

Abstract
Developing selective enzyme inhibitors allows for the expansion of molecular toolboxes to investigate functions and activities of target enzymes. The histone acetyltransferase 1 (HAT1) is among the first histone acetyltransferase (HAT) enzymes that were discovered in the mid-1990s; however, it remains one of the poorly studied enzymes in comparison to the other HATs. Although HAT1 has been linked to various disease states, no inhibitors have been reported to target HAT1. Here we designed a set of peptide-CoA conjugates as bisubstrate inhibitors of HAT1 with submicromolar potency. In particular, the bisubstrate inhibitor H4K12CoA exhibited a low Ki value of 1.1 nM for HAT1. In addition, H4K12CoA was shown to be a competitive inhibitor with respect to both AcCoA and H4 peptide, suggesting a unique kinetic mechanism of HAT1 catalysis. Creating these submicromolar inhibitors offers a mechanistic tools to better understand how HAT1 recognizes substrates and cofactors, as well as provides chemical leads to further develop therapeutic agents to target this important enzyme for disease therapy.
Keywords: Histone acetyltransferase, HAT, HAT1, Bisubstrate inhibitor, Epigenetics, Chemical probes
Graphical Abstract
HAT1 is an important histone acetyltransferase in eukaryotic cells but its chemical probes are lacking. We designed bisubstrate conjugates as the first set of HAT1 inhibitors. In particular, the most potent inhibitor, H4K12CoA, exhibited a Ki of 1.1 nM for HAT1. Generating these nanomolar inhibitors provides mechanistic tools for investigating functions of HAT1.
Introduction
Developing and identifying potent and selective enzyme inhibitors allow scientists to expand molecular toolbox to investigate activity and function of target enzymes. The histone acetyltransferase1 (HAT1) belongs to the GCN5-related N-acetyltransferase (GNAT) superfamily together with other canonical members such as general control nonderepressible 5 (GCN5) and p300/CBP associated factor (PCAF). Members of this superfamily are conserved in organisms spanning from yeasts to humans and are known to share four distinct regions spanning across 100 residues (1). HAT1 was among the first set of HAT enzymes that were discovered in the mid-1990s (2); however, it is a poorly studied enzyme in comparison to the other HAT members (3, 4). This enzyme was originally categorized as a type B HAT based on its ability to acetylate newly synthesized histones H4 at Lys5 and Lys12, as well as H2A at Lys5 in the cytoplasm, but not histones that are already incorporated into a nucleosome (2, 5–7).
Cancer formation and progression can occur due to the dysregulation of histone modifying enzymes that regulate gene transcription, cell differentiation, proliferation and apoptosis (8–10). HAT1 has been linked to various pathological conditions, such as cardiovascular diseases (11), colorectal cancer (12), liver cancer (13), lung cancer (14), human immunodeficiency virus (HIV) (15), and the dysregulation of immune responses (14). Besides the aberrant expression, dys-localization of HAT1 can also aid in disease progression (3, 12). It has been reported in an HIV study that HAT1 can be a biomarker of the disease state (15). These disease studies shed light on the roles that HAT1 plays in pathology and suggest that HAT1 could be a potential molecular target in the design of new therapeutics treatment.
Reported inhibitors for HATs include peptides, small molecules, natural products, as well as synthetic molecules (16–18). Peptide-CoA bisubstrate inhibitors were first introduced to the HAT field by the Cole group with the p300-selective inhibitor, Lys-CoA (IC50 0.5 µM), and the PCAF-selective inhibitor, H3-CoA-20 (IC50 0.3 µM) (19). These inhibitors were designed to mimic the ternary complex formation of the substrate and cofactor binding to the HAT enzyme. We have previously designed several bisubstrate inhibitors targeting the MYST family of HATs (20). In the search for inhibitors targeting Esa1 and Tip60, we tested and compared the potency of various bisubstrate peptide inhibitors, small molecules, as well as natural products on Esa1, Tip60, p300, and PCAF (20, 21). Of the inhibitors tested H4K16CoA was found to be the most potent against both Esa1 and Tip60 with IC50 of 5.5 µM and 17.7 µM, respectively (20). Furthermore, analysis with Esa1 demonstrated that the mode of inhibition was competitive against acetyl coenzyme A (AcCoA) and noncompetitive against H4-20 substrate (20). Moreover, we rationally designed a set of multivalent peptide inhibitors containing methylated modifications in the peptide chain of the bisubstrate compound to inhibit Tip60, and found that the added methylated marks improved the potency of the bisubstrate inhibitors towards Tip60 by several folds (22).
