

Abstract
Fibrous dysplasia (FD) is a genetic, noninheritable rare bone disease caused by a postzygotic activating mutation of the α subunit of the stimulatory G‐protein causing increased abnormal bone formation leading to pain, deformity and fractures. To date, no cure has been identified for FD/McCune–Albright syndrome (MAS) and treatment is symptomatic and aimed at decreasing pain and/or local bone turnover. Various drugs have been used to achieve clinical improvement in FD/MAS patients including bisphosphonates and denosumab, however further translational studies are also warranted to address unresolved pathophysiological issues and explore novel pharmacological targets for the management of FD/MAS.
In this article, we review literature on the medical treatment of FD/MAS, discuss the unresolved pathophysiological issues and explore novel pharmacological targets for the management of FD/MAS.
Keywords: bisphosphonates, bone tumours, fibrous dysplasia, McCune–Albright syndrome, RANK‐L
Introduction
Fibrous dysplasia/McCune–Albright syndrome (FD/MAS) is a genetic, noninheritable rare bone disease caused by a postzygotic activating mutation of the α subunit of the stimulatory G‐protein (Gsα) 1. In the skeleton, this results in the overproduction of cAMP in affected cells of the osteogenic lineage, leading to the accelerated production of bone marrow stromal cells (BMSC), while inhibiting the differentiation of these progenitor cells into mature osteoblasts 2. Despite expressing early osteoblast markers such as alkaline phosphatase (ALP), these immature cells are dysfunctional, leading to the laying down of fibro‐osseous tissue that is under‐mineralized, of poor quality and of disturbed micro‐architecture. In extensive disease, the mineralization defect is further exacerbated by increased fibroblast growth factor 23 (FGF‐23) expression by the mass of osteogenic mutated cells leading to renal phosphate wasting and impaired 1.25 vitamin D production 3. The disturbed bone microarchitecture and mineralization defect decrease bone strength, resulting in increased risk for deformities and fractures 4 (Figure 1).
Figure 1.
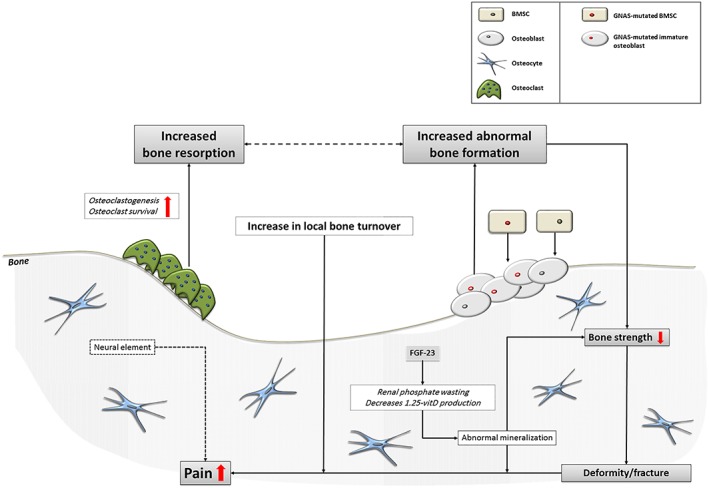
Pathophysiology of fibrous dysplasia. FGF‐23, fibroblast growth factor‐23; BMSC, bone marrow stromal cells
A further pathophysiological characteristic of FD lesions is the increased number of osteoclasts present in and around FD lesions, leading to a local increase in bone resorption 5. The local increase in osteoclastogenesis is believed to be due to the significant up‐regulation of the receptor activator of nuclear factor κ‐B ligand (RANKL) by GNAS‐mutated osteogenic cells 2, 6 and increased production of interleukin‐6 (IL‐6) by these cells 7. The observed focal increase in number and clustering of osteoclasts exhibits a tunneling resorption pattern and endosteal fibrosis analogous to changes characteristically observed in hyperparathyroidism 8. It has been suggested that local increase in bone turnover would be associated with increased deposition of abnormal, under‐mineralized fibro‐osseous tissue, leading to pain, further expansion of FD lesions and increased risk for deformities and fractures.
It has also been suggested that the local increase in osteoclastogenesis may be further promoted by increased PTHrP expression, amplifying the effects of mutated osteogenic cells to upregulate RANK‐L and down‐regulate OPG 9. Increased bone resorption leads to increased abnormal bone formation by the mutated immature osteoblasts, perpetuating the vicious circle that leads to continuing formation of abnormal bone, lesion expansion, pain, and increasing risk for deformities and fractures (Figure 1). The somatic mosaicism associated with the postzygotic nature of the GNAS mutation results in a wide spectrum of clinical manifestations, ranging from the completely asymptomatic, to the severely invalidated patient with multiple skeletal lesions, sometimes in combination with hyperfunctioning endocrinopathies as seen in the context of MAS 1.
Pitfalls and challenges in the pharmacological management of FD
There is no cure for FD/MAS, and to date there is no approved pharmacological treatment for the disorder's ubiquitous manifestations. Pain in particular remains a significant challenge for the treating physician. Pain at the site of FD lesions may occur as a result of a complete fracture, an impending fracture or microfractures, due to mineralization defects or abnormal mechanical forces in case of deformities. Pain may also be related to extent, severity or activity, although it has been suggested that there may be no correlation between skeletal pain and total disease burden as seen in patients with bone metastases 10, 11, 12. This suggests that bone remodelling and disease burden may not be the only contributors to bone pain, and that an additional factor such as neuropathic involvement may also play a role 13, 14. It has thus been proposed that analogous to the pain associated with bone tumours, pain arising from FD lesions may also be induced or exacerbated by sensory nerve involvement and/or the formation of neuromas in or around these lesions 10.
Traditionally, the main therapeutic approach to the management of FD/MAS is a surgical one, ranging from curettage of FD lesions in the early 60s 15, to customized blade plates more recently 16. The notion of using pharmacological agents in the management of FD/MAS has evolved in the 70s, based on the accumulation of evidence for increased bone resorption in FD lesions. This has opened the way for the use of antiresorptive agents in FD/MAS, aiming at decreasing the local increase in bone turnover, in the management of FD thereby potentially decreasing or preventing expansion of lesions, controlling symptoms and decreasing the risk for deformities and fractures. A number of antiresorptive drugs have been used off label, of which the most frequently prescribed have been the bisphosphonates, followed more recently by the anti‐RANK ligand antibody denosumab 17.
An essential first step before considering the use of any antiresorptive agent, however, is the identification and treatment of FGF23‐mediated hypophosphataemia, potentially leading to increased pain, deformity and increased fracture risk 18, 19, 20, 21, 22. Treatment of FGF23‐mediated hypophosphataemia requires the use of active vitamin D metabolites or analogues due to the inhibitory effects of increased levels of FGF23 on the renal α‐hydroxylase enzyme resulting in low circulating levels of 1,25‐dihydoxy vitamin D, as also observed in X‐linked hypophosphataemic rickets 23. Additional phosphate supplementation may be required, particularly in children in whom sufficient phosphate levels are essential for normal growth and the maintenance of bone quality. Attention should also been given to the presence of vitamin D deficiency or insufficiency, which should also be corrected before starting treatment with antiresorptive agents.
