

Summary
We previously discovered that palmitic acid methyl ester (PAME) is a potent vasodilator first identified and released from the superior cervical ganglion and remain understudied. Thus, we investigated PAME’s role in modulating cerebral blood flow (CBF) and neuroprotection after 6 minutes of cardiac arrest (model of global cerebral ischemia). Our results suggest that PAME can enhance CBF under normal physiological conditions, while administration of PAME (0.02 mg/kg) immediately after cardiopulmonary resuscitation can also enhance CBF in vivo. Additionally, functional learning and spatial memory assessments (via T-maze) 3 days after asphyxial cardiac arrest (ACA) suggest that PAME-treated rats have improved learning and memory recovery versus ACA alone. Furthermore, improved neuronal survival in the CA1 region of the hippocampus were observed in PAME-treated, ACA-induced rats. Altogether, our findings suggest that PAME can enhance CBF, alleviate neuronal cell death, and promote functional outcomes in the presence of ACA
Keywords: Cardiac arrest, global ischemia, neuroprotection, hypoperfusion, palmitic acid methyl ester, cerebral blood flow
1. Introduction
Cardiopulmonary arrest (CA) remains one of the leading causes of death and disability in the U.S. affecting more than 350,000 patients/year[1]. Risk factors for CA includes smoking, coronary heart diseases, high blood pressure, high blood cholesterol, obesity, and diabetes[2], while coronary artery disease is thought to be the leading risk factor for CA[3]. The major therapeutic challenge of CA is the inherent whole-body ischemia that causes disruption of cerebral blood flow (CBF), known as hypoperfusion (indicative of depressed CBF). This prolonged hypoperfusion last from hours to days following CA, and is believed to be the major contributor to brain injury and neurological deficits [4, 5]. Thus, CA survivors suffer cognitive dysfunction varying from mild neurological deficits to a more severe vegetative state[6–8] indicating that development of novel therapies to alleviate hypoperfusion after CA can provide therapeutic benefits against CA[5].
Numerous studies have suggested that dietary consumption of unsaturated fatty acids (with one of more double bonds, i.e. docosahexaenoic acid) can increase blood high-density lipoprotein reducing the risk of coronary artery disease (i.e. atherosclerosis) and CA[9, 10]. This is contrary to the consumption of saturated fatty acids (fatty acids with predominantly single bonds between carbon molecules) increasing blood low-density lipoprotein (i.e. cholesterol) resulting in coronary heart disease and CA[11]. Thus, replacement of dietary saturated fats with unsaturated fatty acids reduces the risk of CA. Although saturated fatty acids are traditionally considered as “a detrimental” class of fatty acids causing CA, we previously identified palmitic acid methyl ester (PAME), a C16:0 saturated fatty acid, released from the superior cervical ganglion (SCG, the largest cervical ganglion innervating cerebral arteries and brain regions)[12–15], is a novel and potent vasodilator[12]. Direct application of PAME onto the rat/rabbit thoracic aorta caused potent vasodilation (EC50=0.19 nM) more potent than nitric oxide (NO) donors such as sodium nitroprusside (EC50=34.8 nM), and other vasoactive peptides such as vasoactive intestinal peptide (EC50=7 nM) [12, 16–18]. Since a vasodilatory response in cerebral arteries generally enhance CBF[5], PAME’s potent vasodilatory property may have a potential role in the regulation of vessel tonicity and brain circulation in the context of CA-induced hypoperfusion.
We sought to investigate the neuroprotective effects of PAME on asphyxial cardiac arrest (ACA, an animal model of CA)-induced hypoperfusion and subsequent brain injury. We found that: 1) PAME enhanced cortical CBF under normal and pathological conditions (i.e. ACA). 2) Post-treatment of PAME after oxygen-glucose deprivation or ACA inhibited neuronal cell death in the CA1 region of the hippocampus. 3) Learning/memory deficits can be observed 3 days after ACA, while post-treatment of PAME alleviated ACA-induced learning/memory deficits. Our findings suggest that PAME is a novel vasoactive substance that can enhance post-resuscitation CBF, all-the-while protecting neurons promoting functional learning/memory after ACA.
2. Materials and Methods
2.1. Chemicals
PAME (P5177, Sigma-Aldrich, St. Louis, MO) was dissolved in 100% ethanol (E7023, Sigma-Aldrich, St. Louis, MO) and diluted with prewarmed (37ᴼC) sterile saline (final ethanol concentration of 0.0026%) to required concentrations for injection. For laser Doppler flowmetry (LDF) experiments, PAME/Vehicle (0.0026% ethanol) were injected every 10 mins for 40 mins via the femoral vein (IV, 500 µl total injection volume) with a final dose of 0.9 µg/kg. We repeatedly injected PAME in LDF experiments to study the effects of PAME on CBF and systemic blood pressure during normal physiological conditions. For ACA, animals received a single bolus injection of PAME at 0.02 mg/kg intraperitoneally (IP) immediately after ACA/sham surgery. Furthermore, PAME’s vasodilatory effects caused a slightly decrease mean blood pressure (MABP) by ~10 mmHg. Based on our past experiences, this slight decrease in MABP after resuscitation as presented with IV injection of PAME prolonged the time to return to spontaneous circulation (ROSC) and may not aid in achieving ROSC readily. Importantly, achieving ROSC in a timely fashion can result in more favorable functional outcomes (i.e. enhanced CBF, less pathology, better neurological outcomes). Thus, IP injection was chosen in the present study.
2.2. Animal Preparation
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (Louisiana State University Health Sciences Center in Shreveport and West Virginia School of Osteopathic Medicine in Lewisburg). Adult male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 270–350 grams were housed for 1 week at the Louisiana State University Health Sciences Center Shreveport’s animal facilities and fasted overnight before surgery[5, 19]. Anesthesia was induced with 4% isoflurane and a 30:70 mixture of O2 and N2O respectively, followed by endotracheal intubation. Normal respiration was maintained with a ventilator (VentElite Small Animal Ventilator Harvard Apparatus, Holliston, MA). After endotracheal intubation, isoflurane was reduced from 4% to 1.5%. The right femoral artery and vein were cannulated using a single-lumen (PE-50) catheter for the monitoring of mean arterial blood pressure (MABP), arterial blood gas analyses, glucose assessment, and IV injection of drugs. The rats were immobilized with either rocuronium bromide (LDF experiments, 6.7 mg/kg, IV every 15–20 minutes) or vecuronium bromide (ACA experiments, 0.67 mg/kg, IV every 10 minutes) throughout the experiments[4, 20]. Head and body temperatures were continuously maintained at 36.5–37.3°C using heating pads and lamps.
2.3. LDF
The rat was placed on a stereotaxic frame after anesthesia. A longitudinal midline incision over the head was made exposing the scalp using microsurgical tools. A 2 mm2 thinned skull was drilled at the left frontoparietal cortex (1 mm lateral to the bregma) under constant irrigation with sterile saline to avoid overheating. To monitor CBF, the LDF fiber-optic probe (1 mm2) was placed on the thinned-skull window. Regional cerebral blood perfusion in a 1 mm3 tissue region was measured with a sampling frequency of 2 Hz via a PeriFlux 4001 Master laser Doppler blood perfusion system (Perimed, Jarfalla, Sweden) and analyzed with acquisition software (Perisoft for Windows, Jarfalla Sweden). PAME (0.9 µg/kg), or ethanol (0.0026%) were injected IV every 10 minutes for 40 minutes with continuously monitoring CBF and MABP (Fig. 1). The CBF was presented as peak blood flow values during the 10-minute drug injection. Baseline CBF was defined as CBF 40 minutes prior to drug injections.
Fig. 1. PAME enhanced cortical CBF without reducing MABP in vivo.
