

SUMMARY
Displacement loops (D-loops) are pivotal intermediates of homologous recombination (HR), a universal DNA double strand break (DSB) repair pathway. We developed a versatile assay for the physical detection of D-loops in vivo, which enabled studying the kinetics of their formation and defining the activities controlling their metabolism. Nascent D-loops are detected within 2 h of DSB formation and extended in a delayed fashion in a genetic system designed to preclude downstream repair steps. The majority of nascent D-loops are disrupted by two pathways: one supported by the Srs2 helicase and the other by the Mph1 helicase and the Sgs1-Top3-Rmi1 heli-case-topoisomerase complex. Both pathways operate without significant overlap and are delineated by the Rad54 paralog Rdh54 in an ATPase-independent fashion. This study uncovers a layer of quality control of HR relying on nascent D-loop dynamics.
Graphical Abstract
In Brief
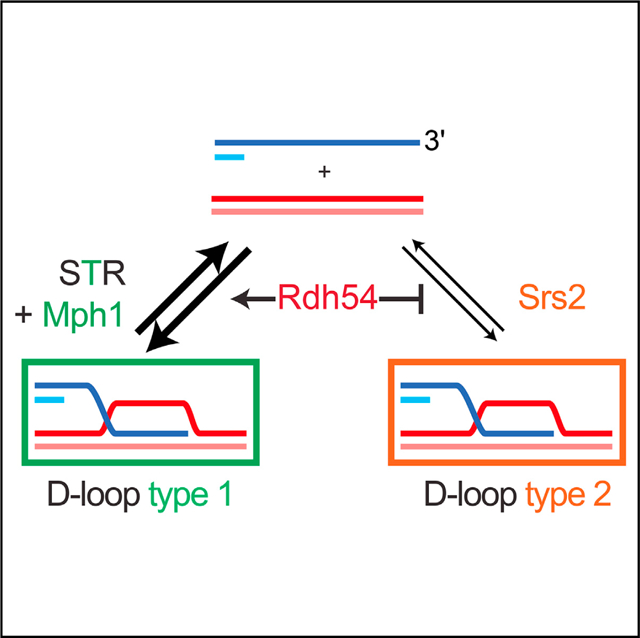
Displacement loops (D-loops) are central intermediates of homologous recombination, a universal DNA repair pathway. Piazza et al. developed a versatile assay for the physical detection of D-loops in cells showing that they exist in a dynamic equilibrium in vivo. Two reversal pathways (Srs2, Mph1 with Sgs1-Top3-Rmi1) operate under control by Rdh54.
INTRODUCTION
Homologous recombination (HR) repairs DNA double-strand breaks (DSBs) by exploiting an intact homologous double-strand DNA (dsDNA) molecule as a template. It proceeds through a succession of metastable intermediates, conferring flexibility and kinetic proof-reading at many steps of the pathway (Heyer, 2015; Kanaar et al., 2008; Zinovyev et al., 2013). Central to the process is the helical filament of Rad51, a member of the RecA family. Together with associated proteins, it assembles onto the 3′-protruding single-strand DNA (ssDNA) generated upon DSB resection, which can be several kilobases long (Symington, 2014). This multivalent filament harnesses the ssDNA sequence information to interrogate nearby dsDNA molecules (Bell and Kowalczykowski, 2016) as it dynamically weaves through the nuclear volume (Seeber et al., 2018). Upon successful identification of homology, Rad51-Rad54-catalyzed DNA strand invasion results in a nascent displacement loop (D-loop) intermediate of possibly varied architecture (Wright and Heyer, 2014 and see below). The salient features of D-loops consist of at least a partially Rad51-free heteroduplex DNA (hDNA), displaced ssDNA, and junctions at both extremities of the hDNA tract (Figure 1A). Initiation of recombination-associated DNA synthesis by Pold primed from the 3′-OH end of the invading strand restores the sequence information disrupted by the DSB. While disruption of nascent D-loops is a mechanism of anti-recombination, disruption of extended D-loops represents a mechanism of crossover avoidance enforcing a non-crossover (NCO) outcome through synthesis-dependent strand annealing (SDSA). Indeed, HR pathway choice (Gangloff et al., 2000), accuracy (Putnam and Kolodner, 2017), and outcome (Ira et al., 2003; Mazón and Symington, 2013; Mitchel et al., 2013; Prakash et al., 2009) are believed to rely, at least in part, on D-loop reversal (reviewed in Daley et al., 2014 and Heyer, 2015).
Figure 1. The DLC Assay Detects D-Loops in S. cerevisiae.

(A) Rationale of the DLC assay.
(B) DSB-inducible construct (chrV) and ectopic donor (chrII) in haploid S. cerevisiae. The restriction sites (EcoRI) and unique regions used for DLC measurement are indicated. The “A” sequence corresponds to the first half of the LYS2 gene.
(C) DLC requires DSB induction.
(D) DLC requires homology between the broken molecule and the donor. “A” and “B” represent different sequences of the same length (see STAR Methods).
(E) DLC is HR dependent.
(F) DLC requires inter-strand DNA crosslink with psoralen and restoration of the resected EcoRI site on the broken molecule.
In C)–(F), bars represent mean ± SEM of at least a biological triplicate.
Several proteins have been implicated in joint molecule/D-loop turnover, whose defects specifically cause repeat-mediated genomic instability through HR (Putnam and Kolodner, 2017). The 3′−5′ Srs2 helicase (putative human counterparts are FBH1, PARI, RECQ5, FANCJ, and RTEL1) is a regulator of HR likely acting at various steps of the pathway. In vitro Srs2 dismantles Rad51 filaments left unprotected by Rad55-Rad57 (Kaniecki et al., 2017; Krejci et al., 2003; Liu et al., 2011; Veaute et al., 2003) and disrupts Rad51-Rad54-mediated nascent and extended D-loops (Liu et al., 2017). Srs2-deficient cells exhibit recombination-dependent lethality and genomic instability (Gangloff et al., 2000; Putnam et al., 2009). They also fail to mature NCO products (Ira et al., 2003; Mitchel et al., 2013) and are less prone to template switch during break-induced replication (BIR) (Ruiz et al., 2009), suggesting that Srs2 disrupts extended D-loops. The 3′−5′ Mph1 helicase (human FANCM) funnels HR toward the NCO repair outcome (Mazón and Symington, 2013; Mitchel et al., 2013; Prakash et al., 2009; Sun et al., 2008; Tay et al., 2010), inhibits BIR (Jain et al., 2016; Luke-Glaser and Luke, 2012), and promotes template-switching during BIR (Stafa et al., 2014). Consistently, purified Mph1 disrupts short synthetic or Rad51-Rad54-mediated nascent and extended D-loops in vitro (Prakash et al., 2009; Sebesta et al., 2011; Zheng et al., 2011). The helicasetopoisomerase complex Sgs1-Top3-Rmi1 (STR, human BLMTOPO3α-RMI1/2) limits formation and/or accumulation of various joint molecules including double Holliday Junction (dHJ) in somatic and meiotic cells (Bzymek et al., 2010; Kaur et al., 2015; Oh et al., 2007; Tang et al., 2015), inhibits CO and/or promotes the NCO repair outcome of HR (Ira et al., 2003; Lo et al., 2006; Mazón and Symington, 2013; Mitchel et al., 2013; Tay et al., 2010), and inhibits BIR and long gap-repair (Jain et al., 2009, 2016; Lydeard et al., 2010). In vitro, STR dissolves dHJs (Cejka et al., 2010; Wu and Hickson, 2003) and D-loops (Fasching et al., 2015). dHJ dissolution requires both the Sgs1 helicase and the type 1A topoisomerase activity of Top3, while disruption of Rad51-Rad54-bound D-loops only requires the topoisomerase activity of the complex (Fasching et al., 2015).
This large body of joint biochemical and genetic evidence established these factors as HR regulators enforcing the accuracy of the pathway (Daley et al., 2014; Heyer, 2015; Piazza and Heyer, 2018a). Yet, the mechanisms by which they do so, including their precise substrates, interactions, and pathway organization remain elusive. Furthermore, the function of certain Rad51-ssDNA-associated proteins involved in regulating the DNA strand exchange reaction are not straightforwardly addressed in vitro, as the substrate and the conditions in which the reaction takes place are unknown. These limitations derive mostly from the technical challenge to physically detect D-loops in somatic cells, unlike dHJ intermediates that can be detected by two-dimensional gel electrophoresis in meiotic and somatic cells (Bzymek et al., 2010; Schwacha and Kleckner, 1995). Here, we developed the D-loop capture (DLC) assay, a proximity ligation technique that enables physical detection of these structures in S. cerevisiae. Using this and a related D-loop extension (DLE) assay (Piazza et al., 2018), we investigated the role of multiple HR regulators on D-loop metabolism in cells.
