

Abstract
Purpose:
The mechanisms underlying doxorubicin cytotoxicity and cardiotoxicity were broadly explored but remain incompletely understood. A multiscale physiologically-based pharmacokinetic (PBPK) model was developed to assess doxorubicin dispositions at levels of system, tissue interstitial, cell, and cellular organelles. This model was adopted to explore the mechanisms-of-action/toxicity of doxorubicin in humans.
Methods:
The PBPK model was developed by analyzing data from mice and the model was verified by scaling up to predict doxorubicin multiscale dispositions in rats and humans. The multiscale dispositions of doxorubicin in human heart and tumors were explicitly simulated to elucidate the potential mechanisms of its cytotoxicity and cardiotoxicity.
Results:
The developed PBPK model was able to adequately describe doxorubicin dispositions in mice, rats and humans. In humans, prolonged infusion, a dosing regimen with less cardiotoxicity, was predicted with substantially reduced free doxorubicin concentrations at human heart interstitium, which were lower than the concentrations associated with oxidative stress. However, prolonged infusion did not reduce doxorubicin-DNA adduct at tumor nucleus, consistent with clinical observations that prolonged infusion did not compromise anti-tumor effect, indicating that one primary anti-tumor mechanism was DNA torsion.
Conclusions:
A multiscale PBPK model for doxorubicin was developed and further applied to explore its cytotoxic and cardiotoxic mechanisms.
Keywords: Physiologically-based pharmacokinetic model, doxorubicin, multiscale, cardiotoxicity, cytotoxicity, mechanisms
INTRODUCTION
Doxorubicin is one of the most widely prescribed antineoplastic agents for solid carcinoma and leukemia (1, 2). However, the high anti-tumor efficacy of doxorubucin is always compromised by its high risk of fatal cardiotoxicity. About 7% to 26% of patients would develop congestive heart failure when the cumulative dose is over 550 mg/m2 (3). This cumulative dose-associated cardiotoxicity has posted a great challenge to doxorubicin broad prescription (4). Potential mechanisms underlying doxorubicin cytotoxicity and cardiotoxicity have been broadly investigated, but it remains an incompletely understood issue.
Most previous investigations on the mechanisms of doxorubicin action were performed in cellular contexts. Cell-based assays are usually adopted to mimic the tissue interstitial condition, where cells directly interact with the drugs. Drug concentrations at tissue interstitium are known to be essential in predicting cytotoxicity. However, directly measuring doxorubicin in human tissues interstitium is not only technically difficult but also ethically challenging. Plasma doxorubicin concentrations were usually adopted to correlate with in vitro studies to predict the toxic mechanisms (5). DNA synthesis inhibition (> 2 μM) was often excluded from the possible mechanisms of its cytotoxicity because a surreally high dose required to achieve a related concentration (6). Other mechanisms of cytotoxicity for doxorubicin were also found concentration-dependent, which include oxidative stress (> 0.1 μM), topoisomerase II inhibition (> 0.4 μM), DNA torsion (> 0.025 μM), and cardiomyocyte apoptosis (> 0.1 μM) (6).
Because of massive nucleotide intercalation, there is a remarkable concentration gradient between plasma and tissue. Doxorubicin concentrations at tissue interstitial fluid, intracellular nucleus, plasma are of multifold difference. For instance, tissue average doxorubicin concentrations is nearly 50-fold higher than that in plasma (7, 8), intracellular doxorubicin concentration is about 200-fold higher than extracellular concentration (9). Therefore, predictions of doxorubicin concentrations in different biological spaces is extremely important to help explore its potential cytotoxic mechanisms in humans.
When only animal data is available and human tissue cannot be experimentally accessible, physiologically-based pharmacokinetic (PBPK) models are of high value to predict drug dispositions at human tissues and even sub-tissues (10, 11). Thus, PBPK models would be a valuable tool to better evaluate these concentration-dependent cytotoxic mechanisms in human physiological contexts. Several PBPK models had been reported in the literature to characterize doxorubicin systemic dispositions (12–14). Unlike previous studies, one purpose of the present study is to predict multiscale disposition of doxorubicin in humans, and then with it to explore the potential mechanisms underlying doxorubicin cytotoxicity and cardiotoxicity. To achieve such a goal, the developed PBPK model entailed doxorubicin dispositions at scales of system, tissue interstitium, tissue cells, and cell nucleus, especially in heart and tumor, two toxicity and efficacy associated tissues, respectively.
MATERIALS AND METHODS
PK Data Collection
Literature search was conducted to collect doxorubicin PK and tissue distribution data in mice, rats and humans. The inclusion and exclusion criteria are: (1) the dosing approach should be i.v. bolus of doxorubicin solution, (2) no other drug was simultaneously administered, and (3) studies that only reported blood concentrations were excluded by considering the concentration difference between blood and plasma (15, 16). For each included paper, plasma and/or tissue concentration-time profiles were collected. The plasma concentration-time profiles were extracted using Digitizer (17). All studies used HPLC with fluorescent detectors to measure both plasma and tissue concentrations.
Doxorubicin concentration-time profiles in plasma and tissues were extracted from 9 studies for mice, 11 studies for rats, and 7 studies for humans (references in supplemental materials, 1–27). All studies reported plasma concentration-time profiles and most studies had tissue concentration-time profiles. Nine sets of tissue data were collected for mice, including tumor, heart, liver, spleen, kidney, lung and gut; 6 sets of tissue data were collected in rats, including heart, liver, spleen, kidney and lung; and 2 sets of tissue data were collected in humans, including liver and tumor. Considering doxorubicin exhibits linear PK, concentrations were then normalized to 10 mg/kg for mice, 6 mg/kg for rats and 50 mg/m2 for humans for comparisons across doses.
PBPK Model
A PBPK model was developed to include 7 tissues: tumor, heart, liver, spleen, lung, kidney, and gut (Fig. 1A). Other tissues were collectively lumped as “others” compartment. To explore the multiscale kinetics, each tissue was further divided into four sub-compartments: vascular, interstitial, intracellular, and nucleus (Fig. 1B). Tissues were connected through venous and arterial blood. Considering doxorubicin is greatly accumulated in blood cells and doxorubicin concentration in blood is about 200–500 times higher than plasma (16, 18), the blood compartment was further divided into blood cells and plasma sub-compartments.