These substrate-based inhibitors are valuable chemical probes for mechanistic and structural studies of HAT activities. Several bisubstrate inhibitors have been applied to understand the substrate recognition mechanisms in HATs (23, 24). It is known that HAT1 prefers to acetylate the N-terminal tail of histone H4 at Lys5 and Lys12 (2, 7). Thus, to develop inhibitors targeting HAT1 in this study, we rationally synthesized various histone H4-CoA conjugates containing the first 20 amino acids of histone H4 (H4-20) with the CoA moiety conjugated to different lysine residues. For the first time, inhibitors were identified that displayed potent inhibition activity towards HAT1. Two of the best inhibitors, H4K5CoA and H4K12CoA, had nanomolar inhibition potency towards HAT1 with Ki values at 22 nM and 1.1 nM, respectively. Steady-state kinetic analysis found that the most potent inhibitor, H4K12CoA, was competitive towards both AcCoA and H4-20 peptide.
Methods
Synthesis of bisubstrate inhibitors
Various peptides containing the first N-terminal 20aa of histone H4 (i.e., ac-SGRGKGGKGLGKGGAKRHRK, H4-20) were synthesized using the N-(9-fluorenyl) methoxycarbonyl (Fmoc)-based solid-phase peptide synthesis protocol. The Fmoc-Lysine(Boc) preloaded Wang resin was used at a 0.1 mmol scale. 0.4 mmol of each Fmoc-protected amino acid was weighed out and was individually coupled to the resin using 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (144 mg; 3.8 equiv) and N-Hydroxybenzotriazole (HOBt) (51 mg; 3.8 equiv). 20% piperidine in DMF was used to remove the Fmoc protecting group. To deprotect peptides containing the dimethyldioxocyclohexylidene group (Dde) on either Lys5, Lys8, Lys12, or Lys16, 8 mL of 65% hydrazine monohydrate was added and the peptide was shaken for 1 h. This process was repeated once more for another hour. Next, 324 mg (30 equiv) of bromoacetic acid was mixed with 366 uL (30 equiv) of N, N’-diisopropylcarbodiimide (DIC) in 4 mL of dimethylformamide (DMF) and incubated with peptide-resin for 4 h to create a bromoacetyl linker. The peptide was cleaved off the resin using 95% trifluoroacetic acid (TFA), 2.5% H2O, and 2.5% triisopropylsilane (TIS) for 4 h. The peptides were purified using a C-18 reverse phase column on a high-performance liquid chromatography (HPLC) instrument, and each sample was analyzed by matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS) on the Bruker Daltonics, model Autoflex with a 0.02% error. CoA was conjugated to the peptide by mixing the bromoacetyl-peptide with 2 equiv of CoA in sodium phosphate buffer (0.1 M, pH 8). The reaction mixture was then shaken overnight in the dark at room temperature. Purification and verification of the CoA-peptide compounds were done following the previous procedure. The concentrations of the CoA-peptide conjugates were determined by measuring the absorbance of the serial diluted compounds at 260 nm and using the molar extinction coefficient (ɛ260) of 16045 M−1cm-1. Lastly, concentrated NaOH solution was used to neutralize the pH of the compound solutions prior to enzymatic assays.
HAT1 expression and purification
The expression and purification of the catalytic domain of HAT1 (20-341) was performed following the method described by Wu et al. (Addgene plasmid # 25239) (25). Briefly, this protein was expressed in Escherichia coli and purified using the Ni-NTA resin. Transformation was done in E coli BL21-CodonPlus (DE3)-RIL competent cells using the heat-shock method, and then the cells were spread on agar plates containing the antibiotics kanamycin and chloramphenicol. Protein expression was induced by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the flask was shaken for 16 hours at 16°C. The cells were collected and suspended in the lysis buffer (50 mm Na-phosphate (pH 7.4), 250 mm NaCl, 5 mm imidazole, 5% glycerol, 2 mM β-mercaptoethanol, and 1 mm phenylmethanesulfonyl fluoride (PMSF)) then disrupted using the Microfluidics cell disruptor. The supernatant was passed through a column containing Ni-NTA resin equilibrated with column washing buffer (20 mM HEPES pH8, 250 mM NaCl, 5% glycerol, 30 mM imidazole, 1 mM PMSF) and the resin was washed with column washing buffer. Next, the resin was washed with the buffer containing a higher concentration of imidazole (20mM HEPES pH 8, 250 mM NaCl, 5% glycerol, 50 mM imidazole, 1 mM PMSF). Lastly, HAT 1 was eluted with elution buffer (20 mM HEPES pH 8, 250 mM NaCl, 5% glycerol, 500 mM imidazole, 1 mM PMSF). The eluted protein was dialyzed against the dialysis buffer (25 mM HEPES pH 8, 150 mM NaCl, 1 mM dithiothreitol (DTT) and 10% glycerol) for overnight. The HAT1 protein was concentrated using the GE Healthcare Vivaspin, and lastly was aliquoted and stored at −80⁰C. Protein concentrations were measured with the Bradford assay.
