

Abstract
Aims
Trifluridine/tipiracil (FTD/TPI) prolongs survival in refractory metastatic colorectal cancer, but limited data exist on its use in patients with hepatic impairment. This Phase I, open‐label, nonrandomized study investigated the safety, tolerability and pharmacokinetics of FTD/TPI in patients with advanced solid tumours (except breast cancer) and varying degrees of hepatic impairment, to provide dosing recommendations.
Methods
Patients aged ≥18 years with advanced solid tumours and normal hepatic function, or mild, moderate or severe hepatic impairment according to National Cancer Institute criteria, were planned to be enrolled. Patients received FTD/TPI 35 mg/m2 orally twice daily on days 1–5 and 8–12 of each 28‐day cycle.
Results
Twenty‐four patients were enrolled to the normal hepatic function (n = 8) and mild (n = 10) and moderate (n = 6) hepatic impairment cohorts. Overall, 12 patients (50.0%) had at least 1 adverse event leading to study discontinuation. In the moderate hepatic impairment cohort, 5 of 6 patients experienced grade ≥ 3 elevation in bilirubin. No patients with severe hepatic impairment were enrolled. FTD area under the curve at steady state decreased by 18% and 22% in the mild and moderate cohorts, respectively; however, no clear change was observed in TPI area under the curve.
Conclusions
FTD/TPI can be safely administered in patients with normal hepatic function and mild hepatic impairment, with no initial dose adjustment. FTD/TPI is not recommended for use in patients with moderate hepatic impairment because of findings of grade 3 or 4 increased blood bilirubin. Therefore, FTD/TPI is not recommended for patients with moderate or severe hepatic impairment.
Keywords: chemotherapy, drug development, gastroenterology, liver, oncology, pharmacokinetics, phase I
What is already known about this subject
There are limited data on trifluridine/tipiracil (FTD/TPI) use in patients with advanced cancer and hepatic impairment, which may alter the pharmacokinetics of FTD/TPI
It is yet to be understood if the dose of FTD/TPI will require adjustments to maintain appropriate exposure in patients with varying degrees of hepatic impairment
What this study adds
Exposure to FTD or TPI was not increased by hepatic impairment in patients with advanced solid tumours (excluding breast cancer)
FTD/TPI can be safely administered to patients with normal hepatic function or mild hepatic impairment
FTD/TPI treatment is not recommended for patients with moderate or severe hepatic impairment
1. INTRODUCTION
Trifluridine/tipiracil (FTD/TPI) is a novel oral chemotherapy combination of a cytotoxic thymidine‐based nucleoside analogue, FTD, and a thymidine phosphorylase inhibitor, TPI, at a molar ratio of 1:0.5. After phosphorylation, FTD is incorporated into DNA, which leads to cell‐cycle arrest and cell death.1 Degradation of FTD is inhibited by TPI, which results in sustained plasma concentrations of FTD after oral administration.2 Preclinical xenograft studies in mice have demonstrated cytotoxic effects of FTD/TPI on tumours that have relapsed after treatment with fluoropyrimidine therapy.1, 3
In humans, after oral administration of FTD/TPI, both FTD and TPI are rapidly absorbed and eliminated from the plasma, with an apparent terminal‐phase elimination half‐life of 1.5–4 hours.4 FTD is primarily eliminated via metabolism in the intestinal tract and liver by thymidine phosphorylase to trifluoromethyluracil, the major metabolite. Administered TPI is excreted as its unchanged form, mainly in faeces and also in urine.5
In several Phase I and II clinical trials, FTD/TPI has shown promising safety and efficacy results when dosed in 28‐day cycles, each comprising 5 days of treatment followed by a 2‐day rest period each week for 2 weeks, and then a 14‐day rest period.4, 6, 7, 8, 9, 10, 11 The global, Phase III RECOURSE trial (NCT01607957) in patients with refractory metastatic colorectal cancer who were previously treated with standard and targeted chemotherapy demonstrated significant improvements with FTD/TPI vs placebo in overall survival (7.2 vs 5.2 months, respectively [hazard ratio 0.69; P < .001]) and progression‐free survival (2.0 vs 1.7 months, respectively [hazard ratio 0.48; P < .001]).12, 13
There are limited data surrounding the use of FTD/TPI in patients with advanced cancer and hepatic impairment. Impaired hepatic function, which can be caused by metastatic liver disease and direct hepatic toxicity from chemotherapy or chemotherapy‐associated hypersensitivity reactions,14 may alter the pharmacokinetics of FTD/TPI. As a result, drug metabolism can be affected and nonhepatic toxicity can occur.14 The dose of FTD/TPI may require adjustments to maintain appropriate exposure for efficacy and minimize changes to the safety profile. Further research was needed in order to provide specific dosing recommendations of FTD/TPI for patients with varying degrees of hepatic impairment.
The objective of this study was to evaluate the safety, tolerability and pharmacokinetic profiles of the components of FTD/TPI in patients with advanced solid tumours (except breast cancer) and varying degrees of hepatic impairment, in order to provide specific FTD/TPI dosing recommendations for these patients.
2. METHODS
2.1. Study design
This was a Phase I, open‐label, nonrandomized study (NCT02301104) conducted at 7 study centres in the USA. The study had two parts: the pharmacokinetic part and the extension part (Figure S1). In the pharmacokinetic part, patients were planned to be enrolled into the normal hepatic function cohort or the mild, moderate or severe hepatic impairment cohort based on National Cancer Institute Hepatic Impairment Classification Criteria (Table 1). For study inclusion, patients had to fulfil both the total bilirubin and the aspartate aminotransferase criteria. Patients received the same FTD/TPI dosing regimen as in the RECOURSE trial of 35 mg/m2 orally twice daily, and administration of FTD/TPI occurred on days 1–5 (recovery period on days 6 and 7) and days 8–12 (recovery period on days 13–28) of a 28‐day treatment cycle (Figure S1).12 The pharmacokinetic results from the mild and moderate cohorts would be used to determine dosing for the enrolment of patients with severe hepatic impairment.