RESULTS
The DLC Assay
DLC assay (Figure 1A) was applied on a pair of ectopic and inter-chromosomal loci in haploid Saccharomyces cerevisiae cells (Figures 1B and S1; STAR Methods). As the donor lacks homology to the right side of the break, our genetic design purposefully restricts joint molecules to nascent and extended D-loops (Figure 1B; see discussion in STAR Methods). The DSB-inducible and donor constructs are located at untethered chromosomal loci that have been extensively used by others (Agmon et al., 2013; Inbar and Kupiec, 1999; Mazón and Symington, 2013; Miné-Hattab and Rothstein, 2012). Following site-specific DSB induction (step 1) and DNA strand invasion at the donor, in vivo inter-strand DNA crosslinking with psoralen was performed. Psoralen is a B-DNA intercalator that covalently links the hDNA within the D-loop to preserve it during subsequent steps (step 2) (Oh et al., 2009). The restriction site ablated by DSB resection is restored by annealing a long complementary oligonucleotide (step 3). Following restriction digestion (step 3), the ligation reaction performed in dilute conditions leads to preferential ligation of ends from tethered DNA molecules, i.e., those held together by the cross-linked hDNA (step 4), a rationale common to all chromosome conformation capture approaches (Dekker et al., 2002). The resulting unique chimeric ligation product is quantified by qPCR (step 5) (details in STAR Methods; Figure S1) and is referred to as DLC signal.
As expected, the DLC signal depends on DSB formation (Figure 1C), homology between the broken and donor molecules (Figure 1D), and the central HR proteins required for filament assembly (Rad52) and DNA strand invasion (Rad51, Rad54), all consistent with biochemical data (Petukhova et al., 1998; Wright and Heyer, 2014) (Figure 1E). This is in contrast with assays aiming to track DNA strand invasion using Rad51 chromatin immunoprecipitation (ChIP), which did not require Rad54 nor DNA sequence homology, and likely reflects the formation of nonspecific contacts between the Rad51-ssDNA filament and the surrounding dsDNA (Renkawitz et al., 2013; Sugawara et al., 2003). Finally, DLC relies on D-loop stabilization by psoralen crosslinking and restoration of the restriction site eliminated by resection (Figure 1F). These results demonstrate that the DLC assay detects D-loops and not non-specific contacts between the broken and the donor molecule.
Limitations of the DLC Assay
A first limitation of the DLC assay resides in the psoralen-mediated inter-strand crosslink density (≈ 1/500 bp, not shown; see also Oh et al., 2009). Because the in vivo hDNA length distribution is unknown, the DLC assay cannot distinguish between a single long and several shorter D-loops comprising the same total length of hDNA (Figure S2A). Consequently, a change in DLC can reflect either a change of the average hDNA length or a change in the number of D-loops in the cell population.
Second, upon long-range DNA synthesis, the D-loop will move away from the homologous donor loci and thus from the upstream restriction site. If it migrates past a downstream EcoRI site (located 11.1 kb away in our design), it will cause a physical uncoupling between the hDNA (i.e., the crosslink point between invading and donor molecules) and the upstream restriction site used for DLC chimera formation, thus precluding proximity ligation of both partners (Figure S2B). For the analysis of early time points (1–2 h), which show no to little appreciable extension (Figure 2C), this limitation is irrelevant.
Figure 2. D-Loop Formation and Extension Kinetics.

(A) Rationale of the DLE assay (Piazza et al., 2018). Like the DLC assay, the DLE assay exploits the proximity ligation principle of physically tethered DNA extremities, which in this case is being acquired by the broken molecule upon D-loop extension off the ectopic donor. (R) denotes restriction sites made uncuttable as ssDNA.
(B) Same DSB-inducible construct (chrV) and ectopic donor (chrII) as for the DLC assay (Figure 1B). The restriction sites (HindIII) and unique regions used to quantify DLE are indicated.
(C) Kinetics of DSB formation, DLC, and D-loop extension in wild-type cells. DLC and DLE represent the mean ± SEM of 11 biological replicates. (D) DLC50 and DLE50 represent the time between 50% of DSB formation and 50% of the maximum DLC and DLE signal, respectively. The mean ± SEM is shown in red.
Nascent and Extended D-Loops Are Temporally Resolved
The fast and synchronous DSB induction upon HO expression (≈90% molecules cleaved within 30 min) enables kinetic study of the subsequent repair steps (White and Haber, 1990). While monitoring D-loop formation kinetics with the DLC assay, D-loop extension (DLE) is quantified using a related assay (Figures 2A and S3; Piazza et al., 2018). DLE quantifies acquisition by the broken molecule of a restriction site located in the unique region downstream of the homologous donor on chromosome (chr) II (396 bp from the invading end; Figure 2B). Consequently, it does not require psoralen-mediated hDNA crosslinking and enables absolute quantification of the extended molecules in the cell population (Piazza et al., 2018). D-loop formation is first detected 1 h post DSB induction and increases 40-fold at 3–4 h when it peaks before declining slightly (Figure 2C). D-loop extension occurs in a delayed fashion: first detected at 2 h, it increases 31-fold until 8 h (Figure 2C). In absolute terms, the plateau corresponds to ~100% of broken molecule being extended at 8 h (see STAR Methods; Piazza et al., 2018). The meantime, between half the DSB formed (~20 min) and half the maximum DLC and DLE signal is reached is DLC50 = 122 ± 13 min and DLE50 = 269 ± 21 min, respectively (Figure 2D), corroborating the delay between D-loop formation and extension previously proposed from Rad51-ChIP data in a similar system (Jain et al., 2009). This ~2 h delay enables independent investigation of the regulation of nascent and extended D-loops at 2 and 6 h post DSB induction, respectively.
Role of Srs2, Mph1, and STR in the Dynamics of Nascent and Extended D-Loops
We addressed the role of the Mph1 and Srs2 helicases as well as the helicase-topoisomerase STR complex in nascent and extended D-loop metabolism. Deletion of either MPH1 or SRS2 results in a significant 2- to 3-fold increase in DLC signal at all time points (Figures 3A and 3C, respectively). The ATPasedeficient mph1-D209N and srs2-K41A mutants also exhibit elevated DLC levels compared to wild-type, not significantly different from the corresponding deletion mutants (Figures 3B and 3D, respectively). Thus, both helicases inhibit the DLC signal, and hence steady-state levels of D-loops in wild-type cells depend on their catalytic activity. These results are consistent with earlier biochemical evidence showing that Srs2 as well as Mph1 disrupt both Rad51-Rad54-mediated nascent and extended D-loops in an ATPase-dependent fashion (Liu et al., 2017; Prakash et al., 2009; Sebesta et al., 2011).
Figure 3. D-Loop Regulation by Mph1, Srs2, and STR.

(A) Mph1 inhibits DLC at all time points.
(B) DLC inhibition depends on the helicase activity of Mph1.
(C) Srs2 inhibits DLC at all time points. The decrease observed between 2 and 4–6 h in the srs2Δ mutant is not statistically significant.
(D) DLC inhibition depends on the helicase activity of Srs2.
(E and F) Sgs1 inhibits DLC only 2 h post-DSB induction (E) and in an ATPase-independent manner (F).
(G) DLC inhibition by STR depends on Top3 catalytic activity. This inhibitory activity is epistatic to SGS1 but independent of its helicase activity. Data represent mean ± SEM of at least biological triplicates. Asterisk indicates statistical significance (*p < 0.05). ns, not significant.
In contrast, STR significantly inhibits DLC only at the earliest time point: a sgs1Δ mutant exhibits a significant 2-fold DLC increase at 2 h post-DSB induction but not at 4 and 6 h (Figure 3E). Unlike Mph1 and Srs2, DLC in the ATPase-deficient sgs1-K706A mutant is significantly lower than in the deletion mutant and not significantly different from wild-type (Figure 3F), indicating that the STR inhibitory effect requires the physical presence of Sgs1 but is largely independent of its helicase activity. Previous genetic observations showed that a subset of STR functions requires the physical presence of Sgs1 and its ability to interact with Top3 but is independent of its helicase activity (Jain et al., 2009; Lo et al., 2006; Weinstein and Rothstein, 2008). Furthermore, the topoisomerase activity of Top3, but not the helicase activity of Sgs1, is required for STR to disrupt Rad51-Rad54-mediated D-loops in reconstituted biochemical reactions (Fasching et al., 2015). To avoid working with the slow-growing and suppressor-prone top3Δ mutant, we addressed the role of the topoisomerase activity of Top3 upon induced overexpression of the dominant-negative catalytic-deficient top3-Y356F (top3-cd) mutant (Oakley et al., 2002). Overexpression of TOP3 does not affect DLC, indicating that excess Top3 is unlikely partnered with Rmi1 and Sgs1 and degraded or ineffective at DLC inhibition (Figure 3G). However, overexpression of top3-cd leads to an ~2.5-fold DLC increase over the vector control (Figure 3G), an increase similar to that observed in the sgs1Δ mutant. TOP3 and SGS1 are epistatic, as neither TOP3 nor top3-cd overexpression affects DLC levels in sgs1Δ cells (Figure 3G). Overexpression of top3-cd in sgs1-K706A cells yields an intermediate, although non-significant, effect compared to either wild-type or sgs1Δ cells (Figure 3G). This intermediate effect may suggest a subtle contribution of Sgs1 helicase activity to STR-mediated D-loop disruption. These results show that nascent D-loop disruption requires the topoisomerase activity of Top3-Rmi1 and the physical presence of Sgs1. The largely non-catalytic role of Sgs1 could be to promote DNA strand passage during the Top3-Rmi1-mediated decatenation (Cejka et al., 2012), a reaction believed to underlie disruption of relatively short D-loops by STR (Fasching et al., 2015). Alternatively, Sgs1 may help target Top3-Rmi1 to its substrates in the nuclear context.