Determining , and IC50 values of the bisubstrate inhibitors against HAT1
The potency of all the inhibitors (Lys-CoA, H4K5CoA, H4K8CoA, H4K12CoA, H4K16CoA, and CoA) were determined using the scintillation proximity assay (SPA) (26). SPA experiments were conducted in a 96-well plate (Isolate-96; Perkin Elmer) at 30°C using a reaction buffer containing 50 mM HEPES (pH 8), and 0.1 mM EDTA. The cofactor used as an acetyl donor was [3H]-Ac-CoA (PerkinElmer) and the substrate was a biotinylated H4-20 peptide (Ac-SGRGKGGKGLGKGGAKRHRK(Biotin)-NH2 (abbreviated as H4-20 BTN)). The 30 µL reaction volume typically consisted of various concentration of bisubstrate inhibitor, 2.5 µM of substrate, followed by 1 µM [3H]-AcCoA. After 5 min of incubation, 0.04 µM of HAT1 was added and samples were re-incubated for 20 min. The reaction was quenched with 30 µL of guanidine HCl (0.5 M). Lastly, 10 µL of suspended 20 mg/mL streptavidin-coated SPA beads (Perkin Elmer) were added to each well and thoroughly mixed. The plate was placed in the MicroBeta2 scintillation counter (Perkin Elmer) in total darkness for one minute before the samples were quantified. Samples were performed in duplicate and were typically within 20% of each other. was determined for each inhibitor by fitting the activity versus the inhibitor concentration data to the following Morrison equation:
| (Equation 1) |
Where vi and v0 are the SPA activity signals of the enzyme in the presence and absence of inhibitor, respectively. [E], [I] and are enzyme concentration, inhibitor concentration, and apparent Ki, respectively (27).
Next, the following equation was used to calculate the dissociation rate constant, Ki, from the value:
| (Equation 2) |
Where [S] is the concentration of AcCoA used in the assay, and Km is the Michaelis-Menten constant of AcCoA (27).
Furthermore, the following equation was used to calculate IC50 value from the value:
| (Equation 3) |
Where [E]T is the total enzyme concentration used in the assay (27).
Determining HAT1 kinetics and mode of inhibition
HAT1 kinetics and the mode of inhibition for the bisubstrate inhibitor, H4K12CoA, were measured using the radiometric filter binding assay (28). The reaction time and enzyme concentration were controlled so that the reaction yield was less than 20%. H4K12CoA was added to the reaction at 0 nM, 20 nM and 100 nM. To determine the activity of HAT1 towards H4-20 peptide, various concentrations of H4-20 peptide (0-100 µM) was mixed with a reaction containing [14C]-AcCoA (3 µM) and reaction buffer (50 mM HEPES (pH 8.0), 0.1 mM EDTA, and deionized water). This mixture was incubated for 5min at 30°C. Next, HAT1 (0.02 µM) was added and the sample was re-incubated at 30°C for 9 min. The mixture was spread onto the P81 filter paper to quench the reaction. Filter papers were left to dry for 45 min before they were washed three times with 50 mM NaHCO3 buffer (pH 9). Lastly, the papers were re-dried, put into vials, and quantified with the addition of scintillation cocktail on the Beckman Coulter LS 6500 multi-purpose scintillation counter.
To determine the activity of HAT1 as a function of AcCoA concentration, various concentrations of [14C]-AcCoA (0-10 µM) was mixed with a reaction containing H4-20 peptide (100 µM) and reaction buffer (25 mM HEPES (pH 8.0), 0.1 mM EDTA, and deionized water). Next, HAT1 (5 nM) was added and the sample was re-incubated at 30°C for 9 min. The samples were quenched and the papers were prepared in the same way as mentioned above. All samples were performed in duplicate and were typically within 20% of each other. Activity—substrate concentration data points were fitted to equation 4 to determine Kis and Kii values.
| (Equation 4) |
Variables Vmax, Km, [S], [I], Kis, and Kii represent the maximum velocity, the Michaelis-Menten constant, substrate concentration, inhibitor concentration, the inhibition constant for the inhibitor binding to the free enzyme, and the inhibition constant for the inhibitor binding to the ES complex, respectively (29).
Results and Discussion
The type B histone acetyltransferase, HAT1, has been shown to acetylate newly synthesized histone H4 at Lys5 and Lys12 (2, 7). We rationalized that bisubstrate inhibitors with the CoA moiety attached at those respective lysine residues may possess strong inhibitory property towards HAT1. To test this hypothesis, we synthesized several 20-aa H4 peptides with a CoA motif conjugated at either Lys5, Lys8, Lys12, or Lys16. Lys-CoA and CoA were used as control compounds. The Fmoc-based solid phase peptide synthesis methodology was applied to obtain each CoA-peptide bisubstrate inhibitor as previously reported (20). The synthesis route is depicted in Scheme 1. Briefly, the amino acids were consecutively conjugated to the Fmoc-Lysine(Boc) preloaded Wang resin. Lysine containing a Dde group was used to conjugate with CoA at selected positions. A bromoacetyl linker was constructed on the targeted lysine residue which was then used to conjugate CoA to the peptide. All the compounds were purified using C-18 reversed phase HPLC. The identity and the purity of the final compounds were confirmed with MALDI-MS that showed the observed mass of each inhibitor matched their expected mass, (Table 1; Figure S1) and with analytical HPLC that showed single major peaks in the chromatograms (Figure S2).
