

Abstract
Cathepsin K (CatK) is a cysteine protease abundantly expressed by osteoclasts and localized in the lysosomes and resorption lacunae of these cells. CatK is the principal enzyme responsible for the degradation of bone collagen. Odanacatib is a selective, reversible inhibitor of CatK at subnanomolar potency. The pharmacokinetics of odanacatib have been extensively studied and are similar in young healthy men, postmenopausal women and elderly men, and were qualitatively similar throughout Phase 1 development and in‐patient studies. Following 3 weeks of 50 mg once weekly dosing the geometric mean area under the curve from 0 to 168 hours was 41.1 μM h, the concentration at 168 hours was 126 nM and the harmonic mean apparent terminal half‐life was 84.8 hr. Odanacatib exposure increased in a less than dose proportional manner due to solubility limited absorption. It is estimated that approximately 70% of the absorbed dose of odanacatib is eliminated via metabolism, 20% is excreted as unchanged drug in the bile or faeces, and 10% is excreted as unchanged drug in the urine. The systemic clearance was low (approximately 13 mL/min). Odanacatib decreases the degradation of bone matrix proteins and reduces the efficiency of bone resorption with target engagement confirmed by a robust decrease in serum C‐telopeptides of type 1 collagen (approximately 60%), urinary aminoterminal crosslinked telopeptides of type 1 collagen to creatinine ratio (approximately 50%) and total urine deoxypyridinoline/Cr (approximately 30%), with an increase in serum cross‐linked carboxy‐terminal telopeptide of type 1 collagen (approximately 55%). The 50‐mg weekly dosing regimen evaluated in Phase 3 achieved near maximal reduction in bone resorption throughout the treatment period. The extensive clinical programme for odanacatib, together with more limited clinical experience with other CatK inhibitors (balicatib and ONO‐5334), provides important insights into the clinical pharmacology of CatK inhibition and the potential role of CatK in bone turnover and mineral homeostasis. Key findings include the ability of this mechanism to: (i) provide sustained reductions in resorption markers, increases in bone mineral density, and demonstrated fracture risk reduction; (ii) be associated with relative formation‐sparing effects such that sustained resorption reduction is achieved without accompanying meaningful reductions in bone formation; and (iii) lead to increases in osteoclast number as well as other osteoclast activity (including build‐up of CatK enzyme), which may yield transient increases in resorption following treatment discontinuation and the potential for nonmonotonic responses at subtherapeutic doses.
Keywords: clinical trials statistics and study design, odanacatib, osteoporosis, pharmacodynamics, pharmacokinetics
1. INTRODUCTION
Odanacatib (MK‐0822) is an oral, selective and reversible inhibitor of cathepsin K (CatK) that has been studied as a potential new treatment option for postmenopausal women (PMW) and men with osteoporosis.1 Odanacatib is a potent inhibitor of human CatK (concentration causing 50% inhibition [IC50] = 0.2 nM) and exhibits 300‐fold selectivity to cathepsin S (CatS) and > 1000‐fold selectivity against other cathepsins (cathepsins B, C, F, L, V and Z in humans and bovine H).2 Osteoclastic bone resorption requires demineralization of inorganic bone mineral followed by degradation of organic bone matrix.3, 4, 5 These processes occur sequentially via 2 separate mechanisms. Demineralization requires acid secretion by a vacuolar proton pump into extracellular resorption lacunae on the bone surface; followed by CatK‐mediated digestion of demineralized collagen type I and other bone matrix proteins. CatK is a lysosomal cysteine protease that is abundantly expressed in osteoclasts. Under acidic conditions, CatK exhibits potent collagenolytic activity, cleaving the collagen helical region releasing N‐ and C‐terminal telopeptides of collagen type I (NTx and CTx), the principal bone matrix protein. Confirmation of the pivotal role of CatK in skeletal growth and remodelling in humans and its importance as a tractable osteoporosis target is underscored by the human null mutation for CatK, pycnodysostosis, a rare hereditary bone disorder manifesting a high bone mass phenotype.6
The clinical pharmacology programme that characterized the initial safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of odanacatib included 27 clinical pharmacology studies (835 subjects with 625 receiving odanacatib) as described in Table 1 and model‐based analyses of exposure and exposure–response based primarily on data from Phase 2 and 3 studies in men and women with osteoporosis.20, 21, 22, 23 The range of doses evaluated includes single oral doses of 2 mg to 600 mg (a 1‐mg intravenous [IV] dose was also evaluated). Numerous multiple‐dose regimens were assessed including multiple‐dose weekly administration of up to 100 mg/week, and multiple‐dose daily administration regimens up to 25 mg/day. The odanacatib programme completed multiple Phase 2 and 3 studies.24, 25, 26, 27, 28, 29, 30, 31, 32 Further clinical development of odanacatib was stopped following an observed increase in the risk of stroke in the Phase 3 Long‐term Odanacatib Fracture Trial (LOFT, NCT00529373).28, 29, 33 Although the odanacatib development programme did not proceed to submission for marketing approval, this programme represents the largest and most advanced programme targeting CatK to date and as such a review of this programme provides insight into the understanding of bone biology and impact of CatK perturbation. This review will focus on the odanacatib clinical pharmacology programme, drawing on published manuscripts and meeting abstracts.