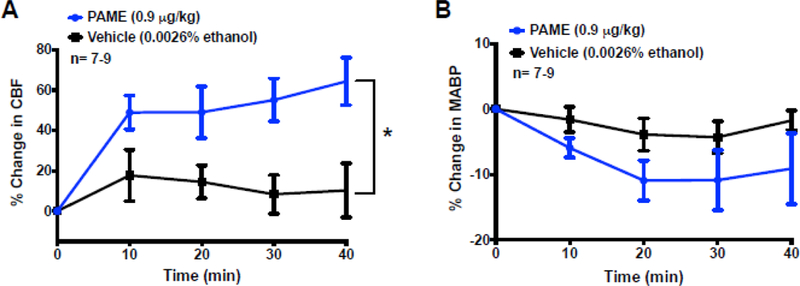
Time-response curves in panel (A) suggest that repeated IV injections of vehicle had minimal influence on cortical CBF, while multiple injections of PAME increased CBF under normal physiological conditions. Repeated injection of PAME, and vehicle had no effect on MABP. A single bolus injection of PAME (0.9 µg/kg), or vehicle (0.0026% ethanol) via femoral vein was applied every 10 mins for 40 mins. Cortical CBF was measured via LDF, while MABP was simultaneously measured via femoral artery throughout the 40-min-experiment. Results were expressed as mean ± SEM. *p≤0.05 indicates significantly different from vehicle, evaluated by linear mixed models: fixed effects. n indicates number of experiments.
2.4. ACA
After anesthesia, endotracheal intubation, and femoral artery/vein catheterization, vecuronium bromide (0.67 mg/kg, IV) was administered every 10 mins throughout the surgery to immobilize the rat. To induce ACA, asphyxia was initiated by disconnecting the ventilator from the endotracheal intubation tube. Cardiopulmonary resuscitation was initiated 6 mins after asphyxia by administering bolus IV injections of epinephrine (0.005 mg/kg) and sodium bicarbonate (1 meq/kg) followed by mechanical ventilation with 100% O2 at 80 breaths/min. Manual chest compressions (200/min) were performed until the MABP reached 60mmHg. PAME (0.02 mg/kg) was administered IP immediately after resuscitation. Control/sham animals received similar surgical procedures without the induction of asphyxia and ischemia.
2.5. Laser speckle contrast imaging (LSCI)
The rat was placed on a stereotaxic frame after anesthesia. A longitudinal midline incision over the head was made exposing the whole scalp using microsurgical tools. A 10 mm diameter circular cranial window was drilled at the frontoparietal cortex (1 mm lateral to the bregma) under constant irrigation with sterile saline to avoid overheating. Regional CBF 30 mins before (served as baseline CBF) and 24 hrs after ACA/sham was measured[21]. To measure CBF, the PeriCam PSI laser apparatus (Perimed, Jarfalla, Sweden) was placed 10 cm above the cranial window. Brain tissue was illuminated by a 785 nm laser for blood perfusion measurements. Regional perfusion was recorded for 5 min and calculated based on pixel intensity.
2.6. Oxygen Glucose Deprivation (OGD)
Organotypic hippocampal slices were obtained from postnatal day 9–12 and were maintained for 12–14 days in vitro prior to experimentation as previously described[22]. In brief, rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg, ~80 to 100 mg/kg) and xylazine (0.1 mg, ~10mg/kg) and the brains were rapidly removed. Transverse slices (400 μm) were dissected from the hippocampi and placed in Grey’s balanced salt solution that was supplemented with 6.5 mg/ml glucose at 4°C. Slices from two rat pup hippocampi will be placed onto one membrane insert that is 30 mm diameter (Millicell-CM, Millipore). Individual inserts will be placed into a six-well culture dish containing 1 mL per well of culture medium (50% MEM, 25% HBSS, 25% heat-inactivated horse serum supplemented with 6.5 mg/mL glucose, and 1 Mm glutamine). The slice cultures will be maintained at 37ºC, in 5% CO2. To simulate ischemia, cultures were exposed to 45 min of oxygen and glucose deprivation (OGD) as previously described[22]. Organotypic slices were washed three times with glycemic Hank’s Balanced Salt Solution (in mM: 1.26 CaCl2•2H2O; 5.37 KCl; 0.44 KH2PO4; 0.49 MgCl2; 0.41 MgSO4•7H2O; 136.9 NaCl; 4.17 NaHCO3; 0.34 Na2HPO4•7H2O; 15 sucrose; pH 7.4) and exposed to an oxygen free environment (90% nitrogen, 5% hydrogen, and 5% CO2, 37°C) using a Biospherix C-Chamber (Parish, NY, USA). Slices were treated after OGD with vehicle or PAME (100 nM) for 1 hour. Organotypic hippocampal cell death was determined by propidium iodide (PI) staining, where slices were incubated in culture medium supplemented with 2 μg/mL of PI for 1 hour prior to imaging. Micrographs of the organotypic slice PI staining were captured: (1) baseline prior to treatment, (2) 24 hours after lethal ischemia or sham procedures to assess neuronal damage, and (3) 24 hours after a 1-hour treatment with N-methyl-D-aspartate (NMDA; 500 μM) to determine maximum neuronal cell death. Fluorescence images were obtained using a 1.4-megapixel Peltier cooled fluorescent CCD camera and were digitized using IS Capture software (AM Scope, Irvine, CA, USA). Percentage of relative optical intensity was used as an index of cell death using Image-J (NIH).
2.7. Immunohistochemistry and Histopathology
For detail methods on histopathology of hippocampal slides, pleases see Lin.HW et al., 2015[20]. Animals were sacrificed 7 days after ACA because ACA-induced neuronal cell death occurred 3–7 days after the onset of ischemia[4]. Animals were perfused with physiological saline (0.9% for 2 min), following a mixture of 36% formaldehyde, glacial acetic acid and methanol (1:1:8, FAM) for 15 min at a constant pressure of 110–120 mmHg. The head was removed and immersed in FAM overnight at 4°C immediately after perfusion. Brain tissue was then removed from the skull, processed in a tissue processor (Leica ASP300S, Wetzlar, Germany), and embedded in paraffin (Leica EG1150, Wetzlar, Germany). Coronal sections (6 μm thickness) were cut using a microtome blade (VWR, Radnor, PA) on a rotary microtome (Leica RM2245, Wetzlar, Germany). Brain slices were collected at 100 μm intervals and placed on a glass slide (M1000W StatLab, McKinney, TX), paraffin was removed by incubating overnight in an oven at 60 ᴼC. Slides were stained with hematoxylin and eosin (H&E) (ThermoFisher, Waltham, MA). Neuronal cell death was assessed via morphological changes, including severe cellular shrinkage, cytoplasmic eosinophilia, pyknotic triangular-shaped nucleus with dark basophilic staining, and eosinophilic staining nucleolus[23]. In a separate set of experiments, brain slices were stained with 0.0004% Fluoro-Jade C (FJC) (AG325, Millipore, Burlington, MA) and co-stained with DAPI (TR-100-FJT, Biosensis, Thebarton, Australia) (to determine degenerative neurons in the CA1 region of the hippocampus[24]. All investigators were blinded to the study. The entirety of the CA1 region was counted along the medial to the lateral extent of the hippocampus. CA1 normal neurons and FJC-postive neurons were manually counted from 4 different slides at 40X magnification (Leica aperio versa, Wetzlar, Germany) using ImageScope software (Leica, Wetzlar, Germany). Rats were excluded from immunohistochemistry study if their hippocampus contained no dead neurons (due to animal variability) or FJC-positive neurons indicating that ACA failed to cause sufficient brain injury. According to the above-mentioned criteria: 1 out of 7 ACA-treated animals was excluded from H&E staining study, while 3 out of 7 ACA-treated and 3 out of 8 ACA+PAME-treated animals were excluded from FJC staining study.