A second important step to be taken before initiating treatment with antiresorptive agents is the identification and treatment of potentially associated endocrinopathies, especially in the more severe forms of FD/MAS. This is especially the case for GH‐excess, in which the stimulatory effects of excess of growth hormone on bone turnover may compete or negate the inhibitory effects of antiresorptive treatment, calling for higher doses of bisphosphonates. Thyroid abnormalities are not uncommon in patients with FD/MAS, including T3 thyrotoxicosis due to a shifted T3/T4 ratio suggesting an increase in intrathyroidal conversion of T4 into the active metabolite T3, nearly always in the presence of echographic structural changes in the thyroid gland. Thyroid pathology should be identified and treated as required as potentially also increasing FD morbidity 24, 25.
The unravelling of further aspects of the pathophysiology of FD/MAS has led to exploring potential new pharmacological avenues (Figure 2), although the lack of adequate translational tools such as appropriate animal models to test new agents represents the main challenge for the development of novel therapies in the management of FD/MAS 2, 6.
Figure 2.
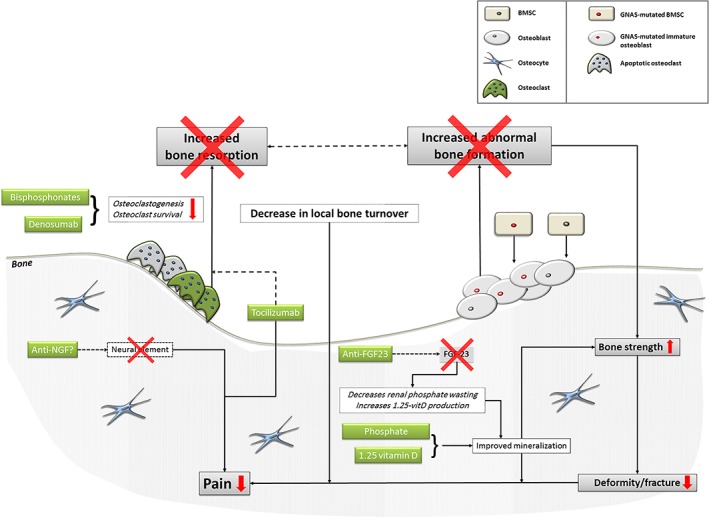
Current and future targets in the treatment of fibrous dysplasia. FGF‐23, fibroblast growth factor‐23; anti‐NGF, anti‐nerve growth factor; BMSC, bone marrow stromal cells
Antiresorptives
The purpose of using antiresorptives in FD/MAS would be to revert the local abnormality in bone turnover, decrease or prevent the deposition of abnormal, under‐mineralized fibro‐osseous tissue and associated lesion expansion, and thereby to control the clinical manifestations of the disorder.
The first publication on the use of an antiresorptive agent in the pharmacological management of FD/MAS appeared in 1970 and described the beneficial effects of using calcitonin in a patient with polyostotic FD 26. Experience with the use of this agent in the management of FD/MAS is very scarce and limited to a few case reports and small case series, with mixed outcomes 26, 27, 28. More than 50 years after its discovery, the choice of calcitonin as antiresorptive has been completely superseded by that of bisphosphonates. The use of calcitonin is therefore not recommended in FD/MAS.
Bisphosphonates
The most common antiresorptive drugs used in the pharmacological management of FD/MAS are the nitrogen‐containing bisphosphonates, which exert their antiresorptive effect by inhibiting the mevalonate pathway by preventing post‐translational prenylation of GTP‐binding proteins, resulting in osteoclast apoptosis 29. All published studies on the use of bisphosphonates have been observational open studies, except for a single randomized controlled trial with the use of oral alendronate 30.
In the early 1990s in a small case series of nine adult patients with FD/MAS was the first to demonstrate the beneficial effect of intravenously administered pamidronate (APD) at a dose of 60 mg daily for 3 consecutive days, every 6 months for 18 months on the clinical manifestations of FD/MAS 31. All patients reported a decrease in pain and none of the patients developed new fractures during treatment or over follow‐up which ranged from 18–48 months. Serum alkaline phosphatase and urinary hydroxyproline normalized on treatment in all patients, and filling and cortical thickening of FD lesions were observed on plain radiology in four patients. Side‐effects included transient diffuse bone pain in two patients, transient fever in three patients and symptomatic hypocalcaemia in two patients.
Since then, a number of publications reported variable beneficial effects of different bisphosphonates on pain, bone turnover markers and radiological features of FD/MAS lesions largely in adults and children with polyostotic FD/MAS. These publications include 18 case reports 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 20 observational case series 11, 30, 31, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66 and a single randomized controlled study (RCT) using oral alendronate 30.
Case reports were predominantly on women (11/18), seven on children and six reporting the effects of treatment with zoledronate and pamidronate in patients with isolated craniofacial FD (CFD) 33, 35, 36, 37, 40, 44. Positive outcome of treatment in the form of decreased pain was reported in all patients, associated by a concomitant normalization in bone turnover markers in six of the 17 patients in whom these markers were measured before and after treatment 37, 38, 41, 44, 47, 48. A total of 462 patients were included in the 20 published case series. Six of these case series included MAS patients, and one study included 106 patients with sinonasal CFD largely as part of polyostotic FD/MAS 55. In these case series, indication of treatment with bisphosphonates was based on pain, (n = 13/20), 31, 50, 51, 54, 55, 56, 57, 58, 59, 61, 64, 65, increased bone turnover markers (BTMs), (n = 9/20), 11, 30, 52, 53, 60, 62, 63, 66, 67, or a combination of both (n = 8/20). Several publications have reported a decrease in bone pain associated with a decrease in bone turnover with the use of intravenous pamidronate 52, 53, 59, 68. Symptoms of pain were not always a prerequisite for starting treatment with a bisphosphonate, in some studies this was based on the presence of increased bone turnover markers. Notwithstanding, the only two studies evaluating pain did not demonstrate it to be affected beneficially by bisphosphonates: the RCT using alendronate in children and adults with polyostotic FD/MAS compared to placebo 30 and the open study using bisphosphonates (type not reported) in patients with craniofacial disease as part of polyostotic FD/MAS 55.
The most commonly used bisphosphonate in the management of FD/MAS in both adults and children is intravenous pamidronate 57, 59, 60, 64, 66. Other bisphosphonates have also been used, such as intravenous zoledronate 45, intravenous and oral olpadronate 69, oral alendronate 30, and oral risedronate 70. The sequential use of different bisphosphonates 50, 61, 65 and their long‐term use resulting in high cumulative doses have also been reported 11. Only one RCT using oral alendronate or placebo in adults and children has been published 30. In this study, alendronate was administered in daily oral doses ranging from 10 to 40 mg daily based on body weight (40 mg daily for subjects >50 kg, 20 mg for 30–50 kg, 10 mg for 20–30 kg) in 6‐month cycles for 2 years. Treatment with this oral bisphosphonate led to a reduction in the bone resorption marker urinary NTx‐telopeptide, and to improvement in areal BMD in normal bone at the lumbar spine and in predetermined regions of FD, but had no significant effect on the bone formation marker serum osteocalcin, on functional parameters nor on pain at the end of 2 years of treatment. This observed difference between bone resorption and formation may hypothetically represent an important factor in the failure of the agent.