Table 1.
List of odanacatib clinical pharmacology studies
| Study type | PN | Protocol short title | n = 837 | clinicaltrial.gov/reference |
|---|---|---|---|---|
| Bioavailability | P006 | Food effect | 8 | NCT008635257 |
| P015 | Formulation assessment #1 | 32 | NA | |
| P019 | Formulation assessment #2 | 40 | NA | |
| P045 | Intravenous bioavailability | 24 | 7 | |
| P071 | Formulation assessment #3 | 14 | NA | |
| Healthy subject PK and initial tolerability | P001 | First in human single dose | 36 | NCT008635908 |
| P002 | Once daily multiple dose | 62 | NCT007694189 | |
| P021 | Supratherapeutic multiple dose safety | 12 | Accepted for publication | |
| P005 | Once weekly multiple dose | 82 | NCT007701599 | |
| P013 | ADME | 6 | 10 | |
| Intrinsic factors | P010 | Single dose Japanese | 19 | 11 |
| P014 | Multiple dose Japanese | 50 | 11 | |
| P034 | Single/multiple dose Chinese female | 12 | 12 | |
| P026 | Hepatic insufficiency (25 mg) | 40 | NCT0151269313 | |
| P070 | Hepatic insufficiency (50 mg) | 17 | NCT0151269313 | |
| P027 | Renal insufficiency (25 mg) | 41 | NA | |
| P067 | Renal insufficiency (50 mg) | 25 | NCT0151266714 | |
| Extrinsic factors | P007 | Ketoconazole interaction | 15 | NA |
| P075 | Clarithromycin interaction | 12 | NA | |
| P023 | Diltiazem interaction | 12 | NA | |
| P055 | Rifampin interaction | 12 | 15 | |
| P056 | Prednisone interaction | 15 | 16 | |
| P024 | Digoxin interaction | 12 | 17 | |
| P025 | Warfarin interaction | 13 | 18 | |
| Healthy subject PD and PK/PD | P017 | Thorough QTc | 116 | Accepted for publication |
| P059 | Multiple dose older (50–75 yr) male | 44 | NCT0106826219 | |
| P039 | Low dose pharmacology in postmenopausal women | 66 | NA |
NA, not Available.
1.1. Pharmacokinetics
The odanacatib pharmacokinetic (PK) profile was qualitatively similar throughout Phase 1 development including in PMW and older men who are more representative of populations in which osteoporosis is prevalent.7, 8, 9, 19 Following 3 weeks of 50 mg once weekly dosing in healthy subjects, the geometric mean steady state area under the curve from 0 to 168 hours (AUC0‐168) was 41.1 μM h, maximum plasma concentration was 393 nM, concentration at 168 hours was 126 nM and the harmonic mean apparent terminal half‐life was 84.8 hours.9 Following oral administration, odanacatib typically exhibits an initial concentration peak at 4–8 hours postdose, followed by a secondary peak of varying magnitude at 24 hours postdose (Figure 1 Panel A).7 Individual odanacatib concentration‐time profiles occasionally exhibit secondary peaks at 48 hours postdose and later. Secondary peaks were also observed following intravenous dosing, indicating that they are related to systemic effects rather than absorption processes. Absorption modelling indicated that the secondary peaks could be consistent with enterohepatic recycling or circadian variations in volume mediated by binding in plasma.7 The combination of secondary peaks with a relatively long duration of absorption results in considerable individual variation in Tmax (median values ranged from 2 to 32 hours across all doses and studies) but with little meaningful effect on the overall shape of the concentration–time profile.7
Figure 1.