2.8. Behavioral Trials (T-Maze)
A modified spontaneous alternation protocol[25, 26] with exclusion criteria[4] was used to investigate the neuroprotective effects of PAME on ACA-induced learning/memory deficits. The T-maze apparatus was constructed from a 3-dimensional printer 5th generation Replicator (Makerbot, New York, NY) using black polylactic acid filament. The outside dimensions of the T-maze are as follows (cm): Goal arm (X2): 50 X 10, Start box/arm: 50 X 16, Central partition: extend 10 into start box, Wall height: 30[25]. These dimensions are modified based on the Deacon et al., 2006 protocol paper[25]. Rats were handled for 5 mins in a dimly light room one day prior to ACA or sham surgery to acclimate to human touch. 3 days after ACA/sham surgery, rats were transfer into a dimly illuminated room for 10 min to allow for acclimatization. The rat was then placed at the start area in the beginning of the trial to explore either right or left goal arm (first run). The investigator gently blocks the opposite door immediately after the rat selected one of the goal arms (this criterion is considered when all four paws of the rat have entered the goal arm). The rat was allowed to stay in the goal arm for 30 sec. The investigator then gently placed the rat back at starting area again. Allow the rat to choose between left and right goal arms once again (second run, end of first trial). Rats performed 2 trials/day at 20 mins intervals for 3 days. Each trial contains 2 separate runs with a time limit of 2 mins/run. A thin layer of cage bedding (1 cm thick) placed on the maze floor was changed between trials to remove scent bias. The T-maze was wiped clean thoroughly with 70% ethanol and distilled water at the end of experiments. Spontaneous alternation was calculated as the number of alternations (defined as the rat chose the novel arm on the second run) divided by the total number of trials (6 trials with 12 runs). Side bias ratio was calculated as the total number of choices for the overall preferred side (right or left) divided by the total number of runs (12 total).
2.9. Statistical Analysis
Results were expressed as means ± S.E.M. Statistical analysis were evaluated by linear mixed models: fixed effects (for LDF and blood pressure studies) and one-way ANOVA (Tukey’s post-hoc test) as appropriate with SPSS (Chicago, IL). p≤0.05 level of probability is accepted as significance.
3. Results
Physiological parameters
For physiological parameters please refer to Tables 1–5. Higher PCO2 after resuscitation was due to inadequate ventilation during ACA, while lower pH after resuscitation was associated with slight acidosis after ischemia. Elevated PO2 and/or MAP after resuscitation were due to increased ventilation rate (60 to 80 breaths/min), delivery of 100% O2, and administration of epinephrine (0.005 mg/kg) during resuscitation.
Table 1.
Physiological parameters from LDF
| Group | Variable | |
|---|---|---|
|
Vehicle
(n=7) |
Body weight (g) | 359 ± 18.0 |
| pH | 7.41 ± 0.06 | |
| pCO2 (mm Hg) | 35 ± 3.5 | |
| pO2 (mm Hg) | 108 ± 15.6 | |
| MABP (mm Hg) | 104 ± 16.2 | |
| Glucose (mg/dL) | 146 ± 58.1 | |
|
PAME
(n=9) |
Body weight (g) | 303 ± 4.96 |
| pH | 7.42 ± 0.07 | |
| pCO2 (mm Hg) | 34 ± 5.0 | |
| pO2 (mm Hg) | 107 ± 12.0 | |
| MABP (mm Hg) | 101 ± 13.5 | |
| Glucose (mg/dL) | 156 ± 32.4 |
n, number of rats; pO2 and pCO2, partial pressure of oxygen and carbon dioxide, respectively; MABP, mean arterial blood pressure; N/A, not applicable.
Table 5.
Physiological parameters from T-maze
| Group | Variable | Before ACA | After ACA |
|---|---|---|---|
|
ACA
(n=7) |
Body weight (g) | 309 ± 13.1 | n/a |
| pH | 7.46 ± 0.02 | 7.41 ± 0.01* | |
| pCO2 (mm Hg) | 35 ± 0.9 | 49 ± 4.3* | |
| pO2 (mm Hg) | 118 ± 9.60 | 209 ± 30.9* | |
| MABP (mm Hg) | 97 ± 2.2 | 111 ± 2.60* | |
| Glucose (mg/dL) | 105 ± 13.9 | ||
|
ACA+PAME
(n=5) |
Body weight (g) | 267 ± 10.8 | n/a |
| pH | 7.42 ± 0.01 | 7.41 ± 0.02 | |
| pCO2 (mm Hg) | 33 ± 1.0 | 33 ± 0.9 | |
| pO2 (mm Hg) | 120 ± 5.90 | 252 ± 40.9* | |
| MABP (mm Hg) | 103 ± 3.71 | 104 ± 2.15 | |
| Glucose (mg/dL) | 103 ± 5.24 |
p ≤ 0.05, significant difference from before ACA; n, number of rats; pO2 and pCO2, partial pressure of oxygen and carbon dioxide, respectively; MABP, mean arterial blood pressure; N/A, not applicable.
Exogenous administration of PAME in vivo enhanced cortical CBF without affecting MABP under normal physiological conditions.
We first measured CBF via LDF to define the physiological importance of PAME on cerebral circulation during normal physiological conditions. Results from LDF indicates that CBF from rats-treated with Vehicle (0.0026% ethanol) was unchanged during the 40-min recording as compared to baseline CBF, while repeatedly IV bolus injection of PAME (0.9 µg/kg) in the rats significantly enhanced cortical CBF, as compared to rats-treated with Vehicle (0.0026% ethanol) (Fig. 1A). Furthermore, repeatedly IV bolus injection of Vehicle (0.0026% ethanol) or PAME (0.9 µg/kg) had no influence on MABP (Fig. 1B). These results suggest that PAME as a vasodilator can enhance CBF in vivo under normal physiological conditions.
PAME can enhance CBF after ACA
We further studied the impact of PAME on CBF regulation in pathological condition such as ACA. CBF 24 hours after ACA was investigated via LSCI. Rats received a bolus injection (IP) with PAME/Vehicle immediately after ACA. Post-treatment of PAME immediately after ACA (denoted as: ACA+PAME) significantly enhanced cortical CBF (21.07%±1.02%) 24 hours after ACA suggesting that PAME can alleviate ACA-induced hypoperfusion (Fig.2).
Fig. 2. PAME can enhance CBF after ACA.
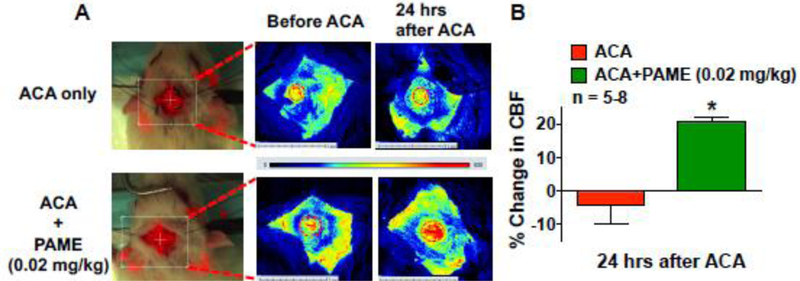
(A) Regional CBF measurement of in vivo cortical microvessels via LSCI in the anesthetized rat. Rats were post-treated with PAME (0.02 mg/kg), or vehicle (IP bolus immediately after ACA). (B) Summary of CBF changes before ACA and 24 hours after ACA. Each animal served as its’ own control. Cortical CBF was expressed as percent change in flow from baseline (CBF 30 mins before ACA). Results were expressed as mean ± SEM. *p≤0.05 indicates significantly different from vehicle, evaluated by ANOVA with Tukey’s post hoc test. n indicates number of experiments. The dashed oval indicates the region of interest where cortical blood flow was measured via the cranial window.