The bulk of evidence for an effect of bisphosphonates on pain in FD/MAS is derived from results of observational studies conducted over the past 3 decades, which report variable improvement in pain after initiation of treatment with an intravenously administered bisphosphonate. These studies are lacking a control arm and the use of validated tools in the evaluation of pain, and may thus have been influenced by the placebo effect. Therefore, more weight should be given to RCTs, which are very scarce in FD/MAS. The only published RCT showed no significant effect on pain of an oral agent 30. Data from the Profidys study (NCT00445575), the only other European multicentre RCT evaluating the efficacy of bisphosphonates in FD, which also used an oral bisphosphonate: risedronate at a dose of 30 mg daily for 2 months every 6 months, for 1 year in symptomatic patients and 3 years in adult asymptomatic patients, are eagerly awaited.
Notwithstanding, conclusions derived from RCTs on the efficacy on pain of a bisphosphonate agent compared to placebo can only be applied to the bisphosphonate used, its mode of administration and the dose used in the particular RCT. Caution should be exerted in extrapolating the findings to other bisphosphonates with a different mode of administration or total dose used. While awaiting the results of the Profidys study, we would not currently recommend the use of oral bisphosphonates in children or adults with FD/MAS.
There are no RCT data to date on the efficacy of intravenous bisphosphonates compared to placebo to reduce FD/MAS‐related pain and, to our knowledge, there are no plans to conduct such a study in the near future. However, based on data from observational studies 11, 71, 72, the consensus recommendation is to consider a trial of short‐term treatment with intravenous bisphosphonates in the management of FD‐related pain.
Effect of bisphosphonates on bone turnover markers
In adults, studies using bisphosphonates in FD/MAS in which BTMs were assessed report a decrease in the various bone markers used within few weeks of initiating treatment; however, in children, this decrease was not observed 60. However as in the case of evaluating the effect of bisphosphonates on pain, one of the main limitations of evaluating the effect of a bisphosphonate on BTMs is that various BTMs have been measured at various time‐points of treatment, and various bisphosphonates at various doses and schedules have been used so that it remains very difficult to compare outcome of studies. The relationship between increased bone turnover markers and pain and concomitant changes in both following treatment with a bisphosphonate has not been systematically examined in all studies, so that no firm conclusion can be drawn on this aspect of efficacy of bisphosphonates.
Effect of bisphosphonates on radiological features
Several publications demonstrate beneficial radiological changes, potentially associated with improved bone quality and decreased risk of complications 31, 52, 53, 56, 57, 58, 65. Beneficial changes in radiological features have thus included cortical thickening of FD lesions as observed after 2–6 years of follow up in three of 11 children with MAS treated with pamidronate 57; a decrease in the size of a monostotic FD lesion observed on computed tomography in three out of six patients treated with pamidronate 58; or progressive filling of osteolytic lesions in 54% of 58 adult patients with MFD (n = 22) and PFD (n = 36) treated with pamidronate 53. Data on changes in radiological features are, however, scarce as these have not been consistently reported in most of the published studies. Of importance when interpreting studies on radiographic changes in skeletal FD lesions, is that longitudinal changes observed may be due, at least in part, to the natural history of the disease, particularly as burn‐out of FD lesions has been described with increasing age 72.
The most common side‐effects observed with the use of a bisphosphonate in FD/MAS have been an acute phase reaction including transient fever after infusion 31, 50, 52, 53, 58, 63, followed by transient bone pain 31, 56, 63, 65 and transient hypocalcaemia 31, 50, 52, 53, 63. Concerns about the effects of bisphosphonates on linear growth in children has not been substantiated also in children with FD/MAS treated with bisphosphonates 11, 73. There have been scarce reports of the recently identified rare risks of osteonecrosis of the jaw (ONJ) and atypical femoral fractures in bisphosphonate‐treated FD/MAS patients, despite their prolonged use at high cumulative doses in some cases 11, 53. It is of note that ONJ has been reported in four FD patients, three of whom had craniofacial FD lesions located at the site of the developing ONJ and the fourth had documented poor dental health before starting treatment 74.
Based on the above literature findings and our Centre's long‐standing experience on efficacy and safety of treatment of patients with FD/MAS with bisphosphonates, we do advocate the use of intravenous bisphosphonates in these patients, although we have so far only done so in patients with measurable increases in BTMs, with or without associated pain symptoms. However, other centres do treat patients with bisphosphonates primarily to decrease pain, regardless of bone turnover. Some patients however, particularly those with high skeletal burden scores, initial growth hormone excess and high FGF‐23 levels, all reflecting extensive and severe disease, may be refractory to treatment with these agents 44, 65, which calls for alternative approaches for their management.
The anti‐RANKL antibody denosumab
The human skeletal progenitor cells expressing the mutated Gsα protein have been shown to produce excess receptor activator of nuclear factor κ‐b ligand (RANKL) 6. The documented increased expression of RANKL by cells of the osteogenic lineage in FD lesions provided the rationale for exploring the use of the anti‐RANKL denosumab as a therapeutic option in the pharmacological treatment of FD/MAS. Using this agent was also supported by in vitro studies showing that BMSCs, transfected with the specific R201‐C or ‐H mutation and exposed to an anti‐RANKL antibody, demonstrated a decrease in cAMP expression. RANKL plays an essential role in osteoclastogenesis by binding to its receptor on the membrane of osteoclast progenitors. The binding of RANKL to RANK induces osteoclast differentiation, activation and survival, leading to increased bone resorption. However, the observed effect of denosumab on GSα mutated BMSCs suggests that in FD/MAS, RANKL inhibition may have an additional direct effect on BMSCs, independently of the established role of anti‐RANKL to inhibit osteoclastogenesis. The creation of GsαR201C‐transgenic mice that develop an FD skeletal phenotype has made it possible to explore anti‐RANKL as a therapeutic option in the management of FD 6. A decrease in the size of osteolytic FD lesions, fewer deformities and fewer FD lesions were reported in abstract form in mice treated with an anti‐RANKL antibody compared to nontreated controls 73. In treated mice, discontinuation of treatment was associated with histological evidence of rebound of the disease. There was no radiographic difference observed at 3 months between anti‐RANKL treated mice and nontreated control mice 73, 75.
Denosumab, a humanized anti‐RANKL antibody, has been approved for the treatment of osteoporosis and skeletal tumour metastases, and has been shown to induce tumour reduction and bone formation in patients with giant cell tumours of bone 76, 77. Over the past 6 years, five case reports have been published on the effect of denosumab in patients with FD 17, 78, 79, 80, 81. A decrease of pain was reported in all patients, and BTMs normalized within a few hours 79 to 3 weeks 78 in all five cases.
