

Abstract
Enhanced vascular permeability is associated with inflammation and edema in alveoli during the exudative phase of acute respiratory distress syndrome (ARDS). Mechanisms leading to the endothelial contribution on the early exudative stage of ARDS are not precise. We hypothesized that modulation of endothelial stromelysin1 expression and activity by Akt1-forkhead box-O transcription factors 1/3a (FoxO1/3a) pathway could play a significant role in regulating pulmonary edema during the initial stages of acute lung injury (ALI). We utilized lipopolysaccharide (LPS)-induced mouse ALI model in vivo and endothelial barrier resistance measurements in vitro to determine the specific role of the endothelial Akt1-FoxO1/3a-stromelysin1 pathway in ALI. LPS treatment of human pulmonary endothelial cells resulted in increased stromelysin1 and reduced tight junction claudin5 involving FoxO1/3a, associated with decreased trans-endothelial barrier resistance as determined by electric cell-substrate impedance sensing technology. In vivo, LPS-induced lung edema was significantly higher in endothelial Akt1 knockdown (EC-Akt1−/−) compared to wild-type mice, which was reversed upon treatment with FoxO inhibitor (AS1842856), stromelysin1 inhibitor (UK356618) or with shRNA-mediated FoxO1/3a depletion in the mouse lungs. Overall, our study provides the hope that targeting FoxO and styromelysin1 could be beneficial in the treatment of ALI.
Keywords: stromelysin1, MMP3, Akt1, FoxO, claudin5, lung injury, ARDS
Graphical abstract
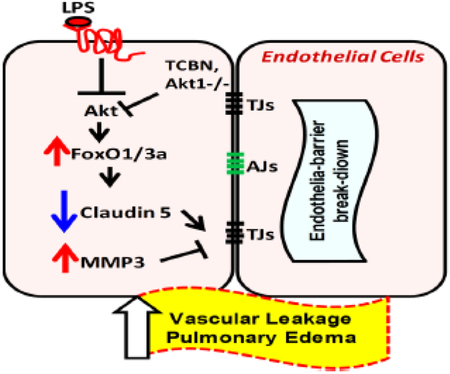
The suppression of Akt1 activity by LPS in pulmonary endothelial cells results in increased FoxO1/3a activation, in turn, leading to increased stromelysin1 expression/activity and reduced expression of tight junction proteins, particularly claudin5. Pharmacologically targeting FoxO or stromelysin activity inhibits LPS-induced pulmonary vascular injury and inflammation
INTRODUCTION
Disruption of the alveolar-capillary unit symbolizes the exudative phase of acute respiratory distress syndrome (ARDS) [1-3]. ARDS is an important cause of acute respiratory failure that is often associated with multiple organ failure and high mortality among ICU patients. ARDS incidence ranges from 10 to 86 cases per 100,000, with the highest rates reported in Australia and the United States [2]. Histologically, the slightly thicker part of blood-air barrier is also composed of the extracellular matrix (ECM) [4]. Endothelial injury and consequent vascular permeability ensuing influx of protein-rich fluid into the alveolar air spaces is a well-established pathological event occurring in the acute/exudative stage of ARDS [5]. Therefore, targeting disruption of the capillary endothelial barrier could provide a potential therapy for ARDS.
Capillary vascular permeability regulated by paracellular pathways play a prominent role in lung edema [6, 7]. This is largely regulated by adherens junction (AJ) proteins such as VE-cadherin and tight junction (TJ) proteins such as claudins [8, 9]. Pulmonary endothelium expresses higher levels of claudin5 than the alveolar epithelium and a decrease in claudin5 expression is associated with aberrant vascular permeability and severe ALI in patients with pneumonia [9, 10]. Although vascular leakage has not been studied in the lungs of Claudin5−/− mice that die within 10 hours after birth, these mice have disrupted blood-brain permeability indicating its importance in maintaining blood-tissue barrier integrity [11]. Whereas TJs are more developed in capillaries, AJs are dominant in post-capillary venules [12]. Lung edema results from the capillary leak, and hence TJs play a prominent role in the maintenance of alveolar-capillary integrity [13-15].
In addition to the damage to the capillary endothelium, disruption of ECM significantly contributes to vascular leak [16]. Matrix metalloproteinases (MMPs) are a diverse family of ECM proteinases that have recently been implicated in destructive pulmonary pathologies [4]. Clinical studies have demonstrated an increase in MMPs in general with more severe ARDS accompanied by an increase in distal organ failure and mortality [17, 18]. Gene knockout studies have shown the importance of stromelysin1 (MMP3) in tissue injury [19, 20]. While it has been shown that the presence of stromelysin1 is associated with worsened ALI, specific involvement of stromelysin1 in mediating ALI, its source of synthesis, mechanisms regulating its expression and activity, its utility as a biomarker and its therapeutic potential are in need of further investigation. In the current study, using intra-tracheal (i.t.) instillation of LPS into the mouse, we test our hypothesis that EC Akt1-FoxO-Stromelysin pathway would play a major role in pulmonary edema during the initial exudative stage of ALI. Animal models for ALI do not completely reproduce all of the complex characteristics of ALI. We chose i.t. instillation of LPS model because this model has been widely used to examine lung injury, moreover this represents pathogen induced lung injury that is commonly observed in clinic [21]. Here we report that the pulmonary ECs are a vital source of stromelysin1 in damaged lungs and that its expression is regulated by the Akt1-FoxO pathway subsequently resulting in TJ turnover. Furthermore, stromelysin1 expression/activity in ECs, mouse lungs and broncho-alveolar lavage fluid (BALF) in LPS-induced ALI model is a reliable marker for ARDS. Finally, we demonstrate that FoxO and stromelysin1 are druggable targets for ARDS therapy.
METHODS AND MATERIALS
Cell culture and preparation of ShAkt1 stable cell lines
Human (telomerase-immortalized) microvascular endothelial cells (HMEC) (CRL-4025; ATCC, Manassas, VA) were maintained in Endothelial Cell Basal Medium-2 (EBM2) with a Growth Medium-2 Bullet Kit (Lonza; Walkersville, MD). Primary Human pulmonary artery endothelial cells (HPAECs) (Part # 0055; Lifeline cell technology) were used as a model to study the in vitro effects of LPS (100ng/ml) [22] and to confirm the results observed in HMECs (which is an immortal endothelial cell line). All cultures were maintained in a humidified 5 % CO2 incubator at 37 °C, and routinely passaged when 80–90 % confluent. Stable ShControl, ShAkt1 (ACGCTTAACCTTTCCGCTG) HMEC cells were generated using SMART vector 2.0 lentivirus particles (109 pfu) (Thermo Scientific, Waltham, MA). Lentivirus particles were mixed in 1 ml Hyclone SFM4Transfx-293 (Fisher, Hanover Park, IL) and added along with 1 μl Polybrene (10 mg/ml, American Bioanalytical, Natick, MA). Three days later, transfection efficiency was tested through Turbo-GFP expression and subjected to 4 μg/ml puromycin (Life Technologies, Grand Island, NY) selection until all cells expressed GFP.