In sum, these results provide direct evidence for three nascent D-loop disruption activities in yeast supported by STR in a topoisomerase-dependent and mostly helicase-independent fashion and by the Srs2 and Mph1 helicase activities. The DLC levels at the later time point (6 h) suggest that Srs2 and Mph1, but not STR, disrupt extended D-loops. However, this interpretation could be complicated by their nascent D-loop disruption activities as well as additional roles played at the nascent-to-extended D-loop transition (see below).
STR and Mph1 Are Part of the Same Nascent D-Loop Disruption Pathway
We next investigated the genetic interactions between these D-loop disruption activities, focusing on nascent D-loop regulation. Cells defective for both Mph1 and STR do not exhibit additional DLC increase: the DLC profile in the mph1Δ sgs1Δ double mutant resembles that of a sgs1Δ single mutant at all time points (Figure 4A). Furthermore, top3-cd overexpression in the absence of Mph1 does not yield further nascent DLC increase (Figure S4A). This epistatic relationship is independent of the heli-case activity of STR (Figure 4B). These results indicate that STR and Mph1 operate in the same nascent D-loop disruption pathway. Regulation of extended D-loops is more complex, as the increased DLC observed at 6 h in the mph1Δ mutant depends on STR (Figure 4A), suggesting an antagonistic role in this context (see below).
Figure 4. Mph1 and STR Belong to the Same Nascent D-Loop Disruption Pathway, which Is Distinct from Srs2.

(A) MPH1 and SGS1 are epistatic in nascent DLC inhibition.
(B) The helicase activity of Sgs1 plays no role in nascent D-loop processing in the absence of Mph1. (C) Overexpression of top3-cd in a srs2Δ mutant causes an additional nascent DLC increase.
(D) Srs2-AID degradation upon auxin addition mimics the srs2Δ mutant.
(E) DLC increases in wild-type, sgs1Δ, and mph1Δ strains upon Srs2-AID degradation.
(F) Absolute extent of DLC increase in wild-type, sgs1Δ, and mph1Δ strains upon Srs2-AID degradation.
(G) Model for nascent D-loop regulation by STR, Mph1 and Srs2. (A–F) Data represent mean ± SEM of at least biological triplicates. Asterisk indicates statistical significance (*p < 0.05). ns, not significant.
STR-Mph1 and Srs2 Are Independent Pathways Targeting Non-overlapping Nascent D-Loop Substrates
We next addressed the genetic interactions of SRS2 with STR and MPH1 on nascent D-loop metabolism. In order to avoid the HR-dependent synthetic sickness of the double-deletion mutants (Gangloff et al., 2000; Prakash et al., 2009), we induced acute inactivation of either STR or Srs2. First, overexpression of top3-cd in a srs2Δ mutant causes a significant 1.9-fold DLC increase over the empty vector control (Figure 4C), suggesting that Srs2 operates in a different D-loop disruption pathway than STR. Second, we used an auxin-inducible degron version of Srs2 (Srs2-AID; STAR Methods). Srs2-AID depletion induced prior to DSB formation (Figures 4D and S4C–S4E) causes a 2.4-fold DLC increase, similar to that observed in a srs2Δ mutant (Figure 4D). This result validates the SRS2-AID system for effectively depleting Srs2 protein and function. Srs2 degradation also leads to a significant DLC increase in the sgs1Δ and mph1Δ mutant backgrounds (Figure 4E). Importantly, the absolute DLC increase observed upon Srs2 depletion in these mutants is not significantly different from what is seen in a wild-type strain (Figure 4F), indicating that the defect imparted by the absence of Srs2 is additive to that of the other mutations. The absence of epistasis or synergy but apparent additivity between the Srs2 and the STR-Mph1 disruption pathways indicate (1) that they target different nascent D-loop substrates, and (2) that these substrates do not interconvert (Figure 4G).
Srs2, Mph1, and STR Differentially Affect D-Loop Extension
The regulatory roles of these factors on extended D-loops could be complicated by the effect they already exert on nascent D-loop and/or could exert at the nascent-to-extended D-loop transition, which are not detected by the DLC assay. To gain insights into the regulation of D-loop extension (i.e., kinetics and efficiency) we analyzed the srs2Δ, sgs1Δ, mph1Δ, and mph1Δ sgs1Δ mutants with the DLE assay (Figures 5A and 5B). Extended D-loops remain a marginal species (<5%) at 2 h in all cases, confirming that the DLC signal previously measured at this time point predominantly corresponds to nascent D-loops in all mutants tested (Figures 3 and 4). The srs2Δ mutant exhibited a DLE50 not different from wild-type cells (270 ± 25 versus 269 ± 21 min) (Figures 5C, and S5). In contrast, the DLE50 indicates that the sgs1Δ, mph1Δ, and mph1Δ sgs1Δ mutants are similarly accelerated by an 40 min compared to wild-type cells (Figures 5C and S5). Hence, the nascent D-loop dynamics imposed by the STRMph1 pathway only modestly delays the onset of D-loop extension, as proposed previously for Sgs1 and Mph1 in a repairable system (Jain et al., 2009, 2016).
Figure 5. Differential Regulation of the Metabolism of Extended D-Loops by Srs2, Mph1, and STR.

(A and B) Time course analysis of D-loop extension in wild-type and srs2Δ strains (A) and in sgs1Δ, mph1Δ, and mph1Δ sgs1Δ strains (B).
(C) DLE50 determined from the DLE time courses presented in (A) and (B).
(D) Single-stranded and double-stranded extension products at 8 h.
(E) Double-stranded extension products at 8 h as a percentage of total extension products.
In (A)–(E), data represent mean ± SEM of at least biological triplicates. Asterisk indicates statistical significance (*p < 0.05).
Besides kinetic information, the DLE assay provides absolute quantitative information and allows determination of the efficiency of D-loop extension. The efficiency of D-loop extension also differs between mutants: while the sgs1Δ and srs2Δ mutants exert no statistically detectable effect, the mph1Δ and mph1Δ sgs1Δ mutants exhibit 2-fold less extended molecules compared to wild-type cells at 8 h, respectively (Figure 5D). These results suggest that Mph1 has an additional role in enabling D-loop extension.
The amount of extension products that became converted to dsDNA (i.e., during BIR) can be quantified as the DLE signal produced independently of restriction sites restoration (Figure S3; Piazza et al., 2018). In wild-type cells, 3.2% ± 1.4% of extended molecules are converted to dsDNA at 8 h (Figure 5D), an estimate corroborated by Southern blot analysis (4.5% ± 0.5%, data not shown). dsDNA products show a significant increase in the sgs1Δ, mph1Δ, and mph1Δ sgs1Δ mutants (3- to 4-fold) (Figure 5D). They account for ~10% of the total extension products in the sgs1Δ and srs2Δ mutants, and up to 25% in the mph1D and mph1D sgs1D mutants, all significantly higher than wild-type (Figure 5E).
Together with the DLC data, these observations show that Srs2 and most prominently Mph1 disrupt extended D-loops, most likely resulting in inhibition of BIR (Ira et al., 2003; Jain et al., 2009, 2016; Luke-Glaser and Luke, 2012; Lydeard et al., 2010; Mazón and Symington, 2013; Mitchel et al., 2013; Prakash et al., 2009; Ruiz et al., 2009; Stafa et al., 2014; Sun et al., 2008; Tay et al., 2010). The extension data validate that the increased DLC signal at 2 h in Mph1-, Srs2-, and STR-deficient cells reports increased nascent D-loop levels unaffected by effects on D-loop extension. It further reveals a differential effect of D-loop dynamics exerted by the STR-Mph1 and Srs2 pathways on the kinetics of D-loop extension initiation.