Scheme1:

The route of bisubstrate inhibitor synthesis.
Table 1:
Inhibitor sequences and masses
| Inhibitor Name | Sequence | Expected Mass (Da) | Observed [M+H] Mass (Da) |
|---|---|---|---|
| (a) Lys-CoA | Ac-Lys(CoA)-NH2 | 994.8 | 995.1 |
| (b) H4K5CoA | Ac-SGRGK(CoA)GGKGLGKGGAKRHRK | 2840.9 | 2841.8 |
| (c) H4K8CoA | Ac-SGRGKGGK(CoA)GLGKGGAKRHRK | 2840.9 | 2841.8 |
| (d) H4K12CoA | Ac-SGRGKGGKGLGK(CoA)GGAKRHRK | 2840.9 | 2841.8 |
| (e) H4K16CoA | Ac-SGRGKGGKGLGKGGAK(CoA)RHRK | 2840.9 | 2841.6 |
Potency measurements of the bisubstrate inhibitors
Dose-dependent inhibition of HAT1 by each compound was determined using the scintillation proximity assay (SPA) with reactions containing 2.5 µM of biotinylated H4-20 peptide (H4-20 BTN), 1 µM of [3H]-AcCoA, and 40 nM of HAT1 incubated at 30°C for 20 min. Signals were read on the MicroBeta2 reader. Activity data points as a function of inhibitor concentration were fitted to the Morrison equation (Equation 1) (Figure S3) to obtain the apparent values. Given the competitive nature of the bisubstrate inhibitors with respect to AcCoA, equations 2 and 3 were used to calculate Ki and IC50 values from the values for each compound. Control compounds, CoA and Lys-CoA had IC50 values at 41.5 µM and 74.9 µM, respectively, while all the H4-CoA bisubstrate inhibitors tested displayed IC50 values below 10 µM. This difference in potency is a suggestion of the innate ability of HAT1 to form a tighter binding by recognizing various moieties of CoA and the amino acids residues of the H4 substrate. Placing the CoA moiety at Lys8 or Lys16 on the H4-20 peptide decreased the IC50 value to 2.6 µM (Ki 0.92 µM) and 7.2 µM (Ki 2.5 µM), respectively. These moderate IC50 values are likely because HAT1 does not demonstrate acetylation activity towards Lys8 or Lys16. Nanomolar potency of the bisubstrate inhibitor was observed when CoA was placed on Lys5 of the H4-20 peptide (IC50 83 nM) (Ki 22 nM). Impressively, the lowest IC50 value of 23 nM (Ki 1.1 nM) was gained when CoA was positioned at Lys12 (Table 2). Such low IC50 values are most likely owing to the property that HAT1 has its preferred acetylation sites at Lys5 and Lys12 on histone H4. In the previous study, Lys-CoA was a potent inhibitor that displayed nanomolar potency for p300 (Ki 19 nM), and H4K12CoA had submicromolar inhibition for p300 (Ki 0.79 µM) which is >700 fold higher than the Ki reported against HAT1 in this study (20, 30). This indicates that among the HATs, H4K12CoA is a highly specific inhibitor for HAT1.
Table 2: , IC50 and Ki of each compound tested with HAT1.
The SPA was used to measure the potency of each inhibitor against HAT1. Reactions containing 40 nM HAT1, 2.5 µM, H4-20 BTN, and 1 µM [3H]AcCoA were incubated at 30°C for 20 min. , IC50, and Ki values were determined using equations 1, 2 and 3, respectively.
| Inhibitor | (µM) | IC50 (µM) | Ki (µM) |
|---|---|---|---|
| H4K5CoA | 0.063 ± 0.006 | 0.083 | 0.022 |
| H4K8CoA | 2.6 ± 0.3 | 2.6 | 0.92 |
| H4K12CoA | 0.0030 ± 0.001 | 0.023 | 0.0011 |
| H4K16CoA | 7.2 ± 1.3 | 7.2 | 2.5 |
| CoA | 41.5 ± 2.6 | 41.5 | 14.6 |
| Lys-CoA | 74.8 ± 8.3 | 74.9 | 26.2 |
Inhibitory mode of the bisubstrate inhibitor
Further studies were carried out to determine the mode of inhibition of H4K12CoA given its high potency in HAT1 inhibition (Figure 1). The radiometric filter binding assay was used to measure the kinetics of HAT1 in the presence of varying H4-20 peptide and AcCoA concentrations, and the data obtained were plotted in the Michaelis-Menten format. Steady state reaction conditions to determine the kinetic parameters of AcCoA included 100 µM of H4-20 peptide, 5 nM of HAT1, and 0-10 µM of [14C]-AcCoA with a 9 min reaction time at 30°C. To determine the mode of inhibition with respect to AcCoA, H4K12CoA was added to the reaction mixture at 0 nM, 20 nM, and 100 nM. The velocity data as a function of substrate concentration were plotted in both Michaelis-Menten format (Figure 2A) and double reciprocal format (Figure 2C). To elucidate the mode of inhibition, the Michaelis-Menten data points were fitted to the mixed inhibition rate equation (Equation 4) which provided Kis of 0.075 ± 0.004 µM and Kii of 0.83 ± 0.47 µM. The fact that the Kii is about 11-fold greater than Kis supports that H4K12CoA is a primarily competitive inhibitor in respect to AcCoA (Table 3) (29). Furthermore, on the double reciprocal plot, three straight lines intersect on the y-axis and vary on the x-axis, further confirming that H4K12CoA behaves as a competitive inhibitor towards AcCoA (Figure 2C).