Table 1.
National Cancer Institute hepatic impairment classification criteria
| Liver function assessment | Normal | Mild [Link] | Moderate | Severe |
|---|---|---|---|---|
| Total bilirubin | ≤ULN |
B1: ≤ULN B2: 1–1.5 × ULN |
1.5–3 × ULN | >3 × ULN |
| Aspartate aminotransferase | ≤ULN |
B1: >ULN B2: Any[Link] |
Any | Any |
Mild liver dysfunction may be defined according to either of the two criteria (B1 and B2), so patients in the mild hepatic impairment cohort may come from either of these groups and are combined for all analyses.
‘Any’ denotes no specific limit of aspartate aminotransferase for a mild, moderate or severe classification assuming that total bilirubin requirements have been met.
ULN, upper limit of normal.
Patients who completed the pharmacokinetic part (28 days) were eligible to enter the extension part, in which patients continued the 28‐day dosing schedule for FTD/TPI until any of the treatment discontinuation criteria were met. Treatment discontinuation criteria included: patient request at any time irrespective of the reason; disease progression; irreversible, treatment‐related, grade 4, clinically relevant adverse event (AE) or the 4th occurrence of a treatment‐related, grade 3, clinically relevant, nonhaematological event; and an unacceptable AE or change in underlying condition such that the patient could no longer tolerate therapy, including the need for more than 3 dose reductions of FTD/TPI, a maximum dose delay of more than 28 days from the scheduled start date of the next cycle of study medication, investigator decision and pregnancy.
2.2. Patient population
Eligible patients had to be aged ≥18 years with histologically, cytologically or radiologically confirmed advanced solid tumours (excluding breast cancer) and have an Eastern Cooperative Oncology Group performance status of 2 or better and hepatic function as described above. Patients must also have failed or been intolerant of standard therapy. The key exclusion criteria were: a serious illness or medical condition, or brain or leptomeningeal metastasis; active infection; pleural effusion; pericardial fluid requiring drainage in the previous 2 weeks; intestinal obstruction; pulmonary fibrosis; acute renal/liver failure; uncontrolled diabetes; myocardial infarction in the previous 12 months; severe or unstable angina; New York Heart Association class III or IV congestive heart failure; gastrointestinal haemorrhage; known human immunodeficiency virus or acquired immunodeficiency syndrome‐related illness; active hepatitis B infection; autoimmune disorder requiring immunosuppressive therapy; psychiatric disease; major surgery; any investigational agent or shunt in the liver in the previous 4 weeks; any anticancer therapy in the previous 3 weeks; extended‐field radiation in the previous 4 weeks or limited‐field radiation in the previous 2 weeks; and any unresolved toxicity over grade 2 from any prior therapies. Pregnant or lactating females and patients who had previously received, or who the investigator considered to be inappropriate for, FTD/TPI treatment were also excluded.
The study was conducted in accordance with ethical principles originating in the Declaration of Helsinki 2008 and in compliance with Good Clinical Practice and all local and national regulatory guidelines. Study approval was obtained from the institutional review board at each site before that site enrolled any patients. All patients provided written, informed consent.
2.3. Safety
Safety end points were assessed by AEs, complete physical examination results, Eastern Cooperative Oncology Group performance status, vital sign measurements and laboratory evaluations. AEs were coded according to Medical Dictionary for Regulatory Activities version 17.1 terminology, and the severity of toxicities was graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. AEs were considered to be serious when they were life‐threatening, led to admission or extension of hospital stay, led to death, turned into persistent or significant disabilities or dysfunctions, or triggered congenital abnormalities or other medically important conditions. All serious AEs, treatment‐emergent AEs and AEs leading to treatment discontinuation were reported to Taiho Pharmacovigilance.