Generation of ‘VECad-Cre-Akt1’ (EC-Akt1−/−) transgenic mouse model
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals. All tests were performed with approval by the Charlie Norwood VAMC Institutional Animal Care and Use Committee (approval reference #14-07-073). For our study, we generated an endothelial-specific, tamoxifen-inducible Akt1 knockout mouse model (EC-Akt1−/−) by crossing Akt1 LoxP mice with VE-Cadherin Cre mice in a pure C57BL6 background [23]. The mice were backcrossed six times before use in experiments. Age-and sex-matched 8- to 12-week-old tamoxifen-treated EC-Akt1−/− mice were used in the study. For a direct comparison of control and EC-Akt1−/− mice on basal vascular permeability, controls used were wild-type (WT) C57BL6 mice treated with tamoxifen. Genotyping was performed using specific primers for A13Loxp: TCACAGAGATCCACCTGTGC, and A1-4113R: GCAGCGGATGATAAAGGTGT. Tamoxifen (Sigma, St. Louis, MO, USA) stock solution (100 mg/ml) was prepared by dissolving in absolute ethanol and stored in an aluminum foil covered plastic tubes at −20 °C. Right before injection, sterile corn oil was added to dilute tamoxifen to a final concentration of 10 mg/ml. Tamoxifen (1mg/10g dose) was administered to the mice using a 27G needle via intraperitoneal (i.p.) injection every 24 h for five consecutive days. Following this, the transgene was maintained active by feeding the mice with a custom-made Tamoxifen diet (Harlan, Madison, WI, USA) for the duration of the experiments. Please refer reference [20] for further details.
Measurement of endothelial-barrier resistance
Endothelial-barrier integrity (measured as the electrical resistance of the endothelial monolayer) was determined using ECIS equipment (Applied Biophysics, Troy, NY) as described previously [24, 25]. Endothelial-barrier resistance was measured at multiple frequency modes. Endothelial cells were grown till a monolayer in full EBM-2 media and then treated with LPS, triciribine, and AS1842856.
Experimental protocol for LPS-induced ALI model
Equal number (6-10) of sex and age-matched mice were randomly divided into 4 groups as follows: Control (saline), Injury (LPS), LPS injury with lung FoxO1/3a knockdown (these mice were intratracheally administered with 107 particles of LentiShFoxO1/3a 5 days prior to the LPS injury and LPS injury in the presence of stromelysin1 inhibitor [i.p. UK-356618 15mg/kg]. Inhibitor was administered in 3 doses starting from 24 hrs. prior to injury followed by 2 more doses on day of injury and 24 hrs. after injury). Alternatively, FoxO1/3a inhibitor (100mg/kg AS1842856) was also used to determine the therapeutic efficacy of pharmacological FoxO inhibitor for ARDS. Both MMP3 and FoxO1/3a inhibitors were prepared in DMSO vehicle and hence DMSO was used in control group of experiments which involved the use of these inhibitors. LPS (5 mg/kg dissolved in PBS) [22] was instilled intratracheally to induce ALI, and the control group was injected with the same quantity of PBS alone. All the mice in each group were euthanized and sacrificed after 48 hrs. of LPS injury. The BALF, lung tissue specimens were collected at the time of sacrifice. Lower lobes of lungs were snap frozen for further molecular analysis, and the upper lobes of the left lung were used for sectioning and staining, while the upper lobes of the right lung were used for lung wet/dry (W/D) weight ratio.
Lung injury Area measurement, injury scoring and lung wet/dry weight ratio analysis
Lung injured area was measured on 10 μm size lung sections using NIH Image J software as described previously [26, 27]. Briefly, particle size was set, images were converted to black and white. The injured area measured as the black area was normalized to the uninjured area and compared between the groups. The white area represents the absence of lung tissue including alveolar spaces and bronchial spaces while the black area represents lung tissue along with inflammatory cells infiltrated and lung consolidation due to ECM deposition as a result of the injury. Therefore, greater the black area indicates worsened injury due to enhanced ECM deposition, inflammatory cell infiltration and lung consolidations [28]. ALI was scored based on predefined criteria [22] by 3 blinded reviewers. Average of the three reviewer scores were considered for the analysis. Briefly, all lung fields at ×20 magnification were examined for each sample. Assessment of histological lung injury (from mice lung inferior lobes, post-caval lobes and left lobes were used for lung histology analysis using H&E staining) was performed by scoring from 1 to 5, with 1 being the best (normal lung) and 5 being the worst (severe most ALI). Scoring was performed as follows: 1- normal; 2- focal (<50% lung section) interstitial congestion and inflammatory cell infiltration; 3- diffuse (>50% lung section) interstitial congestion and inflammatory cell infiltration; 4- focal (<50% lung section) consolidation (Combining into a solid mass without the alveoli structure) and inflammatory cell infiltration and 5- diffuse (>50% lung section) consolidation and inflammatory cell infiltration.
The Wet/Dry (W/D) weight ratio was assayed in the upper 2 lobes (the superior lobe and middle lobes) of mouse right lung in every single mouse to maintain consistency and to assess lung edema. The upper lobes of the right lung were excised, and the wet weights were determined. Then, the lungs were placed on sterile paper and incubated. After incubation at 80°C for 96 hours to remove all moisture, the dry weights were measured, and the W/D ratios were calculated.
Analysis of BALF
At the time of sacrifice, animals were anesthetized using i.p. ketamine/xylazine (100 mg/kg & 10mg/kg respectively). When deep anesthesia was attained, the abdominal content and thorax were exposed through an abdominal midline incision, and exsanguination was performed through excision of the inferior vena cava. A tracheostomy tube was subsequently inserted and secured in the trachea, the diaphragm cut and the chest wall opened via a midline incision to expose the lungs. Lungs were then lavaged with 3 × 1 mL aliquots of saline, and each aliquot was instilled and withdrawn three times. The total volume of lavage fluid collected from each mouse was recorded. Lung lavage fluid was immediately centrifuged at 380 g for 10 minutes at 4°C to isolate the cell pellet; the supernatant was collected, and aliquots were frozen at −80°C for subsequent analyses. The supernatant containing protein was used to measure BAL protein using Bio-Rad protein assay (catalog #23208).
Western blot analysis and Immunofluorescence staining
Western blotting and immunohistochemistry were performed as described previously [25, 29, 30]. Antibodies include Akt1 (Cat # CST2938), pS473Akt (Cat # CST4060), pT308Akt (Cat # CST13038), stromelysin1 (Cat # CST14351), pFoxO1/3a (Cat # CST 9464) and FoxO3a (Cat # CST2497) from Cell Signaling (Danvers, MA), β-actin (Cat # A5441) from Sigma (St. Louis, MO), claudin5 (Cat # 15106) and ZO-1 (Cat #5406) from Abcam (Cambridge, MA). Human Pulmonary Artery Endothelial cells (HPAECs) were treated with 100ng/ml LPS or an equal volume of PBS for 12-24 hrs.
Immunofluorescent staining of HMEC monolayers was performed using the chamber slides. Cells were then washed twice with PBS, fixed using 2 % paraformaldehyde for 30 min, permeabilized with 0.1 % Triton X-100 for 15 min, and blocked with 2 % BSA in sterile PBS. Cell monolayers were then incubated with antibodies against Claudin5 (1:100, Rabbit antibody, Cell Signaling, Danvers, MA, USA) at 4°C overnight. Immunofluorescence was revealed using AlexaFlour secondary antibodies (1:2000 dilution of goat anti-rabbit 488 and goat anti-mouse 594) obtained from Life Technologies, Grand Island, NY, USA. Cells on chamber slides were mounted on to a glass slide using DAPI containing mounting medium (Vector Laboratories). Samples were observed under a confocal microscope equipped with argon and helium/neon lasers (LSM510, Zeiss, Germany). Controls were performed by omitting either one or both primary antibodies. All controls gave negative results with no detectable non-specific labeling.
Stromelysin1 activity assay
The enzymatic activity of stromelysin1 was determined using a fluorescence resonance energy transfer (FRET) peptide and immunocapture assay as previously described elsewhere [31] with minor modifications. Briefly, 50 μg total protein of endothelial cells or lung lysates were incubated at 4°C for 2 hours with rabbit polyclonal anti-MMP3 antibody (cat no. sc-6839-R; Santa Cruz Biotechnology, Dallas, TX). A/G agarose beads were then added and allowed to incubate overnight at 4°C. The beads were then washed, and samples were transferred to black 96well plate and 100 μL of 2 μmol/L 5-FAM/QXL 520 FRET peptide (cat no. 60580-01; AnaSpec, San Jose, CA) in assay buffer was added per well. Plates were incubated for 15 hours at 37°C; then relative fluorescence units were read and monitored at excitation/emission wavelengths of 485 of 528 nm in a Synergy HT multimode microplate fluorescence reader (BioTek, Winooski, VT) running Gen5 data analysis software.