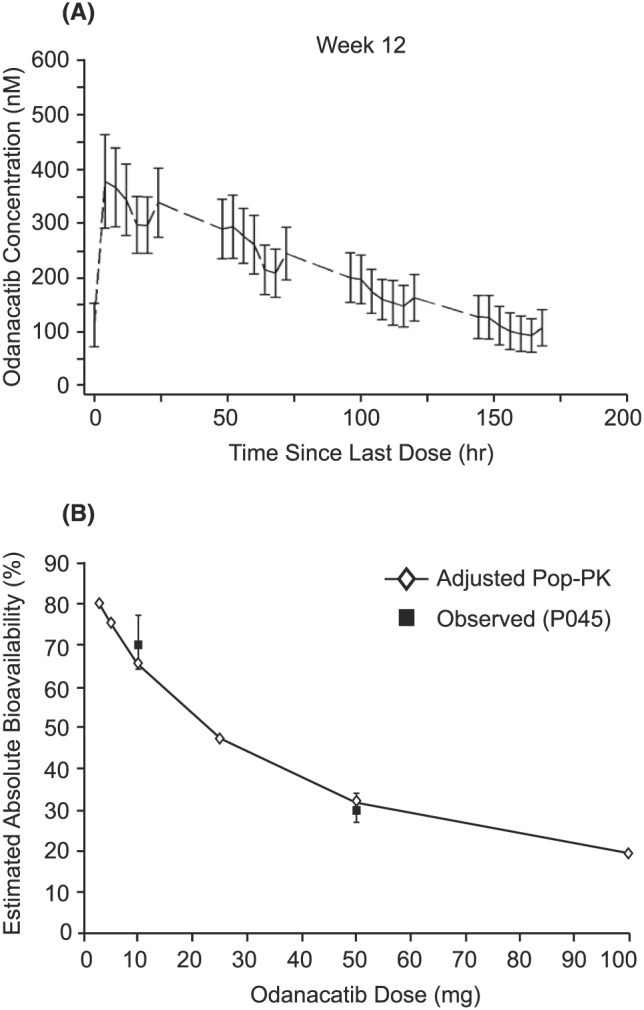
Odanacatib pharmacokinetics (PK). Panel A: representative odanacatib PK profile following 50 mg once weekly (mechanism study; n = 22); panel B: The absolute bioavailability of odanacatib (estimated and observed) decreases with increasing dose. Relative bioavailability results from the population (Pop) PK model21 were adjusted to reflect the absolute values through a scaling factor derived from the 10 and 50 mg absolute data from reference 7
As a result of the relatively long apparent terminal half‐life (approximately 80 hours) and the broad absorption peak observed in most individuals, the terminal elimination phase is not typically apparent until approximately 48 to 72 hours postdose. At the 50‐mg dose studied in Phase 3, this pharmacokinetic profile maintains plasma concentrations at levels associated with near maximal reduction of bone resorption biomarkers throughout the entire weekly dosing interval at steady‐state.9, 22, 23 Odanacatib exposure increases in a less than dose‐proportional manner (Figure 1, panel B).7, 8, 9 Relative to an IV reference dose, the mean oral bioavailability of odanacatib was 70% at 10 mg and 30% at 50 mg when administered to fasted PMW.7 When 50‐mg odanacatib was administered to PMW with low‐fat and high‐fat meals vs administered in the fasted state, the bioavailability of the 50‐mg dose increased to 35% and 49%, respectively, with corresponding increases in AUC0‐∞ of 15% and 63%.7 This is consistent with the hypothesis that odanacatib absorption, and thus bioavailability, is limited by solubility, and that administration with meals containing dietary lipids increases solubility of odanacatib, which is a lipophilic molecule.