In 2012, Boyce and colleagues reported on a 9‐year‐old boy with MAS and an FD lesion in the femur, which was rapidly expanding despite pamidronate treatment causing pain and functional impairment 17. Denosumab was initiated at a dose of 1 mg kg–1 monthly, increasing stepwise every 3 months to 1.25, 1.5 and 1.75 mg kg–1. Tumour growth had slowed down from 4.2% to 0.56% per month after 9 months of treatment, and BTMs normalized already after the first dose, remaining suppressed thereafter for the duration of treatment. However, the patient had become hypophosphataemic and hypocalcaemic and had developed secondary hyperparathyroidism soon after the first injection so that he needed supplementation with phosphorus, calcitriol and calcium 17. Denosumab treatment was stopped before his seventh dose as he had sustained a femoral fracture at the site of an FD lesion after falling out of bed. Within 2 months of discontinuing treatment with denosumab, the child developed severe symptomatic hypercalcaemia requiring treatment with high dose bisphosphonates: pamidronate followed by zoledronate to normalize. Similar to the case with the use of bisphosphonates in children, sclerotic bands were observed in the radius and ulna of the child, parallel to the growth plates, reflecting periods of inhibition of resorption followed by periods of normal bone formation at the time intervals of administration, rather than a persistent suppression of skeletal activity.
A rapid positive outcome of denosumab treatment has been reported by others 78, 79, 80, 81, also using the dosage of 60 mg administered subcutaneously at 3–9 months intervals. Both pain and BTMs are reported to respond quite rapidly following each dose, although the beneficial effect on bone pain seems to wane within weeks to a maximum of 4 months after a dose. In all case reports, patients were switched to denosumab because of insufficient effect of bisphosphonate therapy on pain and/or BTMs.
A case series from our Centre, published as an abstract, demonstrates a positive outcome of treatment with denosumab in 12 adult patients with polyostotic FD/MAS with persistent pain and persistently increased bone turnover markers despite long‐term treatment with bisphosphonates 82. Bone turnover markers normalized in all patients after initiation of denosumab therapy, and all reported an improvement in pain symptoms.
Although these early findings with the use of denosumab in FD/MAS appear to be promising, especially in the management of patients with severe skeletal burden who are refractory to treatment with bisphosphonates, there are a number of safety issues that remain to be addressed before this potent antiresorptive agent could be advocated for use in all patients with FD/MAS. One of these questions clearly relate to the rebound effect observed after inadvertently discontinuing treatment. This question is in the process of being formally addressed in a study about to start at the National Institutes of Health, NCT03571191. In this study, patients will receive a monthly dose of denosumab 120 mg for 6 consecutive months, including a loading dose on days 7 and 14 of the first month. The primary outcome of this study will be the change in BTMs, with secondary endpoints pain, lesion intensity and effect of discontinuation of therapy as patients will be followed for 21 months. In addition to safety, the second question to be addressed is the evidence for the efficacy of this drug as obtained by means of a multicentre RCT including sufficient numbers of patients, which is currently being designed by the International FD/MAS Consortium. Until data from these two studies become available, caution should be exerted with the indiscriminate use of this potent antiresorptive agent in FD/MAS.
Over the past decade, there has been increasing awareness about the potential risks associated with the long‐term use of antiresorptive agents, such as ONJ. Better knowledge about these risks and measures to be adopted to prevent them (such as securing adequate dental hygiene, identifying and treating vitamin D deficiency and FGF23‐mediated hypophosphatemia and FD/MAS‐associated hyperactive endocrinopathies, particularly GH‐excess) before initiating treatment with antiresorptives should be extended to all health care professionals involved in the management of patients with FD/MAS.
Other therapeutic options
Other than the antiresorptives, there have only been a handful of potential therapeutic options in the pharmacological management of FD/MAS, all largely based on increasing insight into the pathophysiology of the disorder.
Anti‐IL6 (tocilizumab)
IL‐6 is one of several cytokines implicated in stimulating bone resorption 83. IL‐6 expression is upregulated by PTH and acts via Gsα and the cAMP‐mediated signalling pathway 84. It has been hypothesized that since cAMP is a known stimulant of IL‐6 gene activity 85, increased cAMP production (as seen in FD/MAS) could lead to IL‐6 overexpression, a proposed mechanism of FD related bone pain 5, 10. To test this hypothesis in vitro, increased IL‐6 secretion was induced by transfecting MC3T3 cells with Gsα, reproducing the same activating mutation as seen in FD/MAS 86. Cells isolated from FD lesions were found to secrete higher levels of IL‐6 87. FD lesions with a high number of osteoclasts showed the presence of a direct link between GNAS mutations in BMSCs and IL‐6 production, but also defined the complex role of IL‐6 in regulating osteoclastogenesis in FD in vivo 7. Patterns of osteoclastogenesis and bone resorption do not only reflect the cell‐autonomous effects of GNAS mutations in osteogenic cells (including IL‐6 production), but also the local and systemic context to which non‐osteogenic cells, local proportions of wild‐type vs. mutated cells, and systemic hormones contribute. In addition, it was observed that IL‐6 knockout mice were protected from bone loss 7. Combining these findings, de Boysson and colleagues 88 hypothesized that Il‐6 inhibition might lead to reduction of bone pain in FD/MAS and went on to treat a 35‐year‐old woman with severe CFD and persisting headaches, with a score of 4–5/10 despite bisphosphonate treatment, with the anti‐IL‐6 antibody tocilizumab at a dose of 600 mg monthly, with a complete disappearance of pain after the second month of treatment. Treatment was continued at the lower dose of 400 mg two‐monthly for a further 6 months after which it was discontinued. There were no adverse effects associated with the treatment or its discontinuation 89. At this point, one single trial is registered (NCT1791842) using tocilizumab in patients with FD/MAS related pain resistant to bisphosphonates; however, the status of this study is currently unknown.
The identification of potential novel treatment targets
Despite the significant advances in understanding the pathophysiology of the skeletal and extra skeletal manifestations of FD/MAS, a number of molecular and cellular consequences of a GNAS mutation remain unresolved. This is largely due to the difficulties encountered by the mosaicism of the disease and this hampers the development of mouse models, which would mimic the full clinical picture of the FD/MAS human disease. Several models have been developed; however, none have been able to fully capture all the characteristics of the human disease. Specific models mimicking the skeletal disease or the extraskeletal manifestations came to development 75, 90 allowing the possibility to not only evaluate the anti‐RANKL antibody denosumab, but also the speculation on new therapies. Zhao and colleagues [2] recently developed a conditional tetracycline‐inducible animal model expressing the Gsα mutation (R201C) in the skeletal stem cells. Here, typical FD bone lesions develop in both embryos and adult mice in <2 weeks following doxycycline administration 2. The mutation resulted in the increased expression of RANKL by mutated osteogenic cells leading to the typical FD‐related histomorphometric changes. Gsα expression ablation by doxycycline withdrawal resulted in FD‐like lesion regression, supporting the rationale for Gsα‐targeted drugs to pursue curation. Such a model, which develops FD‐like lesions that can form rapidly and revert on cessation of mutant Gsα expression, may provide a breakthrough in expanding the possibilities for medical treatment for FD/MAS such as gene therapy using suramin sodium (SS), that can inhibit Gsα activation 91. Using a knock‐out mouse model in which the human FD R201H mutation was conditionally knocked into the corresponding mouse GNAS locus, Khan and colleagues [90] found upregulated Wnt/β‐catenin signalling in the FD mutant mouse which exhibited human FD features. They also showed significant rescue of the FD phenotype by removing one lipoprotein receptor‐related protein (LRP)‐6 copy 90. These findings open new avenues in the management of FD/MAS by showing that Wnt/β‐catenin signalling may represent an interesting new target for drug development in Gsα activation bone disorders. Although this is an exciting development, much more work is required in this field before gene therapy is considered in the clinical management of FD/MAS.