Figure 1: Potency of H4K12CoA towards HAT1.
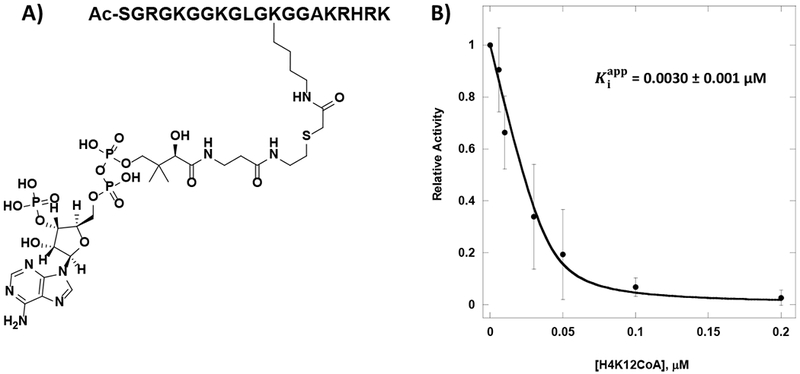
A) Structure of H4K12CoA. B) Dose-dependent inhibition of HAT1 by H4K12CoA.
Figure 2: Michaelis-Menten data points fitted to the mixed inhibition rate equation and Double reciprocal plots.
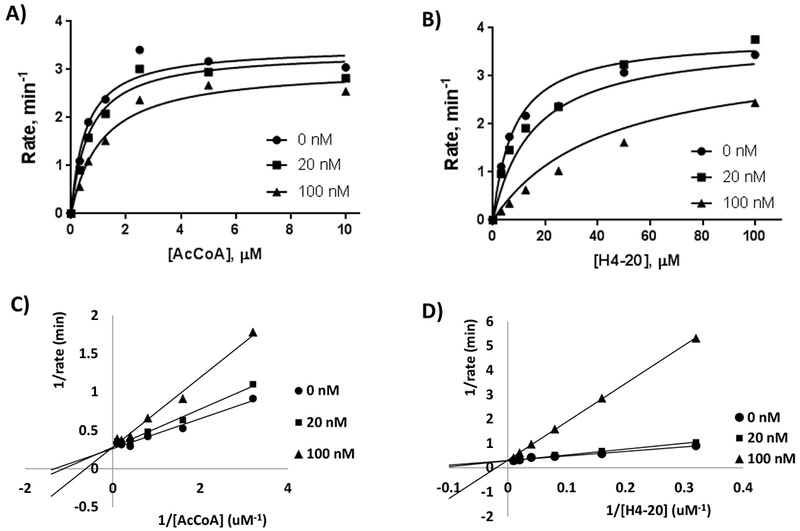
To determine the mode of inhibition in respect to AcCoA and H4-20 peptide, H4K12CoA was added to the reaction mixture at 0 nM, 20 nM, and 100 nM. A) Conditions to determine HAT1 kinetics in respect to AcCoA included 100 µM H4-20 peptide, 5 nM HAT1, and 0-10 µM AcCoA, with a reaction time of 9 min. B) Conditions to determine HAT1 kinetics in respect to H4-20 peptide included 3 µM AcCoA, 20 nM HAT1, and 0-100 µM AcCoA, with a reaction time of 9 min. Double reciprocal plots of H4K12CoA with respect to AcCoA (C) and H4-20 (D).
Table 3: Kinetic parameters of HAT1 with respect to AcCoA and H4-20 peptide when inhibited by H4K12CoA.
Kinetics in the presence of varying concentrations of H4K12CoA was obtained by fitting the Michaelis-Menten data points to the mixed inhibition rate equation.
| Varying [AcCoA] | Varying [H4-20] | |
|---|---|---|
| Vmax | 3.5 ± 0.2 min−1 | 3.8 ± 0.2 min−1 |
| Km | 0.54 ± 0.11 µM | 8.1 ± 1.9 µM |
| Kis | 0.075 ± 0.004 µM | 0.020 ± 0.002 µM |
| Kii | 0.83 ± 0.47 µM | 1.3 ± 0.1 µM |
We also determined the mode of inhibition of H4K12CoA with respect to H4-20 peptide. A similar steady state measurement from above was conducted with different concentrations of H4K12CoA (0 nM, 20 nM, and 100 nM) as well as with 3 µM [14C]-AcCoA, 20 nM HAT1, and 0-100 µM of H4-20 peptide. Michaelis-Menten data points were fitted to Equation 4 to verify the mode of inhibition. The values obtained for Kis and Kii were 0.020 ± 0.002 µM and 1.3 ± 0.1 µM, respectively. Since Kii is about 65-fold greater than Kis, this supports that H4K12CoA is a competitive inhibitor in respect to H4-20 ( Figure 2B; Table 3) (29). Further, the double reciprocal graph shows a single intersection point on the y-axis and a change in the Km value; thus further approving that this bisubstrate inhibitor is a competitive inhibitor towards H4-20 peptide (Figure 2D).