MPO activity assay
MPO activity assay was performed using Mouse MPO ELISA kit from Hycult biotech (The Netherlands) according to the manufacturer's protocol. Briefly, 10 mg of the lungs were homogenized in 200 μl lysis buffer as the following composition: 200 mM NaCl, 5 mM EDTA, 10% glycerin, 1 mM PMSF, 1 μg/ml leupeptin, 28 μg/ml aprotinin and Tris–HCl (pH 7.4). Sample aliquots were applied onto microtiter wells pre-coated with capture antibody. After washing, biotinylated tracer antibody was added to each well. After the incubation for binding of biotin to streptavidin-peroxidase conjugate, the color development with tetramethylbenzidine was performed. The color reaction was stopped by the addition of oxalic acid. The absorbance at 450 nm was measured with a spectrophotometer. The mouse MPO concentration of each sample was calculated from the standard curve with various concentrations of mouse MPO by serial dilution.
Ingenuity Pathway analysis
Ingenuity Pathway Analysis (IPA, Qiagen Bioinformatics) is a system that transforms a list of genes into a set of relevant networks associated with pathology based on extensive records maintained in the Ingenuity Pathways Knowledge Base [32]. Highly interconnected networks are predicted to represent significant biological function [33]. IPA was used to connect 132 genome-wide association study (GWAS)- implicated ARDS genes [34, 35] along with Akt-FoxO-MMP3 pathway. Only those genes that were directly affected by the pathway of interest and ARDS are shown.
Statistical Analysis
All the data are presented as mean ± SD and were calculated from multiple independent experiments performed in triplicates. The ‘n’ value for each figure implies the multiple independent experiments we performed. All the data were analyzed by parametric testing using the Student's unpaired t-test or one-way ANOVA, followed by the post-hoc test using the GraphPad Prism 6.01 software. Data with P<0.05 were considered significant.
RESULTS
LPS treatment inhibits Akt, activates FoxO1/3a and suppresses claudin5 expression leading to paracellular barrier leakage in primary human pulmonary endothelial cells
To determine whether LPS-induced ALI alters endothelial Akt activity and modulates FoxO pathway, claudin5 expression, and endothelial barrier resistance, we investigated the direct effect of LPS on phosphorylation of Akt and FoxO and their role in the claudin5 expression and endothelial barrier resistance in response to LPS treatment. In our study, LPS significantly decreased Akt phosphorylation in primary human pulmonary endothelial cells (HPAECs) in a time-dependent manner (Figure S1) and a significant reduction in the pS473Akt and pT308Akt were observed after 12 hours of treatment (Figure 1A-B). As anticipated, LPS treatment significantly reduced phosphorylation of both FoxO1 and FoxO3a in HPAECs (Figure 1C). To mimic the microvascular nature of capillary endothelial cells that surround the alveoli forming alveolar epithelial and endothelial barrier in the lung, we used human microvascular endothelial cells (HMECs) to examine the effect of LPS on claudin5 expression. Since we hypothesized that LPS induced inhibition of endothelial Akt1 activates downstream FoxO1/3a, which are transcriptional suppressor for claudin5, we observed the expression of claudin5 at a later time point (24 hrs). Moreover, it has also been observed that LPS induced ALI peaks between 24-48 hrs [22]. LPS treatment significantly decreased claudin5 expression in HMECs at 24 hrs. (Figure 1D-E). This was also confirmed in primary HPAECs (Figure 1F). To further investigate the involvement of Akt-FoxO signaling in LPS-induced claudin5 suppression, we co-treated HPAECs with a pan FoxO inhibitor AS1842856. Prior treatment (4 hrs) with AS1842856 blunted LPS-induced claudin5 suppression in HPAECs confirming the role of FoxOs in the suppression of claudin5 expression (Figure 1F). Next, we studied the effect of LPS on endothelial barrier integrity in vitro using an ECIS assay that measures real-time transendothelial barrier resistance in an HPAEC monolayer. A single dose of LPS (100 ng/ml) and Akt inhibitor triciribine (10 μM) disrupted endothelial barrier integrity, which was gauged in terms of decreased resistance in an HPAEC monolayer with LPS or triciribine treatments compared to PBS. This was significantly blunted when FoxO inhibitor AS1842856 was used along with LPS treatment (Figure 1G and S2A). Together, these results indicated the integral role of Akt-FoxO pathway in LPS-induced endothelial barrier injury.
FIGURE 1: LPS treatment disrupts EC-barrier integrity via the FoxO1/3a mediated suppression of claudin5 in primary HPAECs.
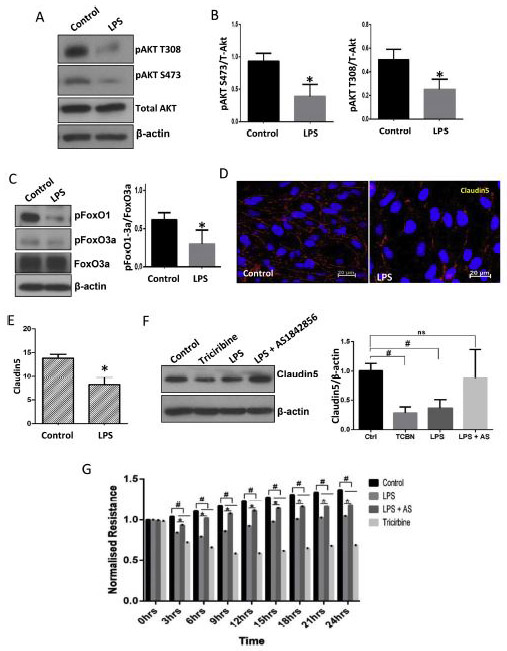
(A-B) Representative Western blot images and the densitometry analysis indicating the inhibitory effect of LPS treatment (100 ng/ml) on Akt phosphorylation in primary HPAECs after 12 hrs (n=3). (C) Representative Western blot images and the densitometry analysis showing the effect of LPS on FoxO1/3a phosphorylation in HPAECs after 12 hrs treatment (n=3). (D-E) Representative images and bar graph showing percentage of claudin5 positive area in the HMEC monolayers probed with Claudin5 antibodies in the absence and presence of LPS treatment for 12 hrs. (F) Representative Western blot images and the densitometry analysis of HPAECs indicating the effect of Akt inhibitor triciribine (10 μM), LPS and with LPS in combination with FoxO inhibitor AS1842856 (10 μM) for 24hrs on claudin5 expression (n=5). (G) Graph showing the real-time changes in resistance by the HPAEC monolayers upon treatment with LPS, triciribine, and LPS in combination with FoxO inhibitor AS1842856 (10 μM; pretreated) compared to DMSO control as recorded using the ECIS assay (n=3). HPAEC: Human Pulmonary Artery Endothelial Cells; HMEC: Human Microvascular Endothelial cells. * (p<0.05); # (p<0.01); ns = non-significant, unpaired Student’s t-test for two groups and One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
Silencing Akt1 in HMECs mimics the effect of LPS treatment on human primary lung ECs
We previously showed that Akt1 is the predominant isoform of Akt in endothelial cells [36]. To determine if Akt1 suppression in ECs will have a similar effect of LPS treatment, we silenced Akt1 in HMECs using lentivirus-based shRNAs followed by generation of a stable shAkt1 HMEC cell line through antibiotic selection. ShAkt1 HMECs had ~80% reduction in their Akt1 expression and total Akt phosphorylation (Figure 2A-C) and significantly reduced FoxO1/3a phosphorylation (Figure 2D-E) as compared to the shControl (shCtrl) HMECs. This was accompanied by reduced expression of TJ proteins ZO-1 (zona occludens 1) and claudin5 (Figure 2F-G), an effect that was similar to what was observed with LPS treatment of HMECs and HPAECs (Figure 1). ShAkt1 HMECs compared to shCtrl HMECs exhibited a significant reduction in the normalized transendothelial barrier resistance in ECIS (Figure 2H and S2B). Interestingly, whereas LPS treatment decreased resistance in shCtrl HMECs, it did not have any further effect on already resistance compromised shAkt1 HMECs compared to PBS treatment (Figure 2H and S2B). These results indicated that LPS, through Akt1 inhibition and FoxO1/3a activation suppresses claudin5 expression in ECs thus disrupting endothelial barrier resistance.