2.4. Pharmacokinetic assessments
The pharmacokinetic population included all patients in the as‐treated population with evaluable pharmacokinetic profiles on either day 1 or day 12, or both, of cycle 1. Pharmacokinetic analyses were conducted on plasma samples following administration of FTD/TPI on days 1 and 12. Blood samples were taken within 30 minutes before the dose (0 hours) and 0.5, 1, 2, 4, 6, 8, 10 and 12 hours after the dose to measure the concentrations of FTD and TPI. The concentrations of FTD and TPI in plasma were measured using validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) as previously described.5 For FTD analysis, samples were extracted with 1 mL t‐butyl methyl ether, dried and reconstituted with a mobile phase consisting of 0.1% acetic acid–methanol (75:25 v/v). Chromatographic separation was achieved on a Capcell Pak C18 AQ column (2.0 × 150 mm; Phenomenex Inc, Torrance, CA, USA) using isocratic elution with the mobile phase at a flow rate of 0.24 mL/min. An API 4000 LC–MS/MS System (SCIEX, Framingham, MA, USA) operating in negative ion mode was utilized to detect FTD. For TPI analysis, samples were extracted using Bond Elut PRS extraction cartridges (Agilent Technologies, Santa Clara, CA, USA), dried and reconstituted with a mobile phase consisting of 10 mmol ammonium acetate in water–methanol (90:10 v/v). Chromatographic separation was achieved on an Inertsil ODS‐3 column (2.1 × 150 mm; GL Sciences Inc, Tokyo, Japan) using isocratic elution with the mobile phase at a flow rate of 0.20 mL/min. An API 4000 LC–MS/MS System operating in positive ion mode was utilized to detect TPI. Stable isotope internal standards were added to each sample, calibration standard and quality‐control sample. Samples were analysed in multiple‐reaction monitoring mode by monitoring ion transitions at m/z 295.1 → 42.1 for FTD and m/z 243.2 → 183.0 for TPI, with collision energy adjusted between −40 and 25 V. The range of quantification for FTD was 5.00 ng/mL to 5000 ng/mL and dilutions up to 1:4 (v/v) were accurately quantitated. The range of quantification for TPI was 0.200 ng/mL to 200 ng/mL. Accuracy for FTD and TPI ranged from 88.2% to 107% and from 92.9% to 111.8%, respectively. Noncompartmental analysis using actual sampling times was applied to derive the pharmacokinetic parameters for FTD and TPI. Estimation of pharmacokinetic parameters was performed using Phoenix WinNonlin version 6.4 software (Certara USA, Inc., Princeton, NJ, USA). The FTD and TPI pharmacokinetic parameter for comparison of the normal liver function cohort with the hepatic impairment cohorts (mild and moderate) was maximum observed plasma concentration (Cmax). In addition, comparisons were performed for area under the plasma concentration–time curve from time 0 to the last measurable plasma concentration estimated by linear trapezoidal rule (AUC0‐last) and extrapolated to infinity (AUC0‐inf) by dividing the last quantifiable concentration by the terminal disposition rate constant (λz), which was estimated from at least the last 3 observations in the elimination phase by uniform weighting, area under the plasma concentration–time curve from time 0 to the end of the dosing interval (AUCtau), terminal‐phase elimination half‐life (t1/2) calculated by dividing ln(2) by λz, oral clearance (CL/F) and steady‐state oral clearance (CLss/F) calculated from the dose divided by AUC0‐inf for day 1 and AUCtau for day 12, respectively, accumulation ratio of geometric mean Cmax (Rc max) at day 12 vs day 1 and the lowest concentration of the drug before the next dose (Ctrough).
2.5. Statistical analyses
The study was designed as an open‐label, nonrandomized study with a sample size of 6 evaluable patients per cohort. Taking into account the individual variability in pharmacokinetic parameters and assuming a 25% drop‐out rate, approximately 8 patients per cohort were enrolled to obtain approximately 6 evaluable patients per cohort. Time of collection and plasma concentrations below the lower limit of quantification (LLOQ) were set to zero. Plasma concentrations below the LLOQ in the middle of the curve that were flanked by measurable concentrations were assigned a value of missing. Mean plasma concentrations were not presented if 50% or more of the values at any 1 time point in the terminal phase were below the LLOQ. The number of analysis results was defined as the number of actual, reported values as received from the bioanalytical laboratory before any transformations were made. Any missing value due to causes such as sample not drawn, sample not received, sample not suitable or result not reported did not contribute to the count of number of analysis results. Descriptive statistics for plasma concentrations, including number of observations (n), mean and standard deviation, were calculated and presented by cohort and analyte. The pharmacokinetic parameters AUC0‐inf, AUCtau, Cmax, CL/F, CLss/F, RCmax and Ctrough for FTD and TPI were analysed by analysis of variance using the categorical hepatic impairment cohorts as class variables. The mean ratios (each hepatic impairment cohort to the control cohort) for AUC0‐inf, AUCtau, CL/F and CLss/F were calculated. Comparisons were made between the control group (normal hepatic function) and each hepatic impairment cohort (mild or moderate). Point estimates with their corresponding 90% confidence interval (CI) were constructed and then back‐transformed from the log‐scale to express the estimates as ratios.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology.15
3. RESULTS
3.1. Patient disposition and baseline characteristics
Twenty‐four patients were enrolled into the normal hepatic function (n = 8), mild hepatic impairment (n = 10) and moderate hepatic impairment (n = 6) cohorts (Figure S2). The majority of patients had colorectal, pancreatic or biliary cancer (Table S1). All patients, with the exception of 1 who had advanced cancer, had metastatic cancer and reported prior treatment with anticancer therapy. Patients received metastatic, adjuvant, neoadjuvant or a combination of therapies, ranging from 1 to 7 regimens. All other baseline patient demographics and characteristics were balanced across cohorts (Table 2). Four, 7 and 5 patients in the normal hepatic function, mild hepatic impairment and moderate hepatic impairment cohorts, respectively, had liver metastases.
Table 2.
Patient demographics and baseline disease characteristics (treated population)
| Parameter | Normal hepatic function cohort (n = 8) | Mild hepatic impairment cohort (n = 10) | Moderate hepatic impairment cohort (n = 6) |
|---|---|---|---|
| Age, median (range), y | 64.0 (47–77) | 48.5 (37–65) | 62.0 (56–70) |
| Male | 4 (50.0) | 5 (50.0) | 4 (66.7) |
| Race, n (%) | |||
| Asian | 1 (12.5) | 1 (10.0) | 0 |
| Black or African American | 2 (25.0) | 0 | 0 |
| White | 5 (62.5) | 9 (90.0) | 6 (100) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 0 | 0 | 1 (16.7) |
| Not Hispanic or Latino | 8 (100) | 10 (100) | 5 (83.3) |
| Height, mean (SD), cm | 165.80 (10.75) | 168.15 (7.76) | 167.87 (9.57) |
| Weight, mean (SD), kg | 73.20 (27.51) | 75.49 (17.79) | 76.63 (14.64) |
| Body surface area, mean (SD), m2 | 1.79 (0.34) | 1.85 (0.22) | 1.88 (0.21) |
| Bilirubin, mean (SD), μM | 8.55 (3.17) | 13.51 (6.42) | 45.32 (12.65) |
| Aspartate aminotransferase, mean (SD), U/L | 25.63 (7.96) | 55.60 (15.24) | 64.17 (24.21) |
| Number of prior regimens meana (range) | 3.4 (1–7) | 3.5 (1–7) | 3.3 (0–7) |
All prior regimens in neoadjuvant, adjuvant and metastatic settings.
