruction of intrinsic neurons in the lateral hypothalamus disrupts the classical conditioning of autonomic but not behavioral emotional responses in the rat. Brain Res. 368, 161–166 (1986). [DOI] [PubMed] [Google Scholar]
- 58.LeDoux JE, Iwata J, Cicchetti P & Reis DJ Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J. Neurosci. 8, 2517–2529 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DiMicco JA, Samuels BC, Zaretskaia MV & Zaretsky DV The dorsomedial hypothalamus and the response to stress: part renaissance, part revolution. Pharmacol. Biochem. Behav 71, 469–480 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Stotz-Potter EH, Willis LR & DiMicco JA Muscimol acts in dorsomedial but not paraventricular hypothalamic nucleus to suppress cardiovascular effects of stress. J. Neurosci. 16, 1173–1179 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tigerstedt R & Bergmann PG Niere und Kreislauf. Skandinav Arch Physiol 8, 223 (1898). [Google Scholar]
- 62.Bader M Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu. Rev. Pharmacol. Toxicol 50, 439–465 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Ferrario CM New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 55, 445–452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santos RA & Ferreira AJ Angiotensin-(1–7) and the renin-angiotensin system. Curr. Opin. Nephrol. Hypertens 16, 122–128, doi: 10.1097/MNH.0b013e328031f362 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Fischer-Ferraro C, Nahmod VE, Goldstein DJ & Finkielman S Angiotensin and renin in rat and dog brain. J. Exp. Med 133, 353–361 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganten D et al. Angiotensin-forming enzyme in brain tissue. Science 173, 64–65 (1971). [DOI] [PubMed] [Google Scholar]
- 67.de Morais SDB, Shanks J & Zucker IH Integrative Physiological Aspects of Brain RAS in Hypertension. Curr. Hypertens. Rep 20, 10, doi: 10.1007/s11906-018-0810-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grobe JL, Xu D & Sigmund CD An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) 23, 187–193, doi: 10.1152/physiol.00002.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jackson L, Eldahshan W, Fagan SC & Ergul A Within the Brain: The Renin Angiotensin System. Int. J. Mol. Sci 19, doi: 10.3390/ijms19030876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lavoie JL & Sigmund CD Minireview: overview of the renin-angiotensin system--an endocrine and paracrine system. Endocrinology 144, 2179–2183, doi: 10.1210/en.2003-0150 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Wright JW & Harding JW The brain renin-angiotensin system: a diversity of functions and implications for CNS diseases. Pflugers Arch. 465, 133–151, doi: 10.1007/s00424-012-1102-2 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Johnson AK The periventricular anteroventral third ventricle (AV3V): its relationship with the subfornical organ and neural systems involved in maintaining body fluid homeostasis. Brain Res. Bull 15, 595–601 (1985). [DOI] [PubMed] [Google Scholar]
- 73.Lind RW & Johnson AK in The Renin Angiotensin System in the Brain (eds Ganten D, Printz M, Phillips MI, & Scholkens BA) 353–364 (Springer-Verlag, 1982). [Google Scholar]
- 74.Lind RW & Johnson AK Subfornical organ-median preoptic connections and drinking and pressor responses to angiotensin II. J. Neurosci 2, 1043–1051 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smith PM & Ferguson AV Circulating signals as critical regulators of autonomic state--central roles for the subfornical organ. Am. J. Physiol. Regul. Integr. Comp. Physiol 299, R405–415, doi: 10.1152/ajpregu.00103.2010 (2010). [DOI] [PubMed] [Google Scholar]
- 76.de Kloet AD, Liu M, Rodriguez V, Krause EG & Sumners C Role of neurons and glia in the CNS actions of the renin-angiotensin system in cardiovascular control. Am. J. Physiol. Regul. Integr. Comp. Physiol 309, R444–458, doi: 10.1152/ajpregu.00078.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marina N, Teschemacher AG, Kasparov S & Gourine AV Glia, sympathetic activity and cardiovascular disease. Exp. Physiol 101, 565–576, doi: 10.1113/ep085713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Felder RB Mineralocorticoid receptors, inflammation and sympathetic drive in a rat model of systolic heart failure. Exp. Physiol 95, 19–25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M & Demkow U Brain inflammation and hypertension: the chicken or the egg? J. Neuroinflammation 12, 85, doi: 10.1186/s12974-015-0306-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi P, Raizada MK & Sumners C Brain cytokines as neuromodulators in cardiovascular control. Clin. Exp. Pharmacol. Physiol 37, e52–57, doi: 10.1111/j.1440-1681.2009.05234.