Anti‐FGF23
Patients with severe polyostotic disease are likely to have high FGF‐23 levels due to high mutated osteogenic cell disease burden often associated with renal phosphate wasting 3. High levels of FGF‐23 are also associated with inhibition of the one‐α hydroxylase enzyme and a decrease in 1.25‐dihydroxy vitamin D levels (Figure 1). The abnormalities in mineral metabolism can be readily reverted by treatment with active vitamin D metabolites and phosphate supplements; however, especially during growth, high doses might be needed, which can lead to complications such as nephrocalcinosis, renal stones and tertiary hyperparathyroidism. The use of a human monoclonal antibody to FGF‐23 is another option that was shown to be effective in X‐linked hypophosphataemic rickets, in which a mutation in the PHEX gene leads to high FGF‐23 levels, renal phosphate wasting and hypophosphataemia. The human monoclonal antibody to FGF‐23 binds to and directly inhibits the activity of FGF‐23, thereby increasing renal tubular reabsorption of phosphate, increasing circulating levels of 1.25‐OH2 levels and improving clinical outcome 92. However, the number of secreting mutated osteogenic cells due to a high skeletal burden of the disease is not affected and the use of anti‐FGF‐23 would only be beneficial in case of untreatable symptomatic hypophosphataemia despite optimal active vitamin D metabolites and phosphate supplementation. There are currently no literature data on outcome of treatment of FD/MAS with anti‐FGF‐23.
Anti‐nerve growth fact
Nerve growth factor (NGF) was discovered as a tumour tissue‐produced soluble factor that promotes growth and differentiation of sensory and sympathetic ganglia 93. NGF plays a significant role in the generation of pain and hyperalgesia in acute and chronic pain in rodents 94. Injury and inflammation increase NGF expression, since activated macrophages express NGF and are thus able to directly sensitize primary afferent neurons expressing NGF receptors. Since the discovery of NGF, a number of studies have been conducted in models of bone metastasis to address the potential use of NGF‐modifying drugs in reducing cancer‐related pain. Administration of a blocking antibody to NGF in a rodent model for prostate metastasis has thus been shown to result in a significant reduction in pain 95. There is as yet no evidence for a contributory role for NGF in the pathophysiology of pain in FD/MAS and no data available in animal models of FD/MAS to suggest a beneficial effect of their use in controlling pain. However, more translational work addressing the mechanism of pain in FD/MAS and its potential management is certainly warranted in view of the severe and sometimes crippling pain, which may be very difficult to control that not only can affect patients with the most severe forms of FD/MAS, but also sometimes those with monostotic disease.
The past 3 decades since the discovery of the mutation responsible for the manifestations of FD/MAS have witnessed significant advances in the understanding of the pathophysiology of the skeletal and extraskeletal manifestations of this ubiquitous disorder. Whereas bisphosphonates remain to date the cornerstone of the medical treatment of symptomatic patients with FD/MAS, opinion is still divided on which bisphosphonate to use, at which dose and at which dosing interval. Most commonly used schedules are pamidronate at a dose of 90 mg 3‐monthly or 180 mg 6‐monthly in adults, and 1 mg kg–1 with a maximum of 30 mg per day for 3 consecutive days in children. Denosumab is emerging as a useful alternative in patients refractory to treatment with bisphosphonates, but caution should be exerted with their use because of concern about a deleterious rebound effect on discontinuation of treatment. There is a clearly unmet need for multicentre RCTs including sufficient patients to confirm the efficacy and safety of currently used drugs in controlling pain and decreasing the risk of expansion of FD lesions, and thereby the risk of deformities and fractures. Further translational studies are warranted to address unresolved pathophysiology issues and explore novel pharmacological targets for the management of FD/MAS.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 96, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 97.
Competing Interests
There are no competing interests to declare.
We are very grateful to Mr Jan Schoones, from the University of Leiden's Walaeus Library for his invaluable help with the literature search used in this review.
M.R.: Bontius Foundation, N.M.A.‐D.: Dutch advisory board for denosumab.
Rotman M., Hamdy N. A. T., and Appelman‐Dijkstra N. M. (2019) Clinical and translational pharmacological aspects of the management of fibrous dysplasia of bone, Br J Clin Pharmacol, 85, 1169–1179. doi: 10.1111/bcp.13820.