FIGURE 2: Silencing Akt1 in HMECs mimics the effect of LPS treatment on HMECs.

(A) Representative Western blot images showing changes in the expression of HMEC Akt1, pS473Akt, pFoxO1/3a and tight junction proteins ZO-1 and claudin5 in ShAkt1 compared to ShCtrl HMECs. (B-G) Bar graphs showing quantification of the band densitometry indicating a significant reduction in the expression levels of Akt1, pS473Akt, pFoxO1/3a and tight junction proteins ZO-1 and claudin5 in ShAkt1 compared to ShCtrl HMECs normalized to the loading control, β-actin (n=5). (H) Graph showing the real-time changes in resistance by the ShControl and ShAkt1 HMEC monolayers in the presence and absence of LPS treatment compared to PBS control as recorded using the ECIS assay (n=4). * (p<0.05); # (p<0.01); ns = non-significant, unpaired Student’s t-test for two groups and One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
Endothelial Akt1 loss exacerbates LPS-induced ALI in EC-Akt1−/− mice via FoxO1/3a activation
To further investigate the involvement of Akt1-FoxO1/3a pathway in LPS-induced ALI in vivo, we utilized a previously characterized tamoxifen-inducible VE-cadherin promoter driven endothelial specific Akt1 knockdown (EC-Akt1−/−) mice. Intra-tracheal (i.t.) instillation of LPS (5mg/kg) resulted in ALI in both WT and EC-Akt1−/− mice, however, LPS induced injury was exacerbated in EC-Akt1−/− mice compared to injury observed in WT as confirmed by the blinded analysis of the H&E stained mouse lung sections (Figure 3 A-B). Lung wet/dry weight ratio, used as a measure of lung edema was significantly higher in LPS injured EC-Akt1−/− mouse lungs compared to the LPS injured WT mouse lungs (Figure 3D). Similarly, vascular leakage was significantly enhanced in LPS injured EC-Akt1−/− mouse lungs compared to the LPS injured WT mouse lungs in an Evans blue dye extravasation assay, thus confirming enhanced pulmonary injury and edema in LPS injured EC-Akt1−/− mouse lungs (Figure 3C-E). Next, we investigated the effect of FoxO1/3a inhibition in vivo on LPS induced ALI in WT and EC-Akt1−/− mouse lungs. Lung-targeted knockdown of FoxO1/3a was achieved by intratracheal instillation of LentiShFoxO1/3a (107 virus particles) 5 days prior to LPS administration (Figure S3 B-D) and systemic inhibition of FoxO achieved by treatment with FoxO inhibitor AS1842856 (30mg/kg), both of which blunted the increased lung wet/dry weight ratio and Evans blue extravasation in EC-Akt1−/− mouse lungs compared to untreated controls (Figure 3C-E). Thus, both lung-specific FoxO1/3a knockdown, as well as pharmacologic FoxO inhibition, were found to be inhibiting LPS induced ALI. Genome-wide association studies (GWAS) using the Ingenuity software implicated that there is no existing report on whether or not FoxO1/3a is involved in ARDS disease pathology. Even though a couple of studies have suggested a role for FoxO in ALI [37, 38], its interaction with a few known ARDS regulating genes in humans and rodents along with our results support the importance of FoxO1/3a in ARDS pathology (Figure 3F).
FIGURE 3: Endothelial Akt1 loss exacerbates LPS-induced ALI in EC-Akt1−/− mice, which is partially rescued by FoxO1/3a inhibition.

(A-B) Representative H&E images and bar graph showing the area of injury in the mouse lung sections indicating enhanced LPS induced ALI in EC-Akt1−/− mice compared to WT mice (n=6). (C) Representative lung images showing enhanced lung vascular leak (Evans blue extravasation) in EC-Akt1−/− mice, which were rescued by pulmonary FoxO1/3a inhibition using LentiShFoxO1/3a or FoxO1 inhibitor (AS1842856) i.p. 30mg/kg. (D) Bar graph showing lung wet/dry weight ratio as a measure of lung edema in LPS injured EC-Akt1−/− mice compared to WT mice and its partial reversal with pulmonary FoxO1/3a inhibition using LentiShFoxO1/3a or FoxO1 inhibitor (AS1842856) i.p. 30mg/kg (n=6). (E) Bar graph showing Evans blue extravasation confirming enhanced lung vascular leakage in EC-Akt1−/− mice and its partial rescue with FoxO inhibition using LentiShFoxO1/3a or FoxO1 inhibitor (AS1842856) i.p. 30mg/kg (n=6). (F) FoxO1/3a regulates ARDS genes and pathology in humans and rodents as identified from genome-wide association studies using ingenuity pathway analysis (IPA). * (p<0.05); # (p<0.01), One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01). Green lines indicate direct interaction with concerned pathway; Pink lines indicate indirect interaction with the concerned pathway; Blue lines indicate the interaction between genes of concerned pathway.
Treatment with LPS or Akt1 deletion in endothelial cells, both increases stromelysin1 expression and activity involving FoxO1/3a
Since the involvement of MMPs in the disruption of alveolar epithelial-endothelial barrier in ALI is prominent [4], we determined whether LPS treatment and/or Akt1 knockdown in endothelial cells will lead to increased stromelysin1 expression, which has the potential to break down the TJ proteins such as claudins and ZO-1 with its proteolytic activity in addition to its ability to digest the ECM [39-41]. We observed that LPS treatment for 24hrs on HPAECs (as well as HMECs) significantly increased stromelysin1 expression (Figure 4A). To further investigate the involvement of endothelial Akt1 signaling in LPS-induced stromelysin1 expression, both ShCtrl and ShAkt1 HMECs were treated with LPS or PBS. Loss of Akt1 activity by itself was sufficient to increase stromelysin1 expression in ShAkt1 HMECs compared to ShCtrl HMECs. Interestingly, while LPS significantly enhanced stromelysin1 expression in ShCtrl HMECs, it did not have any additive effect in ShAkt HMECs (Figure 4B-C). To confirm that increased stromelysin1 expression correlates with its activity, we performed stromelysin1 activity assay in HMEC lysates. To further investigate the signaling downstream of Akt1 that is involved in the LPS-induced stromelysin1 expression, we used inhibitors of FoxO and β-catenin, two major downstream signaling pathways regulated by Akt1 in ECs. HMECs treated with LPS alone or in combination with inhibitors of FoxO (AS1842856 10μM) or β-catenin (10 μM of ICG001 or IWR1) signaling, and stromelysin1 expression was assessed. While LPS treatment of HMECs enhanced stromelysin1 expression, prior treatment with FoxO inhibitor (AS1842856 10μM), but not β-catenin inhibitors reversed LPS-induced stromelysin1 expression (Figure 4D-E). Silencing Akt1 significantly resulted in increased stromelysin1 activity in HMEC lysates (Figure 4F). Suppressive effect of FoxO inhibitor on LPS-induced stromelysin1 expression was also observed in its reduced activity in LPS, and AS1842856 treated HMEC lysates compared to LPS only treated controls (Figure 4G). The effect of LPS on stromelysin1 activity and its suppression by FoxO inhibitor was also confirmed in HPAECs (Figure S3 A-D). Interestingly, saline instilled EC-Akt1−/− mouse lung lysates exhibited higher stromelysin1 activity (Figure 4H) and expression compared to WT lungs, which was further elevated upon LPS treatment (Figure 4I). These results indicate the role of Akt1-FoxO1/3a mediated stromelysin1 expression and activity in ECs on LPS-induced vascular injury.