SD, standard deviation.
3.2. Safety
All patients were evaluable for safety, and AEs were reported in all patients (Table 3). Twelve patients (50.0%) had at least 1 AE leading to study discontinuation, with 1 AE outcome of death. In this patient, the cause of death was a small intestinal obstruction caused by disease progression and was not considered to be related to FTD/TPI. In the moderate hepatic impairment cohort, 5 of 6 patients experienced grade ≥ 3 elevations in bilirubin (Figure 1); therefore, no patients were enrolled into the severe hepatic impairment cohort as initially planned.
Table 3.
Summary of adverse events (AEs, treated population)
| Patients with AEs, n (%) | Normal hepatic function cohort (n = 8) | Mild hepatic impairment cohort (n = 10) | Moderate hepatic mpairment cohort (n = 6) |
|---|---|---|---|
| AEs | 8 (100) | 10 (100) | 6 (100) |
| Serious AEs | 4 (50.0) | 6 (60.0) | 4 (66.7) |
| Grade ≥ 3 AEs | 7 (87.5) | 9 (90.0) | 6 (100) |
| Treatment‐emergent AEs | 7 (87.5) | 10 (100) | 6 (100) |
| AEs leading to discontinuation of study drug | 2 (25.0) | 5 (50.0) | 5 (83.3) |
| AEs with outcome of death | 0 | 1 (10.0) | 0 |
AE, adverse event.
Figure 1.
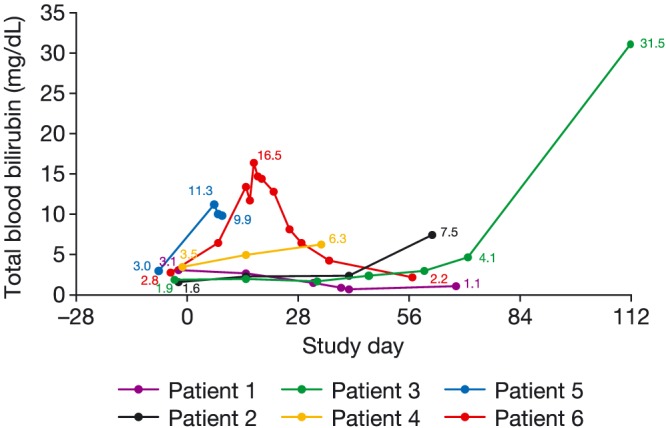
Total blood bilirubin levels in the moderate hepatic impairment cohortPatient 3 had a grade 3 bile duct obstruction which was not related to the study treatment. No reason was given for the bile duct obstruction and it was not due to disease progression
The most common treatment‐emergent AEs included nausea (45.8%), diarrhoea (25.0%), fatigue (25.0%), increased blood bilirubin (20.8%), anaemia (20.8%), constipation (16.7%) and vomiting (16.7%). Neutropenia (all grades) occurred in 20.8% of patients, and a further 12.5% of patients reported decreased neutrophil count. Two patients reported treatment‐emergent serious AEs of increased bilirubin, 1 patient reported bacteraemia in the moderate hepatic impairment cohort, and 1 patient reported hyponatraemia in the mild hepatic impairment cohort.
3.3. Pharmacokinetics
Twenty‐three patients (1 patient in the mild hepatic impairment cohort had no data available) were included in the pharmacokinetic assessment population. The reasons for exclusion of patients from the pharmacokinetic population for each cohort are listed in Supplementary Table S2.
Mean plasma concentration–time profiles by cohorts are shown in Figures 2 and 3 for FTD and TPI, respectively, after the first administration (cycle 1, day 1) or after multiple‐dose administrations (cycle 1, day 12). For all analytes, the mean concentrations were generally comparable across the 3 cohorts. However, the mean plasma concentrations of FTD were slightly lower in the elimination phase in the mild and moderate hepatic impairment cohorts at both days 1 and 12 in cycle 1. By contrast, the mean TPI plasma concentration was slightly higher in the moderate hepatic impairment cohort at both day 1 and day 12 in cycle 1.
Figure 2.
