

Abstract
This research work was directed toward the development of highly active, stable, and reusable functionalized polymeric membrane domains for enzymatic catalysis. Functionalized membranes were created by two different approaches. In the first approach, which involved alternative attachment of cationic and anionic polyelectrolytes, functionalization was performed using a layer-by-layer (LBL) assembly technique within a nylon-based microfiltration (MF) membrane. In the second approach, a hydrophobic polyvinylidene fluoride (PVDF) MF membrane was functionalized by the in situ polymerization of acrylic acid. The enzyme, glucose oxidase (GOX), was then electrostatically immobilized inside the functionalized membrane domains to study the catalytic oxidation of glucose to gluconic acid and H2O2. Characterization of the functionalized membranes, in terms of polyelectrolyte/polymer domains and permeate flux, was also conducted. The kinetics of H2O2 formation was discussed, along with the effects of residence time and pH on the activity of GOX. The stability and reusability of the electrostatically immobilized enzymatic system were also investigated.
1. Introduction
Traditional membranes are considered to be a separation media based solely on the size (ultrafiltration, microfiltration) or solubility (reverse osmosis, pervaporation) of the species. However, with the advancement of functionalization chemistry and the availability of novel materials, further versatility can be obtained in the form of functionalized membranes. Functionalized membranes can be tailor-made to impart the desired functionality by a variety of active groups/molecules/moieties, depending on the applications.1–6 This research work involves extensive studies on the development of functionalized polymeric membranes that contain electrostatically immobilized enzymes and their application in enzymatic catalysis.
Enzymatic catalysis by free enzyme possesses the advantage of higher activity; however, its use is limited, because of lower stability, inhibition from product, and a lack of reusability.7 On the other hand, the traditional covalently immobilized enzyme is more stable, because of the protective action of the solid matrix, and can easily be reused. However, its activity is significantly low,8 because of the conformation change of the enzyme by multipoint binding and the accessibility issues arising due to the proximity to the solid matrix. Site-directed immobilization of enzymes has been performed to restrict the conformation change and increase the accessibility.9,10 However, site-directed immobilization requires complicated techniques and is considerably expensive.
To improve the situation, new techniques of enzyme immobilization have been explored. Table 1 summarizes some of the recent approaches reported in the literature, along with the results of kinetic study. One approach has been to keep the mode of attachment the same (i.e., covalent) but modify the solid matrix, such as activated beads/particles,11–13 nanoparticles,14 membranes,15,16 and films.17 The other approach is the nonco-valent attachment of enzyme in the solid matrix. These include electrochemical polymerization,18 Layer-By-Layer (LBL) deposition along with gold nanoparticles,19 electrostatic immobilization on anionic polystyrene beads,20 electrostatic immobilization on LBL assembled nylon membrane,21 and LBL assembly, along with multiwalled carbon nanotubes on gold electrodes.22
Table 1.
Comparative Study of the Types of Solid Matrix Used and Kinetic Parameters Obtained for Different GOX Immobilization Techniques Reported in the Literature
| reference | type of solid matrixa | mode of GOX immobilization | Kinetics Study of Immobilized GOX |
|
|---|---|---|---|---|
| Km (mM) | υmax | |||
| Ekinci et al.18 | Pt-electrode coated with copolymer of pyrrole and polystyrene | entrapment | 11.8 | 0.04μmol/(min mL) |
| Tomotani and Vitolo20 | anionic polystyrene beads + UF membrane | electrostatic | - | 34 × 10−4 mmol/(min mg) |
| Ahuja et al.11 | polystyrene beads activated with hydrazide + glutaraldehyde | site-directed | 29 | 0.248 mmol/(min mg) |
| Ahuja et al.11 | polystyrene beads activated with hydrazide + glutaraldehyde | covalent | 39.3 | 0.052 mmol/(min mg) |
| Pandey et al.14 | gold nanoparticle + MUDA + EDC-NHS | covalent | 3.74 | 14.2 × 10−4 mmol/(min mg) |
| Rauf et al.15 | CA-PMMA rnembrane + glutaraldehyde | covalent | 41.65 | 1.17 mM/min |
| Hou et al.12 | PSt-GMA microsphere | covalent | 35.2 | 15.5 mM/min-mg) |
| Ozyilmaz et al.13 | magnesium silicate + APTES | covalent | 259 | 0.217 mmol/(min mg) |
| Ying et al.16 | grafted PAA on PVDF membrane | covalent | 10.5 | 14 × 10−4 mmol/(min mg) |
| Li et al.17 | grafted AAc on polyaniline film | covalent | 80.6 | 0.62 mM/min |
Abbreviation legend: MUDA, mercaptoundecanoic acid; EDC, N-ethyl-N’-(3-dimethylaminopropyl)carbodiimide;, NHS, N-hydroxysuccinimide; PMMA, polymethyl methacrylate; PSt, polystyrene; GMA, glycidolmethacrylate; APTES, 3-aminopropyl trimethoxysilane; PAA, polyacrylic acid; PVDF, polyvinylidene fluoride; AAc, acrylic acid; CA, cellulose acetate.
An efficient alternative to traditional covalent immobilization of enzymes is to create a charged domain of polyelectrolytes inside a membrane pore, and then immobilize the enzyme using electrostatic interaction. The advantages of electrostatic immobilization are (i) a higher activity, close to that of free enzyme, and (ii) the stability and reusability of an immobilized enzyme. There also exists the potential of reusing the membrane matrix by detaching the enzyme, when needed, and attaching a fresh enzyme.
1.1. Layer-By-Layer (LBL) Assembly.
The alternative attachment of polycations and polyanions is a simple but robust technique to create a layered assembly in a solid support with high capacity and versatile functionality. The method was first introduced by Decher,23–25 based on a study by Iler26 that indicated the possible existence of such a technique. It is popularly known as the layer-by-layer (LBL) technique. A detailed literature review that covered the mechanisms of layer formation, applications in various fields, and scope of future studies was recently published.27 The LBL technique is used to create functional domains that contain various types of species, such as polyelectrolytes,21,28–31 proteins,32–34 dyes,35,36 nanoparticles,37–41 thiols,42 metal ions,43,44 viruses,45 DNA,46 and lipids.47 It is also used in different fields of applications, such as separation,48–50 biosensors,19,22,51 and biocatalysis.21,52–54
Our research group has developed polyelectrolyte domains within the membrane pores using the LBL technique.4 Multiple layers of charged polyelectrolytes were created inside the pores of gold-coated polycarbonate membranes for ion exclusion. The formation of an LBL assembly within the membrane pores was further extended for enzymatic catalysis.21 Enzymes, glucose oxidase (GOX), and alkaline phosphatase (AP) were electro-statically immobilized within polyelectrolyte domains created inside a nylon membrane by the LBL assembly. The kinetics of enzymatic catalysis was also studied. Convective flow was used for all of these studies, to enhance the mass-transfer coefficient.
1.2. Functionalized PVDF Membranes.
PVDF membranes are inherently hydrophobic in nature; however, studies have been performed to incorporate hydrophilicity into it. Polyacrylic acid (PAA), because of its hydrophilic nature and ion exchange property, has been extensively used for the formation of nanoparticles,55 the adsorption of nonionic surfactants,56 and the capture of metal ions57 in the PVDF membranes. Gabriel et al.58 reported a method to functionalize hydrophobic PVDF membranes via the in situ polymerization of acrylic acid. This technique has already been used for the formation of nanoparticles6 and the immobilization of Fe(II) cations59 inside the PVDF membrane pore. Another study has also been conducted on the covalent immobilization of GOX on acrylic acid-graft-copolymerized-PVDF membrane.16 However, to the best of our knowledge, PAA-functionalized PVDF membranes have never been used for the electrostatic immobilization of GOX.
1.3. Glucose Oxidase (GOX).
In this study, GOX was used as a model enzyme and glucose was used as the substrate. GOX is a well-known enzyme from the 19th century and has been used since Müllar reported its activity in Aspergillus niger.60 GOX specifically catalyzes61 the oxidation of β-D-glucose to δ-gluconolactone and hydrogen peroxide in a two-step mechanism (see Figure 1). In the first step, the co-factor of the enzyme, flavin adenine dinucleotide (FAD), oxidizes glucose to δ-gluconolactone and, itself, converts to the reduced form (FADH2). In the second step, FADH2 is oxidized back to FAD by O2 and O2 itself reduces to H2O2. δ-Gluconolactone is an unstable compound, and it immediately forms gluconic acid. The mechanism and kinetics of glucose oxidation by GOX has been extensively investigated by several researchers62–64 and has been summarized in the literature.65 The kinetics of free and immobilized GOX can be generally studied and compared using the well-known Michaelis-Menten equation, which is discussed later. The structure of GOX,65–67 and the factors that affect the kinetics of glucose oxidation by GOX, such as pH,17,68,69 temperature,17 and inhibition by the product H2O2,70 has also been studied thoroughly.
Figure 1.
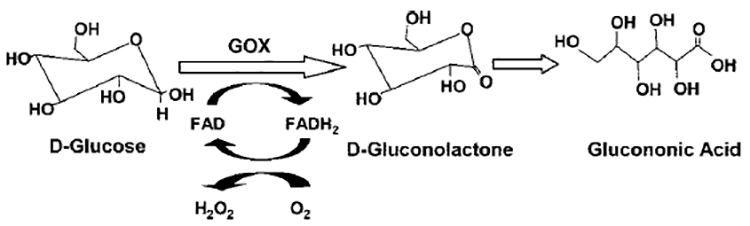
Enzymatic oxidation of the substrate (glucose) by the enzyme (glucose oxidase (GOX)), following a two-step reaction mechanism to produce gluconic acid and hydrogen peroxide.
1.4. Research Objectives.
The overall objective of the present study was to develop highly active, stable, and reusable GOX immobilized (electrostatically) functionalized polymeric membrane domains for the enzymatic oxidation of glucose to produce gluconic acid and H2O2. Two different types of domains were developed. A functionalized nylon-based membrane matrix was created by the alternative electrostatic attachment of anionic and cationic polyelectrolytes using LBL assembly technique. In another approach, a hydrophobic PVDF membrane was functionalized via the in situ polymerization of acrylic acid. Both functionalized membranes were used for the electrostatic immobilization of GOX (PI = 4.2). Fundamental experiments were performed to understand the kinetics of enzymatic oxidation of glucose in the aforementioned membrane domains, under convective flow conditions. The effects of pH and residence time on the activity of the enzyme were studied. The stability of the enzyme and reusability of the enzymatic system were also investigated. To the best of our knowledge, this is the first time that an attempt was made to detach and reattach the enzyme to explore the reusability of the membrane matrix.