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sriramula S, Haque M, Majid DS & Francis J Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51, 1345–1351, doi: 10.1161/hypertensionaha.107.102152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shi P et al. Brain microglial cytokines in neurogenic hypertension. Hypertension 56, 297–303, doi: 10.1161/hypertensionaha.110.150409 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shen XZ et al. Microglia participate in neurogenic regulation of hypertension. Hypertension 66, 309–316, doi: 10.1161/hypertensionaha.115.05333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei SG, Yu Y & Felder RB Blood-borne interleukin-1beta acts on the subfornical organ to upregulate the sympathoexcitatory milieu of the hypothalamic paraventricular nucleus. Am. J. Physiol. Regul. Integr. Comp. Physiol 314, R447–r458, doi: 10.1152/ajpregu.00211.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei SG, Yu Y, Zhang ZH & Felder RB Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension 65, 1126–1133, doi: 10.1161/hypertensionaha.114.05112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wei SG et al. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62, 118–125, doi: 10.1161/HYPERTENSIONAHA.113.01404 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DeFelipe J Brain plasticity and mental processes: Cajal again. Nat. Rev. Neurosci 7, 811–817, doi: 10.1038/nrn2005 (2006). [DOI] [PubMed] [Google Scholar]
- 88.Hebb D The Organization of Behavior. (Wiley & Sons, 1949). [Google Scholar]
- 89.Bliss TV & Lomo T Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol 232, 331–356 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Malenka RC & Bear MF LTP and LTD: an embarrassment of riches. Neuron 44, 5–21, doi: 10.1016/j.neuron.2004.09.012 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Nicoll RA A Brief History of Long-Term Potentiation. Neuron 93, 281–290, doi: 10.1016/j.neuron.2016.12.015 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Alkadhi KA, Alzoubi KH & Aleisa AM Plasticity of synaptic transmission in autonomic ganglia. Prog. Neurobiol 75, 83–108, doi: 10.1016/j.pneurobio.2005.02.002 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Cifuentes F, Arias ER & Morales MA Long-term potentiation in mammalian autonomic ganglia: an inclusive proposal of a calcium-dependent, trans-synaptic process. Brain Res. Bull 97, 32–38, doi: 10.1016/j.brainresbull.2013.05.011 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Kandel ER, Dudai Y & Mayford MR The molecular and systems biology of memory. Cell 157, 163–186, doi: 10.1016/j.cell.2014.03.001 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Rahn EJ, Guzman-Karlsson MC & David Sweatt J Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: implications for pain and memory. Neurobiol. Learn. Mem 105, 133–150, doi: 10.1016/j.nlm.2013.06.008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Latremoliere A & Woolf CJ Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926, doi: 10.1016/j.jpain.2009.06.012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ren K & Dubner R Central nervous system plasticity and persistent pain. J. Orofac. Pain 13, 155–163 (1999). [PubMed] [Google Scholar]
- 98.Ren K & Dubner R Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol. Neurobiol 35, 224–235 (2007). [DOI] [PubMed] [Google Scholar]
- 99.Hyman SE, Malenka RC & Nestler EJ Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci 29, 565–598 (2006). [DOI] [PubMed] [Google Scholar]
- 100.Robinson MJ, Fischer AM, Ahuja A, Lesser EN & Maniates H Roles of “Wanting” and “Liking” in Motivating Behavior: Gambling, Food, and Drug Addictions. Curr. Top. Behav. Neurosci 27, 105–136, doi: 10.1007/7854_2015_387 (2016). [DOI] [PubMed] [Google Scholar]
- 101.Steketee JD & Kalivas PW Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol. Rev 63, 348–365, doi: 10.1124/pr.109.001933 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wolf ME The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog. Neurobiol 54, 679–720 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Hurley SW, Thunhorst RL, Johnson AK in Neurobiology of Body Fluid Homeostasis: Transduction and Integration (ed De Luca LA Jr, Menani JV, and Johnson AK) 279–301 (Taylor and Francis, 2013). [PubMed] [Google Scholar]
- 104.Na ES, Morris MJ, Johnson RF, Beltz TG & Johnson AK The neural substrates of enhanced salt appetite after repeated sodium depletions. Brain Res. 1171, 104–110 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kline DD Plasticity in glutamatergic NTS neurotransmission. Respir. Physiol. Neurobiol 164, 105–111, doi: 10.1016/j.resp.2008.04.013 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mifflin SW Short-term potentiation of carotid sinus nerve inputs to neurons in the nucleus of the solitary tract. Respir. Physiol 110, 229–236 (1997). [DOI] [PubMed] [Google Scholar]
- 107.Pinsker HM, Hening WA, Carew TJ & Kandel ER Long-term sensitization of a defensive withdrawal reflex in Aplysia. Science 182, 1039–1042 (1973). [DOI] [PubMed] [Google Scholar]