FIGURE 4: Treatment of primary HPAECs with LPS induces increased expression and activity of stromelysin1 via Akt1-FoxO1/3a signaling.

(A) Western blot images and densitometry analysis of HPAEC lysates after treatment with 100ng/ml LPS showing enhanced stromelysin1expression after 24 hrs (n=4). (B-C) Western blot images and densitometry analysis of ShControl and ShAkt1 HMEC lysates showing enhanced stromelysin1 expression upon treatment with LPS in ShControl, but not in ShAkt1 cells and that expression levels of Stromelysin1 is already high in ShAkt1 HMECs compared to ShControl cells (n=5). (D-E) Western blot images and densitometry analysis of HMEC lysates showing increased expression of stromelysin1 with LPS treatment and a significant reversal of stromelysin1 expression upon co-treatment with FoxO1/3a inhibitor (10 μM AS1842856), but not with β-Catenin inhibitors (10 μM each of ICG001 and IWR-1) (n=4). (F) Bar graph showing increased activity of stromelysin1 in ShAkt1 HMEC lysates compared to ShControl (n=5). (G) Bar graph showing Stromelysin1 activities in LPS treated HMEC lysates compared to PBS treated controls and its reversal upon co-treatment with FoxO1/3a inhibitor AS1842856 (n=3). (H) Bar graph showing Stromelysin1 activities in LPS treated WT and EC-Akt1−/− mouse lung lysates compared to saline-treated controls and its reversal upon co-treatment with stromelysin1 inhibitor UK-356618 (n=5-6). (I) Western blot images and densitometry analysis of WT and EC-Akt1−/− mouse lung lysates probed for stromelysin1. The bar graph shows a significant increase in stromelysin1 expression in EC-Akt1−/− mouse lung lysates compared to WT, which is further elevated upon LPS injury (n=6). * (p<0.05); # (p<0.01), unpaired Student’s t-test for two groups and One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
Both FoxO1/3a and stromelysin1 are druggable targets in LPS-induced ALI in mice
Next, we sought to investigate whether targeting Akt1-FoxO1/3a signaling would be beneficial to inhibit LPS-induced ALI in vivo. To do this, 8-10-weeks old C57BL/6 mice were randomly divided into four groups of i.t. Saline (sham) injury with i.p. DMSO (control), LPS injury with i.p. DMSO, LPS injury with FoxO1/3a inhibition locally in the lung (i.t. Lenti-ShFoxO1/3a) and LPS injury with stromelysin1 inhibition using i.p. UK-356618 (15 mg/kg). H&E staining of mouse lung tissues obtained 48 hours after injury exhibited enhanced inflammatory cell infiltration, interstitial congestion and lung consolidation with LPS injury (Figure 5A-B). Lung-targeted FoxO1/3a inhibition was used to prevent adverse effects associated with systemic inhibition of this important transcription factor. To a significant extent, local FoxO1/3a inhibition, as well as systemic stromelysin1 inhibition, reduced LPS-induced ALI (Figure 5A-B). Lung wet/dry weight ratio depicted a significant decrease in lung edema with FoxO1/3a and stromelysin1 inhibition (Figure 5C). Examination of BALF from mice revealed reduced LPS-induced protein and cell counts upon FoxO1/3a or stromelysin1 inhibition (Figure 5D-E).
FIGURE 5: Inhibition of FoxO1/3a or stromelysin1 blunts LPS-induced ALI in Mice.

(A) Representative H&E staining (20X) of mouse lung sections showing LPS (5mg/kg) induced ALI that is significantly inhibited upon inhibition of FoxO1/3a (Lentiviral expression of ShFoxO1/3a) or stromelysin1 (15 mg/kg of UK-356618). (B-C) Bar graphs showing blinded analysis of ALI score (by three different reviewers), and lung wet/dry weight ratio analysis depicting a significant reduction in ALI and lung edema with FoxO1/3a or stromelysin1 inhibition, respectively (n=6). (D-E) Bar graph showing elevated BALF total cell counts and BALF total protein in LPS-injured mouse lungs that is reversed upon inhibition of FoxO1/3a or stromelysin1 (n=6). * (p<0.05); # (p<0.01); BALF: Bronchoalveolar lavage fluid, One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
Changes in tissue and BALF stromelysin1 activity is a reliable diagnostic and prognostic marker for mouse ALI
Since stromelysin1 is a secreted enzyme and its activity can be measured in a laboratory setup, we determined whether changes in stromelysin1 activity can be utilized as a screening and/or prognostic marker for ALI. Intriguingly, FoxO1/3a inhibition reduced stromelysin1 activity in the mouse lungs and airways confirming an in vivo role of FoxO1/3a contribution in stromelysin1 expression and activity (Figure 6A-B). Neutrophil infiltration and activity is a marker of ALI in the exudative phase [4, 39]. Neutrophil MPO activity was also significantly decreased with FoxO1/3a and stromelysin1 inhibition (Figure 6C). Next, we assessed stromelysin1 expression in the whole mouse lung lysates. While LPS-induced injury enhanced stromelysin1 expression in mouse lungs, prior inhibition of FoxO1/3a activity in the lungs significantly prevented LPS-induced stromelysin1 expression in the mouse lung tissue lysates (Figure 6D-E). Increased mouse lung stromelysin1 expression and activity with LPS-induced injury and its reversal with FoxO inhibition once again confirmed the contribution of FoxO1/3a signaling in the regulation of stromelysin1 expression. Whereas stromelysin1 inhibitor UK356618 did not alter stromelysin1 expression in mouse lungs, it significantly inhibited its activity (Figure 6B). In vitro results indicated that LPS-treatment resulted in the reduced expression of claudin5 in ECs leading to decreased barrier resistance as measured by TEER. Assessment of claudin5 in mouse lungs in injured mice further confirmed these results from in vitro studies, where LPS-induced decrease in claudin5 expression in the mouse lungs was partially rescued with FoxO1/3a or stromelysin1 inhibition (Figure 6F-G). Congruent with our results demonstrating an integral link between stromelysin1 expression and activity in LPS-induced ALI, Ingenuity pathway analysis of sromelysin1 indicated its increased presence in ARDS pathology in humans and rodents (Figure 6H). Moreover, here our results indicate an integral link between stromelysin1 expression, its activity in lung/body fluids and ARDS. Here we also show the integral role of endothelial cells as a source for stromelysin1 downstream of Akt1-FoxO1/3a signaling.
FIGURE 6: Pharmacological inhibition of FoxO1/3a or stromelysin1 has beneficial effects on LPS- induced lung injury and edema.