Post-treatment of PAME enhanced neuronal survival in the CA1 region of the hippocampus after ACA
CA-induced hypoperfusion is one of the major contributors to neuronal cell death and neurological deficits [5, 20, 27, 28]. Thus, augmentation of CBF after ACA can enhance neuronal survival [4, 5, 20, 29, 30]. Since PAME can enhanced CBF after ACA (Fig. 2), we investigated if PAME could reduce ACA-induced neuronal cell death via histopathological analysis. Since ACA causes delayed neuronal cell death in the vulnerable CA1 region of the hippocampus 7 days after the onset of ischemia[5, 27, 28, 31], hippocampal brain slices were processed and stained with FJC and H&E 7 days after ACA/sham surgery for detection of neurodegenerative and dead neurons, respectively. Our results from immunohistochemistry suggest that ACA resulted in an increase in FJC-positive (neurodegenerative) neurons (564.10±75.36) (Fig. 3), while ACA+PAME had alleviated neurogenerative neurons (170.70±10.24) in the CA1 region of the hippocampus (Fig. 3). Additionally, our histopathological analysis indicated the number of normal neurons in the CA1 region of the hippocampus in ACA animals (492.30±95.65) was significantly lower as compared to control (909.60±20.16) (Fig. 4) and ACA+PAME-treated animals.
Fig. 3. Post-treatment of PAME reduced neuronal degeneration in the CA1 region of the hippocampus after ACA.
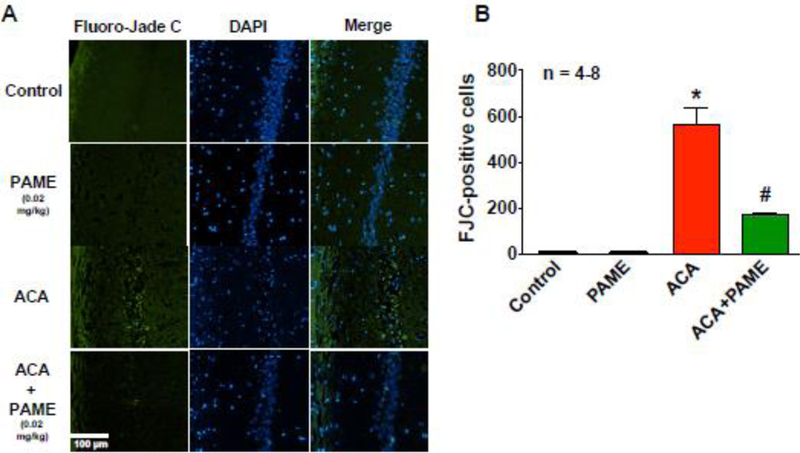
(A) Representative images of FJC staining in the CA1 region of the hippocampus. Rats received an IP bolus injection of PAME (0.02 mg/kg) or vehicle (0.0026% ethanol) immediately after ACA. Rats that were subjected to ACA surgery were sacrificed 7 days after ACA for brain histopathology of the hippocampus. (B) The number of FJC positive neurons from the CA1 region of the hippocampus were counted and expressed in the bar graph. n number of animals used per group. Short horizontal solid bars represent 100 µm in length in the field of view of each representative image. *p≤0.05 indicates significantly different from control and PAME only groups. #p≤0.05 indicates significantly different from ACA only group, evaluated by one-way ANOVA with Tukey’s post-hoc test.
Fig. 4. Post-treatment of PAME enhanced neuronal survival in the CA1 region of the hippocampus after ACA.
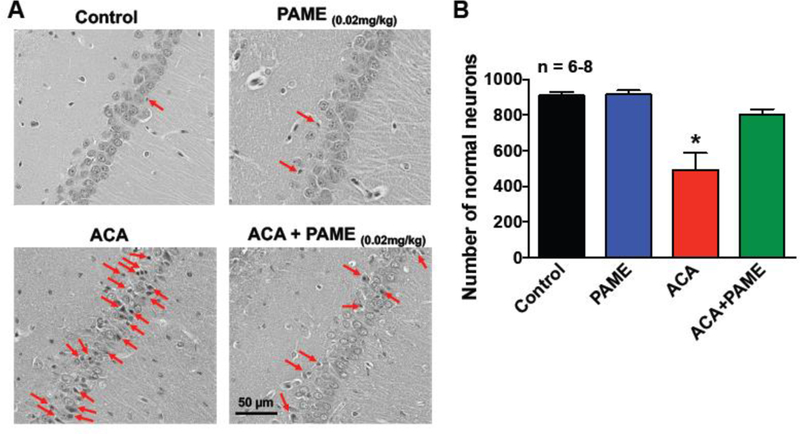
(A) Representative images of H&E in the CA1 region of the hippocampus. Rats received an IP bolus injection of PAME (0.02 mg/Kg) or vehicle (0.0026% ethanol) immediately after ACA. Animals were sacrificed 7 days after ACA for brain histopathology of the hippocampus. Control (no ischemia, no drug) and PAME (vehicle control) groups were performed as internal controls. A lightly stained nucleus with a dark-stained nucleolus and a red-stained cytoplasm can be observed in healthy neurons, while ischemic neurons exhibit shrunken cytoplasm and pyknotic nuclei. Arrows denote typical neuronal cell death in the CA1 region of the hippocampus. Short horizontal solid bars represent 50 µm in length in the field of view of each representative image. (B) The number of healthy neurons from the CA1 region of the hippocampus were counted and expressed in the bar graph. n number of animals used per group. *p≤0.05 indicates significantly different from all groups, evaluated by one-way ANOVA with Tukey’s post-hoc test.
PAME reduced neuronal cell death following in vitro OGD
Fatty acids have been reported to have anti-inflammatory effects against oxidative stress [32]. It is unclear if PAME has additional neuroprotective effects other than CBF modulation. We further utilized organotypic hippocampal slices coupled with OGD (an in vitro model of cerebral ischemia without CBF involvement) to investigate if PAME has any neuroprotective properties independent of CBF modulation. Hippocampal slices were treated with PAME or vehicle after OGD for 1 hr. Extensive neuronal cell death can be observed in the hippocampus of vehicle-treated slices 24 hr after OGD (53.97%±2.76%), while post-treatment with PAME reduced neuronal cell death (23.20%±5.56%) (Fig. 5) suggesting dual effects (independent of CBF modulation) of PAME in ischemic brain injury.
Fig. 5. Post-treatment of PAME (100 nM) provided neuroprotection against 45 mins of OGD-induced injury in the rat hippocampal slice.
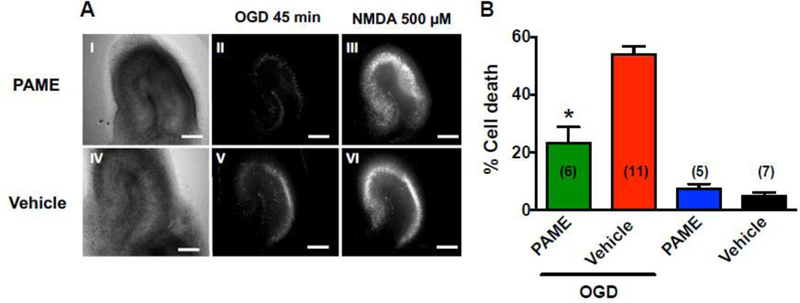
OGD-induced neuronal cell death was determined by PI fluorescence. (A) Representative images of slices: Bright-field images following OGD in PAME- (I) and vehicle-treated slices (IV); PI fluorescence images 24 hours following OGD in PAME- (II) and vehicle-treated (V) slices. Effects of PAME, and vehicle on OGD-induced neuronal cell death were normalized with NMDA (500 μM)-induced maximum neuronal cell death (III and VI) and summarized in panel B. Post-treatment of PAME inhibited OGD-induced neuronal cell death in the CA1 region of the hippocampus. The data were expressed as percent cell death. *p<0.05 indicates significantly different from OGD + vehicle, evaluated by one-way ANOVA with Tukey’s post-hoc test. Scale bars = 500 µm.