- 108.Barnett WH et al. Chemoreception and neuroplasticity in respiratory circuits. Exp. Neurol 287, 153–164, doi: 10.1016/j.expneurol.2016.05.036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cunningham JT, Knight WD, Mifflin SW & Nestler EJ An Essential role for DeltaFosB in the median preoptic nucleus in the sustained hypertensive effects of chronic intermittent hypoxia. Hypertension 60, 179–187, doi: 10.1161/hypertensionaha.112.193789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dempsey JA et al. Role of chemoreception in cardiorespiratory acclimatization to, and deacclimatization from, hypoxia. J Appl Physiol (1985) 116, 858–866, doi: 10.1152/japplphysiol.01126.2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lovett-Barr MR et al. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J. Neurosci 32, 3591–3600 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Herman JP Regulation of Hypothalamo-Pituitary-Adrenocortical Responses to Stressors by the Nucleus of the Solitary Tract/Dorsal Vagal Complex. Cell. Mol. Neurobiol 38, 25–35, doi: 10.1007/s10571-017-0543-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McCarty R Learning about stress: neural, endocrine and behavioral adaptations. Stress 19, 449–475, doi: 10.1080/10253890.2016.1192120 (2016). [DOI] [PubMed] [Google Scholar]
- 114.Michelini LC & Stern JE Exercise-induced neuronal plasticity in central autonomic networks: role in cardiovascular control. Exp. Physiol. 94, 947–960, doi: 10.1113/expphysiol.2009.047449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mueller PJ Exercise training and sympathetic nervous system activity: evidence for physical activity dependent neural plasticity. Clin. Exp. Pharmacol. Physiol 34, 377–384 (2007). [DOI] [PubMed] [Google Scholar]
- 116.Johnson AK et al. The roles of sensitization and neuroplasticity in the long-term regulation of blood pressure and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol 309, R1309–1325, doi: 10.1152/ajpregu.00037.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Houk JC Control strategies in physiological systems. FASEB J. 2, 97–107 (1988). [DOI] [PubMed] [Google Scholar]
- 118.Korner P Essential Hyeprtension and Its Causes: Neural and Non-Neural Mechanisms. (Oxford University Press, 2007). [Google Scholar]
- 119.Dickinson CJ & Yu R The progressive pressor response to angiotensin in the rabbit. J. Physiol 190, 91–99 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dickinson DM, Lawrence JR & Adelaide MB A slowly developing pressor response to small concentrations of angiotensin: its bearing on the pathogenesis of chronic renal hyeprtension. Lancet 281, 1354–1356 (1963). [DOI] [PubMed] [Google Scholar]
- 121.McCubbin JW, DeMoura RS, Page IH & Olmsted F Arterial hypertension elicited by subpressor amounts of angiotensin. Science 149, 1394–1395 (1965). [DOI] [PubMed] [Google Scholar]
- 122.Brown AJ, Casals-Stenzel J, Gofford S, Lever AF & Morton JJ Comparison of fast and slow pressor effects of angiotensin II in the conscious rat. Am. J. Physiol 241, H381–388 (1981). [DOI] [PubMed] [Google Scholar]
- 123.Kawada N, Imai E, Karber A, Welch WJ & Wilcox CS A mouse model of angiotensin II slow pressor response: role of oxidative stress. J. Am. Soc. Nephrol 13, 2860–2868 (2002). [DOI] [PubMed] [Google Scholar]
- 124.Hood SG, Cochrane T, McKinley MJ & May CN Investigation of the mechanisms by which chronic infusion of an acutely subpressor dose of angiotensin II induces hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol 292, R1893–1899, doi: 10.1152/ajpregu.00803.2006 (2007). [DOI] [PubMed] [Google Scholar]
- 125.Ames RP, Borkowski AJ, Sicinski AM & Laragh JH Prolonged Infusions of Angiotensin II and Norepinephrine and Blood Pressure, Electrolyte Balance, and Aldosterone and Cortisol Secretion in Normal Man and in Cirrhosis with Ascites. J. Clin. Invest 44, 1171–1186 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bohr DF in Angiotensin: Handbook of Experimental Pharmacology Vol. 37 (ed Bumpus FM Page IH) 424 (Springer-Verlag, 1974). [Google Scholar]
- 127.Godfraind T Angiotensin auto-potentiation. Br. J. Pharmacol 40, 542P–543P (1970). [PMC free article] [PubMed] [Google Scholar]
- 128.Skulan TW, Brousseau AC & Leonard KA Accelerated induction to two-kidney hypertension in rats and renin-angiotensin sensitivity. Circ. Res 35, 734–741 (1974). [DOI] [PubMed] [Google Scholar]
- 129.ten Berg R & de Jong W Mechanism of enhanced blood pressure rise after reclipping following removal of a renal artery clip in rats. Hypertension 2, 4–13 (1980). [DOI] [PubMed] [Google Scholar]
- 130.Aoki K & Masson GM Pressor responsiveness to renin and angiotensin in renal hypertensive rats. Nephron 6, 484–497 (1969). [DOI] [PubMed] [Google Scholar]
- 131.Xue B et al. Central Renin-Angiotensin System Activation and Inflammation Induced by High-Fat Diet Sensitize Angiotensin II-Elicited Hypertension. Hypertension 67, 163–170, doi: 10.1161/hypertensionaha.115.06263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xue B et al. Post-traumatic stress sensitizes the angiotensin II-elicited hypertensive response. FASEB J. , 866.862 (2017). [Google Scholar]