Absorption modelling indicates that the majority of the compound is absorbed by 6–10 hours postdose (i.e. in 97% of individuals, >50% of the amount of drug that will be absorbed has been absorbed by 10 hours postdose) with almost complete absorption within 24 hours (i.e., in 88% of individuals, >80% of the amount of drug that will be absorbed has been absorbed by 24 hours postdose).7 This absorption behaviour for odanacatib is consistent with a low solubility BCS II compound; odanacatib has been shown to have very low solubility (<1 μg/mL) in both aqueous buffers and simulated intestinal fluids. All the Phase 2 and Phase 3 studies were conducted with dosing without regard to food as the magnitude of the food effect in Phase 1 was not assessed as clinically relevant as exposures were maintained within the range of clinical experience.7
Odanacatib is highly bound (97.5%) to human plasma proteins and does not preferentially distribute into red blood cells.10 A whole‐body autoradiography study in rats indicated that odanacatib‐related material was widely distributed in tissues with the exception of ocular, central nervous system and reproductive tissues.34 In addition, odanacatib‐related material was undetectable by 28 days postdose, suggesting low potential for longer‐term retention.10 The mean volume of distribution of odanacatib is approximately 100 L in humans,7 which is moderate in size and, given that it exceeds total body water (60 L), suggests that odanacatib distributes into tissues.
Odanacatib is metabolized mainly via oxidative pathways, with the principal pathway being fluoroleucine methyl hydroxylation.10 The oxidative metabolism of odanacatib is predominantly catalyzed by cytochrome P450 3A. Metabolites of odanacatib do not circulate in plasma at detectable levels. In addition to being a substrate of the oxidative metabolism enzyme cytochrome P450 3A, odanacatib is also a substrate of the transporters P‐glycoprotein and breast cancer resistance protein.34 As a result, these transporters potentially play a role in the elimination of unchanged parent compound. The systemic clearance of odanacatib is low, 0.8 L/h or approximately 13 mL/min,7 which, based on a typical blood:plasma ratio of 0.7 from in vitro testing, corresponds to a blood clearance of approximately 9 mL/min (<1% of total hepatic blood flow), consistent with low metabolic turnover in in vitro metabolism studies. In the human absorption, distribution, metabolism and excretion (ADME) study,10 following administration of an oral dose of radiolabelled odanacatib, approximately 17 and 74% of the radioactivity were excreted over 34 days in urine and faeces, respectively. The radioactivity in urine was mostly composed of metabolites, while faecal radioactivity was predominantly due to odanacatib. Based on an integration of those ADME data10 with data from the absolute bioavailability study,7 it is estimated that 68% of the absorbed dose of odanacatib is eliminated via metabolism, 21% is excreted as unchanged drug in the bile or faeces, and 12% is excreted as unchanged drug in the urine (as detailed in34; based on estimated bioavailability in the ADME study of 34% using a historical reference intravenous AUC of 2.3 μM h7; percent sum to 101% due to rounding). Renal clearance was directly assessed in the once‐daily multiple dose study,9 and renal clearance values were in general low, and were similar between men and women, ranging from 0.07 to 0.08 L/h in men and 0.08 to 0.15 L/h in women, generally consistent with the assessment above based on the bioavailability and ADME studies. The percent of administered dose that was renally excreted ranged from 3.4 to 5.7% in men and 7.6 to 15.2% in women.
Following multiple weekly doses, little (up to 1.5‐times) accumulation in AUC0‐168hr and maximum plasma concentration occurred with repeated dosing.9, 19 Daily administration in several Phase 1 dosing studies resulted in greater accumulation (up to 5‐times).9 The systemic pharmacokinetics of odanacatib (as directly assessed in the IV/oral absolute bioavailability study) are linear with accumulation predictable from single‐dose data.7 Steady‐state concentrations of odanacatib were achieved within 3–4 weeks of weekly or daily dosing, consistent with the apparent elimination half‐life of odanacatib.9, 19
The effects of age, sex, race/ethnicity, renal and hepatic impairment on odanacatib PK (and in some cases PD) have been characterized in Phase 1 studies and population PK/PD analyses.11, 12, 13, 14, 19, 20 The effects of these factors on odanacatib PK are generally small (Figure 2), and unlikely to be clinically meaningful, thus Phase 3 was conducted with limited restrictions based on intrinsic factors (hepatic insufficiency and end‐stage renal dysfunction). There were no clinically important differences in PK in healthy subjects compared to patients with osteoporosis.21 No clinically important sex differences in the PK and exposure–response (resorption biomarkers, bone mineral density) of odanacatib were identified in a Phase 1 study in PMW and elderly men,19 a pooled Phase 1 PK/PD analysis,35 and a pooled Phase 2/3 analysis.20 These results supported the use of the same 50‐mg odanacatib weekly dosing regimen in men and PMW in the Phase 3 trials.28, 29, 31
Figure 2.