Post-treatment of PAME after ACA alleviated short-term memory deficits
Neurons in the CA1 region of the hippocampus play an essential role in learning/memory formation. Post-treatment of PAME (ACA+PAME) reduced CA1 neuronal cell death (Figs. 3 and 4) indicating that PAME can enhance functional learning/memory after ACA. We thus utilized T-maze spontaneous alternation to study the effects of PAME on learning/memory formation after ACA. Results from the T-maze study suggest that ACA-treated rats had lower spontaneous alternation ratio (26.29%±4.87) and a higher side-preference ratio (79.71%±5.81%) as compared to control (63.30%±6.47% and 55.00%±1.36%, respectively), and PAME only-treated rats 56.50%±6.17% and 64.93%±2.43%, respectively) (Fig. 6). Interestingly, posttreatment of PAME reduced side-preference ratio (65.78%±2.33%), while enhanced spontaneous alternation ratio (51.78%±4.05%) after ACA (Fig. 6) suggesting that the PAME can enhance CBF, neuronal survival, and functional learning/memory after ACA.
Fig 6. Post-treatment of PAME (0.02 mg/Kg, IP) immediately after ACA enhanced functional learning/memory.
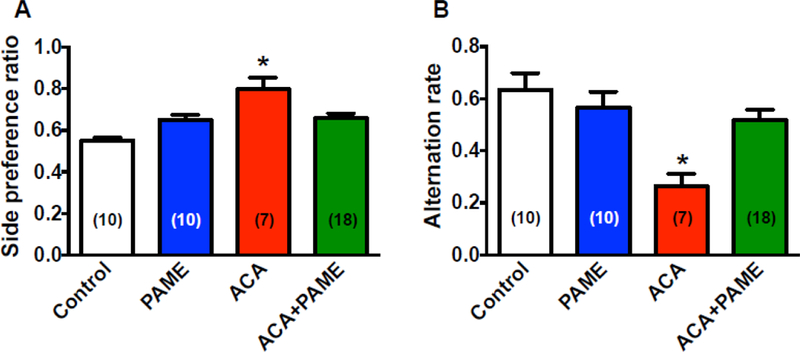
We utilized the modified spontaneous alternation protocol to detect possible hippocampal lesions associated with ACA [26, 59]. (A) A significant increase in side-preference ratio was observed in rats treated with ACA, which suggests that rats with hippocampal injury developed a side-preference[26]. Post-treatment of PAME (ACA+PAME, 0.02 mg/kg, IP) alleviated ACA-induced side-preference ratio suggesting that PAME can reduce hippocampal injury after ACA. Spontaneous alternation ratio was decreased in ACA-treated rats as compared to normal control and PAME only-treated rats, while post-treatment of PAME (ACA+PAME) at 0.02 mg/kg enhanced spontaneous alternation rate after ACA indicating that PAME can alleviate ACA-induced short-term memory deficits. Results were expressed as mean ± SEM. *p≤0.05 indicates significantly different from all groups, evaluated by one-way ANOVA with Tukey’s post-hoc test. n indicates number of experiments.
4. Discussion and Conclusions
One of the major challenges of post-resuscitation care are derangements of CBF following CA, which leads to brain injury and neurological deficits. CA-induced CBF derangements are initiated by hyperemia (increase in CBF) followed by prolonged hypoperfusion. Although hyperemia is thought to be involved in neuronal cell injury[33, 34], CA-induced hypoperfusion lasting hours to days after ischemia plays a vital role in ischemia-mediated neuronal cell death and neurological deficits (learning/memory)[34–37]. Thus, development of novel therapies that can enhance CBF while counteracting hypoperfusion following CA is greatly needed.
Cerebral blood vessels are tightly regulated by many vasoactive substances, where there is a delicate balance among vasoconstriction and dilation factors[13, 38–41]. Of extreme importance are the cerebral arteries densely innervated by perivascular nerves from the sympathetic and parasympathetic out-flows of the autonomic nervous system[14, 38–42]. These nerves play a central role in vascular tone regulation by releasing neurotransmitters either presynaptically on the same or neighboring nerves or postsynaptically on vascular smooth muscle[13].
From the sympathetic chain, the SCG provides sympathetic out-flows to the brain innervating cerebral arteries. We previously discovered a novel vasodilator, PAME, released from the SCG. Since revival of CBF after CA has been shown to enhance neuronal survival in the brain[5, 43], PAME’s vasodilatory properties offer potential therapeutic opportunities in the treatment against CA-induced hypoperfusion and subsequent brain injury and neurological deficits. In our previous publication, we have reported that pre-treatment of PAME can enhance hippocampal CA1 neuronal survival after ACA. Additionally, we also reported that posttreatment of PAME 2 hours after middle cerebral artery occlusion enhances hippocampal CA1 neuronal survival [20]. Since assessment of neurological outcomes is the primary endpoint in pre-clinical trials of anti-stroke/ischemia therapy, in the present study we focused on the more valuable “post-treatment” paradigms of PAME in a clinically relevant global model of cerebral ischemia. Our findings suggest that post-treatment of PAME alleviates ACA-induced hypoperfusion, neuronal cell death, and in particular neurological deficits (i.e. learning/memory deficits).
We first used LDF to evaluate the role of PAME in regulating cortical microcirculation during normal physiological conditions. We used multiple injections of PAME (0.9 µg/Kg IV, every 10 mins) as presented in Fig. 1 to determine if PAME had an additive effect on CBF. Although results from LDF indicate that PAME enhanced cortical CBF under normal physiological conditions (Fig. 1), multiple injections of PAME had no additive effect on CBF. We further utilized in vivo imaging of regional CBF via LSCI to determine if PAME is a novel CBF modulator against hypoperfusion after ACA. The dose (0.02 mg/Kg IP) of PAME used in the present study was derived from our previous publication that PAME caused maximum aortic vasodilation at 1 μM ex vivo[12], which is equivalent to ~ 0.02 mg/kg in vivo in a 300 g rat (approximately 20 mL of blood by volume). Results from LSCI suggest that PAME enhanced cortical CBF under pathological conditions such as ACA (Fig. 2), which suggests that PAME as a novel CBF modulator can alleviate ACA-induced hypoperfusion.
One of the major disadvantages of the use of broad vasodilators in the treatment against CA is their ability to dilate vessels in multiple vascular beds, which results in hypotension during/after resuscitation. It is interesting to note that PAME had minimal influence on systemic blood pressure under both normal and pathological conditions such as ACA (Figs. 1 and 2, Tables 1 and 2), which suggest PAME’s specific/unique mechanism of action on cerebral vasculatures. Mechanisms underlying PAME-induced vasodilation remains to be elucidated. However, it has been shown that voltage-gated potassium (Kv) channel blockers (i.e. 4-aminopyridine and tetraethylammonium) concentration-dependently inhibited aortic vasodilation elicited by PAME[44], which suggests that Kv channels on the smooth muscle cells play an essential role in PAME-induced vasodilation. Since more than 40 Kv channel subunits have been identified throughout the body, studies have shown that the diversity of Kv channel expression can cause differential responses of Kv channels to different drugs/stimulations[45–47]. This may explain PAME’s specific action on cerebral vasculatures, but not peripheral arteries as we observed in the present study.
Table 2.