Fig 1.
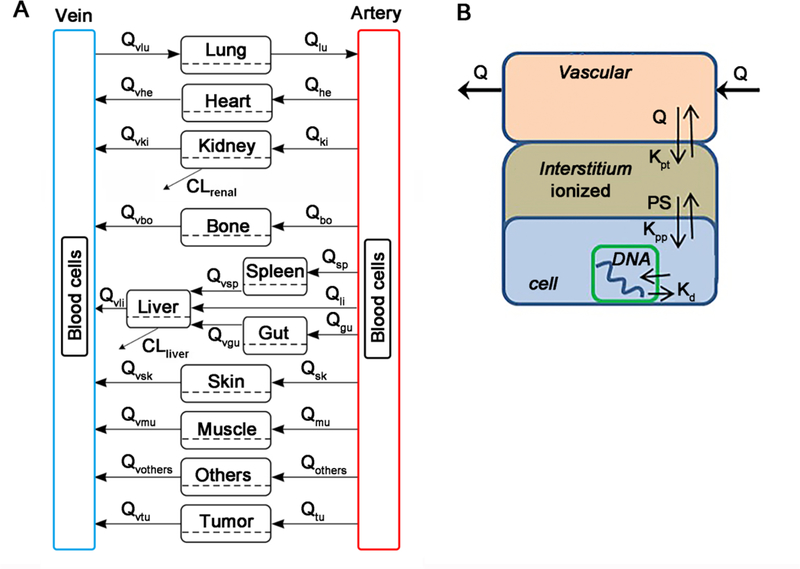
Schematic diagram of the developed multiscale PBPK model for doxorubicin. (A) The whole-body PBPK model is comprised of 7 tissues and 2 blood compartments. Blood compartment are further divided into plasma and blood cell sub-compartments. (B) Tissue model. Each tissue is divided into vascular, interstitial, intracellular, and nucleus DNA bound sub-compartments.
In the PBPK model, Fick’s law of perfusion was used to describe the diffusion of doxorubicin across vascular membrane. Albumin is known as the major binding protein in interstitial fluid and plasma. The concentration ratios of albumin between interstitial fluid and plasma were used to calculate the interstitial fluid/plasma partition coefficient, Kpt_org:
| (1) |
where E/P is the concentration ratio of albumin between interstitial fluid and plasma, and E/P value for each tissue was obtained from the literature (19). fup is the free fraction of doxorubicin in plasma, which was set to literature value 0.26 (20).
Fick’s law of diffusion was applied to describe the distribution process of doxorubicin across cytoplasmic membrane. PER is the cytoplasmic membrane permeability coefficient and Sorg is the total surface area of cell membrane in each tissue. The cross membrane distribution of doxorubicin involves diffusion and transporter-mediated process. The diffusion rate was estimated to be 0.000756 by analyzing doxorubicin cellular uptake data from the literatures (21, 22). Fra, a parameter reflects the joint effect of influx/efflux transporters, which is inversely proportional to the activities of efflux transporter, such as P-gp. Fra was optimized around 100 to well capture the quick cellular uptake profiles of doxorubicin in cellular uptake studies. The product of PER and Sorg, represents the diffusion rate of doxorubicin across cytoplasmic membrane. Sorg was calculated as:
| (2) |
where Vcell is the volume of a cell, Scell is the membrane surface area of a cell. The intracellular to interstitial partition coefficient was determined by two factors: pH gradient and nonspecific protein binding, which were respectively denoted by Kpp and Kp. Kpp, the partition coefficient associated with pH gradient, was calculated:
| (3) |
where pKa (= 8.15) is doxorubicin acid dissociation constant. pHi (= 7.4) and pHe (= 7.0) are interstitial and intracellular pH. For solid tumors, interstitial pH is usually much lower than 7.0, often in the range of 6.5 ~ 7.0 (23), which would support a lower Kpp value in tumors. Kp, partition coefficient due to nonspecific protein binding, was optimized.
Considering extremely fast binding process of doxorubicin to DNA (24), a quasi-equilibrium model was assumed to describe kinetics of doxorubicin-DNA intercalation. The folded structure of DNA makes doxorubicin only bind to a portion of nucleotides and the fraction was set to a literature value of 0.18 (22).
Hepatic metabolism and bile excretion are the main elimination routes for doxorubicin, with a small portion of doxorubicin excreted by kidney (20). In the model, hepatic-biliary elimination was assumed to account for 92% of total clearance and the rest 8% was assumed as renal clearance according to the literature (25, 26).
All symbols and parameters used in the model are shown in the glossary (Table S1, supplemental materials). The general differential equations for plasma and tissue compartments are:
Blood compartment:
| (4) |
| (5) |
| (6) |
where the abbreviations represent doxorubicin concentrations in artery (Ca), vein (Cp), interstitial fluid (Ci_org), and lung (Ci_lung). Cet_blood is doxorubicin concentration in cytoplasm of blood cells. Ce_blood is DNA-bound complex concentration in blood cells. Qorg and Qlung are blood flows in each organ and lung. Vp and Va are venous and artery volumes. Ve_blood and Sblood are volume and surface area of blood cells. Kpt_org indicates interstitial fluid/plasma partition coefficient in each tissue, and Kp_blood is blood cell/plasma partition coefficient. CLrenal is renal clearance. In Eq (4) to (6), org represents all the tissues except lung.
Tissue compartment:
Nucleus sub-compartment (27):
| (7) |
Interstitial sub-compartment:
| (8) |
Cytoplasmic sub-compartment:
| (9) |
Doxorubicin concentration in the interstitial fluid (ISF) of liver:
where the abbreviations represent interstitial fluid (Ci_org), intracellular total (Cet_org), intracellular free concentrations (Ce_org), DNA concentrations (CNorg), equilibrium dissociation constant of DNA intercalation (Kd), volume of interstitial fluid (Vi_org), intracellular volume (Ve_org) and hepatic intrinsic clearance (CLhepatic).