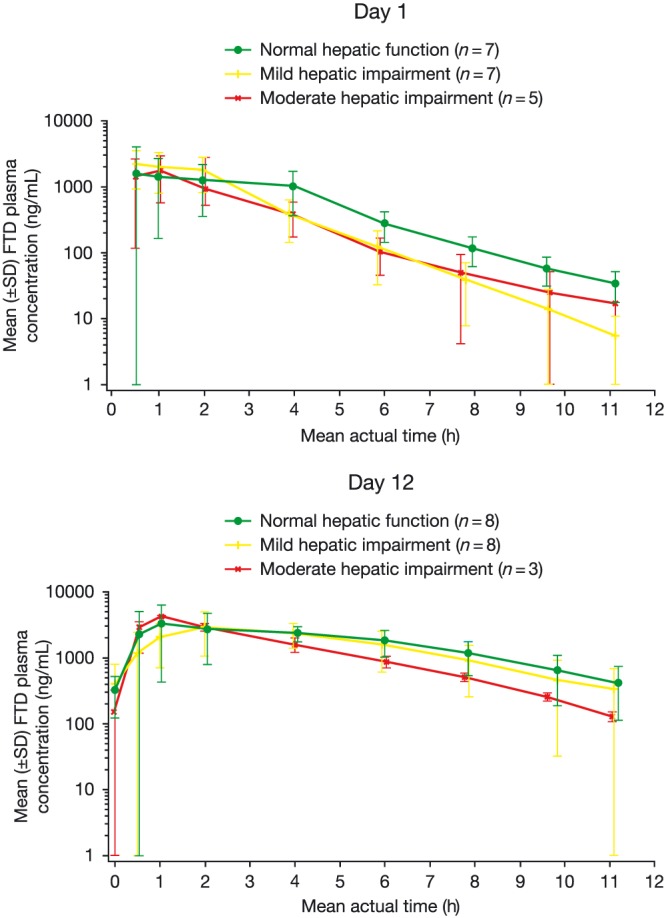
Mean plasma concentration–time profile: FTD (pharmacokinetic population) FTD, trifluridine; SD, standard deviation
Figure 3.
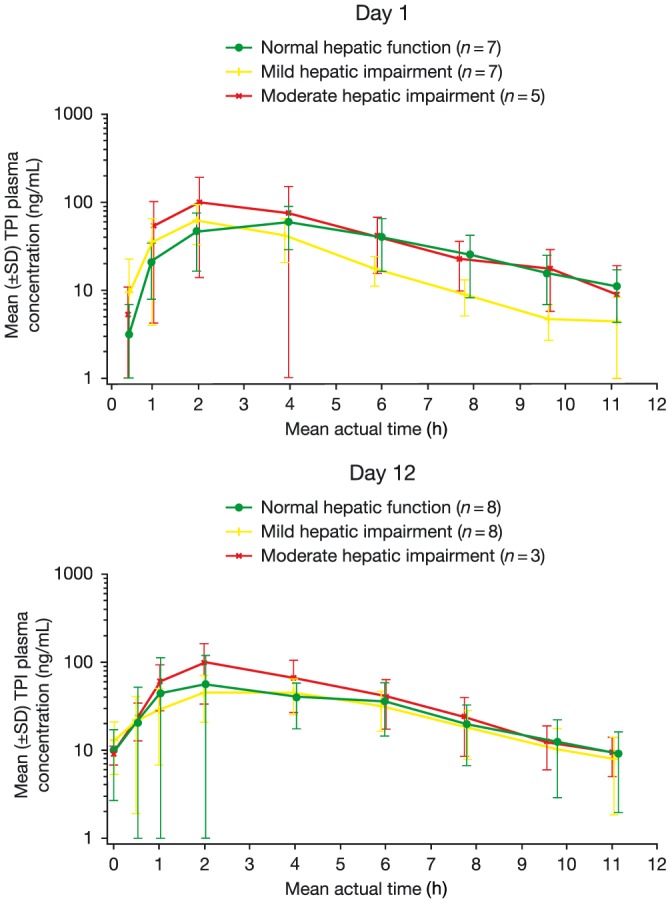
Mean plasma concentration–time profile: TPI (pharmacokinetic population) SD, standard deviation; TPI, tipiracil
At day 1 for Cmax, the geometric mean ratio for mild hepatic impairment/normal was 1.43 (90% CI 0.84, 2.43; P = .26) for FTD and 0.89 (90% CI 0.51, 1.58; P = .73) for TPI; moderate hepatic impairment/normal was 0.78 (90% CI 0.43, 1.39; P = .46) for FTD and 1.23 (90% CI 0.66, 2.30; P = .57) for TPI. At day 12 for Cmax, the geometric mean ratio for mild hepatic impairment/normal was 0.87 (90% CI 0.64, 1.19; P = .44) for FTD and 0.99 (90% CI 0.59, 1.65; P = .97) for TPI; moderate hepatic impairment/normal was 1.00 (90% CI 0.66, 1.52; P > .999) for FTD and 1.45 (90% CI 0.72, 2.90; P = .37) for TPI.
Overall, no significant differences in AUC values for FTD were observed between the normal, mild and moderate cohorts. However, the mean AUC0‐inf values for FTD at day 1 tended to decrease with severity of hepatic impairment, which was consistent with the trend observed at day 12 (Table 4). At day 1 for AUC0‐inf, the geometric mean ratio for mild hepatic impairment/normal was 0.92 (90% CI 0.66, 1.26; P = .67) and moderate hepatic impairment/normal was 0.67 (90% CI 0.46, 0.97; P = .08). At day 12 for AUCtau, the geometric mean ratio for mild hepatic impairment/normal was 0.82 (90% CI 0.61, 1.11; P = .27); moderate hepatic impairment/normal was 0.78 (90% CI 0.52, 1.16; P = .29).
Table 4.