(A) Bar graph showing increased stromelysin1 activity in mouse BALF upon LPS injury and its significant inhibition upon FoxO1/3a knock down or co-treatment with stromelysin1 inhibitor (n=6). (B) Bar graph showing increased stromelysin1 activity in mouse lung tissues upon LPS injury and its significant inhibition upon FoxO1/3a knock down or co-treatment with inhibitor stromelysin1 (n=6). (C) Bar graph showing significantly increased myeloperoxidase (MPO) activity in mouse lungs after LPS injury and a significant inhibition with FoxO1/3a and stromelysin1 inhibition (n=6). (D-E) Representative Western blot images and band densitometry analysis showing increased stromelysin1 expression in lung tissue lysates with LPS injury and its significant reversal upon FoxO1/3a knock down or co-treatment with an inhibitor of stromelysin1 (n=6). (F-G) Representative Western blot images and band densitometry analysis showing decreased claudin5 expression in lung tissue lysates with LPS injury and its significant reversal upon FoxO1/3a knockdown or co-treatment with an inhibitor of stromelysin1 (n=6). (H) Stromelysin1 (MMP3) regulates ARDS genes and pathology in humans and rodents as identified from genome-wide association studies using ingenuity pathway analysis. * (p<0.05); # (p<0.01); MPO: Myeloperoxidase, One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
Stromelysin1 inhibition significantly rescues LPS-induced ALI in EC-Akt1−/− mice
Since we observed a significant decrease in LPS-induced ALI with FoxO1/3a inhibition in EC-Akt1−/− mice, we determined if we would also see reduced ALI in EC-Akt1−/− mice with stromelysin1 inhibition. Stromelysin1 inhibition using 15 mg/kg of UK356618 i.p. significantly reduced lung injury score (Figure 7A-B), decreased lung edema (Figure 7C), decreased cell count and total protein in BALF in EC-Akt1−/− mice compared to PBS-treated controls (Fig. 7D & 7E), suggesting that stromelysin1 inhibition is also effective to treat ALI in EC-Akt1−/− mice.
FIGURE 7: Stromelysin1 is an important therapeutic target and a biomarker for ARDS.

(A) Representative H&E images of (20X) of EC-Akt1−/− mouse lungs showing significantly increased LPS-induced ALI compared to WT mouse lungs, and that co-treatment with stromelysin1 inhibitor (15 mg/kg UK-356618) reverses this effect. (B-C) Bar graphs showing lung injury score (blindly scored by 3 different reviewers) and the lung wet/dry weight ratio in EC-Akt1−/− mice, respectively, depicting a significant reduction in lung injury score and edema with stromelysin1 inhibition (n=6). (D-E) Bar graph showing total cell count and total protein content in mouse BALF indicating significantly higher cell count and protein content in EC-Akt1−/− mice compared to WT mice, and its significant reversal with stromelysin1 inhibition (n=6). * (p<0.05); # (p<0.01), One-way ANOVA-Ordinary for more than two groups (GraphPad Prism 6.01).
GWAS indicates the importance of Akt-FoxO-stromelysin1 signaling as a potential therapeutic target for ARDS
Interaction of Akt-FoxO-stromelysin1 pathway with GWAS-implicated ARDS genes along with their functions depicts the impact this pathway bears in the pathology of ARDS (Figure S4). This figure represents only those genes that have been implied in the pathology of ARDS observed in human and rodent studies. Among these various genes found to be altered in ARDS, the impact that Akt-FoxO-stromelysin1 signaling plays (through interactions with other reported genes to form relevant networks responsible for the altered pathological functions) is prominent and thus an attractive therapeutic target.
DISCUSSION
Compromising capillary-alveolar integrity leads to lung edema in the initial exudative phase of ARDS [2, 42, 43]. The mechanisms leading to the disruption of the capillary barrier and a translational approach to prevent this is still an area to be vastly explored. In this study, we investigated the endothelial Akt1-FoxO-stromelysin1 pathway as a potential mechanism mediating aberrant vascular permeability, lung edema and inflammation like in the exudative phase of ARDS in a LPS-induced ALI model and determined whether FoxO and stromelysin1 are putative therapeutic targets for ALI therapy. We also determined whether stromelysin1 expression and activity in lung tissues and biological fluids could be developed into a diagnostic and prognostic marker for ALI.
The Ingenuity Pathway analysis identified a correlation between increased FoxO1/3a expression and ARDS pathology. However, it is interesting to note that many previous studies report the activation of Akt, presumably resulting in FoxO inhibition in the lungs in response to LPS treatment [44-48]. Most of these studies examined Akt activity in lung epithelial cells and macrophages and not in endothelial cells. Our recent studies demonstrated the importance of Akt1-FoxO1/3a signaling in the long-term EC barrier protection in vitro and prevention of vascular leakage in vivo [25, 49]. Although LPS stimulated Akt activity in the total lung lysates has been reported to be higher, our results indicate that LPS stimulation in ECs results in the inhibition of Akt activity leading to FoxO1/3a activation at 12hrs. time point. Hence, the overall increase in Akt activity in the LPS-treated lungs that consists of 40 different cell types [50] could be as a result of collective activation of Akt in non-endothelial cell types including, but not limited to the inflammatory cells and lung epithelial cells. Intriguingly, LPS treatment or Akt1 knockout in EC, disrupted EC barrier through decreased expression of TJ protein claudin5 and ZO1, two molecules that have been previously shown to regulate paracellular EC barrier permeability [12, 51, 52]. Interestingly, Akt1-FoxO1/3a pathway directly regulated LPS-induced endothelial stromelysin1 expression. Ingenuity Pathway inquiry also identified the potential involvement of stromelysin1 in ARDS regarding its increased expression and interaction with GWAS-implicated ARDS genes and pathology, thus prompting us to investigate a link between endothelial Akt1-FoxO-stromelysin1 pathway in LPS-induced ALI.
Increased stromelysin1 expression and activity in the normal, uninjured EC-Akt1−/− mouse lungs compared to WT and the reversal of LPS-induced ALI by treatment with FoxO1/3a and stromelysin1 inhibitors demonstrated the integral role of the endothelial Akt1-FoxO-stromelysin1 pathway in the exudative phase of LPS-induced ALI. No additive effect of LPS stimulation on stromelysin1 expression in Akt1 deficient ECs indicated the indispensable role of the EC Akt1-FoxO pathway in stromelysin1 expression. Even though the basal stromelysin1 expression and activity are higher in EC-Akt1−/− mouse lungs, no ALI was observed in these mice without LPS instillation. There could be several reasons for this such as insufficient levels of stromelysin1 to cause injury by itself and inability of stromelysin1 to promote inflammation in the absence of LPS. It is important to note that EC-Akt1−/− without any injury survived with no altered phenotype. However, LPS instillation aggravated the injury in EC-Akt1−/− lungs compared to WT, indicating that endothelial-derived stromelysin1 performs a priming effect in the lungs to further exaggerate the LPS-induced ALI.
Literature reporting the involvement of various MMPs, with comparatively very little evidence for stromelysin1 in ALI/ARDS pathology has been available since the early 1990s [53, 54]. Until today, none of these MMPs have been directly implied to have any therapeutic or diagnostic value for ALI therapy. Although previous studies have used Stromelysin1−/− mice to study its role in ALI and other tissue injuries [19, 20, 55], there were intrinsic limitations related to the systemic gene ablation such as sex differences in response to LPS-induced injury. In our study, which is the first of its kind in LPS induced ALI, we used stromelysin1 specific inhibitor in both male and female mice to surpass such intrinsic compromises in gene knockout models. We have performed a highly selective and sensitive method to determine stromelysin1 enzymatic activity in ECs, lungs and body fluids. We did not find significant differences between male and female mice in their response to LPS-induced injury, where both the genders responded to treatments similarly. In ischemic stroke, claudin5 loss disrupted the Blood-Brain Barrier [56]. Our in vitro results indicated enhanced stromelysin1 expression along with reduced claudin5 expression in HMECs and HPAECs treated with LPS, both of which were rescued upon genetic or pharmacological inhibition of FoxO1/3a, indicating these are all different pieces of the same puzzle. Thus, the enhanced stromelysin1 expression and its proteolytic activity in ECs with LPS treatment might be a contributing factor to the reduced claudin5 expression in the ECs. Prolonged treatment with LPS can reduce cell viability but also at higher doses (500 μg/ml) and greater timepoint than the ones used in our study [57]. LPS treatment significantly reduced claudin5 expression which was responsible for reduced barrier function.