2. Experimental Section
2.1. Materials.
The nylon-based anhydride activated micro-filtration membranes (Immunodyne ABC, pore diameter of 0.45 μm, thickness of 165 μm, 80% porosity, as supplied by the manufacturer) that were used for all the LBL experiments were purchased from the Pall Corporation. Hydrophobic PVDF membranes (pore diameter of 0.45 μm, thickness of 125 μm, 75% porosity, as supplied by the manufacturer) were purchased from the Millipore Corporation. The enzyme glucose oxidase from Aspergillus niger (GOX, Product No. G0543, MW 160000), β-D(+)-glucose (Product No. G5250, MW 180), and poly-L-lysine-hydrochloride (PLL, Product No. P9404, MW 105500, with the MW of the repeat units being 164.5) were purchased from Sigma. Poly(sodium 4-styrenesulfonate) (PSS, Product No. 243051, MW 70000, with the MW of the repeat units being 206), poly(allylamine-hydrochloride) (PAH, Product No. 283223, MW 56000, with the MW of the repeat units being 93.5), 15% solution of titanium oxysulfate in dilute H2SO4 (Product No. 495379), benzoyl peroxide (Product No. 179981), trimethylolpropane triacrylate (TMPTA, Product No. 246808), anhydrous toluene (Product No. 244511), and acrylic acid (Product No. 147230, MW 72) were purchased from Aldrich Chemical Co. The reagents for Bradford assay were purchased from Bio-Rad Laboratories. Unless noted otherwise, the remaining chemicals, including the deionized ultrafiltered water (DIUF), were purchased from Fisher Scientific.
2.2. Equipment.
Experiments were conducted in a stainless steel membrane cell from Osmonics. The effective external surface area of the membranes in this cell was 13.2 cm2.
2.3. Analytical Procedures.
All spectrophotometric measurements were performed using ultraviolet-visible light (UV–vis) spectrophotometry (Varian, Cary 300). All experiments were performed in triplicate, and the variations in experimental results are represented as the standard deviation.
2.3.1. Analysis of PAH and PLL.
The PLL and PAH were quantified using a total organic carbon (TOC) analyzer (Shimadzu, model TOC 5000 A), using a calibration curve in the range of 5–100 ppm. The analytical error for this analysis was 5%–10%.
2.3.2. Analysis of PSS.
The PSS in the layer was quantified by measuring the absorbance in the feed, permeate, and wash directly at 261 nm, using UV–vis spectrometry. The linear range of detection for PSS was 1–400 ppm, and the analytical error was <1%.
2.3.3. Analysis of GOX.
The amount of GOX in a solution was determined via the well-known Bradford Protein Assay,71 using a calibration curve prepared from GOX at 595 nm. The linear range of the Bradford Protein Assay was 1–24 μg/mL, and the analytical error was <1% for 2–24 μg/mL and 1%–5% for 1–2 μg/mL of GOX in this research work.
2.3.4. Analysis of H2O2.
The H2O2 in solution was analyzed using a colorimetric method that was reported by Clapp et al.,72 using acidified titanium oxysulfate, and the absorbance at 407 nm was measured. The linear range of calibration for H2O2 analysis was 0.03–3 mM (1–100 ppm), and the analytical error was <5% for 1–3 mM and 5%–10% for 0.03–1 mM.
2.4. Experimental Methods.
2.4.1. Formation of Functionalized Membranes.
2.4.1.1. Development of LBL Assembly in Anhydride Activated Nylon Membrane.
Layers of polyelectrolytes, mainly within the membrane pores, was assembled via the alternative electrostatic attachment of cationic and anionic polyelectrolytes (see Figure 2a), under a convective flow and constant stirring conditions. A convective flow was applied to obtain the functionalization mainly within the membrane pores. Membrane pores were preferred, because of the higher pore surface area over external membrane surface area. The immobilization was nonstoichiometric, which means the charges were oversaturated with the oppositely charged polyelectrolyte in the next layer. To ensure nonstoichiometric attachment of subsequent layers, all layer formation steps were performed in the presence of a 0.25 M NaCl solution. The shielding effect of salt increases the availability of free charges of polyelectrolytes for the next layer.
Figure 2.
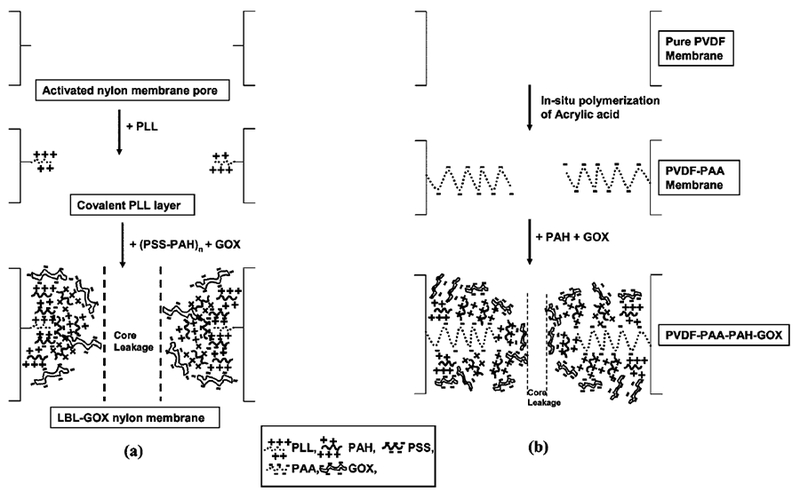
Schematic of the formation of (a) layers of polyelectrolytes inside an anhydride activated nylon-based membrane using layer-by-layer (LBL) assembly, followed by electrostatic immobilization of GOX, and (b) functionalized PVDF membrane by PAA (shown here as a single chain, for illustration purposes) and electrostatic attachment of PAH, followed by electrostatic immobilization of GOX.
To impart stability in the subsequent layers, the first layer of PLL was attached covalently in the membrane matrix via the reaction between the terminal amine group of PLL and the anhydride group of the membrane. This was done by permeating 100 mL of a 40 ppm PLL solution (4 mg or 0.039 μmol of PLL) in DIUF at 0.07 bar (1 psi) pressure and pH 9.3. The solution was recirculated once. The recirculation step also was followed for all other layer formation steps. As the first layer, PLL was preferred over PAH, because the difference of pKa between the terminal amine group (pKa ~9) and amine groups in the side chains (pKa ~10.5) of PLL is known. Hence, at a working pH of 9.3, the terminal amine group is uncharged and, therefore, forms covalent attachment preferentially over the side-chain amine groups, which are positively charged at that pH. This has enhanced the probability of single-point attachment of the first layer, rather than multipoint attachment through the side-chain amine groups. The side-chain amine groups provide free positive charges available for the next layer formation step.
The second layer of PSS was attached by electrostatic interaction between the side-chain amine groups of PLL and sulfonic acid groups (pKa 1.0) of PSS.73 a One hundred millilters of the 400 ppm solution (40 mg of PSS, 0.2 mmol of negative charges based on repeat units) of PSS was permeated in DIUF at pH 6. At a working pH of 6, the net charge on amine groups of PLL is positive, whereas the net charge on sulfonic acid groups of PSS is negative. Therefore, they easily interact with each other.
The third layer of PAH was formed by the electrostatic interaction between the negatively charged sulfonic acid groups of PSS and the positively charged amine groups of PAH. PAH was attached by permeating 100 mL of a 300 ppm solution (30 mg of PAH, 0.3 mmol of negative charges, based on repeat units) in DIUF at pH 6.
Then, two more bilayers of PSS and PAH were attached in the membrane matrix to obtain PLL–(PSS–PAH)3. The goal was to synthesize a net positively charged membrane in the working pH (i.e., pH 6), so that GOX (PI = 4.2) can be attached using its negative charges. More layers have been introduced to increase the enzyme loading and also to decrease the core leakage (see Figure 2a) of the substrate molecules.
The stability of each layer was checked by permeating solutions of varying pH (over a range of 2.7–9). The data of pure water flux, as a function of pressure, were obtained after the formation of each layer.
2.4.1.2. Functionalization of PVDF Membrane.
A method reported in the literature58 was adopted to functionalize a hydrophobic PVDF membrane with PAA via the in situ polymerization of acrylic acid monomer. The polymerization solution consisted of 70% toluene, 30% acrylic acid, 0.5% benzoyl peroxide, and 1.2% TMPTA (by weight). Benzoyl peroxide was used as the initiator. TMPTA was used as the cross-linker to increase the stability and density of the polymer network inside the membrane matrix. The membrane was dipped into the polymerization solution for 30 s, and then it was sandwiched between two Teflon plates and clipped tightly. It was then inserted into an oven at 90 °C for 4 h under a constant N2 environment. The membrane then was washed with a copious amount of DIUF water, under convective flow conditions, to remove the impurities. The membrane obtained at this point was functionalized with the hydrogen form (H-form) of PAA. It was converted to Na-form by permeating 200 mL of a 0.01 M NaOH solution. The Na-form resists any drastic change in pH that generally occurs in the H-form for the following ion exchange steps. Permeate flux of water at different stages of functionalization and effect of pH on water flux through PAA-functionalized PVDF (PVDF–PAA) membrane were studied.
To immobilize GOX in the PVDF-PAA membrane electro-statically, a net positively charged membrane was created by attaching a layer of PAH, using the same principle of electro-static interaction (see Figure 2b) that used earlier for the PSS–PAH layer in the LBL membrane. One hundred millilters of a 300 ppm solution of PAH (30 mg of PAH, 0.3 mmol of negative charges, based on repeat units) in DIUF water at pH 6 was permeated through the PVDF–PAA membrane. The carboxylate anions of PAA have a pKa of 4.7, and, at pH 6, these groups interact with positively charged amine groups of PAH to form PVDF–PAA–PAH.
2.4.2. Electrostatic Immobilization of GOX in functionalized Membranes.
After the formation of PLL–(PSS–PAH)3 layers, GOX was immobilized electrostatically (see Figure 2a) by permeating 100 mL of a 25 ppm solution in DIUF at pH 6. GOX was attached electrostatically in the absence of salt, so that all charges on the membrane matrix could be neutralized by it. For comparison, the covalent attachment of GOX was performed by the reaction between its amine groups and the anhydride groups in the activated nylon (Immunodyne) membrane, as reported in another study.74 GOX was immobilized in the PVDF–PAA–PAH membrane (see Figure 2b), similar to that described for the LBL membrane. The stability of both the systems was checked frequently by analyzing the leakage of GOX in the permeate. The GOX immobilized membranes were stored at 4 °C.