Physiological parameters from LSCI
| Group | Variable | Before ACA | After ACA |
|---|---|---|---|
|
ACA
(n=8) |
Body weight (g) | 319 ± 9.00 | n/a |
| pH | 7.42 ± 0.02 | 7.45 ± 0.03 | |
| pCO2 (mm Hg) | 40 ± 0.9 | 49 ± 2.6* | |
| pO2 (mm Hg) | 116 ± 8.64 | 300 ± 45.6* | |
| MABP (mm Hg) | 95 ± 2.9 | 113 ± 6.18* | |
| Glucose (mg/dL) | 112 ± 12.4 | ||
|
ACA+PAME
(n=5) |
Body weight (g) | 300 ± 4.66 | n/a |
| pH | 7.45 ± 0.02 | 7.47 ± 0.02 | |
| pCO2 (mm Hg) | 40 ± 1.9 | 51 ± 2.7* | |
| pO2 (mm Hg) | 123 ± 9.38 | 232 ± 29.2* | |
| MABP (mm Hg) | 102 ± 4.19 | 120 ± 3.70* | |
| Glucose (mg/dL) | 119 ± 13.0 |
p ≤ 0.05, significant difference from before ACA; n, number of rats; pO2 and pCO2, partial pressure of oxygen and carbon dioxide, respectively; MABP, mean arterial blood pressure; N/A, not applicable.
Since pial arteries give rise to smaller penetrating arterioles that, in turn, transport oxygen and nutrient supplies down to the hippocampus[5, 48–50], CA-induced cortical hypoperfusion mainly causes neuronal cell death in the vulnerable CA1 region of the hippocampus[51, 52]. Results from LSCI indicate that PAME enhanced cortical CBF after ACA, which suggest that PAME can protect CA1 neurons from ACA-induced ischemic injury. Since hypoperfusion-induced delayed neuronal cell death occurs 3–7 days after the initial ischemic insult[5, 27, 28, 31], PAME’s neuroprotective effects on CA1 neurons were further evaluated by H&E and FJC staining 7 days after ACA. Results from H&E and FJC staining indicate that post-treatment of PAME immediately after ACA can alleviate neuronal cell death in the CA1 region of the hippocampus, which suggest that revival of cortical CBF by PAME after ACA can enhance neuronal survival in the CA1 region of the hippocampus (Figs. 3 and 4)[4, 5].
It is unclear if PAME provides other non-vasodilatory-dependent neuroprotective effects against cerebral ischemia. We thus utilized organotypic hippocampal slice cultures coupled with PI staining to examine if PAME offers neuroprotection against ischemia in a CBF-independent manner. Interestingly, results from oxygen-glucose deprivation (OGD) study (an in vitro ischemia-reperfusion injury model) indicate that PAME reduced neuronal cell death in the CA1 region of the hippocampus following OGD (Fig. 5), which suggest that besides enhancement of CBF, mechanisms underlying PAME-induced neuroprotection are multifaceted, affording multiple routes (i.e. anti-apoptosis and anti-inflammation) for neuroprotection.
Hippocampal CA1 neurons play a crucial role in learning/memory formation[53]. Thus, CA survivors suffer from severe cognitive dysfunction including learning/memory deficits[6]. PAME alleviated hypoperfusion and CA1 neuronal cell death after ACA (Figs. 2, 3, and 4) suggesting its possible therapeutic potentials in the treatment against ACA-induced learning/memory deficits. We further utilized behavioral trials to evaluate the neuroprotective effects of PAME on learning/memory deficits after ACA. The Morris water, Barnes, and T mazes are the most widely used behavioral test to examine spatial learning/memory in rodents, while the Morris water and Barnes maze are more prominent for measuring reference (long-term) memory[54, 55]. Since ACA-induced neuronal cell death in the CA1 region of the hippocampus mainly results in working (short-term) memory deficits[56–58], we used the spontaneous alternation T-maze test for assessing the impact of PAME on short-term memory function after ACA[26].
ACA-treated rats with hippocampal injuries developed a side-preference[26]. Therefore, their side-preference ratio was higher than control, and PAME only-treated rats (Fig 6). In addition, the spontaneous alternation ratio in ACA-treated rats were lower than control, and PAME only-treated rats (Fig 6) indicating that ACA-treated rats with memory deficits had difficulties remembering earlier explored goal arms. Post-treatment of PAME decreased side-preference ratio and enhanced spontaneous alternation ratio (Fig. 6) after ACA suggesting that PAME can alleviate ACA-induced hippocampal injuries and memory deficits.
In conclusion, CA remains one of the leading causes of death in U.S. suggesting that development of novel therapies to efficiently enhance post-resuscitation survival rate and functional outcomes is urgently needed. In the present study, we found that post-treatment of PAME immediately after ACA can alleviate ACA-induced hypoperfusion and neuronal cell death in the CA1 region of the hippocampus ultimately improving functional learning/memory. To our knowledge, we are the first to describe that PAME, traditionally considered as “a detrimental” class of fatty acids, can counteract CA-related brain injury. Our study will offer novel insight to further our understanding of saturated fatty acids residing in the brain/CNS as “beneficial” fatty acids in the treatment against cerebral ischemia.
Table 3.
Physiological parameters from FJC staining
| Group | Variable | Before ACA | After ACA |
|---|---|---|---|
|
ACA
(n=4) |
Body weight (g) | 316 ± 6.13 | n/a |
| pH | 7.44 ± 0.04 | 7.41 ± 0.02 | |
| pCO2 (mm Hg) | 35 ± 1.7 | 51 ± 6.0 | |
| pO2 (mm Hg) | 121 ± 16.5 | 202 ± 28.0 | |
| MABP (mm Hg) | 105 ± 3.35 | 115 ± 2.53* | |
| Glucose (mg/dL) | 80 ± 27 | ||
|
ACA+PAME
(n=5) |
Body weight (g) | 316 ± 5.17 | n/a |
| pH | 7.42 ± 0.01 | 7.39 ± 0.01 | |
| pCO2 (mm Hg) | 35 ± 1.7 | 46 ± 2.0* | |
| pO2 (mm Hg) | 100 ± 4.90 | 217 ± 49.3 | |
| MABP (mm Hg) | 103 ± 3.89 | 109 ± 2.90 | |
| Glucose (mg/dL) | 93 ± 18 |
p ≤ 0.05, significant difference from before ACA; n, number of rats; pO2 and pCO2, partial pressure of oxygen and carbon dioxide, respectively; MABP, mean arterial blood pressure; N/A, not applicable.
Table 4.
Physiological parameters from H&E staining
| Group | Variable | Before ACA | After ACA |
|---|---|---|---|
|
ACA
(n=6) |
Body weight (g) | 319 ± 4.15 | n/a |
| pH | 7.44 ± 0.03 | 7.38 ± 0.02* | |
| pCO2 (mm Hg) | 34 ± 1.1 | 48 ± 4.1* | |
| pO2 (mm Hg) | 118 ± 10.7 | 228 ± 41.2 | |
| MABP (mm Hg) | 97 ± 2.7 | 113 ± 3.00* | |
| Glucose (mg/dL) | 84 ± 16 | ||
|
ACA+PAME
(n=8) |
Body weight (g) | 312 ± 4.73 | n/a |
| pH | 7.43 ± 0.01 | 7.42 ± 0.02 | |
| pCO2 (mm Hg) | 34 ± 1.2 | 43 ± 2.5* | |
| pO2 (mm Hg) | 105 ± 5.13 | 224 ± 45.9* | |
| MABP (mm Hg) | 100 ± 2.92 | 115 ± 4.81* | |
| Glucose (mg/dL) | 81 ± 12 |
p ≤ 0.05, significant difference from before ACA; n, number of rats; pO2 and pCO2, partial pressure of oxygen and carbon dioxide, respectively; MABP, mean arterial blood pressure; N/A, not applicable.
Highlights.
We previously discovered palmitic acid methyl ester (a C16:0 saturated fatty acid) is a novel and potent vasodilator.
We investigated the therapeutic potential of palmitic acid methyl ester against cardiac arrest-induced brain injury and neurological deficits.
We found that post-treatment of palmitic acid methyl ester after cardiac arrest can enhance cerebral blood flow and neuronal cell survival ultimately improving functional learning/memory.
5. Acknowledgements
This study was supported by grants from the NIH/NINDS R01-NS096225–01A1 and the AHA 17GRNT33660336 and 17POST33660174.