- 133.Xue B et al. Post-traumatic stress-induced sensitization of angiotensin II hyeprtension is reversed by blockade of angiotensin-converting enzyme or tumor necrosis factor-alpha. Hypertension 404, P389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Xue B et al. Leptin Mediates High-Fat Diet Sensitization of Angiotensin II-Elicited Hypertension by Upregulating the Brain Renin-Angiotensin System and Inflammation. Hypertension 67, 970–976, doi: 10.1161/hypertensionaha.115.06736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang YP et al. Maternal high-fat diet acts on the brain to induce baroreflex dysfunction and sensitization of angiotensin II-induced hypertension in adult offspring. Am. J. Physiol. Heart Circ. Physiol. 314, H1061–h1069, doi: 10.1152/ajpheart.00698.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Barth SW & Gerstberger R Differential regulation of angiotensinogen and AT1A receptor mRNA within the rat subfornical organ during dehydration. Brain Res. Mol. Brain Res 64, 151–164 (1999). [DOI] [PubMed] [Google Scholar]
- 137.Charron G, Laforest S, Gagnon C, Drolet G & Mouginot D Acute sodium deficit triggers plasticity of the brain angiotensin type 1 receptors. FASEB J. 16, 610–612 (2002). [DOI] [PubMed] [Google Scholar]
- 138.Chen Y, da Rocha MJ & Morris M Osmotic regulation of angiotensin AT1 receptor subtypes in mouse brain. Brain Res. 965, 35–44 (2003). [DOI] [PubMed] [Google Scholar]
- 139.Moellenhoff E et al. Effect of repetitive icv injections of ANG II on c-Fos and AT1-receptor expression in the rat brain. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 280, R1095–R1104 (2001). [DOI] [PubMed] [Google Scholar]
- 140.Nunes FC & Braga VA Chronic angiotensin II infusion modulates angiotensin II type I receptor expression in the subfornical organ and the rostral ventrolateral medulla in hypertensive rats. J. Renin Angiotensin Aldosterone Syst 12, 440–445, doi: 10.1177/1470320310394891 (2011). [DOI] [PubMed] [Google Scholar]
- 141.Sanvitto GL, Johren O, Hauser W & Saavedra JM Water deprivation upregulates ANG II AT1 binding and mRNA in rat subfornical organ and anterior pituitary. Am. J. Physiol 273, E156–163 (1997). [DOI] [PubMed] [Google Scholar]
- 142.Wilson KM, Sumners C & Fregly MJ Effects of increased circulating angiotensin II (AII) on fluid exchange and binding of AII in the brain. Brain Res. Bull 20, 493–501 (1988). [DOI] [PubMed] [Google Scholar]
- 143.King SJ, Harding JW & Moe KE Elevated salt appetite and brain binding of angiotensin II in mineralocorticoid-treated rats. Brain Res. 448, 140–149 (1988). [DOI] [PubMed] [Google Scholar]
- 144.Shelat SG, Flanagan-Cato LM & Fluharty SJ Glucocorticoid and mineralocorticoid regulation of angiotensin II type 1 receptor binding and inositol triphosphate formation in WB cells. J. Endocrinol 162, 381–391 (1999). [DOI] [PubMed] [Google Scholar]
- 145.Shelat SG, King JL, Flanagan-Cato LM & Fluharty SJ Mineralocorticoids and glucocorticoids cooperatively increase salt intake and angiotensin II receptor binding in rat brain. Neuroendocrinology 69, 339–351, doi:54436 (1999). [DOI] [PubMed] [Google Scholar]
- 146.Wilson KM, Sumners C, Hathaway S & Fregly MJ Mineralocorticoids modulate central angiotensin II receptors in rats. Brain Res. 382, 87–96 (1986). [DOI] [PubMed] [Google Scholar]
- 147.Laragh JH & Sealey JE The plasma renin test reveals the contribution of body sodium-volume content (V) and renin-angiotensin (R) vasoconstriction to long-term blood pressure. Am. J. Hypertens 24, 1164–1180, doi: 10.1038/ajh.2011.171 (2011). [DOI] [PubMed] [Google Scholar]
- 148.McAreavey D & Robertson JI Angiotensin converting enzyme inhibitors and moderate hypertension. Drugs 40, 326–345 (1990). [DOI] [PubMed] [Google Scholar]
- 149.Castellucci V & Kandel ER Presynaptic facilitation as a mechanism for behavioral sensitization in Aplysia. Science 194, 1176–1178 (1976). [DOI] [PubMed] [Google Scholar]
- 150.Clayton SC, Zhang Z, Beltz T, Xue B & Johnson AK CNS neuroplasticity and salt-sensitive hypertension induced by prior treatment with subpressor doses of ANG II or aldosterone. Am. J. Physiol. Regul. Integr. Comp. Physiol 306, R908–917, doi: 10.1152/ajpregu.00010.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Huang BS, Ahmadi S, Ahmad M, White RA & Leenen FH Central neuronal activation and pressor responses induced by circulating ANG II: role of the brain aldosterone-”ouabain” pathway. Am. J. Physiol. Heart Circ. Physiol 299, H422–430, doi: 10.1152/ajpheart.00256.2010 (2010). [DOI] [PubMed] [Google Scholar]
- 152.Xue B et al. Central interactions of aldosterone and angiotensin II in aldosterone- and angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol 300, H555–564, doi: 10.1152/ajpheart.00847.2010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Git KC & Adan RA Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes. Rev 16, 207–224, doi: 10.1111/obr.12243 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Hall JE, do Carmo JM, da Silva AA, Wang Z & Hall ME Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ. Res. 116, 991–1006, doi: 10.1161/circresaha.116.305697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kalupahana NS & Moustaid-Moussa N The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes. Rev 13, 136–149, doi: 10.1111/j.1467-789X.2011.00942.x (2012). [DOI] [PubMed] [Google Scholar]