Rdh54 Inhibits Nascent D-Loops in an ATPase-Independent Fashion as Part of the STR-Mph1 Pathway
We sought to determine the control of these two nascent D-loop disruption pathways. We surmised that it should involve components of the DNA strand invasion apparatus. Rdh54 (Tid1), a paralog of the Snf2 family Rad54 dsDNA translocase (Nimonkar et al., 2007; Prasad et al., 2007), is presumably associated with the Rad51-ssDNA filament. Indeed, Rdh54 physically interacts with Rad51 in vitro (Busygina et al., 2008; Petukhova et al., 2000) in an ATPase-independent manner (Santa Maria et al., 2013) and catalyzes DNA strand invasion by Rad51 although less efficiently than Rad54 (Nimonkar et al., 2012; Petukhova et al., 2000). Furthermore, Rdh54 is recruited to DSBs in a Rad51-dependent manner (Lisby et al., 2004) and promotes engagement of dsDNA by Rad51 during homology search redundantly with Rad54 (Renkawitz et al., 2013). These observations suggest that Rdh54 is part of the Rad51-ssDNA filament in cells, similarly to Rad54. Notably, its capacity to promote inter-chromosomal template-switch suggests that it is involved in nascent and/or extended D-loop metabolism (Tsaponina and Haber, 2014).
Deletion of RDH54 causes a strong (~3-fold) DLC increase at early time points corresponding to nascent D-loops (Figure 6A). Intriguingly, the ATPase-defective rdh54-K318R mutant did not cause nascent DLC increase, indicating that the early inhibitory effect of Rdh54 is exerted independently of its catalytic activity (Figure 6B).
Figure 6. Rdh54 Demarcates the Two Nascent D-Loop Disruption Pathways.

(A) Rdh54 inhibits DLC at early time points.
(B) Rdh54 inhibits nascent DLC in an ATPase-independent fashion.
(C) RDH54 is epistatic to the STR-Mph1 nascent D-loop disruption axis.
(D) Deletion of both RDH54 and SRS2 causes a synergistic nascent DLC increase.
(E) The ATPase activity of Rdh54 is not required for nascent DLC inhibition in a srs2Δ mutant.
(F) Model for the regulation of the nascent D-loop reversal pathways by Rdh54. Borders thickness reflects contribution to the DLC signal of each D-loop type: the D-loop type 2 contributes more DLC signal than the type 1 (see Discussion and Figure S7).
In (A)–(E), data represent mean ± SEM of at least biological triplicates, except rdh54-K318R srs2Δ (biological duplicate). Asterisk indicates statistical significance (*p < 0.05).
Absence of Rdh54 did not significantly affect the kinetics or efficiency of D-loop extension initiation, akin to a srs2Δ mutant (Figure S6A). The rdh54-K318R mutant exhibited a defect in the efficiency of D-loop extension initiation, indicating that a later function of Rdh54 in D-loop metabolism requires its motor activity (Figure S6A).
RDH54 is epistatic to SGS1 and MPH1, as any mutant combination exhibits the same 3-fold nascent DLC increase over wild-type (Figure 6C). This result was corroborated upon top3-cd overexpression in rdh54Δ and rdh54Δ sgs1Δ cells, which did not exhibit significant nascent DLC increase over the empty vector control (Figure S6B). Consequently, Rdh54 belongs to the STR-Mph1 nascent D-loop disruption pathway and exerts this function independently of its catalytic activity.
RDH54 Exhibits Unique Genetic Interactions with SRS2
Combined defects of Srs2 with either STR or Mph1 causes HR-dependent synthetic sickness (Gangloff et al., 2000; Prakash et al., 2009). Contrary to mph1Δ srs2Δ and sgs1Δ srs2Δ mutants, rdh54Δ srs2Δ cells did not exhibit major growth defects (Figure S6C). Remarkably, the rdh54Δ srs2Δ mutant exhibits a strong nascent DLC increase, 12-fold over wild-type levels and 4-fold higher than in any single mutant (Figure 6D). It indicates that SRS2 and RDH54 synergistically inhibit nascent D-loops. Hence, Rdh54 is epistatic to the STR-Mph1 disruption axis but exhibits unique genetic interactions with the Srs2 pathway, both for DLC (synergy) and viability (no synthetic lethality). The rdh54-K318R srs2Δ mutant does not exhibit further DLC increase over the srs2Δ mutant (Figure 6E), confirming that the catalytic ac tivity of Rdh54 is dispensable for nascent DLC inhibition. Based on these results, we propose that the physical presence of Rdh54 demarcates the two nascent D-loop disruption pathways defined on the one hand by STR and Mph1, and on the other hand by Srs2 (Figures 6F and S7A; see Discussion).
Rdh54 Promotes Maturation of NCO in a Homology Length-Dependent Manner
Mph1, STR, and Srs2 promote conservative HR outcome, either by promoting NCO formation (Srs2) or by inhibiting CO formation (STR and Mph1) (Ira et al., 2003; Mazón and Symington, 2013; Mitchel et al., 2013; Prakash et al., 2009). We addressed the role of Rdh54 in regulating HR outcome using a well-established physical recombination assay exploiting 5,653 bp of homology (4,020 and 1,633 bp on each side of the DSB, respectively) with DSB-inducible and donor sequences conveniently located at the same loci and in the same genetic background as in our DLC system (Inbar and Kupiec, 1999; Inbar et al., 2000; Mazón and Symington, 2013). The DSB repair efficiency decreases significantly (by 34%) in the rdh54Δ mutant compared to wild-type cells (Figures 7A and 7B). This decrease is due to a loss of NCO events, which are not compensated for by an increase in CO events. The rdh54-K318R mutant causes an additional repair defect, which affects the production of both NCO and CO (Figures 7A and 7B), possibly related to its defect in D-loop extension initiation (Figure S6A). This decrease in repair efficiency is accompanied by a commensurate decrease in cell viability (Figure S6D). Similar results, although quantitatively less pronounced, were obtained with an independent physical recombination assay with shorter homologies (≈1.5 kb and 0.5 kb on each side of the DSB, respectively) and located at different loci (Figure S6E; Ira et al., 2003). To address whether homology length could account for these quantitative differences, we measured DSB repair efficiency in the initial recombination assay with substrates bearing 10-fold shorter homology (Inbar et al., 2000). In this context, the rdh54D mutant did not exhibit a DSB repair defect compared to the wild-type strain (Figure 7C).
Figure 7. Rdh54 Promotes Maturation of HR Repair Products.

(A) Southern blot analysis of DSB repair outcome (CO and NCO) between the URA3 and LYS2 loci bearing 5.6 kb of homology (Inbar and Kupiec, 1999) in wild-type, rdh54Δ, and rdh54-K318R strains.
(B) Quantification of DSB repair outcome in (A) at 8 h post-DSB induction.
(C) Quantification of the DSB repair efficiency in rdh54Δ strains bearing either 0.5 kb or 5.6 kb of homology 8 h post-DSB induction, normalized onto the repair of a parallel wild-type strain.
(D) Quantification of the DSB repair outcome in wild-type, rdh54Δ, srs2Δ, rdh54Δ srs2Δ, mph1D, and rdh54Δ mph1Δ strains 8 h post-DSB induction in the same system as in (A).
(E) Model of the regulation of the HR pathway deduced, from physical detection of HR intermediates, steps and products using the DLC, DLE, and Southern blot assays. The components of the regulation of the repair outcome determined by Southern blot are likely a composite of the regulations exerted on nascent and extended D-loops as well as downstream intermediates, but their individual contributions remain undefined.
In (B)–(D), data represent mean ± SEM of at least biological triplicates. Asterisk indicates statistical significance (*p < 0.05).
These results uncover a role for Rdh54 in promoting maturation of NCO repair products that depends on homology length. It suggests that in the absence of Rdh54 D-loops formed at long homologies persist and become dead-end substrates that cannot be processed in NCO or salvaged as CO.
RDH54 Is Epistatic with MPH1 and SRS2 in Promoting HR Completion
The NCO maturation-promoting function of Rdh54 is reminiscent of that reported for Srs2 with physical and genetic assays exhibiting limited homologies (Ira et al., 2003; Mitchel et al., 2013). We addressed the role of Srs2 in HR repair in the initial recombination assay bearing long homologies. We confirm the defect of srs2Δ cells in maturing NCO, and also uncover a slight but significant defect in CO suppression (Figures 7D and S6F). This CO suppression defect remains less pronounced than in a mph1Δ mutant (Figures 7D and S6F; Mazón and Symington, 2013). The DSB repair defects of the rdh54Δ srs2Δ and rdh54Δ mph1Δ double mutants are not significantly different from that of the srs2Δ and mph1Δ single mutants, respectively (Figures 7D and S6F). This indicates that Rdh54 participates in the same mechanism as Srs2 for NCO production while it is synergistic for the DLC signal. Hence, the genetic interactions defined at the nascent D-loop stage remain insufficient to account for the observations made at the final product stage. These results highlight the difficulty in inferring regulation potentially occurring at multiple and inter-dependent steps of this complex pathway from repair products (Figure 7E).