Our results demonstrating that the mode of inhibition for H4K12CoA towards HAT1 is competitive against both the cofactor AcCoA and substrate H4-20 is an interesting finding; because almost all of the previous bisubstrate inhibitors for acetyltransferases were shown to be competitive versus AcCoA but noncompetitive versus peptide substrate (20, 31). Such a kinetic inhibition pattern is pertinent to the ordered Bi-Bi sequential kinetic mechanism and is demonstrated by acetyltransferases: RimI (32), AANAT (33), and PCAF (31) in which AcCoA binds to the active site first followed by substrate binding. Herein, our data on the competitive inhibition pattern of the HAT1 bisubstrate inhibitor versus both AcCoA and H4 peptide substrate indicates that HAT1 does not have a preferred order of AcCoA and H4 peptide binding. Such an inhibition pattern, most possibly, supports that HAT1 catalysis follows a random sequential kinetic mechanism (32).
Although HAT1 is evolutionarily conserved from yeast to humans, the functions of human HAT1 in cells currently remains poorly defined (4, 34). The solved crystal structure of human HAT1 (PDB: 2P0W) does, however, provide hints on the catalytic mechanism and possibly how these bisubstrate inhibitors are recognized and bound (25). A ternary complex composed of HAT1-AcCoA-H4 peptide was depicted in the crystal structure. Residues such as Glu54, Asp62, and Glu64 of HAT1 are responsible for the specificity and anchoring of the substrate through the formation of hydrogen bonds. AcCoA binding requires various interactions with many different residues. Also, it is speculated that HAT1 catalysis requires a general catalytic base from residues Glu187 and Glu276 which aid Asp277 in deprotonating the ɛ-amino group of the lysine residue (25). These combined interactions bring the amino moiety of Lys12 of H4 substrate close to the acetyl group of AcCoA. The distance between the sulfur atom of CoA and the epsilon nitrogen of Lys12 of H4 in the HAT1 complex structure is 4.5 Å (Figure 3A). On the other hand, we estimated the sulfur-nitrogen distance in the H4K12CoA bisubstrate inhibitor to be 3.3 Å. This measurement is based on the reported p300-Lys-CoA complex structure (PDB: 3BIY) (Figure 3B). Overall, the two distances are very close which explains why the bisubstrate inhibitors can bind to HAT1 with nanomolar potency, and it also supports that H4K12CoA binding is located in the active site of HAT1. Although Lys-CoA can bind to HAT1, its potency is very low even compared to CoA (IC50 41.5 µM), indicating that the other amino acid residues of the H4 peptide significantly influence the potency to a great degree and are required for enzyme-substrate binding. In addition, the positioning of the CoA moiety at the proper lysine residue on the H4 peptide can also stimulate and further enhance potency. Further investigation of the interactions between HAT1 and these bisubstrate inhibitors can provide insight into the greater specificity that HAT1 has towards its substrates in comparison to other HATs such as p300 which has been reported to be more promiscuous (35).
Figure 3: Distance between AcCoA and the lysine residue and measurements of bisubstrate inhibitor.
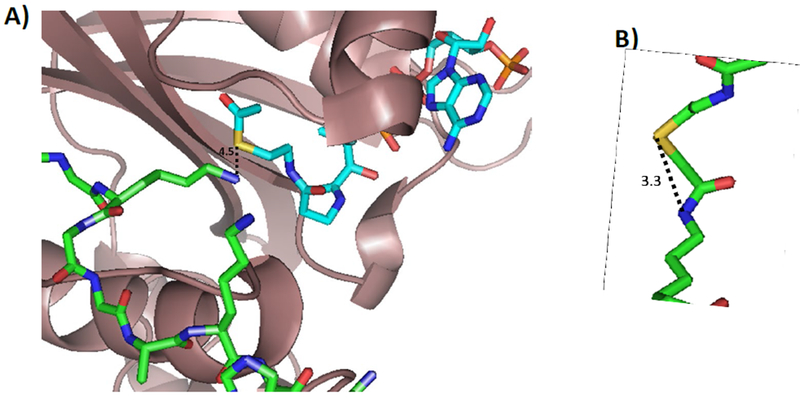
A) Distance measurements between AcCoA and lysine residue. Using the crystal structure of HAT1 bound to AcCoA and H4-20 peptide (PDB: 2P0W) measurements were done to determine the distance between the sulfur of AcCoA and the ɛ-amino group of the lysine residue. B) Distance between sulfur and amine of lysine residue of bisubstrate inhibitor, Lys-CoA. LysCoA was obtained from PDB: 3BIY.