- 156.Sriramula S, Cardinale JP & Francis J Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One 8, e63847, doi: 10.1371/journal.pone.0063847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yu Y et al. Early interference with p44/42 mitogen-activated protein kinase signaling in hypothalamic paraventricular nucleus attenuates angiotensin II-induced hypertension. Hypertension 61, 842–849, doi: 10.1161/hypertensionaha.111.00080 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet 355, 253–259 (2000). [PubMed] [Google Scholar]
- 159.Alonso-Galicia M, Brands MW, Zappe DH & Hall JE Hypertension in obese Zucker rats. Role of angiotensin II and adrenergic activity. Hypertension 28, 1047–1054 (1996). [DOI] [PubMed] [Google Scholar]
- 160.Armitage JA et al. Rapid onset of renal sympathetic nerve activation in rabbits fed a high-fat diet. Hypertension 60, 163–171, doi: 10.1161/hypertensionaha.111.190413 (2012). [DOI] [PubMed] [Google Scholar]
- 161.Lim K, Burke SL & Head GA Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension 61, 628–634, doi: 10.1161/hypertensionaha.111.00705 (2013). [DOI] [PubMed] [Google Scholar]
- 162.Prior LJ et al. Exposure to a high-fat diet alters leptin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 55, 862–868, doi: 10.1161/hypertensionaha.109.141119 (2010). [DOI] [PubMed] [Google Scholar]
- 163.Maric T, Woodside B & Luheshi GN The effects of dietary saturated fat on basal hypothalamic neuroinflammation in rats. Brain. Behav. Immun 36, 35–45, doi: 10.1016/j.bbi.2013.09.011 (2014). [DOI] [PubMed] [Google Scholar]
- 164.Hall JE, Crook ED, Jones DW, Wofford MR & Dubbert PM Mechanisms of obesity-associated cardiovascular and renal disease. Am. J. Med. Sci 324, 127–137 (2002). [DOI] [PubMed] [Google Scholar]
- 165.Tchernof A & Despres JP Pathophysiology of human visceral obesity: an update. Physiol. Rev 93, 359–404, doi: 10.1152/physrev.00033.2011 (2013). [DOI] [PubMed] [Google Scholar]
- 166.Harlan SM et al. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ. Res 108, 808–812, doi: 10.1161/circresaha.111.240226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shi Z, Li B & Brooks VL Role of the Paraventricular Nucleus of the Hypothalamus in the Sympathoexcitatory Effects of Leptin. Hypertension 66, 1034–1041, doi: 10.1161/hypertensionaha.115.06017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Young CN, Morgan DA, Butler SD, Mark AL & Davisson RL The brain subfornical organ mediates leptin-induced increases in renal sympathetic activity but not its metabolic effects. Hypertension 61, 737–744, doi: 10.1161/hypertensionaha.111.00405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gao Y et al. Hormones and diet, but not body weight, control hypothalamic microglial activity. Glia 62, 17–25, doi: 10.1002/glia.22580 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.de Kloet AD et al. Obesity induces neuroinflammation mediated by altered expression of the renin-angiotensin system in mouse forebrain nuclei. Physiol. Behav 136, 31–38, doi: 10.1016/j.physbeh.2014.01.016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hilzendeger AM et al. A brain leptin-renin angiotensin system interaction in the regulation of sympathetic nerve activity. Am. J. Physiol. Heart Circ. Physiol 303, H197–206, doi: 10.1152/ajpheart.00974.2011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang X et al. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73, doi: 10.1016/j.cell.2008.07.043 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Fraser A, Nelson SM, Macdonald-Wallis C, Sattar N & Lawlor DA Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension 62, 614–620, doi: 10.1161/hypertensionaha.113.01513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Himmelmann A, Svensson A & Hansson L Five-year follow-up of blood pressure and left ventricular mass in children with different maternal histories of hypertension: the Hypertension in Pregnancy Offspring Study. J. Hypertens 12, 89–95 (1994). [PubMed] [Google Scholar]
- 175.Himmelmann A, Svensson A & Hansson L Relation of maternal blood pressure during pregnancy to birth weight and blood pressure in children. The Hypertension in Pregnancy Offspring Study. J. Intern. Med 235, 347–352 (1994). [DOI] [PubMed] [Google Scholar]
- 176.Lazdam M et al. Elevated blood pressure in offspring born premature to hypertensive pregnancy: is endothelial dysfunction the underlying vascular mechanism? Hypertension 56, 159–165, doi: 10.1161/hypertensionaha.110.150235 (2010). [DOI] [PubMed] [Google Scholar]
- 177.Staley JR et al. Associations of blood pressure in pregnancy with offspring blood pressure trajectories during childhood and adolescence: findings from a prospective study. J Am Heart Assoc 4, doi: 10.1161/jaha.114.001422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tenhola S, Rahiala E, Halonen P, Vanninen E & Voutilainen R Maternal preeclampsia predicts elevated blood pressure in 12-year-old children: evaluation by ambulatory blood pressure monitoring. Pediatr. Res 59, 320–324, doi: 10.1203/01.pdr.0000196734.54473.e3 (2006). [DOI] [PubMed] [Google Scholar]