DISCUSSION
The DLC Assay: A Versatile Tool for Studying DNA Joint Molecules in Cells
HR is a complex pathway involving multiple protein players possibly acting in an overlapping fashion at the level of several meta-stable intermediates. This complexity is not efficiently tackled by studying only the end-point repair products (Figure 7E). The DLC assay enables physical detection in cells of the central D-loop intermediate in HR, allowing determination of the pathways controlling its formation and processing. More generally, this assay can detect any joint molecule crosslinkable with psoralen. The simple rationale, inspired by the chromosome conformation capture approach (Dekker et al., 2002), and the large dynamic range (over three orders of magnitude) of the qPCR readout, suggests the assay is suitable to experimental set-ups with less frequent DSB formation and/or DNA strand invasion than in our system. For example, we previously used a variant of this assay to detect multi-invasion HR byproducts, which are less frequent than single D-loops (Piazza et al., 2017). Because psoralen has already been extensively used in a variety of organisms (including mammalian cells), and DSB delivery by CRISPR-Cas9 is nearly universally applicable, we expect this versatile approach to open the way for physical study of the HR process in various organisms and cell types.
A Complex Network of Proteins Enforces D-Loop Dynamics
The steady-state DLC increase observed in various mutants reveals that the majority of D-loops involving a perfectly homologous 2 kb-long donor are disrupted in wild-type cells. We provide direct evidence for three activities mediated by STR, Mph1, and Srs2 in promoting nascent and extended D-loop turnover in S. cerevisiae cells. Srs2 and Mph1 disrupt D-loops in a helicase-dependent manner, while STR acts in a topoisomerase-dependent and largely helicase-independent manner. The increase in nascent D-loop levels in Srs2-deficient cells is unlikely related to its Rad51-dissociation activity (Krejci et al., 2003; Veaute et al., 2003). The Rad51 paralogs Rad55-Rad57 appear to completely insulate the Rad51-ssDNA filament from Srs2-mediated disruption during DSB repair, as indicated by the complete suppression of the IR-sensitivity of the rad55 and rad57 mutants by an Srs2 defect (Liu et al., 2011). Moreover, an additional upstream Rad51 filament dissociation defect in the srs2D mutant would be expected to synergize with a D-loop disruption defect in a STR or MPH1 mutant, which was not observed. Hence, we interpret the increase in DLC signal in srs2Δ cells to largely reflect a D-loop disruption defect. Hence, nascent D-loops are subject to multiple regulatory activities resulting in a dynamic equilibrium (Figure 7E), confirming prior conclusions drawn from end-point assays and modeling (Coïc et al., 2011; Zinovyev et al., 2013). These dynamics may at least partly account for the delay observed between D-loop formation and extension (Figures 2C and 5C).
Genetic interaction between these regulatory activities also revealed unexpected complexities in nascent D-loop metabolism (Figures 3 and 4). Indeed, STR with Mph1 and Srs2 define two independent nascent D-loop disruption pathways (Figure 4G). Importantly, combined elimination of any member of both pathways results in an additive increase in D-loop levels. This additivity implies that the two pathways target non-overlapping and non-interconvertible nascent D-loop species. We propose a model for nascent D-loop metabolism by STR-Mph1 and Srs2 in which Rdh54 acts as a gate keeper in delineating the two disruption pathways (Figure 6F). Rdh54 promotes the formation of a substrate for the STR-Mph1 axis, while escapers are exclusively disrupted by Srs2. In particular, this model explains why, despite being part of the Mph1-STR D-loop disruption axis, defects of Rdh54 could synergize with those of a Srs2-defective strain in term of DLC (see Figure S7A for more details).
Demarcation Function of Rdh54 and the Nature of the Two Nascent D-Loop Disruption Pathways
What distinguishes the two nascent D-loop disruption pathways? And how does Rdh54 exert its demarcation role? The ATPase-independent role of Rdh54 rules out chromatin remodeling (Kwon et al., 2008), change in DNA supercoiling (Chi et al., 2006; Petukhova et al., 2000; Prasad et al., 2007), joint molecule disruption (Nimonkar et al., 2007), or Rad51 stripping from dsDNA (Chi et al., 2006) in the pathway demarcation process. Furthermore, the synergistic DLC increase specifically observed in rdh54Δ srs2Δ, but not in cells deficient for any member of the Mph1-STR disruption pathway, suggests that Srs2 substrates potentially contain longer hDNA than the substrates funneled by Rdh54 to the STR-Mph1 disruption pathway (Figure S7A), possibly enhancing the DLC signal due to the higher crosslinking efficiency (Figure S2A). Rdh54 could limit hDNA length by acting as a roadblock for Rad54-mediated hDNA formation (Wright and Heyer, 2014), a function otherwise assumed by heterology blocks such as in the case of short homologies. Whether the hDNA length per se or architectural features such as internal versus end invasion (Adzuma, 1992; Piazza et al., 2017; Wright and Heyer, 2014) distinguishes the two nascent D-loop species and/or guides differential processing by the Srs2 and STRMph1 pathways remains to be addressed (Figure S7B).
Implications of Nascent D-Loops Dynamics for HR Fidelity and Outcome
While extended D-loop disruption is integral to the SDSA pathway, the role of disruption activities targeting perfectly homologous nascent D-loop is less straightforward within the current HR framework. We speculate that it may contribute to HR fidelity in four ways. First, we showed previously that these activities inhibit multi-invasion-induced rearrangements, a tripartite recombination mechanism involving the cleavage of internal and terminal nascent D-loops (Piazza and Heyer, 2018a, 2018b; Piazza et al., 2017). This also likely applies to single D-loop cleavage causing half-CO (Deem et al., 2008; Mazón and Symington, 2013; Pardo and Aguilera, 2012). Second, nascent D-loop turnover is expected to enforce homology search stringency (and thus HR fidelity) by requiring several rounds of donor interrogation prior to initiation of recombinational DNA synthesis (Coïc et al., 2011). Given that homology length stimulates homology search by the Rad51-ssDNA filament (Bell and Kowalczykowski, 2016), this kinetic proofreading strategy is expected to funnel the searching molecule toward the longest homologous donor available. Consequently, the homology search process consists in homology sampling by the Rad51-ssDNA filament in the context of multiple cycles of invasion-disruption, until recombinational DNA synthesis is initiated. Third, reversal of one-ended invasions lowers the probability of two concomitant invasions from both ends of a DSB that would lead to dHJs and increase the potential for crossover. Finally, given that nascent D-loop reversal delays the initiation of D-loop extension, it could disproportionately inhibit mutagenic repair such as BIR, which likely involves multiple rounds of invasion, extension, and disruption cycles (Anand et al., 2014; Ruiz et al., 2009; Stafa et al., 2014).
Conclusion
This work reveals unanticipated dynamics at the nascent joint molecule level, deciphers regulation imposed by several HR regulators at the nascent and extended D-loop levels, and uncovers an unexpected role of the Rdh54 motor protein as a delineating factor between two nascent D-loop disruption pathways.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wolf-Dietrich Heyer (wdheyer@ucdavis.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
The genotype of the haploid Saccharomyces cerevisiae strains (W303 background) used in this study are listed in Table S1. They contain a copy of the HO endonuclease gene under the control of the GAL1/10 promoter at the TRP1 locus on chr. IV (Pannunzio et al., 2008). A point mutation inactivates the HO cut-site at the mating-type locus (MAT) on chr. III (MATa-inc or MATα-inc). The heterozygous DSB-inducible construct replaces URA3 on chr. V (−16 to +855 from the start codon). The DSB-inducible construct contains the 117 bp HO cut-site (Fishman-Lobell and Haber, 1992), a 2086 bp-long “A” sequence (+4 to +2090 of the LYS2 gene), and a 327 bp fragment of the PhiX genome flanked by multiple restriction sites. Alternatively, a same length “B” sequence (+2091 to +4177 of the LYS2 gene) was used to address homology requirement for DLC in Figure 1D. The “A” or “B” donor replaces the LYS2 gene on chr. II. The URA3 locus on chr. V and of the LYS2 locus on chr. II have been chosen because of their interstitial and untethered location (Agmon et al., 2013; Berger et al., 2008; Inbar and Kupiec, 1999; Mazón and Symington, 2013; Miné-Hattab and Rothstein, 2012). Since the region of homology to the ectopic donor “A” does not encompass the DSB site, this system prevents formation of later intermediates involving both ends of the DSB, so as to focus our study on the regulation of D-loop intermediates. The donor sequence is oriented so that BIR (the only available repair pathway) is discouraged by the presence of the centromere (Morrow et al., 1997) and is in any case lethal. This system thus prevents resumption of growth and invasion of the population by cells undergoing early repair. We showed previously that the formation of the BIR product is not detected prior to 8 hr after DSB induction (Piazza et al., 2018). The annotated sequences of the DSB-inducible and donor constructs are available as ape files in Data S1.