Physiological parameters used in this model were listed in Table S2 (supplemental materials) for mice, Table S3 for rats, and Table S4 for humans (28). Drug specific parameters such as clearance, Kp_org, and Kd were jointly optimized using ADAPT 5 (http://bmsr.usc.edu/Software/ADAPT/) with NLEM method. The plasma and tissue data from mice were simultaneously fitted using Maximum likelihood estimation with the following variance model:
| (10) |
where Var(t) is the variance of the concentration at a specific time point, and are the additive and proportional variance, Y(t) is the predicted concentration at time t. The model would be accepted based on a combination of subjective criteria: the predicted profiles is well correlated with literature average profiles and the majority (> 80%) of the observations fall within the model predictive ranges.
Model Across-Species Scaling and Multiscale Simulations
The PBPK model developed based on mouse data was then scaled up to rats and humans to assess the scalability and predictability of the developed PBPK model. Clearance was scaled up according to (29):
| (11) |
where BW is the body weight, and B is the scaling coefficient that was usually fixed to a conventional value 0.75. Monte Carlo simulation was conducted to generate 90% prediction interval of concentration-time profiles using Berkeley Madonna (version 8.3.23; http://www.berkeleymadonna.com/). Kp_liver, Kp_heart and Kp_kidney were optimized around 20, 15 and 8 respectively to account for the distribution differences in these issues between mice and rats, and these differences are suspected to be partly associated with high sample contamination as these tissues usually have high trapped blood. The estimated inter-study variation of parameters was assumed constant across species. 30% variation of blood flow in humans was incorporated to account for the inter-patient variability. To fully optimize the variability of cardiac output, PBPK Bayesian population modeling is required, which is beyond the scope of this study. The simulated concentration-time profiles were overlaid with experimental data from rats and human to evaluate the predictive performance of the PBPK model.
To illustrate sub-tissue kinetics in humans, the concentration-time profiles of doxorubicin in plasma, tissue interstitium, tissue cell cytoplasm, and cell nucleus were simulated. The DNA bound concentration was:
| (12) |
where Ce_org is the free concentration and Cet_org is total concentration of doxorubicin in cytoplasm.
Exploration of Cytotoxic and Cardiotoxic Mechanisms
Several cytotoxic mechanisms have been explored in the literature, mostly in in vitro studies: DNA synthesis inhibition (> 2 μM), oxidative stress (> 0.1 μM), topoisomerase II inhibition (> 0.4 μM), DNA torsion (> 0.025 μM), and cardiomyocyte apoptosis (> 0.1 μM) (6). The most relevant concentrations to cytotoxicity are drug concentrations in cells or nucleus, however, the concentrations concluded in in vitro studies were largely based upon concentrations in cell medium, which are more relevant to concentrations in tissue interstitial fluid. Therefore, the interstitial doxorubicin concentrations were simulated at each tissue and we could compare these cytotoxic concentration ranges with simulated profiles to explore the primary cytotoxic mechanism in human heart and tumors.
In addition, prolonged continuous infusion showed lower incidence of cardiotoxicity and did not compromise anti-tumor efficacy in comparison with bolus dosing (30). We then simulated the concentration-time profiles after 6 hours infusion and compared that with profiles after bolus dosing, in order to examine if the developed PBPK model could predict the reduced cardiotoxicity and comparable efficacy after prolonged infusion.
Sensitivity Analysis
Sensitivity analysis was performed to several drug specific parameters and certain physiological parameters in change of plasma and tissue AUC (area under the curve): KD, S, Kpp, Fr, Fra, CLhepatic, Kp_blood, Kp_lung, Kp_gut, Kp_spleen, Kp_liver, Kp_kidney, Kp_heart, Kp_tumor and Kp_others, where Fr is a fraction of available total nucleotides and Fra is a parameter reflecting transporters-dependent membrane permeability. The dynamic range of these parameters was set from 0.5 to 2-fold of the estimated or original values. AUC ratio (to original AUC) was simulated to test the sensitivity of model parameters. In addition, tissue AUC was normalized by plasma AUC to assess the sensitivity of model parameters to tissue distribution.
RESULTS
The Developed PBPK Model
The developed PBPK model extensively incorporated prior knowledge to improve model identifiability and stability. The optimized parameters were listed in Table I and the fitting curves for mice were shown in Fig. 2. Overall, the developed PBPK model adequately captured the plasma and tissue concentration-time profiles in mice based on VPC, and most of model parameters were estimated with high precision (low relative standard deviation, RSD %). As shown in Fig. 2, the variability of individual study was nicely reproduced by Monte Carlo simulation, with most of the observations falling within the 90% percentile (5 – 95%) of model simulations.
Table I.
Estimated model parameters
| Value (RSD %) | Unit | Source | |
|---|---|---|---|
| Kpp | 2.2836 | - | Calculated |
| Fr | 0.18 | - | (22) |
| Fra | 1 | - | Assumption |
| PER | 0.0756 | cm/h | Unpublished |
| Kd | 0.13 (51.6) | μmol/mL | optimized |
| CLrenal | 18 | mL/h | optimized |
| CLhepatic | 199 (0.193) | mL/h | optimized |
| Kp_lung | 3.38 (10.8) | - | optimized |
| Kp_gut | 1.26 (11.4) | - | optimized |
| Kp_spleen | 5.74 (7.90) | - | optimized |
| Kp_liver | 10.9 (12.9) | - | optimized |
| Kp_kidney | 5.91 (6.15) | - | optimized |
| Kp_heart | 5.63 (11.7) | - | optimized |
| Kp_plasma | 4.07 (30.8) | - | optimized |
| Kp_others | 13.3 (57.6) | - | optimized |
| Kp_tumor | 3.6 (11.3) | - | optimized |
Fig 2.