2.4.3. Activity of the Electrostatically Immobilized GOX for Oxidation of Glucose.
The activity of GOX was determined by measuring the concentration of H2O2 produced. All the activity measurement experiments were performed in a 50-mM sodium acetate–HCl buffer (NaOAC–HCl buffer) saturated with O2. Oxygen is very important in the second step of the two-step reaction mechanism of the catalytic oxidation of glucose. Thus, to maintain the saturation condition of O2 in the media throughout the reaction, the system was designed with oxygen saturated buffer and also kept under a constant pressure of air. NaOH and HCl were used to adjust the pH of the solution. β-D(+)-Glucose is referenced as glucose in the text.
2.4.3.1. Determination of Steady-State H2O2 Production.
The production of H2O2 that was due to the oxidation of glucose by GOX, in this case, was a biocatalytic reaction that was performed in a constant-flow reactor. Hence, steady-state conditions prevails. To get an idea about the time needed to reach the steady state, experiments were performed by permeating 15 and 75 mM of glucose solution (pH 5.5) at a constant flow rate through both types of GOX functionalized membranes. Permeates were collected as a function of time and analyzed for H2O2.
2.4.3.2. Kinetic Study.
For electrostatically immobilized GOX in an LBL membrane (LBL–GOX-convective) and a functionalized PVDF membrane (PVDF–GOX-convective), the glucose concentration was varied over a range of 0.15–75 mM (pH 5.5), while the flow rate was kept constant, so that the condition of equal residence time prevails for all the kinetic experiments. To study the kinetics of the same GOX immobilized LBL membrane in diffusive mode (LBL–GOX-soaking), experiments were performed by soaking the membrane in glucose solution and measuring the activity of GOX in reaction mixture, as a function of time. The kinetics of covalently attached GOX was also studied in convective mode and represented as covalent-GOX-convective.
2.4.3.3. Effect of Residence Time (i.e., Flow Rate).
The effect of residence time on the activity of GOX was studied by varying the permeate flow rate for a fixed concentration of glucose solution at pH 5.5. The flow rate was varied over a range of 2–25 mL/min by varying the pressure over a range of 0.34–3.4 bar (5–50 psi).
2.4.3.4. Effect of pH.
Effect of pH on the activity of GOX immobilized in an LBL membrane was studied by varying the pH over a range of 4.5–7 for a fixed concentration of glucose solution at a constant flow rate. Homogeneous phase experiments for free GOX were also performed, with the pH varying in the same range.
2.4.4. Stability of the Immobilized Enzymatic System in Functionalized Membrane.
The stability of GOX im mobilized in an LBL assembled membrane and a functionalized PVDF membrane was studied by measuring the activity after every 7 days for 28 days. The membrane was stored at 4 °C when it was not being used. A glucose solution (15 mM at pH5.5) was used for all stability studies.
2.4.5. Reusability of the Functionalized Membranes for Enzymatic System.
2.4.5.1. Reusability of the LBL Assembled Membrane for GOX Immobilization.
Immobilized GOX, the activity of which has already been studied, was detached from the membrane matrix by permeating 50 mL of DIUF water of pH 3 at 10–15 psi. After detachment, the pH of the solution was immediately adjusted to 5.5. The activity of any undetached GOX in the membrane matrix was determined by permeating the glucose solution at pH 5.5. The amount of detached GOX was quantified by analyzing the solution using the Bradford Assay. The activity of the detached GOX in free form was determined by homogeneous phase batch reaction at pH 5.5. The remainder of the detached GOX was then reattached in the same membrane matrix, using the principle of electrostatic interaction. The activity of the reattached GOX in the membrane was determined as done previously. The steps of detachment and reattachment were repeated. Attachment of fresh GOX in the same membrane matrix was also performed, followed by the activity study.
2.4.5.2. Reusability of Functionalized PVDF Membrane for GOX Immobilization.
Functionalized PVDF membranes were generated by detaching the used GOX and attaching fresh GOX in the same PVDF–PAA membrane. The detachment was done by permeating DIUF water with pH 4 at 10–15 psi pressure under convective flow mode. The membrane matrix was regenerated by immobilizing fresh GOX using electrostatic interaction. The stability of the domain and the activity of fresh GOX were tested.
3. Results and Discussion
3.1. Characterization of the Functionalized Membranes.
3.1.1. LBL Assembled Nylon Membrane.
The amount of polyelectrolyte attached in each layer was determined from analytical procedures (TOC and/or UV–vis spectroscopy) and is represented in terms of repeat units in Table 2a. A single repeat unit of all the polyelectrolytes contains a unit charge. Based on that, net charges in the subsequent layers are calculated and represented in the same table. The addition of layers was always nonstoichiometric. However, it is important to note here that each layer was associated with excess net opposite charges, which has governed the electrostatic attachment of next layer. The net charge after the seventh layer of polyelectrolyte (PAH3) was positive.
Table 2.
Characterization of (a) LBL Assembled Nylon Membrane and (b) Functionalized PVDF Membrane by Calculating the Number of Repeat Units Immobilized in Subsequent Layers or Domainsa
| layer | domain | number of repeat units immobilized (× 103 mmol) |
net charge (× 103 mmol) |
|---|---|---|---|
| (a) LBL Assembled Nylon Membrane | |||
| PLL | 2.1 | (+) 2.1 | |
| PSS1 | 6.9 | (−)4.8 | |
| PAH1 | 9.1 | (+) 4.3 | |
| PSS2 | 10.4 | (−) 6.1 | |
| PAH2 | 13.1 | (+) 7.0 | |
| PSS3 | 8.2 | (−) 1.2 | |
| PAH3 | 2.5 | (+) 1.3 | |
| GOX | 1.1 | ||
| (b) Functionalized PVDF Membrane | |||
| PAA | 128.7 | (−) 128.7 | |
| PAH | 130 | (+) 1.3 | |
| GOX | 1.3 | ||
The molecular weight of the repeat units of the different polyelectrolytes/polymer is as follows: PLL, 164.5; PSS, 206; PAH, 93.5; PAA, 72. External membrane area = 13.2 cm2, membrane thickness = 165 μm, and pH used for immobilization = 6.
The effective cumulative thickness of the assembly (δn), after the addition of the nth layer, is calculated by applying Hagen-Poiseuille’s equation75 on the permeate water flow rate through the unmodified membrane and through the membrane after the attachment of the nth layer , as shown below.
| (1) |
| (2) |
The permeability after the attachment of the nth layer (Pn), in units of cm3/(cm2 s bar), is calculated as follows:
| (3) |
where the Q terms represent the volumetric flow rates, r0 is the pore radius of the unmodified membrane (r0 = 225 nm), rn is the pore radius of the membrane after the attachment of the nth layer, Am is the external area of membrane (Am = 13.2 cm2), and ΔP is the transmembrane pressure.
The calculated effective cumulative thicknesses of layers are plotted in Figure 3. As expected, cumulative thickness of layers increases as subsequent layers are assembled in the membrane pores. The final cumulative thickness of the functionalized domain after GOX immobilization was 100 nm, which corresponds to a 44% reduction in pore radius.
Figure 3.
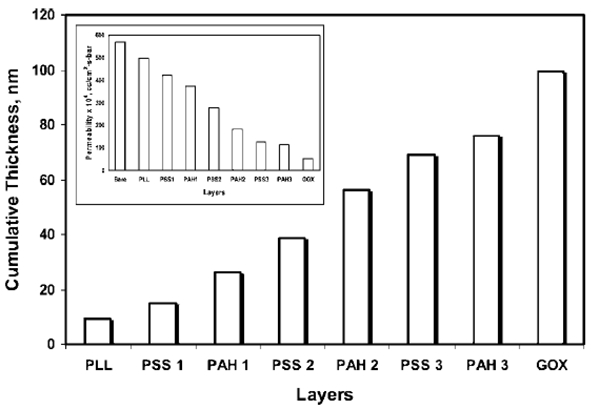
Calculated cumulative layer thickness after electrostatic immobilization inside nylon membrane pore; the Hagen Poiseuiile equation was used to calculate layer thickness. Inset shows the decrease in permeability due to the formation of layers inside the membrane pores. (Conditions: pore radius of pure nylon membrane, 225 nm; water permeability of pure nylon membrane, 570 × 10−4 cm3/(cm2 s bar); permeability after GOX immobilization, 52 × 10−4 cm3/(cm2 s bar); external membrane area, 13.2 cm2, and membrane thickness, 165 μm.)
The inset of Figure 3 represents the decrease in water permeability of the membrane due to the decrease in pore radius with the addition of each layer. The water permeability of the nylon membrane before any functionalization was 570 × 10−4 cm3/(cm2 s bar). After the 7th layer, GOX was immobilized and the water permeability decreased to 52 × 10−4 cm3/(cm2 s bar), which is 4% of the permeability of pure nylon membrane. This shows that the addition of seven layers of polyelectrolytes and a layer of GOX affect the permeability of the membrane significantly.
3.1.2. Functionalized PVDF Membrane.
Characterization of functionalized PVDF membranes was also conducted by determining the amount of functionality incorporated into the membrane (see Table 2b). The amount of PAH and GOX immobilizations were obtained from direct analyses, whereas the amount of PAA immobilization was calculated from the PAH and GOX immobilization values and is explained later. The amount of repeat units of PAH attached in the PVDF–PAA membrane was determined to be 0.13 mmol. The cumulative thickness of the functionalized domain of PVDF–PAA–PAH–GOX membrane was calculated to be 125 nm (using the Hagen-Poiseuille equation), i.e., the effective radius of pore was 100 nm.
The effect of functionalization on the permeability of water was studied for the PVDF–PAA–PAH–GOX membrane and is represented in Figure 4 as a function of pressure at different stages of functionalization. The slope of each curve plotted in Figure 4 is the permeability of water (× 104) through the membrane at that particular stage of functionalization. It can be observed that the water permeability decreases dramatically from the PVDF membrane before functionalization (596 × 10−4 cm3/(cm2 s bar)) to the PVDF-PAA membrane (82 × 10−4 cm3/(cm2 s bar)). This observation can be attributed to the formation of a highly dense PAA network inside the membrane pore. Permeability further decreased as the PVDF–PAA membrane was converted to the PVDF–PAA–PAH membrane (permeability) 50 × 10−4 cm3/(cm2 s bar)) and PVDF–PAA–PAH–GOX membrane (permeability = 24 × 10−4 cm3/(cm2 s bar)), subsequently.