To investigate the genetic interaction of SRS2 with SGS1 and MPH1 we used a conditional protein degradation system induced by auxin (Morawska and Ulrich, 2013). Srs2 is fused to its C terminus to an auxin-inducible degron (AID) tag together with 9-Myc (referred to as Srs2-AID). The Srs2 Strains bearing the uninduced Srs2-AID construct and lacking either the SGS1 or MPH1 genes grow normally (Figure S4B), indicating that the AID tag does not impair the essential Srs2 function in these mutants. The gene encoding the AID-specific E3 ubiquitin ligase OsTIR1 under the control of the pADH1 promoter is constitutively expressed from a centromeric vector (pRS314). Auxin (Sigma I5148) was dissolved in DMSO at 285 mM. In the absence of auxin (equivalent DMSO concentration), Srs2-AID appears slightly more potent in DLC inhibition than untagged Srs2 (Figures 5D and S4C). Srs2 is maximally depleted within 1 h following auxin addition (2 mM final concentration) (Figure S4D). The low level of remaining Srs2 does not significantly contribute to the DLC signal, as shown by the equivalent phenotype of the AID allele compared to the deletion (Figure 4D). Consequently Srs2-AID degradation is induced 1 h before DSB induction (Figure 5B). Auxin did not induce change of DLC, as shown in a treated strain bearing an untagged version of Srs2 (Figures 5C and 5D). The empty, TOP3 and top3-Y356F overexpression vectors were kindly provided by Ian Hickson (Oakley et al., 2002). The genes are under the control of a pGAL1 promoter on a 2-micron multi-copy plasmid (pYES2). The rdh54-K318R (rdh54-K352R in the S288c reference) mutant was kindly provided by Hannah Klein (Chi et al., 2006). Other mutants were generated by traditional gene replacement with antibiotic-resistance or prototrophic genes by regular lithium-acetate transformation.
Strains used for the physical recombination assays are from the W303 (Figures 7A–D and S6E) and JKM backgrounds (Figure S6C). They are isogenic derivatives of MK202 (Inbar and Kupiec, 1999) or OI30 (Inbar et al., 2000), and tGI354 (Ira et al., 2003), respectively (Table S1).
Culture media
Synthetic dropout and rich YPD (1% yeast extract, 2% peptone, 2% dextrose) solid and liquid media have been prepared according to standard protocols. Liquid YEP-lactate (1% yeast extract, 2% peptone, 2% Lactate), Lactate-URA and Lactate-TRP (0.17% Yeast Nitrogen Base, 0.5% Ammonium Sulfate, respective 0.2% amino acids dropout, 2% Lactate) were made using 60% Sodium DL-lactate syrup. All cultures were grown at 30°C.
METHOD DETAILS
DLC assay
Cells were cultured to exponential phase in YEP-lactate and DSB at the HOcs on chr. V was induced by HO expression upon galactose addition at a 2% final concentration (Piazza et al., 2017). A total of 2.108 cells were collected before, or at various time post-DSB induction, pelleted, and re-suspended in 2.5 mL of a Psoralen crosslinking solution (0.1 mg/mL Trioxsalen (Sigma-Aldrich T6137), 50 mM Tris HCl pH 8.0, 50 mM EDTA, 20% ethanol), made immediately before use from a 5X Psoralen stock solution (0.5 mg/mL Trioxsalen in pure ethanol, solubilized overnight with shacking at room temperature). Crosslink of cells was performed in a 60 mm Petri dish upon long wave (365 nm) UV irradiation in a Spectrolinker XL-1500 (Spectroline) for 15 min with permanent orbital agitation (50–70 rpm). Cells were washed in 50 mM Tris HCl pH 8, 50 mM EDTA and the pellet stored at 20°C. Cells were spheroplasted in a zymolyase solution (0.4 M Sorbitol, 0.4 M KCl, 0.5 mM MgCl2, 40 mM Sodium Phosphate buffer pH 7.2, 20 μg/mL Zymolyase 100T (US Biological)) for 15 min at 30°C. Zymolyase was washed 3 times in 1 mL of spheroplasting buffer at 2,500 g and 3 times in 1 mL of 1X Cutsmart Buffer (NEB) at 16000 g. Cells were resuspended at a final concentration of 4.108 cells/mL in 1.4 × Cutsmart buffer containing 6 pM of a long hybridization oligonucleotide (olWDH1770, Table S2) to restore the EcoRI site on the resected broken molecule, and stored at 80°C. Chromatin of 4×107 cells is solubilized upon incubation at 65°C for 10 min with 0.1% SDS. SDS is quenched by addition of 1% Triton X-100. DNA is digested by 20 units of EcoRI-HF (NEB) at 37°C for 1 h. Proteins are denatured by addition of 2% SDS and incubation at 55°C for 10 min. Cells are put in ice and SDS is quenched by addition of 6% Triton X-100. Ligation is performed in 800 μL of a ligation mix (50 mM Tris-HCl pH 8.0, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 0.25 mg/mL BSA, 300 units of T4 DNA ligase (Bayou Biolabs)) at 16°C for 1h30. 25 μg/mL Proteinase K is added and proteins digested for 30 min at 65°C. DNA is extracted following a standard Phenol:Chloroform:Isoamyl Alcohol and isopropanol precipitation procedure. DNA pellets are dried, re-suspended 1 h in 50 μL 10 mM Tris HCl pH 8.0, 1 mM EDTA, 0.4 mg/mL RNase A. The quantitative PCR was performed on a Roche LightCycler 480 machine using the SYBR Green I Master kit (Roche), according to the manufacturer instructions. After an initial denaturation phase, the cycling conditions were 95°C for 15 s, 61°C for 12 s, 72°C for 15 s, repeated 50 times. The nature of the amplified product was confirmed by determination of its melting temperature. Six reactions were performed (Figure S1, primers see Table S2): a loading standard (ARG4) on which the other reactions are normalized (olWDH1760 and 1761); an intra-molecular ligation efficiency control on a 1904 bp fragment at the DAP2 locus (olWDH1762 and 1763); a control to verify DSB formation at HOcs on chr. V (olWDH1766 and 1767); a control of EcoRI restriction site digestion on the broken molecule (olWDH1768 and 1769); a control of EcoRI restriction digestion of a control dsDNA locus (DAP2; olWDH1762 and 1769); a reaction to detect the DLC chimera, i.e., the product of the ligation of the 5′ flanking regions of the broken molecule and the donor (olWDH1764 and 1765). DLC values were normalized on the intra-molecular ligation efficiency and in some instances corrected for the filament restriction digestion efficiency when differences exceeded ≈30%. Data were analyzed using the Light Cycler 480 Software 1.5.0.
For each mutant assayed, a wild-type strain was run in parallel to buffer for inter-experiment variations that are attributed to differences in the crosslinking efficiency. For TOP3 and top3-cd overexpression experiments, an empty vector control was performed in for each experiment. In the case of the Srs2-AID degradation experiments, the control “-Auxin” treatment was run in parallel of the “+Auxin” treatment. Consequently, data are shown normalized onto parallel wild-type values (for deletion mutant or point mutant), on parallel empty vector control (Top3 overexpression experiments) or on parallel DMSO-treated (Srs2-AID degradation experiments) samples.
DLE assay
The DLE assay is performed with the same genetic constructs as the DLC assay. It follows the same cells spheroplasting, DNA digestion, ligation, and purification steps as the DLC assay. However, it does not require crosslinking of the intermediate, and uses different restriction sites (HindIII) and oligonucleotides for restriction sites restoration. A detailed procedure is described in Piazza et al. (2018). The DLE signal is normalized for digestion and intra-molecular ligation efficiency on a chimera produced upon circularization of a 700 bp-long control dsDNA fragment (Piazza et al., 2018). Although important to buffer for overall differences in digestion/ligation efficiencies between samples, this normalization also introduces an approximation related to the comparison of the intramolecular ligation efficiency of a 700 bp-long dsDNA fragment to that of a mostly ssDNA, 3,109 nt-long molecule (the extension product). This approximation may account for the DLE plateauing at ~110% molecules instead of the expected 100% in the sgs1Δ mutant (Figure 5B). Consequently, this normalization may cause a 10% over-estimation of the proportion of cells having initiated D-loop extension.
Western blot
Proteins were extracted from 2.107 cells by regular TCA procedure. Srs2-AID-9Myc and OsTir1–9Myc were detected with a mouse anti-c-Myc 9E11 antibody (Santa Cruz Biotechnology, sc-47694, lot F11 13) used at a 1:1000 dilution, and GAPDH was detected with a mouse anti-GAPDH GA1R from Thermo Scientific (MA5–15738, lot QG215126) at a 1:5000 dilution.