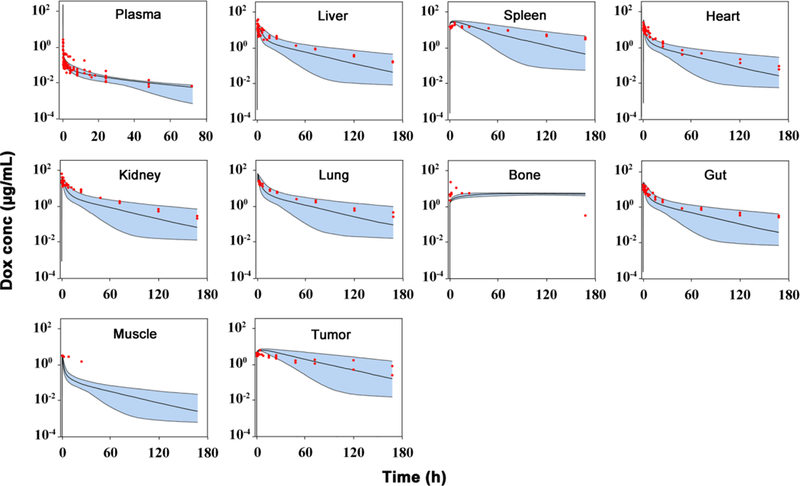
Observed and predicted doxorubicin (Dox) concentration-time profiles in plasma and various tissues in mice. Observed data are shown as dots. Solid lines are the predicted median, dashed lines represent the predicted 5th and 95th percentiles, and the shaded areas depict the 5th−95th percentile population prediction intervals. (Supplemental materials reference 1–9)
Parameter Kpp, the partitioning coefficient due to drug ionization (i.e. pH), was calculated as 2.28, suggesting that approximately 2-fold higher concentrations of doxorubicin in tissue cytoplasm than that in interstitial fluid are associated with pH gradient. The calculated interstitial/plasma partition coefficient (Kpt) in three species were summarized in Table S2-S4. Kpt for gut was calculated as 0.94, and for other tissues was all 0.68. The hepatic intrinsic clearance was estimated as 199 mL/h in mice, which is equivalent to a plasma clearance 48.7 mL/h, in consistence with previous report (12). The estimated partition coefficient Kp was relatively high in spleen (5.74), liver (10.9), heart (5.63), and kidney (5.91), suggesting high nonspecific bindings of doxorubicin to cellular proteins in these tissues. A relatively lower Kp values were obtained in tumors. The model predictions are consistent with general observations that doxorubicin is highly distributed into tissues with higher DNA levels, such as spleens and tumors.
Model across-species scaling and simulations
The clearance of doxorubicin in rats and humans were predicted based on the allometric scaling principle (29). The predicted clearance in rats and humans were 0.27 and 25.5 L/h, respectively, which are close to literature values (12, 27, 31). As shown in Fig. 3 and 4, based on the optimized parameters in mice (Table I), the predicted median concentrations and variable ranges close to the observation in plasma and tissues in rats and humans. Dose-normalized doxorubicin concentrations in human breast, carcinoid, liver, and omental tumors were digitized from the literature and compared with our model predictions (32, 33). In general, our developed model, using xenograft data, roughly predicted doxorubicin concentrations in human tumors. Doxorubicin concentrations in clinical tumors also showed high variability which was found closely related to the tumor blood flow (0.1 ~ 10 mL/min) and DNA concentrations (1.7 ~ 170 μmol/mL) (Table S4). The possible range of tumor concentrations was shown in Fig. 4 with the provided ranges of tumor blood flow and DNA concentrations. Heart, liver, and kidney concentrations in rats and especially liver in human were under-predicted, even though most of determined tissue concentrations were within the 90% prediction interval. Species difference was herein suggested. However, no direct evidence was found in the literature to indicate such species difference. Therefore, we speculated that slightly higher concentrations of doxorubicin in rat liver, heart and kidney were partly associated with high residual bloods in these tissues.
Fig 3.
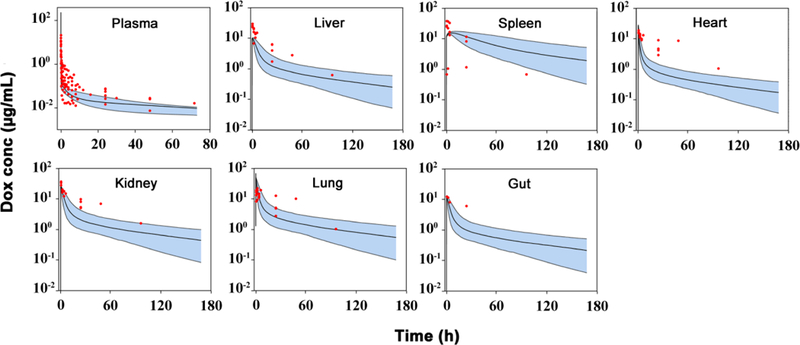
Observed and predicted doxorubicin (Dox) concentration-time profiles in plasma and various tissues in rats. Observed data are shown as dots. Solid lines are the predicted median, dashed lines represent the predicted 5th and 95th percentiles, and the shaded areas depict the 5th−95th percentile population prediction intervals. (Supplemental materials reference 10–20)
Fig 4.