The direct effects of stromelysin1 activity on ALI disease pathology and its importance as a putative therapeutic target for ALI has not been well explored. Inhibition of FoxO or stromelysin1 were effective in treating ALI in both WT and EC-Akt1−/− mice confirming that the changes in Akt and FoxO activities, and changes in claudin5 and stromelysin1 expression/activity leading to ALI follow the same pathway. Interestingly, a modest, but significant additive effect of LPS stimulation in EC-Akt1−/− mouse lungs on stromelysin1 expression and activity was observed in vivo. Decreased claudin5 expression in LPS injured mouse lungs was restored with treatment by either FoxO1/3a or stromelysin1 inhibitor. Stromelysin1 has been reported to play a role in neutrophil migration from pulmonary circulation into the alveolus [4]. We observed a significant reduction in neutrophil activity with stromelysin1 inhibition as well as FoxO1/3a inhibition. Together, we present a novel role for pulmonary ECs in the LPS-induced lung edema and injury by expressing stromelysin1 via the Akt1-FoxO1/3a pathway. In our study, stromelysin1 and FoxO1/3a inhibitors were used prior to ALI indicating a prophylactic approach for patients with high risk of developing ALI/ARDS such as those on a ventilator or suffering from sepsis.
The present diagnostic criteria provided by the Berlin definition of ARDS lacks a sensitive biomarker that can be used to diagnose ARDS [58]. Recent multicenter clinical study showed astonishing ARDS underdiagnoses rates. Only 60.2% of all patients with ARDS were clinician-recognized and the diagnosis of ARDS was frequently delayed [59]. Therefore, there is an imperative need to come up with efficient diagnostic markers for ARDS. A previous study by Ware et al. that examined plasma stromelysin1 levels, which were not predictive of ARDS outcomes indicate the importance of studying stromelysin1 activity and not just their presence [60]. In our study, stromelysin1 activity was enhanced with LPS-induced ALI, which was blunted by FoxO inhibition. Since the elevated stromelysin1 activity in BAL fluid and the mouse lung lysates after the LPS-induced injury was reversed with FoxO inhibitor, our study demonstrated the potential utility of determining stromelysin1 activity in biological samples such as lungs and body fluids as a diagnostic and prognostic marker in ALI via a non-invasive approach. However, our study has limitation with the use of LPS induced lung injury model alone to test our hypothesis. Since this model is not a complete representation of ARDS in clinic, confirmation of the role of EC Akt1-FoxO-stromelysin1 pathway in pathogenesis of ALI in other animal models such as acid induced lung injury and ventilation induced lung injury would further reinforce the potential of targeting FoxO1/3a and/or stromelysin1 to treat ARDS.
In conclusion, our study reports several novel findings pertinent to the etiology and molecular characterization of LPS induced ALI disease progression (Figure S4). Our findings from an inflammation-based murine ALI model indicate that the Akt-FoxO-Stromelysin1 signaling axis provides an interesting diagnostic/therapeutic target that may have an impact in ARDS therapy. With reasonable optimism, we speculate that stromelysin1 activity assay in biological fluids could serve as a simple, non-invasive approach to early diagnosis and maybe prognosis of ALI/ARDS. Although our findings provide some positive evidence, further studies in human ARDS patient samples will confirm the utility of stromelysin1 as a diagnostic marker.
Supplementary Material
ACKNOWLEDGMENTS
Funds were provided by the NHLBI grant R01HL103952, NCATS grant UL1TR002378, Wilson Pharmacy Foundation (intramural) and Translational Research Initiative grant (intramural). This work has been accomplished using the resources and facilities at the VA Medical Center in Augusta, GA. The funders had no role in the study design, data collection, analysis, and decision to publish the data. The contents of the manuscript do not represent the views of the Department of Veteran Affairs or the United States Government.
ABBREVIATIONS
- ARDS
Acute respiratory distress syndrome
- ALI
Acute lung injury
- LPS
Lipopolysaccharide
- FoxO
Forkhead box-O
- MMP
Matrix metalloprotease
- ECM
Extracellular matrix
- AJ
Adherens junction
- TJ
Tight junction
- ZO-1
zonna occluden 1
- EC
Endothelial cell
- BALF
Broncho-alveolar lavage fluid
- HMEC
Human microvascular endothelial cell
- HPAEC
Human pulmonary artery endothelial cell
- WT
Wild-type
- DAPI
4',6-diamidino-2-phenylindole
- FRET
Fluorescence resonance energy transfer
- MPO
Myeloperoxidase
- ELISA
Enzyme-linked immunosorbent assay
- IPA
Ingenuity pathway analysis
- GWAS
Genome-wide association studies
- ANOVA
Analysis of variance
Footnotes
CONFLICT OF INTEREST
Authors declare that there are no financial or any other conflicts of interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Matthay MA, Ware LB, and Zimmerman GA, The acute respiratory distress syndrome. J Clin Invest, 2012. 122(8): p. 2731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson BT, Chambers RC, and Liu KD, Acute Respiratory Distress Syndrome. N Engl J Med, 2017. 377(6): p. 562–572. [DOI] [PubMed] [Google Scholar]
- 3.Bernard G, Acute Lung Failure - Our Evolving Understanding of ARDS. N Engl J Med, 2017. 377(6): p. 507–509. [DOI] [PubMed] [Google Scholar]
- 4.Davey A, McAuley DF, and O'Kane CM, Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J, 2011. 38(4): p. 959–70. [DOI] [PubMed] [Google Scholar]
- 5.Ware LB and Matthay MA, The acute respiratory distress syndrome. N Engl J Med, 2000. 342(18): p. 1334–49. [DOI] [PubMed] [Google Scholar]
- 6.Dudek SM and Garcia JG, Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol (1985), 2001. 91(4): p. 1487–500. [DOI] [PubMed] [Google Scholar]
- 7.Chen W, et al. , Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am J Respir Cell Mol Biol, 2014. 50(2): p. 328–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaarteenaho R, et al. , Divergent expression of claudin −1, −3, −4, −5 and −7 in developing human lung. Respir Res, 2010. 11: p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlingmann B, Molina SA, and Koval M, Claudins: Gatekeepers of lung epithelial function. Semin Cell Dev Biol, 2015. 42: p. 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang AS, et al. , Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am J Respir Cell Mol Biol, 2011. 44(4): p. 483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitta T, et al. , Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol, 2003. 161(3): p. 653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komarova YA, et al. , Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ Res, 2017. 120(1): p. 179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staub NC, Pulmonary edema. Physiol Rev, 1974. 54(3): p. 678–811. [DOI] [PubMed] [Google Scholar]
- 14.Staub NC, Pulmonary edema due to increased microvascular permeability. Annu Rev Med, 1981. 32: p. 291–312. [DOI] [PubMed] [Google Scholar]
- 15.Matthay MA and Zemans RL, The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol, 2011. 6: p. 147–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelosi P, et al. , The extracellular matrix of the lung and its role in edema formation. An Acad Bras Cienc, 2007. 79(2): p. 285–97. [DOI] [PubMed] [Google Scholar]
- 17.Kong MY, et al. , Matrix metalloproteinase activity in pediatric acute lung injury. Int J Med Sci, 2009. 6(1): p. 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fligiel SE, et al. , Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol, 2006. 37(4): p. 422–30. [DOI] [PubMed] [Google Scholar]
- 19.Warner RL, et al. , Role of stromelysin 1 and gelatinase B in experimental acute lung injury. Am J Respir Cell Mol Biol, 2001. 24(5): p. 537–44. [DOI] [PubMed] [Google Scholar]