References
- 1. Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune‐Albright syndrome. N Engl J Med 1991; 325: 1688–1695. [DOI] [PubMed] [Google Scholar]
- 2. Zhao X, Deng P, Iglesias‐Bartolome R, Amornphimoltham P, Steffen DJ, Jin Y, et al Expression of an active Galphas mutant in skeletal stem cells is sufficient and necessary for fibrous dysplasia initiation and maintenance. Proc Natl Acad Sci U S A 2018; 115: E428–E437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, et al FGF‐23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003; 112: 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Collins MT, Riminucci M, Bianco P. Fibrous Dysplasia In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, Eight Edition (pp. 786–793). Edited by Rosen Clifford J. American Society for Bone and Mineral Research. Published 2013. by John Wiley & Sons, Inc. [Google Scholar]
- 5. Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, et al The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site‐specific patterns and recurrent histological hallmarks. J Pathol 1999; 187: 249–258. [DOI] [PubMed] [Google Scholar]
- 6. Piersanti S, Remoli C, Saggio I, Funari A, Michienzi S, Sacchetti B, et al Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res 2010; 25: 1103–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Robey PG. Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL‐6 expression. Bone 2003; 33: 434–442. [DOI] [PubMed] [Google Scholar]
- 8. Riminucci M, Corsi A, Kuznetsov S, Licci S, Robey PG, Bianco P. Angiogenesis and expression of alpha‐SM actin in fibrous dysplasia of bone. Bone 2001; 28: S124. [Google Scholar]
- 9. Fraser WD, Walsh CA, Birch MA, Durham B, Dillon JP, McCreavy D, et al Parathyroid hormone‐related protein in the aetiology of fibrous dysplasia of bone in the McCune Albright syndrome. Clin Endocrinol (Oxf) 2000; 53: 621–628. [DOI] [PubMed] [Google Scholar]
- 10. Chapurlat RD, Gensburger D, Jimenez‐Andrade JM, Ghilardi JR, Kelly M, Mantyh P. Pathophysiology and medical treatment of pain in fibrous dysplasia of bone. Orphanet J Rare Dis 2012; 7 (Suppl. 1): S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Majoor BC, Appelman‐Dijkstra NM, Fiocco M, van de Sande MAJ, Dijkstra PDS, Hamdy NAT. Outcome of long‐term bisphosphonate therapy in McCune‐Albright syndrome and polyostotic fibrous dysplasia. J Bone Miner Res 2017; 32: 264–276. [DOI] [PubMed] [Google Scholar]
- 12. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer 2002; 2: 584–593. [DOI] [PubMed] [Google Scholar]
- 13. Jimenez‐Andrade JM, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, et al A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone 2010; 46: 306–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halvorson KG, Sevcik MA, Ghilardi JR, Rosol TJ, Mantyh PW. Similarities and differences in tumor growth, skeletal remodeling and pain in an osteolytic and osteoblastic model of bone cancer. Clin J Pain 2006; 22: 587–600. [DOI] [PubMed] [Google Scholar]
- 15. Harris WH, Dudley HR Jr, Barry RJ. The natural history of fibrous dysplasia. An orthopaedic, pathological, and roentgenographic study. J Bone Joint Surg Am 1962; 44‐A: 207–233. [PubMed] [Google Scholar]
- 16. Majoor BCJ, Leithner A, van de Sande MAJ, Appelman‐Dijkstra NM, Hamdy NAT, Dijkstra PDS. Individualized approach to the surgical management of fibrous dysplasia of the proximal femur. Orphanet J Rare Dis 2018; 13: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, et al Denosumab treatment for fibrous dysplasia. J Bone Miner Res 2012; 27: 1462–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benhamou J, Gensburger D, Messiaen C, Chapurlat R. Prognostic factors from an epidemiologic evaluation of fibrous dysplasia of bone in a modern cohort: the FRANCEDYS study. J Bone Miner Res 2016; 31: 2167–2172. [DOI] [PubMed] [Google Scholar]
- 19. Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, et al Fracture incidence in polyostotic fibrous dysplasia and the McCune‐rAlbright syndrome. J Bone Miner Res 2004; 19: 571–577. [DOI] [PubMed] [Google Scholar]
- 20. Pan KS, Heiss JD, Brown SM, Collins MT, Boyce AM. Chiari I malformation and basilar invagination in fibrous dysplasia: prevalence, mechanisms, and clinical implications. J Bone Miner Res 2018; 33: 1990–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shafqat H, Alquadan KF, Olszewski AJ. Severe hypocalcemia after denosumab in a patient with acquired Fanconi syndrome. Osteoporos Int 2014; 25: 1187–1190. [DOI] [PubMed] [Google Scholar]
- 22. Cundy T, Michigami T, Tachikawa K, Dray M, Collins JF, Paschalis EP, et al Reversible deterioration in Hypophosphatasia caused by renal failure with bisphosphonate treatment. J Bone Miner Res 2015; 30: 1726–1737. [DOI] [PubMed] [Google Scholar]
- 23. Liu ES, Martins JS, Raimann A, Chae BT, Brooks DJ, Jorgetti V, et al 1,25‐Dihydroxyvitamin D alone improves skeletal growth, microarchitecture, and strength in a murine model of XLH, despite enhanced FGF23 expression. J Bone Miner Res 2016; 31: 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong SC, Zacharin M. Long‐term health outcomes of adults with McCune‐Albright syndrome. Clin Endocrinol (Oxf) 2017; 87: 627–634. [DOI] [PubMed] [Google Scholar]
- 25. Congedo V, Celi FS. Thyroid disease in patients with McCune‐Albright syndrome. Pediatr Endocrinol Rev 2007; 4 (Suppl. 4): 429–433. [PubMed] [Google Scholar]
- 26. Bell NH, Avery S, Johnston CC Jr. Effects of calcitonin in Paget's disease and polyostotic fibrous dysplasia. J Clin Endocrinol Metab 1970; 31: 283–290. [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto K, Maeyama I, Kishimoto H, Morio Y, Harada Y, Ishitobi K, et al Suppressive effect of elcatonin, an eel calcitonin analogue, on excessive urinary hydroxyproline excretion in polyostotic fibrous dysplasia (McCune–Albright's syndrome). Endocrinol Jpn 1983; 30: 651–656. [DOI] [PubMed] [Google Scholar]
- 28. Fighera TM, Spritzer PM. Effect of intranasal calcitonin in a patient with McCune–Albright syndrome, fibrous dysplasia, and refractory bone pain. Case Rep Endocrinol 2017; 2017: 7898713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luckman SP, Hughes DE, Coxon FP, Graham R, Russell G, Rogers MJ. Nitrogen‐containing bisphosphonates inhibit the mevalonate pathway and prevent post‐translational prenylation of GTP‐binding proteins, including Ras. J Bone Miner Res 1998; 13: 581–589. [DOI] [PubMed] [Google Scholar]
- 30. Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, et al A randomized, double blind, placebo‐controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab 2014; 99: 4133–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liens D, Delmas PD, Meunier PJ. Long‐term effects of intravenous pamidronate in fibrous dysplasia of bone. Lancet 1994; 343: 953–954. [DOI] [PubMed] [Google Scholar]
- 32. Aragao AL, Silva IN. Oral alendronate treatment for severe polyostotic fibrous dysplasia due to McCune‐Albright syndrome in a child: a case report. Int J Pediatr Endocrinol 2010; 2010: 432060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aravinda K, Ratnakar P, Srinivas K. Oral manifestations of McCune‐Albright syndrome. Indian J Endocrinol Metab 2013; 17: 170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aycan Z, Onder A, Cetinkaya S. Eight‐year follow‐up of a girl with McCune‐Albright syndrome. J Clin Res Pediatr Endocrinol 2011; 3: 40–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bachelet JT, Mendes LC, Delafond C, Barba T, Gleizal A, Paulus C. Giant hemifacial fibrous dysplasia functional treatment and place of pamidronate. J Craniofac Surg 2017; 28: 706–708. [DOI] [PubMed] [Google Scholar]
- 36. Classen CF, Mix M, Kyank U, Hauenstein C, Haffner D. Pamidronic acid and cabergoline as effective long‐term therapy in a 12‐year‐old girl with extended facial polyostotic fibrous dysplasia, prolactinoma and acromegaly in McCune‐Albright syndrome: a case report. J Med Case Reports 2012; 6: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Di Pede C, Congedi S, Rossin S, Divisic A, De Gregorio A, Agosto C, et al Use of zoledronic acid in paediatric craniofacial fibrous dysplasia. Case Rep Pediatr 2016; 2016: 2329483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Florez H, Peris P, Vidal‐Sicart S, Monegal A, Guañabens N. Lack of scintigraphic response of fibrous dysplasia to bisphosphonate treatment. Rheumatology (Oxford) 2016; 55: 1735. [DOI] [PubMed] [Google Scholar]