Figure 4.
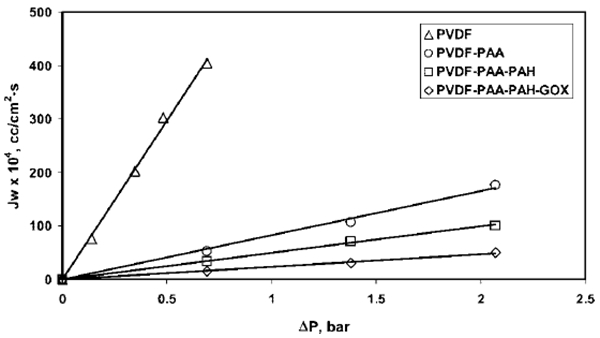
Effect of functionalization on PVDF membrane, as depicted by the curve of permeate flux versus pressure. Pore radius of the pure PVDF membrane = 225 nm, pore radius of PVDF–PAA–PAH–GOX membrane = 100 nm. Water (pH 6) permeability values for different steps of functionalized membrane are given by the slope of the curves. External membrane area = 13.2 cm2, membrane thickness = 125 μm.
The water flux behavior, as a function of pH, is demonstrated in Figure 5 for the PVDF–PAA membrane. It can be observed that, starting from pH 2.5, as the pH increases, the water flux remains constant until 4.5, and then it starts to decrease sharply. At pH 5.5, the flux reduces to 40% and at pH 7, the flux reduces to 20% of the initial value. After pH 7, again it becomes constant. This indicates that, between pH 4.5 and pH 7, some type of conformational change of PAA structure occurs. For a single layer of polyelectrolyte immobilized in a solid matrix, this type of phenomenon is called the”helix-to coil transition”.3 However, PAA, in this case, forms a random network structure inside the PVDF membrane pore, and the existence of helical or coil conformations are unlikely. It can be stated that, throughout this pH range (4.5–7), the PAA network inside the membrane undergoes “retracted-to-elongated” conformational transformation (see Figure 5). Below pH 4.5, PAA remains totally protonated and, hence, retracted. With increasing pH, it starts to deprotonate, and after pH 7, all of its acid groups become negatively charged (deprotonated) and fully elongated (repulsion of like charges to form extended configuration). This caused a substantial decrease in the membrane pore radius.
Figure 5.
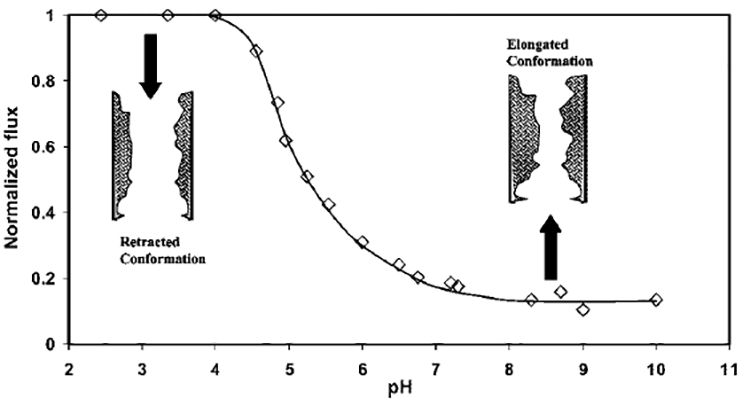
Effect of pH on permeate water flux and functionalized domain of the PVDF–PAA membrane in Na-form. PAA network inside the membrane undergoes a “retracted-to-elongated” conformational change as pH increases. Flux was normalized with maximum flux for this experiment (i.e., 330 × 10−4 cm3/(cm2 s)) at pH 2.5 and a pressure of 1.38 bar. (Conditions: pore radius of the pure PVDF membrane = 225 nm, external membrane area = 13.2 cm2, membrane thickness = 125 μm.)
3.2. Electrostatic Immobilization of GOX.
Results obtained for electrostatic immobilization of GOX in both LBL and PVDF membranes are described below. The Bradford Assay was performed on the permeate obtained from different experiments with the GOX immobilized membranes, and they demonstrated no leakage of GOX.
3.2.1. Immobilization of GOX in LBL Assembled Nylon Membrane.
The amount of GOX immobilized electro-statically in the seven layers of polyelectrolyte assembled nylon membrane was 1.5 ± 0.2 mg (9.37 × 10−5 ± 1.25 × 10−5 mmol). In another set of experiments, the amount of GOX immobilized was 1.1 ± 0.1 mg for three layers of LBL assembled membrane. A comparison of the aforementioned results suggests that four additional layers did not increase the amount of immobilized GOX substantially. However, the additional layers contributed significantly in regard to reducing the core leakage of the reaction mixture through the center region of the membrane pore. The results discussed here are associated with a seven-layer membrane only. The amount of covalently immobilized GOX was 1 ± 0.1 mg (6.25 × 10−6 mmol, or 5 × 10−7 mmol/cm2). In a previous study, the amount of covalent immobilization of avidin in the same type of nylon membranes was reported to be 8 × 10−7 mmol/cm2.74 Considering the fact that GOX is bulkier than avidin, these results show that our experiments with a covalently immobilized protein in the nylon membrane are consistent.
3.2.2. Immobilization of GOX in Functionalized PVDF Membrane.
The amount of GOX attached electrostatically on the functionalized PVDF membrane was 1.8 ± 0.1 mg. This corresponds to 1.12 × 10−5 mmol GOX or 1.33 × 10−3 mmol negative charges, considering that 1 mol of GOX contains 118 mol of negative charges (calculated from the number of aspartic and glutamic acid residues present in GOX). Assuming, for the present membrane system, this is the saturation capacity of GOX attachment by electrostatic immobilization, the excess positive charges of PAH was neutralized by 0.13 − 1.33 × 10−2 = 12.87 × 10−2 mmol (9.3 mg) of PAA functionalized by in situ polymerization.
3.3. Activity Study of Electrostatically Immobilized GOX.
3.3.1. Determination of Steady-State H2O2 Production.
The activity of GOX was measured by determining the production of H2O2 (glucose + O2 → gluconic acid + H2O2) as a function of time. For a fixed substrate concentration and flow rate, the concentration of H2O2 in the permeate reached steady state rapidly and remained almost constant with time (variation is <5%). It was also observed that the amount of H2O2 produced in the feed side of the membrane was <5% of that produced in the permeate after t = 30 min for both LBL assembled nylon and functionalized PVDF membranes. This is because the surface immobilization of GOX was insignificant, compared to pore immobilization. However, if the experiment is conducted for a long time, the production of H2O2 will be significant, because it accumulates on the feed side. Hence, all data were collected within the first 5–10 min of the experiments.
3.3.2. Kinetics of Glucose Oxidation.
Understanding of hydrodynamics within the reactor (membrane pores) is very important to analyze the kinetics of glucose oxidation by immobilized GOX. The hydrodynamics have an important role in determining the residence time (i.e., the ratio of the effective membrane reactor volume to the volumetric flow rate).
There are two regions inside the functionalized membrane pores; polyelectrolyte/polymer region, where GOX is immobilized, and a region void of GOX (see Figure 2). The polyelectrolyte layers were developed from the pore wall toward the pore center. The other region, which is devoid of GOX, was located near the center of the pore and known as the core region. A fraction of glucose molecules permeating through the core region never interacts with enzyme. This phenomenon is known as core leakage3 and has a significant effect on the calculations of enzyme activity.21 It is reasonable to consider only the volume of the membrane pore that contains the enzyme as the effective membrane reactor volume.
3.3.3. Calculation of Effective Membrane Reactor Volume.
For the LBL membrane, the thickness of layers after GOX immobilization is 100 nm, and the pore radius of the pure membrane is 225 nm. Therefore, the core region volume is given as
and the effective membrane reactor volume is given as
i.e.,
where ϵ is the porosity of the Immunodyne membrane (ϵ = 0.8), Am the external area of membrane (Am = 13.2 cm2), and l the length of the membrane (l = 165 × 10−4 cm).
Similarly, for the PVDF-PAA membrane, the effective reactor volume
These effective membrane reactor volumes are used to calculate the residence time. The rate of oxidation of glucose by immobilized GOX is then calculated by considering the continuously stirred tank reactor (CSTR) equation (for low conversion) as follows.
| (4) |
where is the concentration of H2O2 in the permeate and τ is the residence time. Under steady-state conditions, for a fixed residence time and substrate concentration, the concentration of H2O2 produced in the permeate is constant, irrespective of the volume of the reaction mixture permeated.
The rate of oxidation of glucose for GOX immobilized in both the LBL (LBL-GOX-convective) and PVDF (PVDF-GOX-convective) membranes (with convective flow of reaction mixture) are obtained for different substrate concentrations using eq 4 and plotted in Figure 6. The residence time was 0.6 s (flow rate = 11 mL/min) for all convective flow experiments. As observed from the figure, at lower glucose concentrations, the rate increases very sharply with concentration, and then flattens out gradually at higher substrate concentrations. For example, the rate increases from 3 mM/(mg min) to 10 mM/(mg min) as the glucose concentration increases from 0.15 mM to 0.75 mM, whereas the rate increases only to 21 mM/(mg min) as the glucose concentration increases to 75 mM.
Figure 6.
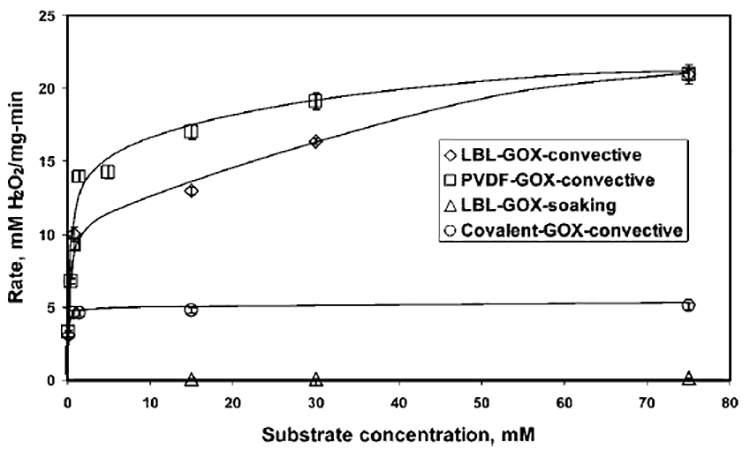
Rate of oxidation of glucose, as a function of the substrate concentration for different GOX immobilization experiments; the amount of GOX immobilized is 1.8, 1.5, and 1 mg for the PVDF membrane, the LBL membrane, and covalent immobilization, respectively. (Conditions: residence time for all experiments, 0.6 s; temperature, 25 °C; pH of the NaOAc–HCl buffer, 5.5.)