Plasma pharmacokinetic parameters for FTD/TPI on days 1 and 12 of cycle 1
| FTD | TPI | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | Mild | Moderate | Normal | Mild | Moderate | ||||||||
| n | Mean ± SD | n | Mean ± SD | n | Mean ± SD | n | Mean ± SD | n | Mean ± SD | n | Mean ± SD | ||
| Day 1 | |||||||||||||
| Cmax, ng/mL | 7 | 2615 ± 2107 | 7 | 3061 ± 781 | 5 | 1933 ± 1175 | 7 | 68.27 ± 27.65 | 7 | 64.89 ± 32.18 | 5 | 105.20 ± 84.80 | |
| AUC0‐inf, ng h/mL | 7 | 6873 ± 2407 | 7 | 6324 ± 2208 | 5 | 4594 ± 1586 | 6 | 421 ± 208 | 6 | 272 ± 120 | 4 | 591 ± 419 | |
| CL/F, L/h | 7 | 9.79 ± 4.49 | 7 | 11.10 ± 4.57 | 5 | 15.02 ± 4.15 | 6 | 84.44 ± 52.01 | 6 | 137.33 ± 83.39 | 4 | 85.69 ± 64.61 | |
| t1/2, h | 7 | 1.78 ± 0.49 | 7 | 1.22 ± 0.28 | 5 | 1.49 ± 0.61 | 6 | 2.42 ± 0.54 | 6 | 2.02 ± 0.48 | 4 | 2.71 ± 1.34 | |
| Ctrough, ng/mL | 7 | 35 ± 17 | 7 | 6 ± 5 | 4 | 17 ± 17 | 7 | 10.82 ± 6.47 | 7 | 4.44 ± 3.81 | 4 | 9.01 ± 10.29 | |
| Day 12 | |||||||||||||
| Cmax, ng/mL | 8 | 4669 ± 1996 | 8 | 3860 ± 1232 | 3 | 4277 ± 153 | 8 | 72.50 ± 60.77 | 8 | 60.18 ± 21.07 | 3 | 98.87 ± 65.27 | |
| AUCtau, ng h/mL | 7 | 20392 ± 5609 | 8 | 17489 ± 7379 | 3 | 15406 ± 1244 | 7 | 335 ± 230 | 7 | 305 ± 112 | 3 | 495 ± 288 | |
| CLss/F, L/h | 7 | 3.15 ± 1.00 | 8 | 4.25 ± 1.81 | 3 | 4.36 ± 0.55 | 7 | 118.27 ± 62.92 | 7 | 110.71 ± 47.47 | 3 | 90.86 ± 71.81 | |
| t1/2, h | 7 | 2.14 ± 0.38 | 8 | 1.97 ± 0.69 | 3 | 1.93 ± 0.35 | 7 | 2.68 ± 0.81 | 7 | 2.35 ± 0.55 | 3 | 2.58 ± 0.49 | |
| Ctrough, ng/mL | 8 | 320 ± 199 | 7 | 334 ± 361 | 3 | 131 ± 22 | 8 | 9.10 ± 7.12 | 7 | 7.98 ± 6.13 | 3 | 9.03 ± 2.23 | |
| Rc max | 7 | 1.89 | 6 | 1.19 | 2 | 2.01 | 7 | 0.94 | 6 | 0.91 | 2 | 1.07 | |
AUC0‐inf, area under the plasma concentration–time curve from time 0 to infinity; AUCtau, area under the plasma concentration–time curve from time 0 to the end of the dosing interval; CL/F, oral clearance; CLss/F, steady‐state oral clearance; Cmax, maximum observed plasma concentration; Ctrough, lowest plasma concentration before next dose; FTD, trifluridine; Rc max, accumulation ratio of geometric mean Cmax at day 12/Cmax at day 1; SD, standard deviation; t1/2, apparent terminal‐phase elimination half‐life; TPI, tipiracil.
The CL/F for FTD tended to increase with the severity of hepatic impairment (Table 4). At day 1 for CL/F, the geometric mean ratio for mild hepatic impairment/normal was 1.16 (90% CI 0.79, 1.68; P = .51); moderate hepatic impairment/normal was 1.63 (90% CI 1.08, 2.45; P = .06). The trend was consistent with the results at day 12 for CLss/F, when the geometric mean ratio for mild hepatic impairment/normal was 1.31 (90% CI 0.95, 1.80; P = .16); moderate hepatic impairment/normal was 1.44 (90% CI 0.94, 2.20; P = .16).
Similar to FTD, no significant difference in AUC values for TPI was observed (Table 4). However, the mean AUC0‐inf values at day 1 and AUCtau values at day 12 for TPI were reduced in the mild hepatic impairment cohort and increased in the moderate hepatic impairment cohort. At day 1 for AUC0‐inf, the geometric mean ratio for mild hepatic impairment/normal was 0.64 (90% CI 0.35, 1.18; P = .22); moderate hepatic impairment/normal was 1.21 (90% CI 0.61, 2.38; P = .63). At day 12 for AUCtau, the geometric mean ratio for mild hepatic impairment/normal was 1.00 (90% CI 0.61, 1.65; p = 1.00); moderate hepatic impairment/normal was 1.46 (90% CI 0.77, 2.78; P = .31). The CL/F for TPI tended to be higher in the mild hepatic impairment cohort and comparable in the moderate cohort at day 1 (the geometric mean ratio for mild hepatic impairment/normal was 1.69; moderate hepatic impairment/normal was 0.95). On day 12, the trend was generally consistent for patients with moderate hepatic impairment, but the pharmacokinetic parameters were comparable in patients with mild hepatic impairment (for AUC0‐last, AUCtau and CLss/F, respectively, the geometric mean ratios for mild hepatic impairment/normal were 0.98, 1.00 and 1.05; moderate hepatic impairment/normal were 1.38, 1.46 and 0.76). Ctrough values for the normal, mild and moderate cohorts were increased on day 12 vs day 1 for FTD, but were similar for TPI. RCmax ranged from 1.19 to 2.01 for FTD and from 0.91 to 1.07 for TPI.