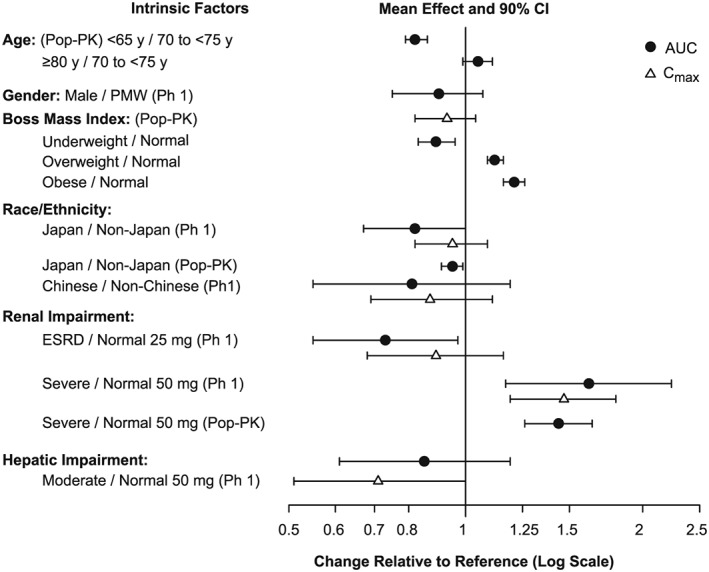
Effect (geometric mean ratio and 90% confidence intervals) of intrinsic factors on the pharmacokinetics (area under the curve, AUC, and maximum plasma concentration, Cmax) of odanacatib. Pop‐PK, population pharmacokinetics; PMW, postmenopausal women
There is no clinically meaningful effect of age on odanacatib PK.19 Within the age range reflecting osteoporosis patients in the clinical trials, exposures increased modestly (~28%) from middle‐aged (50–65 years) to the oldest elderly group examined (>80 years).20, 21
Weight was a statistically significant covariate on both clearance and volume in the population PK model, although the magnitude of effect on exposure was small and not clinically meaningful.20, 21 Body mass index was a statistically significant covariate on both odanacatib potency (IC50) and underlying bone formation rate in the Phase 2 bone turnover model for weight‐bearing bone sites consistent with an expected influence of weight‐bearing on bone remodeling.20, 21, 22, 23
No clinically important effect of race or ethnicity was found on odanacatib pharmacokinetics and exposure–response, including assessments of Hispanic ethnicity, and racial comparisons of White vs Asian populations with subset analyses for Japanese and Chinese ethnicity.11, 12, 21, 30 A lack of covariate effect in the Phase 1 pooled PK/PD analysis was demonstrated for Black subjects (n = 12).35
In Phase 1 studies, no meaningful effect of moderate hepatic impairment on odanacatib pharmacokinetics or PD was found following single doses of either 25 or 50 mg odanacatib, supporting use with no dose adjustment in mild or moderate hepatic impairment13 . Severe hepatic impairment was not studied, and these patients were excluded from Phase 3. In subjects with severe renal impairment14 (defined as a CrCl of 15–29 mL/min and also referred to as Group 4 chronic kidney disease) AUC was increased 60% compared to subjects with normal renal function (>90 mL/min) and odanacatib reduced markers of bone resorption (serum NTx, urine NTx/Cr) comparable to reductions in controls with normal renal function. There were no meaningful differences in the plasma protein binding of odanacatib between Phase 1 renal impairment subjects and healthy subjects. On the basis of these results, patient with renal impairment up to severe degree were included in Phase 3 without dose adjustment. Exposures in LOFT28 patients modestly increased with decreasing estimated creatinine clearance (CrCl) throughout the range of renal function from normal to severe impairment, such that patients with severe renal impairment had only slightly higher exposure (17%) than patients with mild to moderate renal impairment (CrCl 30 to <90 mL/min) who represent 94% of the total patients.21 Although there was a single‐dose PK study performed in subjects with end‐stage renal disease (ESRD) requiring dialysis, patients with osteoporosis and ESRD (CrCl <15 mL/min or on dialysis) were excluded from Phase 3 as ESRD is known to be associated with substantially reduced bone‐turnover36 and instead a specifically designed study in these patients was being considered prior to the discontinuation decision.