For comparison, the rate for the same GOX immobilized LBL membrane in soaking mode (LBL–-GOX-soaking), and GOX covalently immobilized in nylon (Immunodyne) membrane (covalent-GOX-convective), are also presented in Figure 6. For a substrate concentration of 75 mM, the rates are 21, 5, and 0.1 mM/(mg min) for the LBL-GOX-convective, covalent-GOX-convective, and LBL-GOX-soaking modes, respectively. Figure 6 illustrates that the activity of electrostatically immobilized GOX with convective flow of glucose solution is superior to both covalently immobilized GOX with a convective flow of glucose and electrostatically immobilized GOX under a soaking mode of interaction with glucose. Electrostatic immobilization resists the conformation change and accessibility problems associated with covalent immobilization by keeping the enzyme away from the solid support, whereas convective flow increases the accessibility of the substrate for the enzyme, compared to the soaking mode (diffusion).
The well-known Michaelis–Menten rate equation76 is used to describe the kinetics of glucose oxidation by immobilized GOX. The equation, which is expressed as the rate υ, is defined as follows:
| (5) |
where S is the substrate concentration, υmax is the maximum possible rate, and Km is the Michaelis–Menten constant and is defined as the substrate concentration at which the rate becomes half of υmax. Equation 5 can be manipulated to give
| (6) |
Equation 6 is used to calculate the kinetic parameters by a straight-line plot (a Lineweaver-Burke plot) of 1/υ vs 1/S and then determine υmax and Km from the intercept and slope, respectively.
The Michaelis–Menten rate equation is used to describe the immobilized phase reaction kinetics for the substrate concentration range of 0.75–75 mM. It has revealed some interesting facts, which are given as follows. The Lineweaver–Burke plot is constructed from the rate data and is presented in Figure 7. The figure shows that the nature of the Lineweaver–Burke plot is nonlinear for both the LBL-GOX-convective and PVDFGOX-convective modes. It can be speculated that the Michaelis–Menten rate equation (R2 = 0.7) is not efficient to describe the immobilized phase reaction kinetics accurately in the entire range of substrate concentrations studied (S = 0.75–75 mM (i.e., 1/S = 1.33–0.013 mM−1)).
Figure 7.
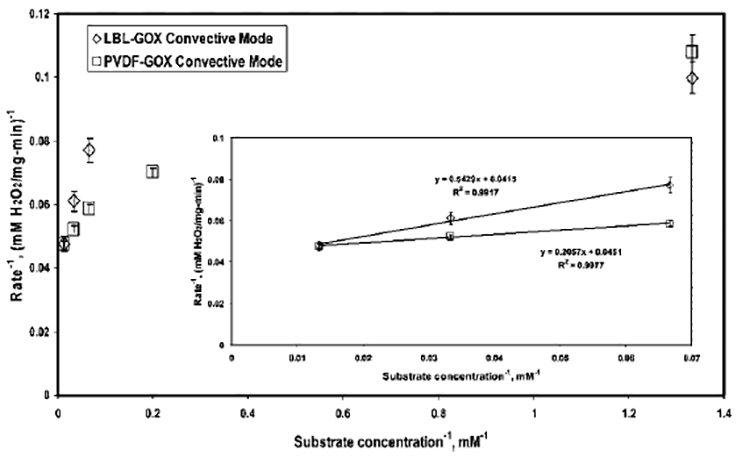
Lineweaver–Burke plot for GOX immobilized in functionalized LBL (LBL-GOX-convective) and PVDF (PVDF-GOX-convective) membranes. Conditions: concentration of glucose solution, 0.75–75 mM (1/S = 1.33–013 mM−1); amount of GOX immobilized, 1.5 and 1.8 mg for LBL and PVDF membranes, respectively; temperature, 25 °C; pH of NaOAc–HCl buffer, 5.5. Inset shows the Lineweaver–Burke plot for a substrate concentration range of 15–75 mM (1/S = 0.067–0.013 mM−1).
To study the kinetics behavior of immobilized GOX in a specific region of substrate concentration, a Lineweaver–Burke plot is constructed for both membranes (shown as the inset in Figure 7) in the substrate concentration range of 15–75 mM (1/S = 0.067–0.013 mM−1). For both cases, the straight- line plots fit the experimental data extremely well, as evident from the correlation values of R2 > 0.99. The values of υmax and Km are determined from the Lineweaver–Burke plots in this concentration range. The value of υmax for the LBL-GOX-convective and PVDF-GOX-convective modes are 24 and 22 mM/(mg min), respectively. The Km value of the LBL-GOX-convective mode is 13 mM, and, for the PVDF-GOX-convective mode, it is 4.6 mM. This observation suggests that Michaelis–Menten kinetics is valid in the higher substrate concentration range of 15–75 mM for our cases of immobilized phase GOX. However, it fails to explain the rate behavior at lower ranges. It can be speculated that, at any given time, for lower substrate concentrations, the amount of immobilized enzyme per unit amount of substrate is significantly higher than that for higher substrate concentrations. This might have a role in the deviation from Michaelis–Menten kinetics.
The Km values obtained here are significantly less than most of the reported Km values for covalently immobilized GOX represented in Table 1. One value of particular interest in Table 1 is that reported by Ying et al. For covalently immobilized GOX in PAA grafted PVDF membrane and soaking mode oxidation of glucose, the value of Km is 10.5. On the other hand, for our case, the Km value is 4.6 for the PVDF-GOX-convective membrane system. Lower Km values signify a higher affinity of the enzyme toward the substrate. This again demonstrates the advantage of electrostatic immobilization and covalent flow over covalent immobilization and diffusive flow for an enzyme-substrate system. The υmax values for different cases cannot be compared, because of the differences in units arising from the difference in the mode of hydrodynamics (i.e., convective and diffusive flow).
3.4. Effect of Residence Time (i.e., Permeate Flow Rate).
Effects of residence time (i.e., permeate flow rate) on conversion and rate of oxidation of glucose (production of H2O2) by immobilized GOX in both LBL and PVDF membranes are described by considering them as model reactors, such as plug-flow reactors (PFRs) and CSTRs. The assumption of a CSTR is valid for low conversions. A material balance is done within the membrane pore, and Michaelis-Menten reaction kinetics is incorporated to obtain a mathematical expression that predicts the exit concentration of H2O2 in the permeate .
For PFRs, the equation is
| (7a) |
Similarly, for CSTRs, the equation is
| (7b) |
For different residence times and a particular concentration of substrate, the model-predicted exit concentration of H2O2 in the permeate can be obtained using either eq 7a or eq 7b. The conversion was deliberately kept low, so that the assumption of a constant concentration of substrate remains valid. Km and υmax for the immobilized GOX, determined earlier for the experiments conducted at a residence time of 0.6 s (flow rate = 11 mL/min) in the substrate concentration range of 15–75 mM, are used for calculations. The conversion is calculated as
| (8) |
where S0 is the initial substrate concentration.
Because the conversion was very low, the predicted values from PFR and CSTR models were observed to be almost identical. Hence, for further discussions, both PFR- and CSTR-predicted values are commonly termed as “model-predicted” values.
From the model-predicted concentration of H2O2 in the permeate, the conversion and rate are determined using eqs 8 and 4, respectively. Both the experimentally obtained and model-predicted values for a substrate concentration of 15 mM are demonstrated in Figures 8 (LBL assembled nylon membrane) and 9 (functionalized PVDF membrane).
Figure 8.
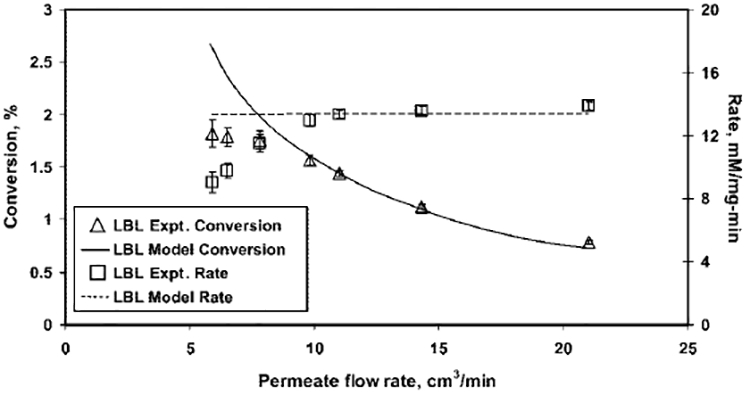
Effect of flow rate (i.e. residence time) on the conversion of glucose and the rate of formation of H2O2 for a 15 mM glucose solution fed in a LBL assembled nylon membrane. Model values are calculated considering both plug-flow reactor (PFR) and continuously stirred tank reactor (CSTR) models. Conditions: amount of GOX immobilized, 1.5 mg; volume of LBL membrane reactor, 0.122 cm3; pH 5.5; temperature, 25 °C; permeate flux, (75–265) × 10−4 cm3/(cm2 s); and residence time, 0.35–1.24 s.
Figure 9.
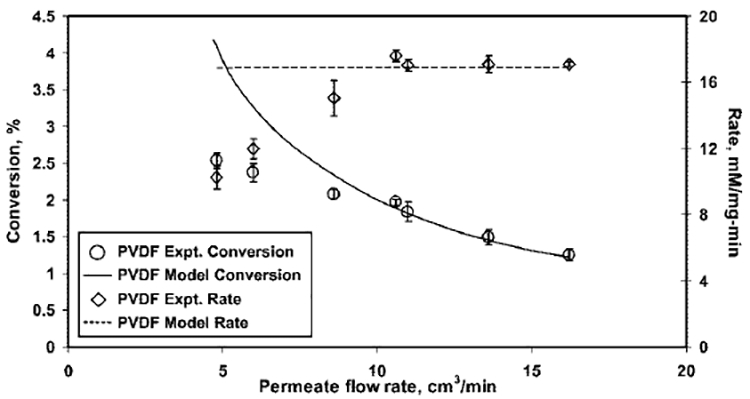
Effect of flow rate (i.e. residence time) on the conversion of glucose and the rate of formation of H2O2 for a 15 mM glucose solution fed in a functionalized PVDF membrane. Model values are calculated considering both PFR and CSTR models. Conditions: amount of GOX immobilized, 1.8 mg; volume of PVDF membrane reactor, 0.1 cm3; pH5.5; temperature, 25 °C; and permeate flux, (60–200) 10−4 cm3/(cm2s); the residence time varied over a range of 0.37–1.25 s.