- 179.Kajantie E, Eriksson JG, Osmond C, Thornburg K & Barker DJ Pre-eclampsia is associated with increased risk of stroke in the adult offspring: the Helsinki birth cohort study. Stroke 40, 1176–1180, doi: 10.1161/strokeaha.108.538025 (2009). [DOI] [PubMed] [Google Scholar]
- 180.Liang M, Cowley AW Jr., Mattson DL, Kotchen TA & Liu Y Epigenomics of hypertension. Semin. Nephrol 33, 392–399, doi: 10.1016/j.semnephrol.2013.05.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lopes HF et al. Increased sympathetic activity in normotensive offspring of malignant hypertensive parents compared to offspring of normotensive parents. Braz. J. Med. Biol. Res 41, 849–853 (2008). [DOI] [PubMed] [Google Scholar]
- 182.Washburn LK et al. The renin-angiotensin-aldosterone system in adolescent offspring born prematurely to mothers with preeclampsia. J. Renin Angiotensin Aldosterone Syst 16, 529–538, doi: 10.1177/1470320314526940 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Alexander BT, Hendon AE, Ferril G & Dwyer TM Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 45, 754–758, doi: 10.1161/01.HYP.0000153319.20340.2a (2005). [DOI] [PubMed] [Google Scholar]
- 184.de Almeida Chaves Rodrigues AF et al. Increased renal sympathetic nerve activity leads to hypertension and renal dysfunction in offspring from diabetic mothers. Am. J. Physiol. Renal Physiol 304, F189–197, doi: 10.1152/ajprenal.00241.2012 (2013). [DOI] [PubMed] [Google Scholar]
- 185.Intapad S et al. Renal denervation abolishes the age-dependent increase in blood pressure in female intrauterine growth-restricted rats at 12 months of age. Hypertension 61, 828–834, doi: 10.1161/hypertensionaha.111.00645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Langley-Evans SC & Jackson AA Captopril normalises systolic blood pressure in rats with hypertension induced by fetal exposure to maternal low protein diets. Comparative biochemistry and physiology. Part A, Physiology 110, 223–228 (1995). [DOI] [PubMed] [Google Scholar]
- 187.Mansuri A, Elmaghrabi A, Legan SK, Gattineni J & Baum M Transient Exposure of Enalapril Normalizes Prenatal Programming of Hypertension and Urinary Angiotensinogen Excretion. PLoS One 10, e0146183, doi: 10.1371/journal.pone.0146183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mizuno M, Lozano G, Siddique K, Baum M & Smith SA Enalapril attenuates the exaggerated sympathetic response to physical stress in prenatally programmed hypertensive rats. Hypertension 63, 324–329, doi: 10.1161/hypertensionaha.113.02330 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mizuno M, Siddique K, Baum M & Smith SA Prenatal programming of hypertension induces sympathetic overactivity in response to physical stress. Hypertension 61, 180–186, doi: 10.1161/hypertensionaha.112.199356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pladys P et al. Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr. Res 55, 1042–1049, doi: 10.1203/01.pdr.0000127012.37315.36 (2004). [DOI] [PubMed] [Google Scholar]
- 191.Xue B, Yin H, Guo F, Beltz T, Thunhorst RL, Johnson AK Maternal gestational hypertension-induced sensitization of angiotensin II hypertension in offspring and its reversal by renal denervation or angiotensin converting enzyme inhibition in rats. Hypertension 69, 669–677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xue B, Beltz TG, Guo F & Johnson AK Sex differences in maternal gestational hypertension-induced sensitization of angiotensin II hypertension in rat offspring: the protective effect of estrogen. Am. J. Physiol. Regul. Integr. Comp. Physiol 314, R274–R281, doi: 10.1152/ajpregu.00216.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Xue B et al. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am. J. Physiol. Heart Circ. Physiol 307, H191–198, doi: 10.1152/ajpheart.01012.2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Alexander BT, Dasinger JH & Intapad S Fetal programming and cardiovascular pathology. Compr Physiol 5, 997–1025, doi: 10.1002/cphy.c140036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dong M, Zheng Q, Ford SP, Nathanielsz PW & Ren J Maternal obesity, lipotoxicity and cardiovascular diseases in offspring. J. Mol. Cell. Cardiol 55, 111–116, doi: 10.1016/j.yjmcc.2012.08.023 (2013). [DOI] [PubMed] [Google Scholar]
- 196.Gademan MG et al. Maternal prepregnancy body mass index and their children’s blood pressure and resting cardiac autonomic balance at age 5 to 6 years. Hypertension 62, 641–647, doi: 10.1161/hypertensionaha.113.01511 (2013). [DOI] [PubMed] [Google Scholar]
- 197.Reynolds RM et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ 347, f4539, doi: 10.1136/bmj.f4539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Prior LJ et al. Exposure to a high-fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 63, 338–345, doi: 10.1161/hypertensionaha.113.02498 (2014). [DOI] [PubMed] [Google Scholar]
- 199.Cesar HC & Pisani LP Fatty-acid-mediated hypothalamic inflammation and epigenetic programming. J. Nutr. Biochem 42, 1–6, doi: 10.1016/j.jnutbio.2016.08.008 (2017). [DOI] [PubMed] [Google Scholar]