- 39. Khadilkar VV, Khadilkar AV, Maskati GB. Oral bisphosphonates in polyostotic fibrous dysplasia. Indian Pediatr 2003; 40: 894–896. [PubMed] [Google Scholar]
- 40. Kodama Y, Ogose A, Oguri Y, Ubaidus S, Iizuka T, Takagi R. Alveolar bone grafting in association with polyostotic fibrous dysplasia and bisphosphonate‐induced abnormal bone turnover in a bilateral cleft lip and palate patient: a case report. J Oral Maxillofac Surg 2012; 70: e500–e508. [DOI] [PubMed] [Google Scholar]
- 41. Kollerova J, Koller T, Zelinkova Z, Kostalova L, Payer J. Treatment of pathological bone fractures in a patient with McCune‐Albright syndrome. Case Rep Endocrinol 2013; 2013: 589872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J, Shin H, Kwon YJ. Treatment of polyostotic fibrous dysplasia of the thoracic spine with intravenous pamidronate: result from 9 months follow up. Korean J Spine 2015; 12: 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li GD, Ogose A, Hotta T, Kawashima H, Ariizumi T, Xu Y, et al Long‐term efficacy of oral alendronate therapy in an elderly patient with polyostotic fibrous dysplasia: a case report. Oncol Lett 2011; 2: 1239–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Makitie AA, Tornwall J, Makitie O. Bisphosphonate treatment in craniofacial fibrous dysplasia‐‐a case report and review of the literature. Clin Rheumatol 2008; 27: 809–812. [DOI] [PubMed] [Google Scholar]
- 45. Ohno I, Higuchi C. Zoledronate therapy for the pathological humeral fracture in polyostotic fibrous dysplasia: a case report. J Clin Med Res 2015; 7: 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rastogi A, Bhadada SK, Bhansali A. Recurrent femur neck fracture and response to bisphosphonates in polyostotic fibrous dysplasia. Indian J Pediatr 2012; 79: 667–669. [DOI] [PubMed] [Google Scholar]
- 47. Weinstein RS. Long‐term aminobisphosphonate treatment of fibrous dysplasia: spectacular increase in bone density. J Bone Miner Res 1997; 12: 1314–1315. [DOI] [PubMed] [Google Scholar]
- 48. Wu D, Ma J, Bao S, Guan H. Continuous effect with long‐term safety in zoledronic acid therapy for polyostotic fibrous dysplasia with severe bone destruction. Rheumatol Int 2015; 35: 767–772. [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto T, Ozono K, Shima M, Yoshikawa H, Okada S. Alendronate and pharmacological doses of 1alpha OHD3 therapy in a patient with McCune‐Albright syndrome and accompanying hypophosphatemia. J Bone Miner Metab 2002; 20: 170. [DOI] [PubMed] [Google Scholar]
- 50. Bhadada SK, Bhansali A, Das S, Singh R, Sen R, Agarwal A, et al Fibrous dysplasia & McCune‐Albright syndrome: an experience from a tertiary care Centre in North India. Indian J Med Res 2011; 133: 504–509. [PMC free article] [PubMed] [Google Scholar]
- 51. Chao K, Katznelson L. Use of high‐dose oral bisphosphonate therapy for symptomatic fibrous dysplasia of the skull. J Neurosurg 2008; 109: 889–892. [DOI] [PubMed] [Google Scholar]
- 52. Chapurlat RD, Delmas PD, Liens D, Meunier PJ. Long‐term effects of intravenous pamidronate in fibrous dysplasia of bone. J Bone Miner Res 1997; 12: 1746–1752. [DOI] [PubMed] [Google Scholar]
- 53. Chapurlat RD, Hugueny P, Delmas PD, Meunier PJ. Treatment of fibrous dysplasia of bone with intravenous pamidronate: long‐term effectiveness and evaluation of predictors of response to treatment. Bone 2004; 35: 235–242. [DOI] [PubMed] [Google Scholar]
- 54. Couturier A, Aumaitre O, Gilain L, Jean B, Mom T, André M. Craniofacial fibrous dysplasia: a 10‐case series. Eur Ann Otorhinolaryngol Head Neck Dis 2017; 134: 229–235. [DOI] [PubMed] [Google Scholar]
- 55. DeKlotz TR, Kim HJ, Kelly M, Collins MT. Sinonasal disease in polyostotic fibrous dysplasia and McCune–Albright syndrome. Laryngoscope 2013; 123: 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gibbons CL, Petra M, Smith R, Athanasou NA. Bisphosphonate treatment of benign multifocal and unifocal osteolytic tumours of bone. Sarcoma 2003; 7: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Isaia GC, Lala R, Defilippi C, Matarazzo P, Andreo M, Roggia C, et al Bone turnover in children and adolescents with McCune–Albright syndrome treated with pamidronate for bone fibrous dysplasia. Calcif Tissue Int 2002; 71: 121–128. [DOI] [PubMed] [Google Scholar]
- 58. Kos M, Luczak K, Godzinski J, Klempous J. Treatment of monostotic fibrous dysplasia with pamidronate. J Craniomaxillofac Surg 2004; 32: 10–15. [DOI] [PubMed] [Google Scholar]
- 59. Lala R, Matarazzo P, Bertelloni S, Buzi F, Rigon F, Sanctis C. Pamidronate treatment of bone fibrous dysplasia in nine children with McCune–Albright syndrome. Acta Paediatr 2000; 89: 188–193. [DOI] [PubMed] [Google Scholar]
- 60. O'Sullivan M, Zacharin M. Intramedullary rodding and bisphosphonate treatment of polyostotic fibrous dysplasia associated with the McCune‐Albright syndrome. J Pediatr Orthop 2002; 22: 255–260. [PubMed] [Google Scholar]
- 61. Ozdemir Kutbay N, Sarer Yurekli B, Kartal Baykan E, Sahin SB, Saygili F. Characteristics and treatment results of 5 patients with fibrous dysplasia and review of the literature. Case Rep Endocrinol 2015; 2015: 670809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Parisi MS, Oliveri B. Long‐term pamidronate treatment of polyostotic fibrous dysplasia of bone: a case series in young adults. Curr Ther Res Clin Exp 2009; 70: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parisi MS, Oliveri B, Mautalen CA. Effect of intravenous pamidronate on bone markers and local bone mineral density in fibrous dysplasia. Bone 2003; 33: 582–588. [DOI] [PubMed] [Google Scholar]
- 64. Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab 2003; 88: 4569–4575. [DOI] [PubMed] [Google Scholar]
- 65. Thomsen MD, Rejnmark L. Clinical and radiological observations in a case series of 26 patients with fibrous dysplasia. Calcif Tissue Int 2014; 94: 384–395. [DOI] [PubMed] [Google Scholar]
- 66. Zacharin M, O'Sullivan M. Intravenous pamidronate treatment of polyostotic fibrous dysplasia associated with the McCune Albright syndrome. J Pediatr 2000; 137: 403–409. [DOI] [PubMed] [Google Scholar]
- 67. Chapurlat RD. Medical therapy in adults with fibrous dysplasia of bone. J Bone Miner Res 2006; 21 (Suppl. 2): P114–P119. [DOI] [PubMed] [Google Scholar]
- 68. Kochar IP, Kulkarni KP. Pamidronate for fibrous dysplasia due to McCune Albright syndrome. Indian Pediatr 2010; 47: 633–635. [PubMed] [Google Scholar]
- 69. Parisi MS, Oliveri MB, Mautalen CA. Bone mineral density response to long‐term bisphosphonate therapy in fibrous dysplasia. J Clin Densitom 2001; 4: 167–172. [DOI] [PubMed] [Google Scholar]
- 70. Chapurlat R. Current pharmacological treatment for fibrous dysplasia and perspectives for the future. Joint Bone Spine 2005; 72: 196–198. [DOI] [PubMed] [Google Scholar]
- 71. DiMeglio LA. Bisphosphonate therapy for fibrous dysplasia. Pediatr Endocrinol Rev 2007; 4 (Suppl. 4): 440–445. [PubMed] [Google Scholar]
- 72. Robinson C, Collins MT, Boyce AM. Fibrous dysplasia/McCune‐Albright syndrome: clinical and translational perspectives. Curr Osteoporos Rep 2016; 14: 178–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brumsen C, Hamdy NA, Papapoulos SE. Osteoporosis and bone marrow mastocytosis: dissociation of skeletal responses and mast cell activity during long‐term bisphosphonate therapy. J Bone Miner Res 2002; 17: 567–569. [DOI] [PubMed] [Google Scholar]
- 74. Metwally T, Burke A, Tsai JY, Collins MT, Boyce AM. Fibrous dysplasia and medication‐related osteonecrosis of the jaw. J Oral Maxillofac Surg 2016; 74: 1983–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Palmisano B, Labella R, Spica E, Remoli C, Corsi A, Robey PG, et al. Anti‐RANKL treatment in a murine model of fibrous dysplasia, in Bone Abstracts (2017) 2017: ICCBH 2017.