The conversion of glucose represents the concentration of H2O2 in the permeates. Figures 8 and 9 show that, as the permeate flow rate increases (i.e., the residence time decreases), the concentration of H2O2 in the permeate decreases and, hence, the conversion. This is because the product formation for a flow reactor is a function of residence time, unlike the rate of reaction. Figures 8 and 9 also demonstrate that the assumption of ideal reactors, such as CSTRs and PFRs, is sufficient to predict the effect of residence time on the rate and conversion of the reaction catalyzed by an immobilized enzyme in a membrane at higher flow rates. However, the ideal reactor model deviates (over predicts) at lower flow rates, because of the mass-transfer issues. At lower flow rates, mass-transfer resistance is significant and glucose molecules cannot access all the active sites of immobilized GOX in the functionalized domain of membranes. In addition, at lower flow rates, the removal of the product from the active sites of the enzyme is also retarded, because of mass-transfer resistances, thereby, further reducing the accessibility. Thus, the rate and conversion is low at lower flow rates. This observation suggests that the incorporation of a mass-transfer resistance term in the mathematical model will be useful for proper prediction.
3.5. Effect of pH.
The effect of pH on the activity of free GOX has been reported in theliterature.13,69 It has been observed that there is a distinct maximum for the activity of free GOX at pH 5.5, and the activity decreases dramatically on either side of that pH. For example, in a study by Ozyilmaz et al.,13 the normalized activity (with respect to the activity at pH 5.5) for a 100 mM glucose solution was reported to be 35% at pH 4.5 and 50% at pH 7. Our study for free GOX, covering a glucose concentration range of 0.75–75 mM, also exhibited a similar trend, as shown in Figure 10. The effect of pH on the activity of the LBL-GOX system is also represented in the same figure. It can be observed that, although the activity curve goes through a maximum, the range of optimum pH is broad. In addition, the decrease in activity on either side of the optimum pH range is less than the free GOX. This proves that GOX immobilized in the membrane can be operated over a wide range of pH without sacrificing the activity significantly.
Figure 10.
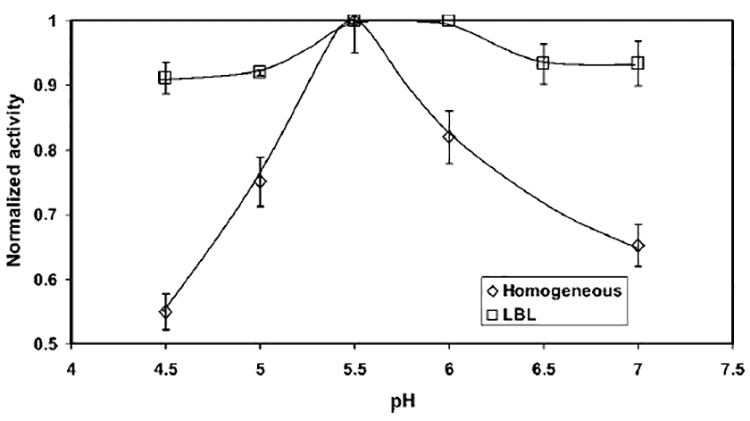
Effect of pH on activity for both free GOX and GOX immobilized in an LBL assembled membrane. Activity is calculated based on the concentration of H2O2 in the permeate and is normalized by dividing by the maximum activity . (Conditions: amount of GOX immobilized, 1.5 mg; volume of LBL membrane reactor, 0.122 cm3; permeate flux, 140 × 10−4 cm3/(cm2 s); residence time, 0.6 s; external membrane area, 13.2 cm2; membrane thickness, 165 μm; pH 5.5; temperature, 25 °C; and concentration of glucose fed, 0.75–75 mM.)
3.6. Stability.
Figure 11 demonstrates the stability of the electrostatically immobilized GOX in both the LBL assembled nylon membrane and the PVDF–PAA–PAH membrane over a period of time. It can be observed from Figure 11 that, even after 28 days, the activity of immobilized GOX reduced to only 80% of the initial activity, whereas, according to Ozyilmaz et al.,13 the activity reduced to 25% after 28 days for free GOX. In another study,77 the activity was reported to be reduced to 30% after the first 7 days. This shows that GOX immobilized within a membrane matrix is well-protected and retains its activity over a longer period of time, compared to that for free GOX.
Figure 11.
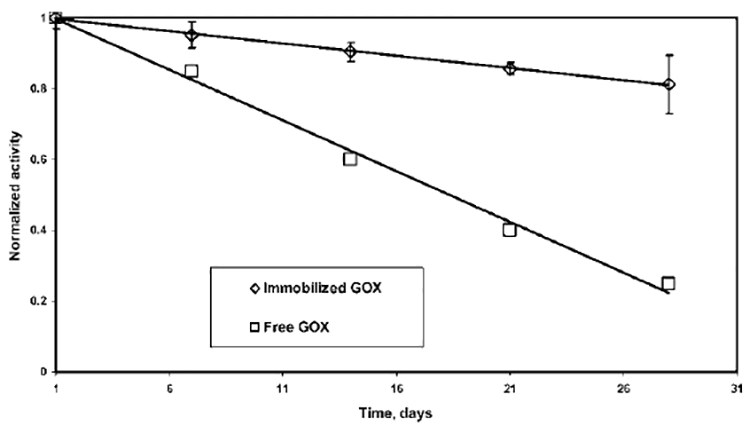
Stability of GOX immobilized in an LBL assembled nylon membrane, as well as in a PVDF–PAA–PAH membrane based on the change in activity over a period of time. The stability is compared with that reported for free GOX.13 Activity is calculated based on concentration of H2O2 and is normalized by dividing by the activity on the first day of immobilization at pH 5.5.
3.7. Reusability of the Membrane Matrix by Detachment and Reattachment of GOX.
3.7.1. LBL-GOX Membrane.
The pKa of amine groups of PAH is 9.5, and the pI of GOX is 4.2. Hence, as the pH decreases below 4.2, the negative charges on GOX are reduced and the attractive interaction with the positively charged nylon–LBL membrane decreases. It was observed, for the nylon–LBL membrane, that the maximum possible pH value at which GOX detaches from the positively charged membrane matrix is 3. The amount of detached GOX was 90% ± 3% of the immobilized GOX. It can be hypothesized that the undetached GOX molecules were either situated in such domains of polyelectrolyte layers that they were inaccessible or they were too strongly bound to detach by changing the pH alone. The detached GOX was unaffected by the drastic change in pH environment and showed 90% activity of the fresh GOX in free form, as revealed by the homogeneous phase experiments. After the homogeneous phase activity studies, the remaining detached GOX was electrostatically reattached in the same LBL membrane matrix. The activity of the reattached GOX was 90% ± 5% of the activity prior to the detachment. The same steps of detachment and reattachment were repeated, and similar results were obtained. GOX then was detached and fresh GOX was attached in the same membrane matrix. The attachment was 90% of the newly made seven-layer LBL membrane, because of the presence of undetached GOX (10%). The activity of the immobilized fresh GOX in the used LBL membrane was identical to the activity obtained for fresh GOX in the newly made seven-layer LBL membrane. The same trend (detachment and reattachment was 90% of the newly made LBL membrane) follows for subsequent detachment/reattachment cycles with freshly attached GOX. This study demonstrates the reusability of the LBL membrane matrix for GOX immobilization. Although the current study involves GOX only, it also can potentially be extended for other enzymes.
3.7.2. PVDF-PAA-PAH-GOX Membrane.
There was a fundamental difference in the regeneration of PVDF–PAA–PAH–GOX membrane, compared to the LBL-GOX membrane. The pKa of amine groups of PAH is 9.5, and the pI of GOX is 4.2. Hence, to reduce the electrostatic interaction and detach GOX, the pH of the detaching solution should be less than 4.2. However, at that pH, the interaction between PAH and PAA also vanishes, because the pKa of carboxylic acid groups of PAA is 4.7. Thus, the detaching solution dislocates the entire PAH-GOX domain from the PVDF–PAA matrix. Therefore, each time, fresh PAH and GOX were attached in the PVDF–PAA membrane. The amount and activity of fresh GOX attached in the same PVDF–-PAA membrane matrix were identical to that obtained for freshly made functionalized PVDF membranes, as observed from multiple studies. This proves that the functionality within the membrane is very consistent and there is no leakage of the components.
4. Conclusions
Functionalized membranes were created by two different approaches. In one case, the alternative layer-by-layer (LBL) assembly of anionic and cationic polyelectrolytes was conducted in a nylon-based membrane. In the other case, in situ polymerization of acrylic acid was conducted in a hydrophobic polyvinylidene fluoride (PVDF) membrane. The enzyme, glucose oxidase (GOX), was then successfully immobilized inside the charged functionalized membrane domains by electrostatic interaction. The functionalized domains in both cases were stable under tested conditions. The activity of GOX was investigated for the oxidation of the substrate (glucose), to produce gluconic acid and H2O2. A detailed kinetic study has been presented with electrostatically immobilized GOX and a convective flow of glucose in the LBL membrane and the polyacrylic acid–polyallylamine hydrochloride (PAA–PAH) functionalized PVDF membrane. The activity was then compared with the covalent mode of GOX immobilization and also for the soaking mode (diffusion) of substrate interaction. It was observed that electrostatic immobilization of the enzyme and pressure-induced convective flow (permeate) of the substrate is beneficial over other combinations for an enzyme–substrate system. The effect of residence time (i.e., permeate flow rate) (through pressure adjustment) and pH on the activity of GOX has also been studied. It was demonstrated that a higher flow rate of reaction mixture through the membrane exposes the maximum possible active sites of the enzyme. It was also established that GOX immobilized in the functionalized membrane has a wide range of operating pH values and was associated with higher stability, compared to free GOX. The potential of reusability of the membrane matrix was an important finding in this research. The reusability has been discussed with an extensive study of the detachment and reattachment of GOX from membrane matrix, along with the activity measurements. This technique also could easily be applied for other enzyme–substrate systems.