Conclusion
To the best of our knowledge, we reported the first set of inhibitors that target HAT1 with submicromolar potency, thus offering a type of chemical tools to study HAT1 mechanism and function. As mentioned previously, HAT1 function is poorly studied, yet it has been linked to multiple diseases. To develop appropriate therapeutic agents to target HAT1, it is crucial to understand the preferential binding regions of the substrate and cofactor, as well as molecular cues to enhance the binding. These bisubstrate inhibitors, particularly H4K12CoA, can be utilized as chemical probes to give mechanistic insight into the catalytic mechanism and structural biology of HAT1. We do acknowledge that peptide-based bisubstrate inhibitors have been reported to not have ideal pharmacokinetic properties such as poor plasma membrane permeability issues (16, 36). Nevertheless, chemical modifications with membrane-penetrating motifs could generate membrane-permeable probes for both in vitro and in vivo applications. Further efforts could also be implemented to develop bisubstrate-based fluorescent probes for enzyme imaging and high throughput inhibitor screening. In all, this study lays a foundation for the discovery and development of new chemical probes targeting HAT1.
Supplementary Material
Figure S1: MALDI-MS spectra of bisubstrate inhibitors.
Figure S2: Analytical HPLC chromatograms of each bisubstrate inhibitor.
Figure S3: Fitting data to Morrison equation to obtain values of each compounds tested with HAT1.
Acknowledgments:
YGZ was supported by NSF grants CHE-1507741 and CHE-1808087 and NIH grant R01GM126154. LN was supported by a Graduate Research Fellowship from the NSF. We acknowledge the Proteomics and Mass Spectrometry facility at UGA for the MS support.
Footnotes
Conflict of Interest:
All authors declare no financial/commercial conflicts of interest.
References
- 1.Neuwald AF, Landsman D (1997) GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. TIBS;22: 154–5. [DOI] [PubMed] [Google Scholar]
- 2.Kleff S, Andrulis ED, Anderson CW, Sternglanz R (1995) Identification of a gene encoding a yeast histone H4 acetyltransferase. J Biol Chem;270: 24674–7. [DOI] [PubMed] [Google Scholar]
- 3.Parthun MR (2007) Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene;26: 5319–28. [DOI] [PubMed] [Google Scholar]
- 4.Parthun MR (2012) Histone acetyltransferase 1: More than just an enzyme? Biochim Biophys Acta;1819: 256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tafrova JI, Tafrov ST (2014) Human histone acetyltransferase 1 (Hat1) acetylates lysine 5 of histone H2A in vivo. Mol Cell Biochem;392: 259–72. [DOI] [PubMed] [Google Scholar]
- 6.Parthun MR, Widom J, Gottschling DE (1996) The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell;87: 85–94. [DOI] [PubMed] [Google Scholar]
- 7.Makowski AM, Dutnall RN, Annunziato AT (2001) Effects of acetylation of histone H4 at lysines 8 and 16 on activity of the Hat1 histone acetyltransferase. J Biol Chem;276: 43499–502. [DOI] [PubMed] [Google Scholar]
- 8.Gelato Kathy A, Fischle W (2008) Role of histone modifications in defining chromatin structure and function. Biol Chem;389: 353. [DOI] [PubMed] [Google Scholar]
- 9.Miglena K, Michael S, Marc D (2016) Role of histone acetylation in cell cycle regulation. Curr Top Med Chem;16: 732–44. [DOI] [PubMed] [Google Scholar]
- 10.Sun X-J, Man N, Tan Y, Nimer SD, Wang L (2015) The role of histone acetyltransferases in normal and malignant hematopoiesis. Front Oncol;5: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu D, Zhang M, Xie W, Lan G, Cheng HP, Gong D, et al. (2016) MiR-486 regulates cholesterol efflux by targeting HAT1. Biochem Biophys Res Commun;472: 418–24. [DOI] [PubMed] [Google Scholar]
- 12.Seiden-Long IM, Brown KR, Shih W, Wigle DA, Radulovich N, Jurisica I, et al. (2005) Transcriptional targets of hepatocyte growth factor signaling and Ki-ras oncogene activation in colorectal cancer. Oncogene;25: 91. [DOI] [PubMed] [Google Scholar]
- 13.Pogribny IP, Tryndyak VP, Muskhelishvili L, Rusyn I, Ross SA (2007) Methyl deficiency, alterations in global histone modifications, and carcinogenesis. J Nutr;137: 216S–22S. [DOI] [PubMed] [Google Scholar]