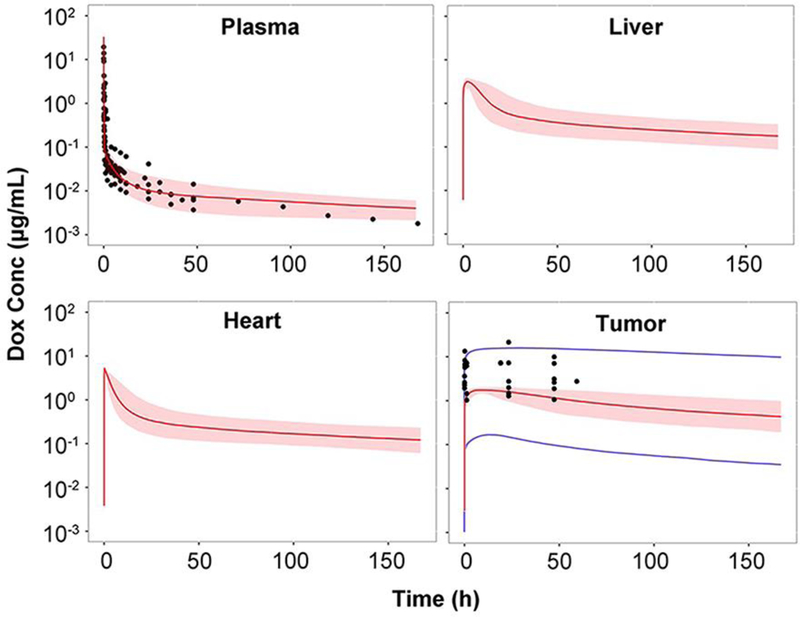
Observed and predicted doxorubicin (Dox) concentration-time profiles in plasma and various tissues in humans. Observed data are shown as dots. Solid red lines depict the predicted median, dashed lines represent the predicted 5th and 95th percentiles, and the shaded areas depict the 5th−95th percentile population prediction intervals. Solid blue lines represent the predicted 5th and 95th percentiles of varied tumor blood flow (0.1 ~ 10 mL/min) and DNA concentrations (1.7 ~ 170 μmol/mL). (Supplemental materials reference 21–27)
Multiscale distribution kinetics of doxorubicin in two target organs-heart and tumors-were specifically simulated. As shown in Fig. 5, doxorubicin rapidly distributes across both vascular and cell membranes, and almost instantaneously achieves distribution equilibrium in heart. Doxorubicin shows high accumulations in DNA-bound form in nucleus, especially in the nucleus of cells in tumors. In accordance with previous experimental study, our simulation suggested 96.2% (95.1% in experiment) of doxorubicin is nucleus binding (34). Plasma concentrations were slightly higher than interstitial concentrations in both heart and tumors, but much lower than that in cytoplasmic and nucleus. Interstitial concentrations are similar between heart and tumors, but nucleus concentrations in tumors are dramatically greater than those in heart, mainly because of excessive abundance of DNA binding sites associated with rapid cell division and proliferation.
Fig 5.
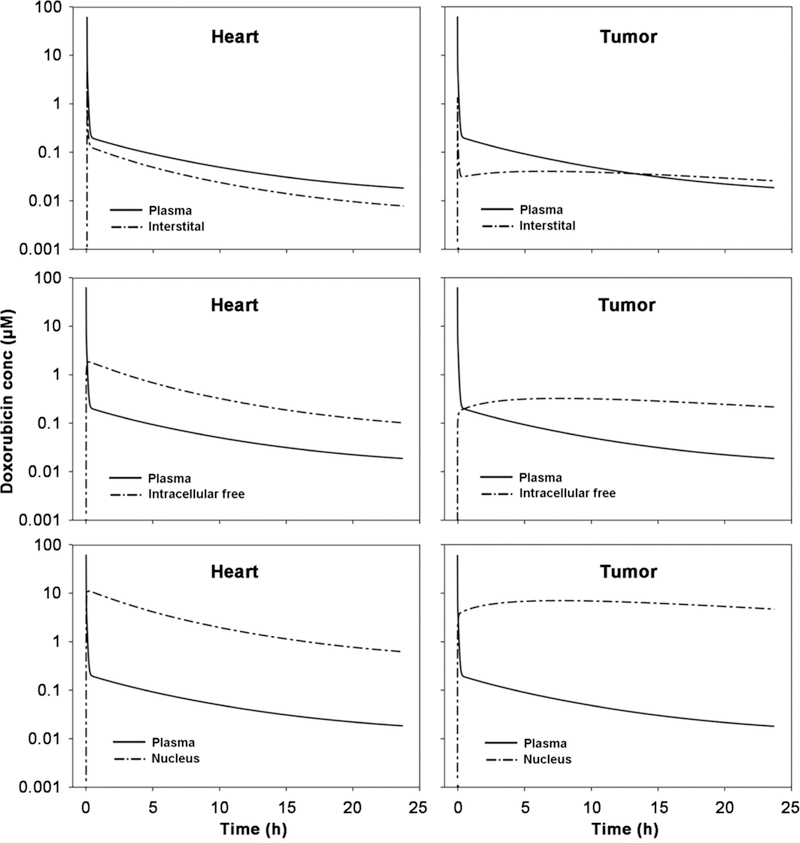
Doxorubicin distributions in heart and tumor after i.v. bolus 50 mg/m2 doxorubicin in 70 kg humans. The plasma, interstitial, intracellular free and nucleus (i.e., DNA-bound) doxorubicin concentration-time profiles are simulated by the developed PBPK model.
Doxorubicin Mechanisms of Cytotoxicity and Cardiotoxicity
Doxorubicin concentrations used in previous cell-based assays were often associated with plasma average concentrations. Actually, the concentrations in tissue interstitial fluid rather that in plasma are equivalent to cell medium concentrations in in vitro assays. Thus, interstitial concentration-time profiles were applied to explore potential cytotoxic mechanisms. As shown in Fig. 6, the cytotoxic concentration ranges for the potential mechanisms were overlaid with simulated profiles. Peak concentrations are about 4.5 μM in heart and 0.74 μM in tumors after bolus dosing (50 mg/m2). Prolonged infusion lowered about 70-fold and 40-fold of peak concentration in heart and tumor interstitial fluid respectively. After prolonged infusion, interstitial concentrations in heart were simulated under 0.1 μM, which might not sufficient to the productions of substantial reactive oxidative species (ROS), as ROS relevant doxorubicin concentrations is usually believed higher than that for other cytotoxic mechanisms. Considering that prolonged infusion could reduce the mean fall in LVEF (left ventricular ejection fraction) from 21% to 6% after 400 mg/m2 doxorubicin dosing (35), our simulation suggested that oxidative stress were considerably related to the mechanisms of doxorubicin cardiotoxicity after bolus administration. Nonetheless, these conclusions are generated based on in silico investigation and further experimental studies are warranted to confirm these in silico findings.