- 20.Nerusu KC, et al. , Matrix metalloproteinase-3 (stromelysin-1) in acute inflammatory tissue injury. Exp Mol Pathol, 2007. 83(2): p. 169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matute-Bello G, Frevert CW, and Martin TR, Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol, 2008. 295(3): p. L379–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Alessio FR, et al. , CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest, 2009. 119(10): p. 2898–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kogata N, et al. , Cardiac ischemia activates vascular endothelial cadherin promoter in both preexisting vascular cells and bone marrow cells involved in neovascularization. Circ Res, 2006. 98(7): p. 897–904. [DOI] [PubMed] [Google Scholar]
- 24.Gao F, Al-Azayzih A, and Somanath PR, Discrete functions of GSK3alpha and GSK3beta isoforms in prostate tumor growth and micrometastasis. Oncotarget, 2015. 6(8): p. 5947–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao F, et al. , Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell Mol Life Sci, 2016. 73(20): p. 3917–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu D, et al. , Human amnion cells reverse acute and chronic pulmonary damage in experimental neonatal lung injury. Stem Cell Res Ther, 2017. 8(1): p. 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, et al. , Small pulmonary vascular alteration and acute exacerbations of COPD: quantitative computed tomography analysis. Int J Chron Obstruct Pulmon Dis, 2016. 11: p. 1965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matute-Bello G, et al. , An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol, 2011. 44(5): p. 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abdalla M, et al. , Akt1 mediates alpha-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. J Biol Chem, 2013. 288(46): p. 33483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fairaq A, et al. , TNFalpha induces inflammatory stress response in microvascular endothelial cells via Akt- and P38 MAP kinase-mediated thrombospondin-1 expression. Mol Cell Biochem, 2015. 406(1-2): p. 227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafez S, et al. , Matrix Metalloprotease 3 Exacerbates Hemorrhagic Transformation and Worsens Functional Outcomes in Hyperglycemic Stroke. Stroke, 2016. 47(3): p. 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calvano SE, et al. , A network-based analysis of systemic inflammation in humans. Nature, 2005. 437(7061): p. 1032–7. [DOI] [PubMed] [Google Scholar]
- 33.Ravasz E, et al. , Hierarchical organization of modularity in metabolic networks. Science, 2002. 297(5586): p. 1551–5. [DOI] [PubMed] [Google Scholar]
- 34.Gao L and Barnes KC, Recent advances in genetic predisposition to clinical acute lung injury. Am J Physiol Lung Cell Mol Physiol, 2009. 296(5): p. L713–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang RT, et al. , Experimental Lung Injury Reduces Kruppel-like Factor 2 to Increase Endothelial Permeability via Regulation of RAPGEF3-Rac1 Signaling. Am J Respir Crit Care Med, 2017. 195(5): p. 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen J, et al. , Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med, 2005. 11(11): p. 1188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White MK and Strayer DS, Survival signaling in type II pneumocytes activated by surfactant protein-A. Exp Cell Res, 2002. 280(2): p. 270–9. [DOI] [PubMed] [Google Scholar]
- 38.Horowitz JC, et al. , Constitutive activation of prosurvival signaling in alveolar mesenchymal cells isolated from patients with nonresolving acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol, 2006. 290(3): p. L415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gurney KJ, Estrada EY, and Rosenberg GA, Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis, 2006. 23(1): p. 87–96. [DOI] [PubMed] [Google Scholar]
- 40.Masciantonio MG, et al. , The Balance Between Metalloproteinases and TIMPs: Critical Regulator of Microvascular Endothelial Cell Function in Health and Disease. Prog Mol Biol Transl Sci, 2017. 147: p. 101–131. [DOI] [PubMed] [Google Scholar]
- 41.Lee JY, et al. , Matrix metalloproteinase-3 promotes early blood-spinal cord barrier disruption and hemorrhage and impairs long-term neurological recovery after spinal cord injury. Am J Pathol, 2014. 184(11): p. 2985–3000. [DOI] [PubMed] [Google Scholar]
- 42.West JB, Thoughts on the pulmonary blood-gas barrier. Am J Physiol Lung Cell Mol Physiol, 2003. 285(3): p. L501–13. [DOI] [PubMed] [Google Scholar]
- 43.Morales MM, et al. , Small airway remodeling in acute respiratory distress syndrome: a study in autopsy lung tissue. Crit Care, 2011. 15(1): p. R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang R, et al. , Aspirin Inhibits LPS-Induced Expression of PI3K/Akt, ERK, NF-kappaB, CX3CL1, and MMPs in Human Bronchial Epithelial Cells. Inflammation, 2016. 39(2): p. 643–50. [DOI] [PubMed] [Google Scholar]
- 45.Abarikwu SO, Kolaviron, a natural flavonoid from the seeds of Garcinia kola, reduces LPS-induced inflammation in macrophages by combined inhibition of IL-6 secretion, and inflammatory transcription factors, ERK1/2, NF-kappaB, p38, Akt, p-c-JUN and JNK. Biochim Biophys Acta, 2014. 1840(7): p. 2373–81. [DOI] [PubMed] [Google Scholar]
- 46.Xu Q, et al. , The inhibition of LPS-induced inflammation in RAW264.7 macrophages via the PI3K/Akt pathway by highly N-acetylated chitooligosaccharide. Carbohydr Polym, 2017. 174: p. 1138–1143. [DOI] [PubMed] [Google Scholar]
- 47.Chen L, et al. , Salidroside suppressing LPS-induced myocardial injury by inhibiting ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang N, et al. , Low Dose of Lipopolysaccharide Pretreatment Preventing Subsequent Endotoxin-Induced Uveitis Is Associated with PI3K/AKT Pathway. J Immunol Res, 2017. 2017: p. 1273940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao F, et al. , Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Src signaling. J Cell Physiol, 2017. 232(10): p. 2599–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franks TJ, et al. , Resident cellular components of the human lung: current knowledge and goals for research on cell phenotyping and function. Proc Am Thorac Soc, 2008. 5(7): p. 763–6. [DOI] [PubMed] [Google Scholar]
- 51.Tornavaca O, et al. , ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol, 2015. 208(6): p. 821–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taddei A, et al. , Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol, 2008. 10(8): p. 923–34. [DOI] [PubMed] [Google Scholar]
- 53.Houghton AM, Matrix metalloproteinases in destructive lung disease. Matrix Biol, 2015. 44-46: p. 167–74. [DOI] [PubMed] [Google Scholar]
- 54.Hendrix AY and Kheradmand F, The Role of Matrix Metalloproteinases in Development, Repair, and Destruction of the Lungs. Prog Mol Biol Transl Sci, 2017. 148: p. 1–29. [DOI] [PubMed] [Google Scholar]
- 55.Puntorieri V, et al. , Lack of matrix metalloproteinase 3 in mouse models of lung injury ameliorates the pulmonary inflammatory response in female but not in male mice. Exp Lung Res, 2016. 42(7): p. 365–379. [DOI] [PubMed] [Google Scholar]
- 56.Yang Y, et al. , Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab, 2007. 27(4): p. 697–709. [DOI] [PubMed] [Google Scholar]
- 57.Zheng X, Zhang W, and Hu X, Different concentrations of lipopolysaccharide regulate barrier function through the PI3K/Akt signalling pathway in human pulmonary microvascular endothelial cells. Scientific Reports, 2018. 8(1): p. 9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Umbrello M, et al. , Current Concepts of ARDS: A Narrative Review. Int J Mol Sci, 2016. 18(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bellani G, et al. , Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. Jama, 2016. 315(8): p. 788–800. [DOI] [PubMed] [Google Scholar]
- 60.Ware LB, et al. , Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care, 2013. 17(5): p. R253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.