Southern blot analysis
The physical recombination assay presented in Figures 7A–7D and S6F has been performed as described in Mazón and Symington (2013) for the strain bearing 5.6 kb of homology, and as in Inbar et al. (2000) for the strain bearing 0.5 kb of homology. It monitors DSB repair at the same location on chr. II (LYS2) and chr. V (URA3) and in the same genetic background (W303) as in our DLC system (Inbar and Kupiec, 1999). The broken locus shares 5.6 kb of homology (4 + 1.6 kb) or 0.5 kb of homology (0.25 + 0.25 kb) to the ectopic donor on chr. II (Inbar and Kupiec, 1999; Inbar et al., 2000). The primers used to generate the URA3 probe centered on the HOcs are listed in Table S2.
The physical recombination assay presented in Figure S6E has been performed as described in Ira et al. (2003). The broken and donor loci share 2.1 kb of homology between chr. III (MAT) and chr. V (ARG5,6). In both cases, the probe encompasses the DSB site so as to equally reveal both DSB ends and both CO products. The primers used to generate the MAT probe are listed in Table S2.
Southern blotting was performed as follow: digested DNA was migrated overnight in Agarose-LE (Affymetrix) 0.8% in TBE 1X at 50 V. The DNA was transferred from the gel onto an Amersham Hybond-XL membrane (GE healthcare) following the manufacturer instructions (alkali protocol). The membrane was blocked with Church buffer (1% BSA, 0.25 M Na2HPO4 pH7.3, 7% SDS, 1 mMEDTA) for 2–3 h at 65°C. The probe (100 ng) together with Phage λ DNA (molecular ladder, 1 ng) were radio-labeled by random priming with P32-αdCTP (6,000 Ci/mmol; Perkin-Elmer) using the Decaprime II kit (Ambion) and incubated with the membrane overnight at 65°C. After 3 – 5 washes for 10 min at 65C (20 mM Na2HPO4 pH 7.3, 1% SDS 1%, 1 mM EDTA), membranes were exposed for 8 to 24 h, and the Storage Phosphor Screen (GE healthcare) scanned on a Storm Phosphorimager (Molecular Dynamics).
Viability analysis
The viability of the wild-type, rdh54Δ and rdh54-K318R strains used for Southern blot analysis (Figure 7A) was determined by plating cells on solid YPD media prior (no DSB) and 2 h after induction of HO expression (+ DSB) in liquid YEP-lactate media. Colonies were counted after 2 days of incubation at 30°C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Southern blot analysis
The exposed Phosphor screen (GE Healthcare) was scanned on a Phosphorimager (Molecular Dynamics) and the Southern blot signal was quantified using ImageJ 1.51k.
Statistical analysis
Mutant DLC values were compared to their paired wild-type or empty vector controls with a Mann-Whitney Wilcoxon test. Other comparisons between normalized DLC values, Southern blot quantifications, and viability estimates were performed using unpaired Mann-Whitney Wilcoxon test. Statistical cutoff was set to α = 0.05 for all tests. All statistical tests were performed under R ×64 3.2.0.
DATA AND SOFTWARE AVAILABILITY
Construct sequences
The annotated sequences of the DSB-inducible and donor constructs used in this study are available as *.ape (ApE Plasmid Editor) files in Data S1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Myc (9E11) | Santa Cruz Biotechnology | sc-47694, lot F11 13; RRID: AB_627266 |
| Mouse monoclonal anti-GAPDH | Thermo Scientific | MA5–15738, lot QG215126; RRID: AB_10977387 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trioxsalen (TMP) | Sigma-Aldrich | Cat#T6137 |
| lndole-3-acetic acid (IAA) sodium salt (Auxin) | Sigma-Aldrich | Cat#l5148 |
| dCTP-alpha-P32 6000 uCi/mmol | PerkinElmer | Cat#BLU-513Z |
| T4 DNA Ligase | New England Biolabs | Cat#M0202 |
| EcoRI-HF | New England Biolabs | Cat#R3101 |
| Hindlll-HF | New England Biolabs | Cat#R3104 |
| Critical Commercial Assays | ||
| LightCycler 480 SYBR Green I Master | Roche Life Science | Cat#04707516001 |
| Ambion DecaPrime II DNA labeling kit | Thermo Fisher | Cat#AM1455 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae; Individual genotypes see Table S1 | This study | N/A |
| Oligonucleotides | ||
| Quantitative PCR primers, see Table S2 | Eurofins Genomics | N/A |
| Hybridization oligonucleotides, see Table S2 | Sigma-Aldrich | N/A |
| Software and Algorithms | ||
| LightCycler 480 Software | Roche Life Science | Cat#04994884001 |
| R ×64 3.2.0 | R Project | https://www.r-project.org/ |
| Ape Plasmid Editor | Wayne Davis | https://biologylabs.utah.edu/jorgensen/wayned/ape/ |
| ImageJ 1.51k | Wayne Rasband | https://imagej.nih.gov/ij/ |
| Other | ||
| Spectrolinker (with 365 nm UV bulbs) | Spectroline | XL-1500 |
Highlights.
Development of a physical assay for D-loop detection in cells
Srs2, Mph1, and Sgs1-Top3-Rmi1 (STR) regulate D-loop levels in vivo
Two distinct pathways (Srs2 and Mph1, STR) target different D-loop species
Rdh54 delineates the two D-loop reversal pathways
ACKNOWLEDGMENTS
We thank members of the Heyer, Hunter, and Kowalczykowski laboratories for helpful discussions, Ian Hickson for providing the TOP3 overexpression vectors, Hannah Klein for the rdh54-K318R mutant, and Lorraine Symington, Martin Kupiec, and Jim Haber for the strains to perform the physical recombination assays. We thank Abou Ibrahim-Biangoro and Noelle Cabral for help with strains construction. We are particularly grateful to Sue Jinks-Robertson and Pallavi Rajput for their critical reading of the manuscript. A.P. was supported by fellowships from the ARC Foundation, the EMBO (ALTF-238–2013), and the Framework Project 7 of the European Union (Marie Sk1odowska-Curie International Outgoing Fellowship 628355) and received financial support from the Philippe Foundation. This research used core services supported by P30 CA93373 and was supported by NIH grants GM58015 and CA92276 to W.-D.H. and the Agence Nationale pour la Recherche grant ANR-13-BSV6-0012-02 to R.K.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, two tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.molcel.2019.01.005.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adzuma K (1992). Stable synapsis of homologous DNA molecules mediated by the Escherichia coli RecA protein involves local exchange of DNA strands. Genes Dev. 6, 1679–1694. [DOI] [PubMed] [Google Scholar]
- Agmon N, Liefshitz B, Zimmer C, Fabre E, and Kupiec M (2013). Effect of nuclear architecture on the efficiency of double-strand break repair. Nat. Cell Biol 15, 694–699. [DOI] [PubMed] [Google Scholar]
- Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, and Haber JE (2014). Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 28, 2394–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JC, and Kowalczykowski SC (2016). RecA: regulation and mechanism of a molecular search engine. Trends Biochem. Sci 41, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AB, Cabal GG, Fabre E, Duong T, Buc H, Nehrbass U, Olivo-Marin JC, Gadal O, and Zimmer C (2008). High-resolution statistical mapping reveals gene territories in live yeast. Nat. Methods 5, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Busygina V, Sehorn MG, Shi IY, Tsubouchi H, Roeder GS, and Sung P (2008). Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev. 22, 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzymek M, Thayer NH, Oh SD, Kleckner N, and Hunter N (2010). Double Holliday junctions are intermediates of DNA break repair. Nature 464, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Plank JL, Bachrati CZ, Hickson ID, and Kowalczykowski SC (2010). Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat. Struct. Mol. Biol 17, 1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Plank JL, Dombrowski CC, and Kowalczykowski SC (2012). Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol. Cell 47, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Kwon Y, Seong C, Epshtein A, Lam I, Sung P, and Klein HL (2006). Yeast recombination factor Rdh54 functionally interacts with the Rad51 recombinase and catalyzes Rad51 removal from DNA. J. Biol. Chem 281, 26268–26279. [DOI] [PubMed] [Google Scholar]
- Coïc E, Martin J, Ryu T, Tay SY, Kondev J, and Haber JE (2011). Dynamics of homology searching during gene conversion in Saccharomyces cerevisiae revealed by donor competition. Genetics 189, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Gaines WA, Kwon Y, and Sung P (2014). Regulation of DNA pairing in homologous recombination. Cold Spring Harb. Perspect. Biol 6, a017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Barker K, Vanhulle K, Downing B, Vayl A, and Malkova A (2008). Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 179, 1845–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromo-some conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Fasching CL, Cejka P, Kowalczykowski SC, and Heyer WD (2015). Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell 57, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, and Haber JE (1992). Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258, 480–484. [DOI] [PubMed] [Google Scholar]
- Gangloff S, Soustelle C, and Fabre F (2000). Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet 25, 192–194. [DOI] [PubMed] [Google Scholar]