Fig 6.
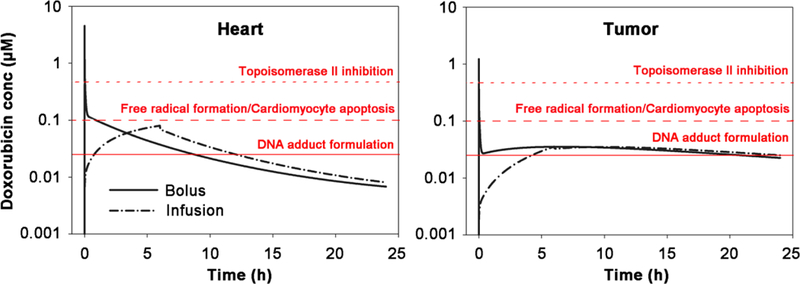
Simulated interstitial doxorubicin concentration-time profiles in heart and tumors after either prolonged infusion (6 hr) or bolus dosing doxorubicin at 50 mg/m2 to a 70 kg human. The concentration ranges for these cytotoxic mechanisms were overlaid: free radical formation/cardiomyocytes apoptosis, topoisomerase II inhibition, and DNA adduct formulation. The literature-supported concentration ranges for these mechanisms are: > 2 μM for DNA synthesis inhibition, > 0.4 μM for topoisomerase II inhibition, > 0.1 μM for oxidative stress and cardiomyocyte apoptosis and > 0.025 μM for DNA torsion. (6)
According to our simulations in Fig. 6, the only possible cytotoxic effect during prolonged infusion is DNA torsion. Heart, not like tumors, have much slower DNA synthesis and cell proliferation, which result in limited accumulations of doxorubicin-DNA adducts. However, in tumors, the total levels of DNA-bound doxorubicin are similar between prolonged perfusion and bolus dosing. This is consistent with clinical observations that prolonged infusion would not compromise anti-tumor effect (35), strongly suggested that DNA torsion largely drive cytotoxicity in tumors.
Sensitivity Analysis
Local sensitivity analysis are shown in Fig. 7. As expected, except for hepatic clearance, most model parameters, such as Kp_org, Kpp, Fr, Fra and KD, showed limited sensitivity to plasma profiles, wherein 0.5 ~ 2-fold changes of parameter values only resulted in around 10% changes of plasma exposures. However, these parameters were quite sensitive to tissue distribution extent (AUCtissue /AUCplasma), thus it is quite critical to have tissue concentration vs time data to develop the multiscale PBPK model and optimize all these model parameters.
Fig 7.
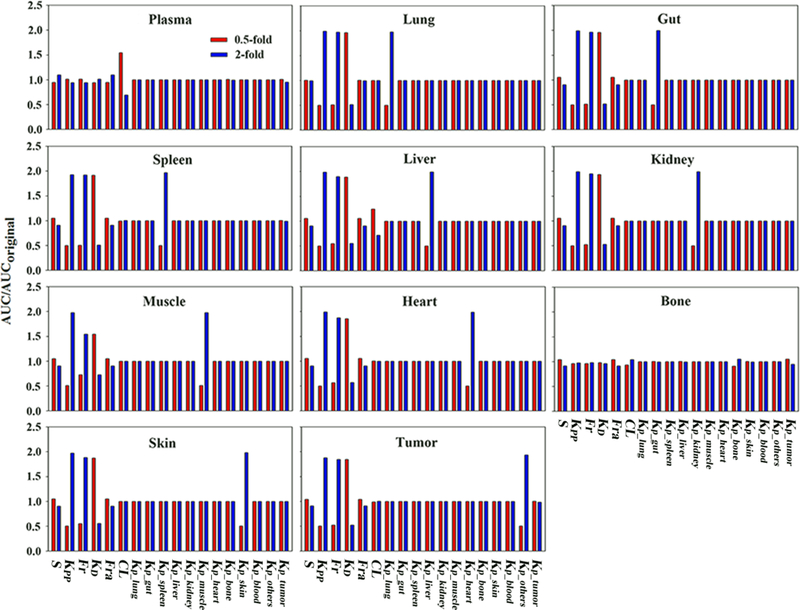
Parameter sensitivity analysis results. Simulations were performed with the parameter varied 0.5-or 2-fold of the original values. The ratio of altered AUC versus original AUC was used to evaluate parameter sensitivity. Tissue AUC was normalized by plasma AUC to indicate changes of tissue distribution extent.
DISCUSSION
A state-of-art PBPK model was developed for doxorubicin to explore its multiscale dispositions at levels of system, tissue interstitium, tissue cytoplasm, and cell nucleus. The PBPK model sufficiently described the pharmacokinetic and tissue distribution of doxorubicin in mice, rats, and humans. Its predictability and scalability were well demonstrated in our analysis. The developed model was then applied to explore the potential cytotoxic and cardiotoxic mechanisms for doxorubicin. The simulated interstitial concentration-time profiles in heart and tumors supported that oxidative stress might be the primary mechanism for doxorubicin cardiotoxicity and DNA torsion might largely drive cytotoxicity in tumors which could be eradicated by dosing schedule modification. Our simulations were in line with clinical observations, which showed that prolonged infusion of doxorubicin would greatly reduce cardiotoxicity, but did not compromise much DNA torsion-induced cytotoxicity in tumors. In comparison with the previously developed PBPK model for doxorubicin (12), a remarkable feature of the present model is its multiscale characterizations of doxorubicin dispositions, which enabled the following evaluations of the mechanisms of actions in heart and tumors.
One challenge in development of complex PBPK models was the lack of stability and reliability to address clinically meaningful questions. Due to model dimensions and complexity, high uncertainties often persist in PBPK model development and predictions. To develop a PBPK with high predictability, we used the mouse data to develop the model and the rat and human data to calibrate and validate the model. Importantly, the developed PBPK model not only well predicted the average but also the varying ranges of plasma and tissue concentration-time profiles in rats and humans. This is in contrast with most previous PBPK models that only used human plasma data to evaluate model predictive performance (36–38).