Individual pharmacokinetic parameters for patients with normal hepatic function and moderate hepatic impairment who had grade 3 or 4 total blood bilirubin are given in Supplementary Tables S3 and S4 (cycle 1, day 1 and cycle 1, day 12, respectively). No trend was seen for individual pharmacokinetic parameters for FTD or TPI in these patients.
4. DISCUSSION
Due to limited data on the use of FTD/TPI in patients with hepatic impairment, this study was designed to investigate the safety, tolerability and pharmacokinetics of FTD/TPI in patients with advanced solid tumours (except breast cancer) and varying degrees of hepatic impairment after single‐ and multiple‐dose oral administrations, in order to provide specific dosing recommendations for these patients.
A review of the exposure (AUC) of both FTD and TPI showed no overexposure in patients with moderate hepatic impairment when compared with normal hepatic function and/or mild hepatic impairment. Rather, the final analysis suggested slightly lower AUC for FTD in patients with moderate hepatic impairment than in patients with normal hepatic function and/or mild hepatic impairment. The geometric mean AUCtau of FTD at steady state was 18% lower in patients with mild hepatic impairment and 22% lower in patients with moderate hepatic impairment than seen in patients with normal hepatic function. By contrast, no consistent trend was observed for the geometric mean AUCtau of TPI at steady state. AUCtau was only slightly higher in the moderate cohort on cycle 1, day 12, which may be a consequence of the large variability in plasma TPI concentrations and the limited data size of only 3 patients; therefore, the occurrences of grade 3 or 4 increased total blood bilirubin were not associated with the level of FTD or TPI exposure.
No correlation was seen for FTD or TPI CL/F and hepatic function parameters, and half‐life times after repeated administration were comparable between patients with normal hepatic function and mild and moderate hepatic impairment. The exposure to FTD or TPI was not increased by hepatic impairment, and the patients who experienced grade 3 or 4 increased total blood bilirubin were not overexposed to FTD or TPI. Based on the findings from this study, reducing the initial starting dose of FTD/TPI is not recommended for patients with moderate hepatic impairment because of the risk of underexposure to FTD. The difference between CL/F at day 1 and CLss/F at day 12 is due to an increase in AUC following repeated dosing of FTD/TPI. The half‐life also becomes larger at day 12 than at day 1 as the elimination is decreased. However, the mechanism by which this occurs is not clear. Ctrough of FTD was higher on day 12 than on day 1, and the accumulation ratios ranged from 1.19 to 2.01, indicating the concentration of FTD increased after repeated doses. By contrast, Ctrough of TPI on day 12 was similar to that on day 1, and the accumulation ratio was approximately 1, indicating the concentration of TPI did not change significantly after repeated doses. These results are in line with the pharmacokinetic results of a previous study.9The bioavailability of FTD and TPI in rats was 31% and 9%, respectively. The primary elimination pathway of FTD was metabolism by thymidine phosphorylase and renal excretion for TPI. In this study, the pharmacokinetics of TPI, an inhibitor of thymidine phosphorylase, did not change in patients with hepatic impairment and would result in no significant increase in the pharmacokinetics of FTD in patients with hepatic impairment.
With the exception of grade ≥ 3 increased blood bilirubin, the safety profile of FTD/TPI was consistent with that seen in previous clinical trials. Both of the serious AEs of increased blood bilirubin in patients in the normal cohort were considered related to disease progression and not to the study drug. In the moderate hepatic impairment cohort, the grade 3 AEs of increased blood bilirubin were nonserious and considered not related to the study drug. The other two AEs were grade 4 suspected, unexpected serious reactions and resulted in discontinuation of the study drug. All of the patients in the moderate cohort who experienced grade 3 or 4 AEs had liver metastases and a high alkaline phosphatase level at baseline, suggesting a cholestasis background. In patients with moderate hepatic impairment, the isolated blood bilirubin elevation findings might be attributed to the effect of 3 factors: liver metastasis status, moderate hepatic impairment status and FTD/TPI. Based on the findings of increased blood bilirubin in this trial, the current label for FTD/TPI does not recommend administration in patients with baseline moderate or severe hepatic impairment. However, while enrolment into the severe hepatic impairment cohort was not performed, the factors listed above may be confounding factors that could affect any causality assessment of FTD/TPI on increased blood bilirubin.
A limitation of this study was the small sample size of only 24 patients; however, this was sufficient to understand the effects of hepatic impairment on FTD/TPI pharmacokinetics.
5. CONCLUSIONS
This Phase I study has confirmed that exposure to FTD or TPI was not increased by hepatic impairment in patients with advanced solid tumours (excluding breast cancer), and the patients who experienced grade 3 or 4 increased total bilirubin were not overexposed to FTD or TPI. Along with the consistent safety profile of FTD/TPI, this study affirms that patients with mild hepatic impairment are able to receive FTD/TPI without the need for any initial dose reduction; FTD/TPI can be safely administered to patients with normal hepatic function or mild hepatic impairment. However, FTD/TPI is not recommended for use in patients with moderate hepatic impairment because of findings of increased blood bilirubin. Patients with severe hepatic impairment could not be studied. Therefore, FTD/TPI treatment is not recommended for patients with moderate or severe hepatic impairment.
COMPETING INTERESTS
M.W.S. has participated in speaker bureaus and received grant funding from Taiho Oncology, Inc. L.R.’s institution has received research funding from Taiho Oncology, Inc. W.S. and C.B. have received speaker fees from Taiho Oncology, Inc. F.Y., P.B. and R.W. are employees of Taiho Oncology, Inc. M.A.R. and D.R.S. have no financial disclosures to report.