- Heyer WD (2015). Regulation of recombination and genomic maintenance. Cold Spring Harb. Perspect. Biol 7, a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar O, and Kupiec M (1999). Homology search and choice of homologous partner during mitotic recombination. Mol. Cell. Biol 19, 4134–4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inbar O, Liefshitz B, Bitan G, and Kupiec M (2000). The relationship between homology length and crossing over during the repair of a broken chromosome. J. Biol. Chem 275, 30833–30838. [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, and Haber JE (2003). Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, and Haber JE (2009). A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 23, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Sugawara N, Mehta A, Ryu T, and Haber JE (2016). Sgs1 and Mph1 helicases enforce the recombination execution checkpoint during DNA double-strand break repair in Saccharomyces cerevisiae. Genetics 203, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaar R, Wyman C, and Rothstein R (2008). Quality control of DNA break metabolism: in the ‘end’, it’s a good thing. EMBO J. 27, 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniecki K, De Tullio L, Gibb B, Kwon Y, Sung P, and Greene EC (2017). Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Rep. 21, 3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, De Muyt A, and Lichten M (2015). Top3-Rmi1 DNA single-strand decatenase is integral to the formation and resolution of meiotic recombination intermediates. Mol. Cell 57, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejci L, Van Komen S, Li Y, Villemain J, Reddy MS, Klein H, Ellenberger T, and Sung P (2003). DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423, 305–309. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Seong C, Chi P, Greene EC, Klein H, and Sung P (2008). ATP-dependent chromatin remodeling by the Saccharomyces cerevisiae homologous recombination factor Rdh54. J. Biol. Chem 283, 10445–10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, and Rothstein R (2004). Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118, 699–713. [DOI] [PubMed] [Google Scholar]
- Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, and Heyer W-D (2011). Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 479, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ede C, Wright WD, Gore SK, Jenkins SS, Freudenthal BD, Todd Washington M, Veaute X, and Heyer WD (2017). Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase d-extending D-loops. eLife 6, e22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YC, Paffett KS, Amit O, Clikeman JA, Sterk R, Brenneman MA, and Nickoloff JA (2006). Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol. Cell. Biol 26, 4086–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, and Luke B (2012). The Mph1 helicase can promote telo-mere uncapping and premature senescence in budding yeast. PLoS ONE 7, e42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Jain S, Eapen VV, and Haber JE (2010). Sgs1 and exo1 redundantly inhibit break-induced replication and de novo telo-mere addition at broken chromosome ends. PLoS Genet. 6, e1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazón G, and Symington LS (2013). Mph1 and Mus81-Mms4 prevent aberrant processing of mitotic recombination intermediates. Mol. Cell 52, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné-Hattab J, and Rothstein R (2012). Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol 14, 510–517. [DOI] [PubMed] [Google Scholar]
- Mitchel K, Lehner K, and Jinks-Robertson S (2013). Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet. 9, e1003340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M, and Ulrich HD (2013). An expanded tool kit for the auxininducible degron system in budding yeast. Yeast 30, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, and Hieter P (1997). “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimonkar AV, Amitani I, Baskin RJ, and Kowalczykowski SC (2007). Single molecule imaging of Tid1/Rdh54, a Rad54 homolog that translocates on duplex DNA and can disrupt joint molecules. J. Biol. Chem 282, 30776–30784. [DOI] [PubMed] [Google Scholar]
- Nimonkar AV, Dombrowski CC, Siino JS, Stasiak AZ, Stasiak A, and Kowalczykowski SC (2012). Saccharomyces cerevisiae Dmc1 and Rad51 proteins preferentially function with Tid1 and Rad54 proteins, respectively, to promote DNA strand invasion during genetic recombination. J. Biol. Chem 287, 28727–28737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley TJ, Goodwin A, Chakraverty RK, and Hickson ID (2002). Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA Repair (Amst.) 1, 463–482. [DOI] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Hwang PYH, Taylor AF, Smith GR, and Hunter N (2007). BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 130, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SD, Jessop L, Lao JP, Allers T, Lichten M, and Hunter N (2009). Stabilization and electrophoretic analysis of meiotic recombination intermediates in Saccharomyces cerevisiae. Methods Mol. Biol 557, 209–234. [DOI] [PubMed] [Google Scholar]
- Pannunzio NR, Manthey GM, and Bailis AM (2008). RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair (Amst.) 7, 788–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, and Aguilera A (2012). Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet. 8, e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova G, Stratton S, and Sung P (1998). Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature 393, 91–94. [DOI] [PubMed] [Google Scholar]
- Petukhova G, Sung P, and Klein H (2000). Promotion of Rad51-dependent D-loop formation by yeast recombination factor Rdh54/Tid1. Genes Dev. 14, 2206–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, and Heyer WD (2018a). Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol. Published online November 26, 2018. 10.1016/j.tcb.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, and Heyer WD (2018b). Multi-invasion-induced rearrangements as a pathway for physiological and pathological recombination. BioEssays 40, e1700249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Wright WD, and Heyer WD (2017). Multi-invasions are recombination byproducts that induce chromosomal rearrangements. Cell 170, 760–773.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A, Koszul R, and Heyer WD (2018). A proximity ligation-based method for quantitative measurement of D-loop extension in S. cerevisiae. Methods Enzymol. 601, 27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, and Ira G (2009). Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 23, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad TK, Robertson RB, Visnapuu ML, Chi P, Sung P, and Greene EC (2007). A DNA-translocating Snf2 molecular motor: Saccharomyces cerevisiae Rdh54 displays processive translocation and extrudes DNA loops. J. Mol. Biol 369, 940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, and Kolodner RD (2017). Pathways and mechanisms that prevent genome instability in Saccharomyces cerevisiae. Genetics 206, 1187–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam CD, Hayes TK, and Kolodner RD (2009). Specific pathways prevent duplication-mediated genome rearrangements. Nature 460, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawitz J, Lademann CA, Kalocsay M, and Jentsch S (2013). Monitoring homology search during DNA double-strand break repair in vivo. Mol. Cell 50, 261–272. [DOI] [PubMed] [Google Scholar]
- Ruiz JF, Gómez-González B, and Aguilera A (2009). Chromosomal trans-locations caused by either pol32-dependent or pol32-independent triparental break-induced replication. Mol. Cell. Biol 29, 5441–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa Maria SR, Kwon Y, Sung P, and Klein HL (2013). Characterization of the interaction between the Saccharomyces cerevisiae Rad51 recombinase and the DNA translocase Rdh54. J. Biol. Chem 288, 21999–22005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwacha A, and Kleckner N (1995). Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83, 783–791. [DOI] [PubMed] [Google Scholar]
- Sebesta M, Burkovics P, Haracska L, and Krejci L (2011). Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair (Amst.) 10, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeber A, Hauer MH, and Gasser SM (2018). Chromosome dynamics in response to DNA damage. Annu. Rev. Genet 52, 295–319. [DOI] [PubMed] [Google Scholar]
- Stafa A, Donnianni RA, Timashev LA, Lam AF, and Symington LS (2014). Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196, 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Wang X, and Haber JE (2003). In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 12, 209–219. [DOI] [PubMed] [Google Scholar]
- Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, and Whitby MC (2008). The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol. Cell 32, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS (2014). End resection at double-strand breaks: mechanism and regulation. Cold Spring Harb. Perspect. Biol 6, a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Wu MKY, Zhang R, and Hunter N (2015). Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Mol. Cell 57, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay YD, Sidebotham JM, and Wu L (2010). Mph1 requires mismatch repair-independent and -dependent functions of MutSalpha to regulate crossover formation during homologous recombination repair. Nucleic Acids Res. 38, 1889–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaponina O, and Haber JE (2014). Frequent interchromosomal template switches during gene conversion in S. cerevisiae. Mol. Cell 55, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, and Fabre F (2003). The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309–312. [DOI] [PubMed] [Google Scholar]
- Weinstein J, and Rothstein R (2008). The genetic consequences of ablating helicase activity and the Top3 interaction domain of Sgs1. DNA Repair (Amst.) 7, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CI, and Haber JE (1990). Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 9, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WD, and Heyer WD (2014). Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Mol. Cell 53, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, and Hickson ID (2003). The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874. [DOI] [PubMed] [Google Scholar]
- Zheng XF, Prakash R, Saro D, Longerich S, Niu H, and Sung P (2011). Processing of DNA structures via DNA unwinding and branch migration by theS. cerevisiae Mph1 protein. DNA Repair (Amst.) 10, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinovyev A, Kuperstein I, Barillot E, and Heyer WD (2013). Synthetic lethality between gene defects affecting a single non-essential molecular pathway with reversible steps. PLoS Comput. Biol 9, e1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Construct sequences
The annotated sequences of the DSB-inducible and donor constructs used in this study are available as *.ape (ApE Plasmid Editor) files in Data S1.