Use of prior knowledge in complex PBPK models has become a common strategy to improve model stability and identifiability. Thereby, the sources and reliability of prior knowledge would be extremely critical, because any bias in prior knowledge would directly influence model predictive performance. Considering that human populations are heterogeneous with high diversity, we used PopGEN to simulate a virtual human population to reveal the general variabilities of several physiological parameters used in the developed PBPK model. We found that cardiac output is a critical parameter to influence doxorubicin disposition. 15% of heart interstitial doxorubicin concentration would be decreased when cardiac output is reduced by 10%. This is mainly because doxorubicin across vascular membrane is perfusion-limited and cardiac output essentially dominates the α-phase in plasma profiles. To recapitulate the high variabilities in the early phase of plasma profiles, we used 30% variability (results in PopGEN) of human cardiac output in Monte Carlo simulations. In addition, DNA concentrations in each tissue had been experimentally measured, but the fraction of available DNA binding sites was largely uncertain. Therefore, we used a literature reported value and our analysis suggested that such a fraction (Fr) was not a sensitive parameter to plasma and tissue concentrations. This is mainly because nucleotide concentrations (multiple binding sites/DNA) are usually much higher than doxorubicin concentrations, and several-folds changes of nucleotide concentrations would not significantly influence DNA-bound doxorubicin concentrations.
Polymorphisms involving doxorubicin metabolic enzymes (e.g. carbonylreductase) and transporters (e.g. ABCB1, SLC22A16) were reported to modulate doxorubicin disposition in humans (39, 40). In this model, CLhepatic describes the elimination of doxorubicin in liver, which essentially reflect the metabolic enzyme activity. The value for CLhepatic can be updated once more information about the enzyme genotypes and activities become available. Fra is a parameter influencing the transporter-dependent permeability across cytoplasmic membrane. The value of Fra should be a function of transporter genotypes and functionality. Therefore, once more PK data at different genetic variants become available, the present model could be applied to explore the polymorphisms of metabolic enzymes and/or transporters.
Once established, PBPK models would be extremely useful to answer many drug development questions. Because of many mechanistic insights, PBPK models have been broadly used in assessments of drug-drug interactions, PK at specific patient population, and complex drug absorption and disposition. To our knowledge, this study was the first to use PBPK model to explore drug mechanism-of-action/toxicity for chemotherapeutic drugs. The multiscale nature of the developed PBPK model enabled us to evaluate and compare the potential mechanisms of doxorubicin cardiotoxicity and cytotoxicity. And the model suggested that oxidative stress was the primary cardiotoxic mechanism and DNA torsion largely drove cytotoxicity in tumors. Further experimental studies are warranted to confirm the in silico findings.
CONCLUSION
In this study, we developed a multiscale and multispecies PBPK model for doxorubicin, which could predict the dispositions of doxorubicin at multiple biological scales (system, tissue, cellular and subcellular). With the developed PBPK model, we explored the potential mechanisms of doxorubicin cardiotoxicity and cytotoxicity and concluded that the mechanisms for its cardiotoxicity primarily involved oxidative stress and DNA torsion may greatly drive its cytotoxic effect in tumors.
Supplementary Material
ACKNOWLEDGEMNETS:
This work was supported by FDA (U01 FD005206) and NIH (GM119661). The views expressed in this article are those of the authors and not necessarily those of the Food and Drug Administration (FDA). We appreciate the discussion and suggestions with Dr. William Jusko from State University of New York at Buffalo.
Footnotes
Conflict of Interest
There is no conflict of interest.
REFERENCES
- 1.Damiani RM, Moura DJ, Viau CM, Caceres RA, Henriques JA, Saffi J. Pathways of cardiac toxicity: comparison between chemotherapeutic drugs doxorubicin and mitoxantrone. Arch Toxicol. 2016;90(9):2063–76. [DOI] [PubMed] [Google Scholar]
- 2.Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp (Warsz). 2009;57(6):435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Senkus E, Jassem J. Cardiovascular effects of systemic cancer treatment. Cancer Treat Rev. 2011;37(4):300–11. [DOI] [PubMed] [Google Scholar]
- 4.Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267–85. [DOI] [PubMed] [Google Scholar]
- 5.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–41. [DOI] [PubMed] [Google Scholar]
- 6.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845(1):84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol. 1981;5(4):267–70. [DOI] [PubMed] [Google Scholar]
- 8.Formelli F, Carsana R, Pollini C. Comparative pharmacokinetics and metabolism of doxorubicin and 4-demethoxy-4’-O-methyldoxorubicin in tumor-bearing mice. Cancer Chemother Pharmacol. 1986;16(1):15–21. [DOI] [PubMed] [Google Scholar]
- 9.Soininen SK, Vellonen KS, Heikkinen AT, Auriola S, Ranta VP, Urtti A, et al. Intracellular PK/PD Relationships of Free and Liposomal Doxorubicin: Quantitative Analyses and PK/PD Modeling. Mol Pharm. 2016; 13(4):1358–65. [DOI] [PubMed] [Google Scholar]
- 10.Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically Based Pharmacokinetic (PBPK) Modeling and Simulation Approaches: A Systematic Review of Published Models, Applications, and Model Verification. Drug Metab Dispos. 2015;43(11):1823–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma V, McNeill JH. To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol. 2009;157(6):907–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gustafson DL, Rastatter JC, Colombo T, Long ME. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J Pharm Sci. 2002;91(6):1488–501. [DOI] [PubMed] [Google Scholar]