- 14.Jin X, Tian S, Li P (2017) Histone acetyltransferase 1 promotes cell proliferation and induces cisplatin resistance in hepatocellular carcinoma. Oncol Res;25: 939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Espíndola MS, Soares LS, Galvão-Lima LJ, Zambuzi FA, Cacemiro MC, Brauer VS, et al. (2018) Epigenetic alterations are associated with monocyte immune dysfunctions in HIV-1 infection. Sci Rep;8: 5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Thompson PR, Cebrat M, Wang L, Devlin MK, Alani RM, et al. (2004) Selective HAT inhibitors as mechanistic tools for protein acetylation. Methods Enzymol;376: 188–99. [DOI] [PubMed] [Google Scholar]
- 17.Zheng YG, Wu J, Chen Z, Goodman M (2008) Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy. Med Res Rev;28: 645–87. [DOI] [PubMed] [Google Scholar]
- 18.Luan Y, Ngo L, Han Z, Wang X, Qu M, Zheng YG (2015) Histone acetyltransferases: enzymes, assays, and inhibitors In: Zheng YG, editor Histone Acetyltransferases: Enzymes, Assays, and Inhibitors. San Diego: Academic Press: p. 291–317. [Google Scholar]
- 19.Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, et al. (2000) HATs off: Selective Synthetic Inhibitors of the Histone Acetyltransferases p300 and PCAF. Mol Cell;5: 589–95. [DOI] [PubMed] [Google Scholar]
- 20.Wu J, Xie N, Wu Z, Zhang Y, Zheng YG (2009) Bisubstrate inhibitors of the MYST HATs Esa1 and Tip60. Bioorg Med Chem;17: 1381–6. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Wang J, Li M, Yang Y, Wang B, Zheng YG (2011) Small molecule inhibitors of histone acetyltransferase Tip60. Bioorg Chem;39: 53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, Ngo L, Zheng YG (2014) Rational design of substrate‐based multivalent inhibitors of the histone acetyltransferase Tip60. ChemMedChem;9: 537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan H, Rossetto D, Mellert H, Dang W, Srinivasan M, Johnson J, et al. (2012) MYST protein acetyltransferase activity requires active site lysine autoacetylation. EMBO J;31: 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poux AN, Cebrat M, Kim CM, Cole PA, Marmorstein R (2002) Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc Natl Acad Sci U S A;99: 14065–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H, Moshkina N, Min J, Zeng H, Joshua J, Zhou M-M, et al. (2012) Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. Proc Natl Acad Sci U S A;109: 8925–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngo L, Wu J, Yang C, Zheng YG (2015) Effective quenchers are required to eliminate the interference of substrate: cofactor binding in the hat scintillation proximity assay. Assay Drug Dev Technol 13: 210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Copeland RA (2013) Tight Binding Inhibition. In Evaluation of Enzyme Inhibitors in Drug Discovery.. Hoboken, NJ: John Wiley & Sons, Inc. [Google Scholar]
- 28.He M, Han Z, Liu L, Zheng YG (2018) Chemical biology approaches for investigating the functions of lysine acetyltransferases. Angew Chem Int Ed Engl;57: 1162–84. [DOI] [PubMed] [Google Scholar]
- 29.Duggleby RG (1988) Determination of inhibition constants, I50 values and the type of inhibition for enzyme-catalyzed reactions. Biochem Med Metab Biol;40: 204–12. [DOI] [PubMed] [Google Scholar]
- 30.Thompson PR, Kurooka H, Nakatani Y, Cole PA (2001) Transcriptional coactivator protein p300: kinetic characterization of its histone acetyltransferase activity. J Biol Chem;276: 33721–9. [DOI] [PubMed] [Google Scholar]
- 31.Lau OD, Courtney AD, Vassilev A, Marzilli LA, Cotter RJ, Nakatani Y, et al. (2000) p300/CBP-associated factor histone acetyltransferase processing of a peptide substrate. Kinetic analysis of the catalytic mechanism. J Biol Chem;275: 21953–9. [DOI] [PubMed] [Google Scholar]
- 32.Yu M, Magalhaes ML, Cook PF, Blanchard JS (2006) Bisubstrate inhibition: theory and application to N-acetyltransferases. Biochemistry;45: 14788–94. [DOI] [PubMed] [Google Scholar]
- 33.Khalil EM, Cole PA (1998) A potent inhibitor of the melatonin rhythm enzyme. J Am Chem Soc;120: 6195–6. [Google Scholar]
- 34.Lu D (2013) Epigenetic modification enzymes: catalytic mechanisms and inhibitors. Acta Pharm Sin B;3: 141–9. [Google Scholar]
- 35.Dancy BM, Cole PA (2015) Protein lysine acetylation by p300/CBP. Chem Rev;115: 2419–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subbaramaiah K, Cole PA, Dannenberg AJ (2002) Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mechanisms. Cancer Res;62: 2522–30. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: MALDI-MS spectra of bisubstrate inhibitors.
Figure S2: Analytical HPLC chromatograms of each bisubstrate inhibitor.
Figure S3: Fitting data to Morrison equation to obtain values of each compounds tested with HAT1.