Acknowledgment
The authors would like to acknowledge the support of the NIEHS–SBRP (P42ES007380) program for this research. C.C. was supported by the NSF-REU Program, through the Center of Membrane Sciences at the University of Kentucky.
Nomenclature
- Am
external surface area of membrane (cm2)
- =
concentration of H2O2 in the permeate (mM)
- Km
Michaelis-Menten constant (mM)
- Pn
permeability through the membrane after attachment of the nth layer of polyelectrolyte (cm3/(cm2 s bar))
- =
volumetric flow rate of water through the unmodified membrane (cm3/s)
- =
volumetric flow rate of water through the membrane after attachment of the nth layer of polyelectrolyte (cm3/s)
- r0
pore radius of the unmodified membrane (nm)
- rn
pore radius of the membrane after attachment of the nth layer (nm)
- S
substrate concentration (mM)
- S0
initial substrate concentration (mM)
- υ
rate of glucose oxidation by GOX (mM/(mg min))
- υmax
maximum rate in Michaelis-Menten kinetics (mM/(mg min))
Greek Letters
- δn
effective cumulative thickness of the nth layer of polyelectrolyte/polymer (nm)
- τ
residence time (s)
Material Abbreviations
- PVDF
polyvinylidene fluoride
- LBL
layer-by-layer
- GOX
glucose oxidase enzyme
- PLL
poly-L-lysine
- PAH
polyallylamine hydrochloride
- PSS
polystyrene sulfonate (Na-form)
- PAA
polyacrylic acid
Literature Cited
- (1).Hestekin JA; Bachas LG; Bhattacharyya D Poly(amino acid)-Functionalized Cellulosic Membranes: Metal Sorption Mechanisms and Results. Ind. Eng. Chem. Res 2001, 40 (12), 2668–2678. [Google Scholar]
- (2).Smuleac V; Butterfield DA; Sikdar SK; Varma RS; Bhattacharyya D Polythiol-functionalized alumina membranes for mercury capture. J. Membr. Sci 2005, 251 (1–2), 169–178. [Google Scholar]
- (3).Hollman AM; Bhattacharyya D Controlled permeability and ion exclusion in microporous membranes functionalized with poly(L-glutamic acid). Langmuir 2002, 18 (15), 5946–5952. [Google Scholar]
- (4).Hollman AM; Bhattacharyya D Pore assembled multilayers of charged polypeptides in microporous membranes for ion separation. Langmuir 2004, 20 (13), 5418–5424. [DOI] [PubMed] [Google Scholar]
- (5).Datta S; Bhattacharyya D; Ray PD; Nath A; Toborek M Effect of Pre-Filtration on Selective Isolation of Tat Protein by Affinity Membrane Separation: Analysis of Flux, Separation Efficiency, and Processing Time. Sep. Sci. Technol 2007, 42 (11), 2451–2471. [Google Scholar]
- (6).Xu J; Bhattacharyya D Fe/Pd Nanoparticle Immobilization in Microfiltration Membrane Pores: Synthesis, Characterization, and Application in the Dechlorination of Polychlorinated Biphenyls. Ind. Eng. Chem. Res 2007, 46 (8), 2348–2359. [Google Scholar]
- (7).Woodward J, Ed. Immobilised Cells and Enzymes: A Practical Approach; IRL: Oxford, U.K., 1985; 177pp. [Google Scholar]
- (8).Cheryan MMMA Membrane Bioreactors In Membrane Separations in Biotechnology; McGregor WC, Ed.; Marcel Dekker: New York, 1986; Vol. 1, pp 255–295. [Google Scholar]
- (9).Liu J; Wang J; Bachas LG; Bhattacharyya D Activity Studies of Immobilized Subtilisin on Functionalized Pure Cellulose-Based Membranes. Biotechnol. Prog 2001, 17 (5), 866–871. [DOI] [PubMed] [Google Scholar]
- (10).Bhattacharyya D, Butterfield DA, Eds. Biofunctional membranes: Site-specifically immobilized enzyme arrays In Membrane Science and Technology: Polymeric and Biofunctional Membranes; Membrane Science and Technology Series 8; Elsevier: Amsterdam, 2003; pp 233–240. [Google Scholar]
- (11).Ahuja DK; Bachas LG; Bhattacharyya D Modified Fenton reaction for trichlorophenol dechlorination by enzymatically generated H2O2 and gluconic acid chelate. Chemosphere 2007, 66 (11), 2193–2200. [DOI] [PubMed] [Google Scholar]
- (12).Hou X; Liu B; Deng X; Zhang B; Chen H; Luo R Covalent immobilization of glucose oxidase onto poly(styrene-co-glycidyl methacry-late) monodisperse fluorescent microspheres synthesized by dispersion polymerization. Anal. Biochem 2007, 368 (1), 100–110. [DOI] [PubMed] [Google Scholar]
- (13).Ozyilmaz G; Tukel SS; Alptekin O Activity and storage stability of immobilized glucose oxidase onto magnesium silicate. J. Mol. Catal., B 2005, 35 (4–6), 154–160. [Google Scholar]
- (14).Pandey P; Singh SP; Arya SK; Gupta V; Datta M; Singh S; Malhotra BD Application of Thiolated Gold Nanoparticles for the Enhancement of Glucose Oxidase Activity. Langmuir 2007, 23 (6), 3333–3337. [DOI] [PubMed] [Google Scholar]
- (15).Rauf S; Ihsan A; Akhtar K; Ghauri MA; Rahman M; Anwar MA; Khalid AM Glucose oxidase immobilization on a novel cellulose acetate-polymethyl methacrylate membrane. J. Biotechnol 2006, 121 (3), 351–360. [DOI] [PubMed] [Google Scholar]
- (16).Ying L; Kang ET; Neoh KG Covalent immobilization of glucose oxidase on microporous membranes prepared from poly(vinylidene fluoride) with grafted poly(acrylic acid) side chains. J. Membr. Sci 2002, 208 (1–2), 361–374. [Google Scholar]
- (17).Li ZF; Kang ET; Neoh KG; Tan KL Covalent immobilization of glucose oxidase on the surface of polyaniline films graft copolymerized with acrylic acid. Biomaterials 1998, 19 (1–3), 45–53. [DOI] [PubMed] [Google Scholar]
- (18).Ekinci O; Boyukbayram AE; Kiralp S; Toppare L; Yagci Y Characterization and Potential Applications of Immobilized Glucose Oxidase and Polyphenol Oxidase. J. Macromol. Sci., Part A 2007, 44 (8), 801–808. [DOI] [PubMed] [Google Scholar]
- (19).Hoshi T; Sagae N; Daikuhara K; Takahara K; Anzai J -i. Multilayer membranes via layer-by-layer depositi on of glucose oxidase and Au nanoparticles on a Pt electrode for glucose sensing. Mater. Sci. Eng., C 2007, 27 (4), 890–894. [Google Scholar]
- (20).Tomotani EJ; Vitolo M Immobilized glucose oxidase as a catalyst to the conversion of glucose into gluconic acid using a membrane reactor. Enzyme Microb. Technol 2007, 40 (5), 1020–1025. [Google Scholar]
- (21).Smuleac V; Butterfield DA; Bhattacharyya D Layer-by-Layer-Assembled Microfiltration Membranes for Biomolecule Immobilization and Enzymatic Catalysis. Langmuir 2006, 22 (24), 10118–10124. [DOI] [PubMed] [Google Scholar]
- (22).Zhao H; Ju H Multilayer membranes for glucose biosensing via layer-by-layer assembly of multiwall carbon nanotubes and glucose oxidase. Anal. Biochem 2006, 350 (1), 138–144. [DOI] [PubMed] [Google Scholar]
- (23).Decher G Fuzzy nanoassemblies: toward layered polymeric multicomposites. Science 1997, 277 (5330), 1232–1237. [Google Scholar]
- (24).Decher G; Hong JD Buildup of ultrathin multilayer films by a self-assembly process: II. Consecutive adsorption of anionic and cationic bipolar amphiphiles and polyelectrolytes on charged surfaces. Ber. Bunsen-Ges 1991, 95 (11), 1430–1434. [Google Scholar]
- (25).Decher G; Lvov Y; Schmitt J Proof of multilayer structural organization in self-assembled polycation-polyanion molecular films. Thin Solid Films 1994, 244 (1–2), 772–777. [Google Scholar]
- (26).Iler RK Multilayers of colloidal particles. J. Colloid Interface Sci 1966, 21 (6), 569–94. [Google Scholar]
- (27).Ariga K; Hill JP; Ji Q Layer-by-layer assembly as a versatile bottom-up nanofabrication technique for exploratory research and realistic application. Phys. Chem. Chem. Phys 2007, 9 (19), 2319–2340. [DOI] [PubMed] [Google Scholar]
- (28).Chen W; McCarthy TJ Layer-by-Layer Deposition: A Tool for Polymer Surface Modification. Macromolecules 1997, 30 (1), 78–86. [Google Scholar]
- (29).Dai J; Jensen AW; Mohanty DK; Erndt J; Bruening ML Controlling the Permeability of Multilayered Polyelectrolyte Films through Derivatization, Cross-Linking, and Hydrolysis. Langmuir 2001, 17 (3), 931–937. [Google Scholar]
- (30).Nguyen QT; Ping Z; Nguyen T; Rigal P Simple method for immobilization of bio-macromolecules onto membranes of different types.J. Membr. Sci 2003, 213 (1–2), 85–95. [Google Scholar]
- (31).Cheng Y; Corn RM Ultrathin Polypeptide Multilayer Films for the Fabrication of Model Liquid/Liquid Electrochemical Interfaces. J. Phys. Chem. B 1999, 103 (41), 8726–8731. [Google Scholar]
- (32).Caruso F; Furlong DN; Ariga K; Ichinose I; Kunitake T Characterization of Polyelectrolyte-Protein Multilayer Films by Atomic Force Microscopy, Scanning Electron Microscopy, and Fourier Transform Infrared Reflection-Absorption Spectroscopy. Langmuir 1998, 14 (16), 4559–4565. [Google Scholar]
- (33).He P; Hu N; Rusling JF Driving forces for layer-by-layer self-assembly of films of SiO2 nanoparticles and heme proteins. Langmuir 2004, 20 (3), 722–729. [DOI] [PubMed] [Google Scholar]