- 13.Dubbelboer IR, Lilienberg E, Sjögren E, Lennernäs H. A Model-Based Approach To Assessing the Importance of Intracellular Binding Sites in Doxorubicin Disposition. Mol Pharm. 2017;14(3):686–98. [DOI] [PubMed] [Google Scholar]
- 14.Gustafson DL, Thamm DH. Pharmacokinetic modeling of doxorubicin pharmacokinetics in dogs deficient in ABCB1 drug transporters. J Vet Intern Med. 2010;24(3):579–86. [DOI] [PubMed] [Google Scholar]
- 15.Reddy LH, Murthy RS. Pharmacokinetics and biodistribution studies of Doxorubicin loaded poly(butyl cyanoacrylate) nanoparticles synthesized by two different techniques. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2004;148(2):161–6. [DOI] [PubMed] [Google Scholar]
- 16.Lu WL, Qi XR, Zhang Q, Li RY, Wang GL, Zhang RJ, et al. A pegylated liposomal platform: pharmacokinetics, pharmacodynamics, and toxicity in mice using doxorubicin as a model drug. J Pharmacol Sci. 2004;95(3):381–9. [DOI] [PubMed] [Google Scholar]
- 17.Rodionov N Graph digitizer version 1.9. 2000. http://www.geocities.com/graphdigitizer
- 18.Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–70. [DOI] [PubMed] [Google Scholar]
- 19.Kawai R, Mathew D, Tanaka C, Rowland M. Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther. 1998;287(2):457–68. [PubMed] [Google Scholar]
- 20.Maniez-Devos DM, Baurain R, Trouet A, Lesne M. Doxorubicin pharmacokinetics in the rabbit. J Pharmacol. 1985;16(2):159–69. [PubMed] [Google Scholar]
- 21.El-Kareh AW, Secomb TW. Two-mechanism peak concentration model for cellular pharmacodynamics of Doxorubicin. Neoplasia. 2005;7(7):705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendriks BS, Reynolds JG, Klinz SG, Geretti E, Lee H, Leonard SC, et al. Multiscale kinetic modeling of liposomal Doxorubicin delivery quantifies the role of tumor and drug-specific parameters in local delivery to tumors. CPT Pharmacometrics Syst Pharmacol. 2012;1:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becelli R, Renzi G, Morello R, Altieri F. Intracellular and extracellular tumor pH measurement in a series of patients with oral cancer. J Craniofac Surg. 2007;18(5):1051–4. [DOI] [PubMed] [Google Scholar]
- 24.Yang M, Chan HL, Lam W, Fong WF. Cytotoxicity and DNA binding characteristics of dextran-conjugated doxorubicins. Biochim Biophys Acta. 1998;1380(3):329–35. [DOI] [PubMed] [Google Scholar]
- 25.Mross K, Maessen P, van der Vijgh WJ, Gall H, Boven E, Pinedo HM. Pharmacokinetics and metabolism of epidoxorubicin and doxorubicin in humans. J Clin Oncol. 1988;6(3):517–26. [DOI] [PubMed] [Google Scholar]
- 26.Benjamin RS, Riggs CE Jr., Bachur NR. Pharmacokinetics and metabolism of adriamycin in man. Clin Pharmacol Ther. 1973;14(4):592–600. [DOI] [PubMed] [Google Scholar]
- 27.Arnold RD, Mager DE, Slack JE, Straubinger RM. Effect of repetitive administration of Doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin Cancer Res. 2005;11(24 Pt 1):8856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10(7):1093–5. [DOI] [PubMed] [Google Scholar]
- 29.Mordenti J Man versus beast: pharmacokinetic scaling in mammals. J Pharm Sci. 1986;75(11):1028–40. [DOI] [PubMed] [Google Scholar]
- 30.van Dalen EC, van der Pal HJ, Kremer LC. Different dosage schedules for reducing cardiotoxicity in people with cancer receiving anthracycline chemotherapy. Cochrane Database Syst Rev. 2016;3:CD005008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speth PA, van Hoesel QG, Haanen C. Clinical pharmacokinetics of doxorubicin. Clin Pharmacokinet. 1988;15(1):15–31. [DOI] [PubMed] [Google Scholar]
- 32.Gunvén P, Theve NO, Peterson C. Serum and tissue concentrations of doxorubicin after IV administration of doxorubicin or doxorubicin-DNA complex to patients with gastrointestinal cancer. Cancer Chemother Pharmcol. 1986;17(2):153–6. [DOI] [PubMed] [Google Scholar]
- 33.Rossi C, Gasparini G, Canobbio L, Galligioni E, Volpe R, Candiani E, et al. Doxorubicin distribution in human breast cancer. Cancer Treat Rep. 1987;71(12):1221–6. [PubMed] [Google Scholar]
- 34.Laginha KM, Verwoert S, Charrois GJ, Allen TM. Determination of doxorubicin levels in whole tumor and tumor nuclei in murine breast cancer tumors. Clin Cancer Res. 2005;11(19 Pt 1):6944–9. [DOI] [PubMed] [Google Scholar]
- 35.Shapira J, Gotfried M, Lishner M, Ravid M. Reduced cardiotoxicity of doxorubicin by a 6-hour infusion regimen. A prospective randomized evaluation. Cancer. 1990;65(4):870–3. [DOI] [PubMed] [Google Scholar]
- 36.Bi Y, Deng J, Murry DJ, An G. A Whole-Body Physiologically Based Pharmacokinetic Model of Gefitinib in Mice and Scale-Up to Humans. AAPS J. 2016;18(1):228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.An G, Morris ME. A physiologically based pharmacokinetic model of mitoxantrone in mice and scale-up to humans: a semi-mechanistic model incorporating DNA and protein binding. AAPS J. 2012;14(2):352–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudachek SF, Gustafson DL. Physiologically based pharmacokinetic model of lapatinib developed in mice and scaled to humans. J Pharmacokinet Pharmacodyn. 2013;40(2):157–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jamieson D, Boddy AV. Pharmacogenetics of genes across the doxorubicin pathway. Expert Opin Drug Metab Toxicol. 2011;7(10):1201–10. [DOI] [PubMed] [Google Scholar]
- 40.Lal S, Wong ZW, Sandanaraj E, Xiang X, Ang PC, Lee EJ, et al. Influence of ABCB1 and ABCG2 polymorphisms on doxorubicin disposition in Asian breast cancer patients. Cancer Sci. 2008;99(4): 816–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.