Funding Source: NIH/NINDS R01-NS096225–02, AHA 17GRNT33660336 and 17POST33660174
Abbreviations:
- PAME
palmitic acid methyl ester
- CBF
cerebral blood flow
- ACA
asphyxial cardiac arrest
- CA
cardiopulmonary arrest
- SCG
superior cervical ganglion
- NO
nitric oxide
- LDF
laser Doppler flowmetry
- MABP
mean arterial blood pressure
- LSCI
laser speckle contrast imaging
- OGD
oxygen and glucose deprivation
- PI
propidium iodide
- NMDA
N-methyl-D-aspartate
- H&E
hematoxylin and eosin
- FJC
Fluoro-Jade C
- Kv
voltage-gated potassium channels
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, C. American Heart Association Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association, Circulation, 135 (2017) e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dunlay SM, Weston SA, Jacobsen SJ, Roger VL, Risk factors for heart failure: a population-based case-control study, Am J Med, 122 (2009) 1023–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Akhabue E, Thiboutot J, Cheng JW, Vittorio TJ, Christodoulidis G, Grady KM, Lerakis S, Kosmas CE, New and emerging risk factors for coronary heart disease, Am J Med Sci, 347 (2014) 151–158. [DOI] [PubMed] [Google Scholar]
- [4].Lee RH, Couto ESA, Lerner FM, Wilkins CS, Valido SE, Klein DD, Wu CY, Neumann JT, Della-Morte D, Koslow SH, Minagar A, Lin HW, Interruption of perivascular sympathetic nerves of cerebral arteries offers neuroprotection against ischemia, American journal of physiology. Heart and circulatory physiology, 312 (2017) H182–H188. [DOI] [PubMed] [Google Scholar]
- [5].Lin HW, Defazio RA, Della-Morte D, Thompson JW, Narayanan SV, Raval AP, Saul I, Dave KR, Perez-Pinzon MA, Derangements of post-ischemic cerebral blood flow by protein kinase C delta, Neuroscience, 171 (2010) 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chiota NA, Freeman WD, Barrett KM, Hypoxic-ischemic brain injury and prognosis after cardiac arrest, Continuum, 17 (2011) 1094–1118. [DOI] [PubMed] [Google Scholar]
- [7].Krause GS, Kumar K, White BC, Aust SD, Wiegenstein JG, Ischemia, resuscitation, and reperfusion: mechanisms of tissue injury and prospects for protection, American heart journal, 111 (1986) 768–780. [DOI] [PubMed] [Google Scholar]
- [8].Nichol G, Thomas E, Callaway CW, Hedges J, Powell JL, Aufderheide TP, Rea T, Lowe R, Brown T, Dreyer J, Davis D, Idris A, Stiell I, Resuscitation Outcomes Consortium I., Regional variation in out-of-hospital cardiac arrest incidence and outcome, Jama, 300 (2008) 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bradberry JC, Hilleman DE, Overview of omega-3 Fatty Acid therapies, P T, 38 (2013) 681–691. [PMC free article] [PubMed] [Google Scholar]
- [10].Van Noolen L, Back M, Arnaud C, Rey A, Petri MH, Levy P, Faure P, Stanke-Labesque F, Docosahexaenoic acid supplementation modifies fatty acid incorporation in tissues and prevents hypoxia induced-atherosclerosis progression in apolipoprotein-E deficient mice, Prostaglandins Leukot Essent Fatty Acids, 91 (2014) 111–117. [DOI] [PubMed] [Google Scholar]
- [11].Siri-Tarino PW, Sun Q, Hu FB, Krauss RM, Saturated fat, carbohydrate, and cardiovascular disease, Am J Clin Nutr, 91 (2010) 502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lin HW, Liu CZ, Cao D, Chen PY, Chen MF, Lin SZ, Mozayan M, Chen AF, Premkumar LS, Torry DS, Lee TJ, Endogenous methyl palmitate modulates nicotinic receptor-mediated transmission in the superior cervical ganglion, Proceedings of the National Academy of Sciences of the United States of America, 105 (2008) 19526–19531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Si ML, Lee TJ, Alpha7-nicotinic acetylcholine receptors on cerebral perivascular sympathetic nerves mediate choline-induced nitrergic neurogenic vasodilation, Circulation research, 91 (2002) 62–69. [DOI] [PubMed] [Google Scholar]
- [14].Lee TJ-F, Putative transmitter in cerebral neurogenic vasodilation, Humana Press, Totowa, New Jersey, 1994. [Google Scholar]
- [15].Lee RH, Wilkins CS, E Silva A. Couto, Valido SE, Wu CY, Lin HW, Fatty acids in vascular health. In Palmitic Acid: Occurrence, Biochemistry and Health Effects, Nova Science Publishers, Inc, (2014) (pp. 1–16). [Google Scholar]
- [16].Brizzolara-Gourdie A, Webb JG, Angiotensin II potentiates vasodilation of rat aorta by cAMP elevating agonists, The Journal of pharmacology and experimental therapeutics, 281 (1997) 354–359. [PubMed] [Google Scholar]
- [17].Vertongen P, Solano RM, Perret J, Langer I, Robberecht P, Waelbroeck M, Mutational analysis of the human vasoactive intestinal peptide receptor subtype VPAC(2): role of basic residues in the second transmembrane helix, Br J Pharmacol, 133 (2001) 1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tian Y, Wu LH, Oxender DL, Chung FZ, The unpredicted high affinities of a large number of naturally occurring tachykinins for chimeric NK1/NK3 receptors suggest a role for an inhibitory domain in determining receptor specificity, J Biol Chem, 271 (1996) 20250–20257. [DOI] [PubMed] [Google Scholar]
- [19].Ibayashi S, Sadoshima S, Ogata J, Yao H, Okada Y, Fujishima M, Effect of blood glucose level in acute cerebral ischemia in spontaneously hypertensive rats--survival and brain pathology, Angiology, 42 (1991) 543–551. [DOI] [PubMed] [Google Scholar]
- [20].Lin HW, Saul I, Gresia VL, Neumann JT, Dave KR, Perez-Pinzon MA, Fatty acid methyl esters and Solutol HS 15 confer neuroprotection after focal and global cerebral ischemia, Translational stroke research, 5 (2014) 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dunn AK, Laser speckle contrast imaging of cerebral blood flow, Ann Biomed Eng, 40 (2012) 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neumann JT, Thompson JW, Raval AP, Cohan CH, Koronowski KB, Perez-Pinzon MA, Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 35 (2015) 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Raval AP, Dave KR, Prado R, Katz LM, Busto R, Sick TJ, Ginsberg MD, Mochly-Rosen D, Perez-Pinzon MA, Protein kinase C delta cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 25 (2005) 730–741. [DOI] [PubMed] [Google Scholar]
- [24].Schmued LC, Hopkins KJ, Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration, Brain Res, 874 (2000) 123–130. [DOI] [PubMed] [Google Scholar]
- [25].Deacon RM, Rawlins JN, T-maze alternation in the rodent, Nature protocols, 1 (2006) 7–12. [DOI] [PubMed] [Google Scholar]
- [26].Wu CYC, Lerner FM, Couto ESA, Possoit HE, Hsieh TH, Neumann JT, Minagar A, Lin HW, Lee RHC, Utilizing the Modified T-Maze to Assess Functional Memory Outcomes After Cardiac Arrest, Journal of visualized experiments : JoVE, (2018). [DOI] [PMC free article] [PubMed]
- [27].Wang JY, Xia Q, Chu KT, Pan J, Sun LN, Zeng B, Zhu YJ, Wang Q, Wang K, Luo BY, Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy, Journal of neuropathology and experimental neurology, 70 (2011) 314–322. [DOI] [PubMed] [Google Scholar]
- [28].Fujioka M, Nishio K, Miyamoto S, Hiramatsu KI, Sakaki T, Okuchi K, Taoka T, Fujioka S, Hippocampal damage in the human brain after cardiac arrest, Cerebrovascular diseases, 10 (2000) 2–7. [DOI] [PubMed] [Google Scholar]