- 200.Deng Y et al. Prenatal inflammation-induced NF-kappaB dyshomeostasis contributes to renin-angiotensin system over-activity resulting in prenatally programmed hypertension in offspring. Sci. Rep 6, 21692, doi: 10.1038/srep21692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Samuelsson AM et al. Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension 55, 76–82, doi: 10.1161/hypertensionaha.109.139402 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Levine S & Mullins RF Jr. Hormonal influences on brain organization in infant rats. Science 152, 1585–1592 (1966). [DOI] [PubMed] [Google Scholar]
- 203.Scott JP Critical periods in behavioral development. Science 138, 949–958 (1962). [DOI] [PubMed] [Google Scholar]
- 204.Viken RJ, Johnson AK & Knutson JF Blood pressure, heart rate, and regional resistance in behavioral defense. Physiol. Behav 50, 1097–1101 (1991). [DOI] [PubMed] [Google Scholar]
- 205.Finnell JE & Wood SK Neuroinflammation at the interface of depression and cardiovascular disease: Evidence from rodent models of social stress. Neurobiol Stress 4, 1–14, doi: 10.1016/j.ynstr.2016.04.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Michopoulos V, Powers A, Gillespie CF, Ressler KJ & Jovanovic T Inflammation in Fear- and Anxiety-Based Disorders: PTSD, GAD, and Beyond. Neuropsychopharmacology 42, 254–270, doi: 10.1038/npp.2016.146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Brudey C et al. Autonomic and inflammatory consequences of posttraumatic stress disorder and the link to cardiovascular disease. Am. J. Physiol. Regul. Integr. Comp. Physiol 309, R315–321, doi: 10.1152/ajpregu.00343.2014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Park J et al. Baroreflex dysfunction and augmented sympathetic nerve responses during mental stress in veterans with post-traumatic stress disorder. J. Physiol 595, 4893–4908, doi: 10.1113/jp274269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Edmondson D et al. The Association of Posttraumatic Stress Disorder With Clinic and Ambulatory Blood Pressure in Healthy Adults. Psychosom. Med 80, 55–61, doi: 10.1097/psy.0000000000000523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kibler JL, Joshi K & Ma M Hypertension in relation to posttraumatic stress disorder and depression in the US National Comorbidity Survey. Behav. Med 34, 125–132, doi: 10.3200/bmed.34.4.125-132 (2009). [DOI] [PubMed] [Google Scholar]
- 211.Paulus EJ, Argo TR & Egge JA The impact of posttraumatic stress disorder on blood pressure and heart rate in a veteran population. J. Trauma. Stress 26, 169–172, doi: 10.1002/jts.21785 (2013). [DOI] [PubMed] [Google Scholar]
- 212.Roy SS, Foraker RE, Girton RA & Mansfield AJ Posttraumatic stress disorder and incident heart failure among a community-based sample of US veterans. Am. J. Public Health 105, 757–763, doi: 10.2105/ajph.2014.302342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Khoury NM et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J. Clin. Psychiatry 73, 849–855, doi: 10.4088/JCP.11m07316 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Levkovitz Y, Fenchel D, Kaplan Z, Zohar J & Cohen H Early post-stressor intervention with minocycline, a second-generation tetracycline, attenuates post-traumatic stress response in an animal model of PTSD. Eur. Neuropsychopharmacol 25, 124–132, doi: 10.1016/j.euroneuro.2014.11.012 (2015). [DOI] [PubMed] [Google Scholar]
- 215.Elijovich F et al. Salt Sensitivity of Blood Pressure: A Scientific Statement From the American Heart Association. Hypertension 68, e7–e46, doi: 10.1161/hyp.0000000000000047 (2016). [DOI] [PubMed] [Google Scholar]
- 216.He FJ & MacGregor GA Salt and sugar: their effects on blood pressure. Pflugers Arch. 467, 577–586, doi: 10.1007/s00424-014-1677-x (2015). [DOI] [PubMed] [Google Scholar]
- 217.MacGregor GA & de Wardener HE Salt, Diet, and Health. (Cambridge University Press, 1998). [Google Scholar]
- 218.Dahl LK, Heine M & Tassinari L Role of genetic factors in susceptibility to experimental hypertension due to chronic excess salt ingestion. Nature 194, 480–482 (1962). [DOI] [PubMed] [Google Scholar]
- 219.Denton D et al. The effect of increased salt intake on blood pressure of chimpanzees. Nat. Med 1, 1009–1016 (1995). [DOI] [PubMed] [Google Scholar]
- 220.Weinberger MH, Miller JZ, Luft FC, Grim CE & Fineberg NS Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 8, II127–134 (1986). [DOI] [PubMed] [Google Scholar]
- 221.Kanbay M, Chen Y, Solak Y & Sanders PW Mechanisms and consequences of salt sensitivity and dietary salt intake. Curr. Opin. Nephrol. Hypertens 20, 37–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Brooks VL, Scrogin KE & McKeogh DF The interaction of angiotensin II and osmolality in the generation of sympathetic tone during changes in dietary salt intake. An hypothesis. Ann. N. Y. Acad. Sci 940, 380–394 (2001). [DOI] [PubMed] [Google Scholar]
- 223.Huang BS, Amin MS & Leenen FH The central role of the brain in salt-sensitive hypertension. Curr. Opin. Cardiol 21, 295–304, doi: 10.1097/01.hco.0000231398.64362.94 (2006). [DOI] [PubMed] [Google Scholar]