CONTRIBUTORS
M.W.S., W.S., F.Y., P.B. and R.W. were involved in the study design. M.W.S., L.R., M.A.R., W.S., D.R.S. and C.B. contributed to data collection. M.A.R., W.S., F.Y., P.B. and R.W. were involved in data analysis and/or interpretation. All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. All authors take full responsibility for the scope, direction, content of and editorial decisions relating to the manuscript, were involved at all stages of development, and have approved the submitted manuscript.
Supporting information
Table S1.
Cancer diagnosis in the total patient population
Table S2. Evaluability of pharmacokinetic population†
Table S3. Individual pharmacokinetic parameters of trifluridine and tipiracil (cycle 1, day 1) of patients who experienced grade 3 or 4 increased total blood bilirubin
Table S4. Individual pharmacokinetic parameters of trifluridine and tipiracil (cycle 1, day 12) of patients who experienced grade 3 or 4 increased total blood bilirubin
Figure S1. Study design
Figure S2. Patient disposition
ACKNOWLEDGEMENTS
This work was supported by Taiho Oncology, Inc. and Taiho Pharmaceutical Co., Ltd. The authors received no compensation related to the development of the manuscript. Medical writing assistance was provided by Fiona Greig, PhD, at Complete HealthVizion, and was contracted and compensated by Taiho Oncology, Inc. Michelle A. Rudek was supported by NIH grant P30CA006973.
Saif MW, Rosen L, Rudek MA, et al. Open‐label study to evaluate trifluridine/tipiracil safety, tolerability and pharmacokinetics in patients with advanced solid tumours and hepatic impairment. Br J Clin Pharmacol. 2019;85:1239–1246. 10.1111/bcp.13856
The authors confirm that the PI for this paper is Dr Muhammad Wasif Saif and that he had direct clinical responsibility for patients.
REFERENCES
- 1. Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS‐102, exhibits antitumor activity in FU‐resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol. 2004;25(3):571‐578. [PubMed] [Google Scholar]
- 2. Fukushima M, Suzuki N, Emura T, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′‐deoxyribonucleosides. Biochem Pharmacol. 2000;59(10):1227‐1236. [DOI] [PubMed] [Google Scholar]
- 3. Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K. A novel antimetabolite, TAS‐102 retains its effect on FU‐related resistant cancer cells. Int J Mol Med. 2004;13:545‐549. [PubMed] [Google Scholar]
- 4. Hong DS, Abbruzzese JL, Bogaard K, et al. Phase I study to determine the safety and pharmacokinetics of oral administration of TAS‐102 in patients with solid tumors. Cancer. 2006;107(6):1383‐1390. [DOI] [PubMed] [Google Scholar]
- 5. Lee JJ, Seraj J, Yoshida K, et al. Human mass balance study of TAS‐102 using 14C analyzed by accelerator mass spectrometry. Cancer Chemother Pharmacol. 2016;77(3):515‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overman MJ, Kopetz S, Varadhachary G, et al. Phase I clinical study of three times a day oral administration of TAS‐102 in patients with solid tumors. Cancer Invest. 2008;26(8):794‐799. [DOI] [PubMed] [Google Scholar]
- 7. Overman MJ, Varadhachary G, Kopetz S, et al. Phase 1 study of TAS‐102 administered once daily on a 5‐day‐per‐week schedule in patients with solid tumors. Invest New Drugs. 2008;26(5):445‐454. [DOI] [PubMed] [Google Scholar]
- 8. Green MC, Pusztai L, Theriault RL, et al. Phase I study to determine the safety of oral administration of TAS‐102 on a twice daily (BID) schedule for five days a week (wk) followed by two days rest for two wks, every (Q) four wks in patients (pts) with metastatic breast cancer (MBC). J Clin Oncol. 2006;24(Suppl 18). abstract 10576 [Google Scholar]
- 9. Doi T, Ohtsu A, Yoshino T, Boku N, et al. Phase I study of TAS‐102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107(3):429‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshino T, Mizunuma N, Yamazaki K, et al. TAS‐102 monotherapy for pretreated metastatic colorectal cancer:a double‐blind, randomised, placebo‐controlled phase 2 trial. Lancet Oncol. 2012;13(10):993‐1001. [DOI] [PubMed] [Google Scholar]
- 11. Bendell JC, Rosen LS, Mayer RJ, et al. Phase 1 study of oral TAS‐102 in patients with refractory metastatic colorectal cancer. Cancer Chemother Pharmacol. 2015;76(5):925‐932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayer RJ, Van Cutsem E, Falcone A, et al. Randomized trial of TAS‐102 for refractory metastatic colorectal cancer. N Engl J Med. 2015;372(20):1909‐1919. [DOI] [PubMed] [Google Scholar]
- 13. Van Cutsem E, Mayer RJ, Laurent S, et al. The subgroups of the phase III RECOURSE trial of trifluridine/tipiracil (TAS‐102) versus placebo with best supportive care in patients with metastatic colorectal cancer. Eur J Cancer. 2018;90:63‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King PD, Perry MC. Hepatotoxicity of chemotherapy. Oncologist. 2001;6(2):162‐176. [DOI] [PubMed] [Google Scholar]
- 15. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Cancer diagnosis in the total patient population
Table S2. Evaluability of pharmacokinetic population†
Table S3. Individual pharmacokinetic parameters of trifluridine and tipiracil (cycle 1, day 1) of patients who experienced grade 3 or 4 increased total blood bilirubin
Table S4. Individual pharmacokinetic parameters of trifluridine and tipiracil (cycle 1, day 12) of patients who experienced grade 3 or 4 increased total blood bilirubin
Figure S1. Study design
Figure S2. Patient disposition