- [29].Lee RHC, Lee MHH, Wu CYC, Couto ESA, Possoit HE, Hsieh TH, Minagar A, Lin HW, Cerebral ischemia and neuroregeneration, Neural Regen Res, 13 (2018) 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lin HW, Gresia VL, Stradecki HM, Alekseyenko A, Dezfulian C, Neumann JT, Dave KR, Perez-Pinzon MA, Protein kinase C delta modulates endothelial nitric oxide synthase after cardiac arrest, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 34 (2014) 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin HW, Saul I, Gresia VL, Neumann JT, Dave KR, Perez-Pinzon MA, Fatty Acid Methyl Esters and Solutol HS 15 Confer Neuroprotection after Focal and Global Cerebral Ischemia, Translational stroke research, (2013). [DOI] [PMC free article] [PubMed]
- [32].Calder PC, Fatty acids and inflammation: the cutting edge between food and pharma, European journal of pharmacology, 668 Suppl 1 (2011) S50–58. [DOI] [PubMed] [Google Scholar]
- [33].Miller CL, Alexander K, Lampard DG, Brown WA, Griffiths R, Local cerebral blood flow following transient cerebral ischemia. II. Effect of arterial PCO2 on reperfusion following global ischemia, Stroke, 11 (1980) 542–548. [DOI] [PubMed] [Google Scholar]
- [34].Sabri M, Lass E, Macdonald RL, Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia, Stroke research and treatment, 2013 (2013) 394036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harukuni I, Bhardwaj A, Mechanisms of brain injury after global cerebral ischemia, Neurologic clinics, 24 (2006) 1–21. [DOI] [PubMed] [Google Scholar]
- [36].Manole MD, Foley LM, Hitchens TK, Kochanek PM, Hickey RW, Bayir H, Alexander H, Ho C, Clark RS, Magnetic resonance imaging assessment of regional cerebral blood flow after asphyxial cardiac arrest in immature rats, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 29 (2009) 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hosomi N, Ohyama H, Ichihara S, Takahashi T, Naya T, Kohno M, Relation of postischemic delayed hypoperfusion and cerebral edema after transient forebrain ischemia, Journal of stroke and cerebrovascular diseases : the official journal of National Stroke Association, 16 (2007) 103–108. [DOI] [PubMed] [Google Scholar]
- [38].Lee TJ, Chiueh CC, Adams M, Synaptic transmission of vasoconstrictor nerves in rabbit basilar artery, European journal of pharmacology, 61 (1980) 55–70. [DOI] [PubMed] [Google Scholar]
- [39].Lee TJ, Hume WR, Su C, Bevan JA, Neurogenic vasodilation of cat cerebral arteries, Circulation research, 42 (1978) 535–542. [DOI] [PubMed] [Google Scholar]
- [40].Lee TJ, Su C, Bevan JA, Nonsympathetic dilator innervation of cat cerebral arteries, Experientia, 31 (1975) 1424–1426. [DOI] [PubMed] [Google Scholar]
- [41].Lee TJ, Su C, Bevan JA, Neurogenic sympathetic vasoconstriction of the rabbit basilar artery, Circulation research, 39 (1976) 120–126. [DOI] [PubMed] [Google Scholar]
- [42].Lee TJ, Kinkead LR, Sarwinski S, Norepinephrine and acetylcholine transmitter mechanisms in large cerebral arteries of the pig, Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 2 (1982) 439–450. [DOI] [PubMed] [Google Scholar]
- [43].Lin HW, Della-Morte D, Thompson JW, Gresia VL, Narayanan SV, Defazio RA, Raval AP, Saul I, Dave KR, Morris KC, Si ML, Perez-Pinzon MA, Differential effects of delta and epsilon protein kinase C in modulation of postischemic cerebral blood flow, Advances in experimental medicine and biology, 737 (2012) 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang N, Kuczmanski A, Dubrovska G, Gollasch M, Palmitic Acid Methyl Ester and Its Relation to Control of Tone of Human Visceral Arteries and Rat Aortas by Perivascular Adipose Tissue, Front Physiol, 9 (2018) 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fu ZJ, Xie MJ, Zhang LF, Cheng HW, Ma J, Differential activation of potassium channels in cerebral and hindquarter arteries of rats during simulated microgravity, American journal of physiology. Heart and circulatory physiology, 287 (2004) H1505–1515. [DOI] [PubMed] [Google Scholar]
- [46].Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E, Yuan JX, Diversity of voltage-dependent K+ channels in human pulmonary artery smooth muscle cells, Am J Physiol Lung Cell Mol Physiol, 287 (2004) L226–238. [DOI] [PubMed] [Google Scholar]
- [47].Brueggemann LI, Haick JM, Cribbs LL, Byron KL, Differential activation of vascular smooth muscle Kv7.4, Kv7.5, and Kv7.4/7.5 channels by ML213 and ICA-069673, Mol Pharmacol, 86 (2014) 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cipolla MJ, The cerebral circulation, in: Integrated systems physiology : from molecules to function #2, Morgan & Claypool Life Sciences,, San Rafael, CA, 2010. [Google Scholar]
- [49].Nishimura J, Endo Y, Kimura F, Increases in cerebral blood flow in rat hippocampus after medial septal injection of naloxone, Stroke, 23 (1992) 1325–1329; discussion 1330. [DOI] [PubMed] [Google Scholar]
- [50].Rusinek H, Brys M, Glodzik L, Switalski R, Tsui WH, Haas F, McGorty K, Chen Q, de Leon MJ, Hippocampal blood flow in normal aging measured with arterial spin labeling at 3T, Magnetic resonance in medicine, 65 (2011) 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Payabvash S, Souza LC, Wang Y, Schaefer PW, Furie KL, Halpern EF, Gonzalez RG, Lev MH, Regional ischemic vulnerability of the brain to hypoperfusion: the need for location specific computed tomography perfusion thresholds in acute stroke patients, Stroke, 42 (2011) 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Neigh GN, Glasper ER, Kofler J, Traystman RJ, Mervis RF, Bachstetter A, DeVries AC, Cardiac arrest with cardiopulmonary resuscitation reduces dendritic spine density in CA1 pyramidal cells and selectively alters acquisition of spatial memory, Eur J Neurosci, 20 (2004) 1865–1872. [DOI] [PubMed] [Google Scholar]
- [53].Bahar AS, Shirvalkar PR, Shapiro ML, Memory-guided learning: CA1 and CA3 neuronal ensembles differentially encode the commonalities and differences between situations, The Journal of neuroscience : the official journal of the Society for Neuroscience, 31 (2011) 12270–12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sunyer B PS, Höger H, Lubec G, Barnes maze, a useful task to assess spatial reference memory in the mice Nature protocols 390, (2007). [Google Scholar]
- [55].Shoji H, Hagihara H, Takao K, Hattori S, Miyakawa T, T-maze forced alternation and left-right discrimination tasks for assessing working and reference memory in mice, Journal of visualized experiments : JoVE, (2012). [DOI] [PMC free article] [PubMed]
- [56].Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J, Basile AS, Alzheimer C, Wess J, M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity, The Journal of neuroscience : the official journal of the Society for Neuroscience, 24 (2004) 10117–10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Olton DS, Feustle WA, Hippocampal function required for nonspatial working memory, Experimental brain research, 41 (1981) 380–389. [DOI] [PubMed] [Google Scholar]
- [58].Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Ohta S, Fukuda K, Hori S, Hydrogen inhalation during normoxic resuscitation improves neurological outcome in a rat model of cardiac arrest independently of targeted temperature management, Circulation, 130 (2014) 2173–2180. [DOI] [PubMed] [Google Scholar]
- [59].Dember WN, Richman CL, Spotaneous alternation behavior, EDS, (1989).