- 224.Oki K, Gomez-Sanchez EP & Gomez-Sanchez CE Role of mineralocorticoid action in the brain in salt-sensitive hypertension. Clin. Exp. Pharmacol. Physiol 39, 90–95, doi: 10.1111/j.1440-1681.2011.05538.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Osborn JW, Fink GD, Sved AF, Toney GM & Raizada MK Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr. Hypertens. Rep 9, 228–235 (2007). [DOI] [PubMed] [Google Scholar]
- 226.Florin M, Lo M, Liu KL & Sassard J Salt sensitivity in genetically hypertensive rats of the Lyon strain. Kidney Int. 59, 1865–1872 (2001). [DOI] [PubMed] [Google Scholar]
- 227.Lo M, Liu KL, Clemitson JR, Sassard J & Samani NJ Chromosome 1 blood pressure QTL region influences renal function curve and salt sensitivity in SHR. Physiol. Genomics 8, 15–21, doi: 10.1152/physiolgenomics.00057.2001 (2002). [DOI] [PubMed] [Google Scholar]
- 228.Netea MG et al. Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098, doi: 10.1126/science.aaf1098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Haley MJ, Brough D, Quintin J & Allan SM Microglial Priming as Trained Immunity in the Brain. Neuroscience, doi: 10.1016/j.neuroscience.2017.12.039 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Beldade P, Mateus AR & Keller RA Evolution and molecular mechanisms of adaptive developmental plasticity. Mol. Ecol. 20, 1347–1363, doi: 10.1111/j.1365-294X.2011.05016.x (2011). [DOI] [PubMed] [Google Scholar]
- 231.Trinkaus E Late Pleistocene adult mortality patterns and modern human establishment. Proc. Natl. Acad. Sci. U. S. A 108, 1267–1271, doi: 10.1073/pnas.1018700108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fournier D, Luft FC, Bader M, Ganten D & Andrade-Navarro MA Emergence and evolution of the renin-angiotensin-aldosterone system. J. Mol. Med. (Berl.) 90, 495–508, doi: 10.1007/s00109-012-0894-z (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Saavedra JM & Benicky J Brain and peripheral angiotensin II play a major role in stress. Stress 10, 185–193, doi: 10.1080/10253890701350735 (2007). [DOI] [PubMed] [Google Scholar]
- 234.Syvalahti E, Lammintausta R & Pekkarinen A Effect of psychic stress of examination on serum growth hormone, serum insulin, and plasma renin activity. Acta Pharmacol. Toxicol. (Copenh.) 38, 344–352 (1976). [DOI] [PubMed] [Google Scholar]
- 235.Sigg EB, Keim KL & Sigg TD On the mechanism of renin release by restraint stress in rats. Pharmacol. Biochem. Behav 8, 47–50 (1978). [DOI] [PubMed] [Google Scholar]
- 236.Golin RM, Gotoh E, Said SI & Ganong WF Pharmacological evidence that the sympathetic nervous system mediates the increase in renin secretion produced by immobilization and head-up tilt in rats. Neuropharmacology 27, 1209–1213 (1988). [DOI] [PubMed] [Google Scholar]
- 237.Blair ML, Feigl EO & Smith OA Elevation of plasma renin activity during avoidance performance in baboons. Am. J. Physiol 231, 772–776, doi: 10.1152/ajplegacy.1976.231.3.772 (1976). [DOI] [PubMed] [Google Scholar]
- 238.Bozovic L & Castenfors J Effect of ganglionic blocking on plasma renin activity in exercising and pain-stressed rats. Acta Physiol. Scand 70, 290–292, doi: 10.1111/j.1748-1716.1967.tb03628.x (1967). [DOI] [PubMed] [Google Scholar]
- 239.Otsuka K, Assaykeen TA, Goldfien A & Ganong WF Effect of hypoglycemia on plasma renin activity in dogs. Endocrinology 87, 1306–1317, doi: 10.1210/endo-87-6-1306 (1970). [DOI] [PubMed] [Google Scholar]
- 240.Grippo AJ, Francis J, Beltz TG, Felder RB & Johnson AK Neuroendocrine and cytokine profile of chronic mild stress-induced anhedonia. Physiol. Behav 84, 697–706, doi: 10.1016/j.physbeh.2005.02.011 (2005). [DOI] [PubMed] [Google Scholar]
- 241.Cox BF & Bishop VS Neural and humoral mechanisms of angiotensin-dependent hypertension. Am. J. Physiol 261, H1284–1291 (1991). [DOI] [PubMed] [Google Scholar]
- 242.Seybold VS The role of peptides in central sensitization. Handb. Exp. Pharmacol, 451–491, doi: 10.1007/978-3-540-79090-7_13 (2009). [DOI] [PubMed] [Google Scholar]
- 243.Citri A & Malenka RC Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41, doi: 10.1038/sj.npp.1301559 (2008). [DOI] [PubMed] [Google Scholar]
- 244.Rudy JW The Neurobiology of Learning and Memory. 2nd edn, (Sinauer Associates, Inc, 2014). [Google Scholar]
- 245.Zhou Y & Danbolt NC Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna) 121, 799–817, doi: 10.1007/s00702-014-1180-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yirmiya R & Goshen I Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain. Behav. Immun 25, 181–213, doi: 10.1016/j.bbi.2010.10.015 (2011). [DOI] [PubMed] [Google Scholar]
- 247.Xue B, Beltz TG, Fuo F & Johnson AK Blockade of glutamate receptors abolishes the sensitization of the angiotensin II-elicited hypertensive response in rats. FASEB J. 32 (Supplement 1):732.1, (2018). [Google Scholar]
- 248.Xue B, Beltz TG, Guo F, Thunhorst RL & Johnson AK Controlled hypotensive hemorrhage sensitizes angiotensin II-elicited hypertension. FASEB J. 30(1 Suppl):1234.2 (2016).26601824 [Google Scholar]