- 76. Branstetter DG, Nelson SD, Manivel JC, Blay JY, Chawla S, Thomas DM, et al Denosumab induces tumor reduction and bone formation in patients with giant‐cell tumor of bone. Clin Cancer Res 2012; 18: 4415–4424. [DOI] [PubMed] [Google Scholar]
- 77. Yamagishi T, Kawashima H, Ogose A, Ariizumi T, Sasaki T, Hatano H, et al Receptor‐activator of nuclear KappaB ligand expression as a new therapeutic target in primary bone tumors. PLoS One 2016; 11: e0154680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Benhamou J, Gensburger D, Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine 2014; 81: 549–550. [DOI] [PubMed] [Google Scholar]
- 79. Eller‐Vainicher C, Rossi DS, Guglielmi G, Beltramini GA, Cairoli E, Russillo A, et al Prompt clinical and biochemical response to denosumab in a young adult patient with craniofacial fibrous dysplasia. Clin Cases Miner Bone Metab 2016; 13: 253–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ganda K, Seibel MJ. Rapid biochemical response to denosumab in fibrous dysplasia of bone: report of two cases. Osteoporos Int 2014; 25: 777–782. [DOI] [PubMed] [Google Scholar]
- 81. Wang HD, Boyce AM, Tsai JY, Gafni RI, Farley FA, Kasa‐Vubu JZ, et al Effects of denosumab treatment and discontinuation on human growth plates. J Clin Endocrinol Metab 2014; 99: 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Appelman‐Dijkstra NM, Majoor BC, Dijkstra PDS, Papapoulos SE, Hamdy NAT. Efficacy and safety of denosumab treatment in bisphosphonate‐resistant fibrous dysplasia: a case series. ASBMR Abstracts 2018, 2018.
- 83. Greenfield EM, Bi Y, Miyauchi A. Regulation of osteoclast activity. Life Sci 1999; 65: 1087–1102. [DOI] [PubMed] [Google Scholar]
- 84. Greenfield EM, Horowitz MC, Lavish SA. Stimulation by parathyroid hormone of interleukin‐6 and leukemia inhibitory factor expression in osteoblasts is an immediate‐early gene response induced by cAMP signal transduction. J Biol Chem 1996; 271: 10984–10989. [DOI] [PubMed] [Google Scholar]
- 85. Sehgal PB. Interleukin‐6: molecular pathophysiology. J Invest Dermatol 1990; 94 (Suppl. 6): 2S–6S. [DOI] [PubMed] [Google Scholar]
- 86. Motomura T, Kasayama S, Takagi M, Kurebayashi S, Matsui H, Hirose T, et al Increased interleukin‐6 production in mouse osteoblastic MC3T3‐E1 cells expressing activating mutant of the stimulatory G protein. J Bone Miner Res 1998; 13: 1084–1091. [DOI] [PubMed] [Google Scholar]
- 87. Yamamoto T, Ozono K, Kasayama S, Yoh K, Hiroshima K, Takagi M, et al Increased IL‐6‐production by cells isolated from the fibrous bone dysplasia tissues in patients with McCune‐Albright syndrome. J Clin Invest 1996; 98: 30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. de Boysson H, Johnson A, Hablani N, Hajlaoui W, Auzary C, Geffray L. Tocilizumab in the treatment of a polyostotic variant of fibrous dysplasia of bone. Rheumatology (Oxford) 2015; 54: 1747. [DOI] [PubMed] [Google Scholar]
- 89. Faruqi T, Dhawan N, Bahl J, Gupta V, Vohra S, Tu K, et al Molecular, phenotypic aspects and therapeutic horizons of rare genetic bone disorders. Biomed Res Int 2014; 2014: 670842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Khan SK, Yadav PS, Elliott G, Hu DZ, Xu R, Yang Y. Induced Gnas(R201H) expression from the endogenous Gnas locus causes fibrous dysplasia by up‐regulating Wnt/beta‐catenin signaling. Proc Natl Acad Sci U S A 2018; 115: E418–E427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lv M, Li X, Huang Y, Wang N, Zhu X, Sun J. Inhibition of fibrous dysplasia via blocking Gsalpha with suramin sodium loaded with an alendronate‐conjugated polymeric drug delivery system. Biomater Sci 2016; 4: 1113–1122. [DOI] [PubMed] [Google Scholar]
- 92. Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, et al Burosumab therapy in children with X‐linked hypophosphatemia. N Engl J Med 2018; 378: 1987–1998. [DOI] [PubMed] [Google Scholar]
- 93. Cohen S, Levi‐Montalcini R, Hamburger V. A nerve growth‐stimulating factor isolated from Sarcom as 37 and 180. Proc Natl Acad Sci U S A 1954; 40: 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shu X‐Q, Mendell LM. Neurotrophins and hyperalgesia. Proc Natl Acad Sci 1999; 96: 7693–7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Halvorson KG, Kubota K, Sevcik MA, Lindsay TH, Sotillo JE, Ghilardi JR, et al A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Res 2005; 65: 9426–9435. [DOI] [PubMed] [Google Scholar]
- 96. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 2017; 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]