- (34).Lvov Y; Ariga K; Ichinose I; Kunitake T Assembly of Multicomponent Protein Films by Means of Electrostatic Layer-by-Layer Adsorption. J. Am. Chem. Soc 1995, 117 (22), 6117–6123. [Google Scholar]
- (35).Tang T; Qu J; Muellen K; Webber SE Molecular Layer-by-Layer Self-Assembly of Water-Soluble Perylene Diimides through π-π and Electrostatic Interactions. Langmuir 2006, 22 (1), 26–28. [DOI] [PubMed] [Google Scholar]
- (36).Tedeschi C; Caruso F; Moehwald H; Kirstein S Adsorption and desorption behavior of an anionic pyrene chromophore in sequentially deposited polyelectrolyte-dye thin films. J. Am. Chem. Soc 2000, 122 (24), 5841–5848. [Google Scholar]
- (37).Dotzauer DM; Dai J; Sun L; Bruening ML Catalytic Membranes Prepared Using Layer-by-Layer Adsorption of Polyelectrolyte/Metal Nanoparticle Films in Porous Supports. Nano Lett. 2006, 6 (10), 2268–2272. [DOI] [PubMed] [Google Scholar]
- (38).Ko H; Jiang C; Tsukruk VV Encapsulating Nanoparticle Arrays into Layer-by-layer Multilayers by Capillary Transfer Lithography. Chem. Mater 2005, 17 (22), 5489–5497. [Google Scholar]
- (39).Kotov NA; Dekany I; Fendler JH Layer-by-Layer Self-Assembly of Polyelectrolyte-Semiconductor Nanoparticle Composite Films.J. Phys. Chem 1995, 99 (35), 13065–13069. [Google Scholar]
- (40).Zhang H; Zhou Z; Yang B; Gao M The Influence of Carboxyl Groups on the Photoluminescence of Mercaptocarboxylic Acid-Stabilized CdTe Nanoparticles. J. Phys. Chem. B 2003, 107 (1), 8–13. [Google Scholar]
- (41).Caruso F; Lichtenfeld H; Giersig M; Moehwald H Electrostatic Self-Assembly of Silica Nanoparticle-Polyelectrolyte Multilayers on Polystyrene Latex Particles. J. Am. Chem. Soc 1998, 120 (33), 8523–8524. [Google Scholar]
- (42).Chun K-Y; Stroeve P External Control of Ion Transport in Nanoporous Membranes with Surfaces Modified with Self-Assembled Monolayers. Langmuir 2001, 17 (17), 5271–5275. [Google Scholar]
- (43).Krass H; Papastavrou G; Kurth DG Layer-by-layer self-assembly of a polyelectrolyte bearing metal ion coordination and electrostatic functionality. Chem. Mater 2003, 15 (1), 196–203. [Google Scholar]
- (44).Khopade AJ; Caruso F Investigation of the Factors Influencing the Formation of Dendrimer/Polyanion Multilayer Films. Langmuir 2002, 18 (20), 7669–7676. [Google Scholar]
- (45).Lvov Y; Haas H; Decher G; Moehwald H; Mikhailov A; Mtchedlishvily B; Morgunova E; Vainshtein B Successive Deposition of Alternate Layers of Polyelectrolytes and a Charged Virus. Langmuir 1994, 10 (11), 4232–4236. [Google Scholar]
- (46).Decher G; Lehr B; Lowack K; Lvov Y; Schmitt J New nanocomposite films for biosensors: layer-by-layer adsorbed films of polyelectrolytes, proteins or DNA. Biosens. Bioelectron. 1994, 9 (9/10), 677–684. [Google Scholar]
- (47).Ichinose I; Senzu H; Kunitake T Stepwise adsorption of metal alkoxides on hydrolyzed surfaces: A surface sol-gel process. Chem. Lett 1996, (10), 831–832. [Google Scholar]
- (48).Malaisamy R; Bruening ML High-Flux Nanofiltration Membranes Prepared by Adsorption of Multilayer Polyelectrolyte Membranes on Polymeric Supports. Langmuir 2005, 21 (23), 10587–10592. [DOI] [PubMed] [Google Scholar]
- (49).Krasemann L; Tieke B Ultrathin self-assembled polyelectrolyte membranes for pervaporation. J. Membr. Sci 1998, 150 (1), 23–30. [Google Scholar]
- (50).Liu X; Bruening ML Size-selective transport of uncharged solutes through multilayer polyelectrolyte membranes. Chem. Mater 2004, 16 (2), 351–357. [Google Scholar]
- (51).Zhou X; Arnold MA Internal enzyme fiber-optic biosensors for hydrogen peroxide and glucose. Anal. Chim. Acta 1995, 304 (2), 147–156. [Google Scholar]
- (52).Matsuno H; Nagasaka Y; Kurita K; Serizawa T Superior Activities of Enzymes Physically Immobilized on Structurally Regular Poly(methyl methacrylate) Surfaces. Chem. Mater 2007, 19 (9), 2174–2179. [Google Scholar]
- (53).Onda M; Lvov Y; Ariga K; Kunitake T Sequential reaction and product separation on molecular films of glucoamylase and glucose oxidase assembled on an ultrafilter. J. Ferment. Bioeng 1996, 82 (5), 502–506. [Google Scholar]
- (54).Su X; Zong Y; Richter R; Knoll W Enzyme immobilization on poly(ethylene-co-acrylic acid) films studied by quartz crystal microbalance with dissipation monitoring. J. Colloid Interface Sci 2005, 287 (1), 35–42. [DOI] [PubMed] [Google Scholar]
- (55).Xu J; Dozier A; Bhattacharyya D Synthesis of Nanoscale Bimetallic Particles in Polyelectrolyte Membrane Matrix for Reductive Transformation of Halogenated Organic Compounds. J. Nanopart. Res 2005, 7 (4–5), 449–467. [Google Scholar]
- (56).Ladhe AR; Radomyselski A; Bhattacharyya D Ethoxylated Nonionic Surfactants in Hydrophobic Solvent: Interaction with Aqueous and Membrane-Immobilized Poly(acrylic acid). Langmuir 2006, 22 (2), 615–621. [DOI] [PubMed] [Google Scholar]
- (57).Rivas BL; Jara M; Pereira ED Preparation and adsorption properties of the chelating resins containing carboxylic, sulfonic, and imidazole groups. J. Appl. Polym. Sci 2003, 89 (10), 2852–2856. [Google Scholar]
- (58).Gabriel EM; Gillberg GE In situ modification of microporous membranes. J. Appl. Polym. Sci 1993, 48 (12), 2081–90. [Google Scholar]
- (59).Li Y; Bachas LG; Bhattacharyya D Selected Chloro-Organic Detoxifications by Polychelate (Poly(acrylic acid)) and Citrate-Based Fenton Reaction at Neutral pH Environment. Ind. Eng. Chem. Res 2007, 46 (24), 7984–7992. [Google Scholar]
- (60).Muller D Studies on the new enzyme glucose oxidase. I. Biochem.Z 1928, 199, 136–170. [Google Scholar]
- (61).Johnson KA; Kroa BA; Yourey T Factors affecting reaction kinetics of glucose oxidase. J. Chem. Educ 2002, 79 (1), 74–76. [Google Scholar]
- (62).Duke FR; Weibel M; Page DS; Bulgrin VG; Luthy J Glucose oxidase mechanism. Enzyme activation by substrate. J. Am. Chem. Soc 1969, 91 (14), 3904–3909. [Google Scholar]
- (63).Gibson QH; Swoboda BEP; Massey V Kinetics and mechanism of action of glucose oxidase. J. Biol. Chem 1964, 239 (11), 3927–3934. [PubMed] [Google Scholar]
- (64).Weibel MK; Bright HJ Glucose oxidase mechanism. Interpretation of the pH dependence. J. Biol. Chem 1971, 246 (9), 2734–2744. [PubMed] [Google Scholar]
- (65).Leskovac V; Trivic S; Wohlfahrt G; Kandrac J; Pericin D Glucose oxidase from Aspergillus niger: The mechanism of action with molecular oxygen, quinones, and one-electron acceptors. Int. J. Biochem. Cell Biol 2005, 37 (4), 731–750. [DOI] [PubMed] [Google Scholar]
- (66).Hecht HJ; Kalisz HM; Hendle J; Schmid RD; Schomburg D Crystal structure of glucose oxidase from Aspergillus niger refined at2.3 Å resolution. J. Mol. Biol 1993, 229 (1), 153–172. [DOI] [PubMed] [Google Scholar]
- (67).Wong DWS Food Enzymes: Structure and Mechanism; Chapman and Hall: New York, 1995; 408pp. [Google Scholar]
- (68).Bright HJ; Appleby M pH dependence of the individual steps in the glucose oxidase reaction. J. Biol. Chem 1969, 244 (13), 3625–3634. [PubMed] [Google Scholar]
- (69).Chen TL; Weng HS A method for the determinations of the activity and optimal pH of glucose oxidase in an unbuffered solution. Biotechnol. Bioeng 1986, 28 (1), 107–109. [DOI] [PubMed] [Google Scholar]
- (70).Bao J; Furumoto K; Yoshimoto M; Fukunaga K; Nakao K Competitive inhibition by hydrogen peroxide produced in glucose oxidation catalyzed by glucose oxidase. Biochem. Eng. J 2003, 13 (1), 69–72. [Google Scholar]
- (71).Bradford MM A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72 (1–2), 248–254. [DOI] [PubMed] [Google Scholar]
- (72).Clapp PA; Evans DF; Sheriff TSS Spectrophotometric determination of hydrogen peroxide after extraction with ethyl acetate. Anal. Chim. Acta 1989, 218 (2), 331–334. [Google Scholar]
- (73).Mao Z, M. L; Gao C; Shen J Preformed microcapsules for loading and sustained release of ciprofloxacin hydrochloride. J. Controlled Release 2005, 104, 193–202. [DOI] [PubMed] [Google Scholar]
- (74).Datta S; Ray PD; Nath A; Bhattacharyya D Recognition based separation of HIV-Tat protein using avidin-biotin interaction in modified microfiltration membranes. J. Membr. Sci 2006, 280 (1 + 2), 298–310. [Google Scholar]
- (75).Ho W; Sirkar KK Membrane Handbook; van Nostrand Reinhold: New York, 1992. [Google Scholar]
- (76).Voet D; Voet JG; Pratt CW Fundamentals of Biochemistry; John Wiley & Sons: New York, 1999. [Google Scholar]
- (77).Ahuja DK Degradation of toxic chloroethylenes and chloroaromatics by vitamin B12-based reductive dechlorination and by hydroxyl radical-based oxidative dechlorination reactions, Ph.D. Thesis, University of Kentucky, Lexington, KY, 2006. [Google Scholar]