1.2. PD
Reductions in bone resorption biomarkers associated with subsequent increases in bone mineral density (BMD) are characteristic of antiresorptive therapies for osteoporosis.37, 38, 39, 40, 41, 42 When bone is resorbed, degradation products of type I collagen are released and can be measured in the serum and/or urine. The N‐terminal (NTx) and C‐terminal (CTx) telopeptides of the cross‐links of type I collagen are generated by enzymatic action of CatK on collagen and the reduction of these markers confirms pharmacological inhibition of CatK (Figure 3). CatK cleaves the N‐telopeptide of collagen to generate NTx. The large fragment of C‐terminal cross‐linked telopeptides of type I collagen (1CTP) is produced by the proteolytic action of matrix metalloproteinases and, therefore, this biomarker increases when CatK is inhibited reflecting an increased proportion of breakdown via this alternate route. Subsequently, 1CTP is cleaved by CatK to generate CTx. However, generation of 1CTP is not required for CatK‐mediated release of CTx, which can also be directly generated from collagen and other fragments. CTx and NTx, frequently used in the clinical research, were used throughout the odanacatib programme (Phase 1, 2, 3). Deoxypyridinoline (DPD), small breakdown fragments of collagen excreted in urine with the attached cross‐linked moiety, represents a CatK‐independent release biomarker of bone turnover. In Phase 1, odanacatib target engagement was confirmed by a robust decrease in serum CTx (sCTx; approximately 60%), urinary (u)NTx/Cr (approximately 50%) and total uDPD/Cr (approximately 30%), with an increase in serum 1CTP (approximately 55%).8, 10, 12, 19, 22, 23, 35 The magnitude of reduction in bone resorption markers (sCTx, uNTx/Cr, uDPD/Cr) are generally consistent with the maximal responses observed with other classes of osteoporosis treatments that target resorption, including bisphosphonates and anti‐RANKL antibody.43, 44, 45 As shown in Table 2, the changes in resorption biomarkers with 50‐mg odanacatib weekly were generally consistent across the Phase 1 and 2 studies, thus demonstrating robustness of the bone resorption marker response across healthy subject and patient populations, including men, Japanese individuals, and subjects with renal and hepatic impairment.8, 9, 11, 19, 24, 30, 31 Marked reductions of bone resorption markers were observed at weekly doses ≥10 mg after several weeks of therapy. Sustained near maximal reduction of resorption biomarkers across the week long dosing interval were demonstrated at doses of ≥50 mg and greater in Phase 1, while at the 5 mg and to some extent 25 mg odanacatib doses, the degree of reduction varied over the dosing interval in line with the PK profile (Figure 4).9
Figure 3.
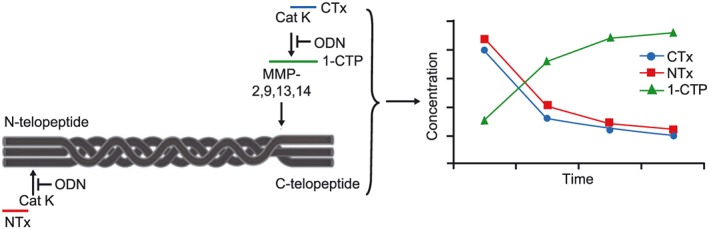
Odanacatib activity in blocking collagen degradation9
Table 2.
Bone resorption markers following 50‐mg odanacatib given once weekly (geometric mean % change from baseline for weeks 1 [at 168 hours], 4, and 12)
| Phase | PN | Time point | Population | n a | sCTx (ng/mL) | uNTx/Cr (nmol BCE/mmol creatinine) | uDPD/Cr (nmol/mmol creatinine) | 1CTP (μg/L) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GM | SE | GM | SE | GM | SE | GM | SE | |||||
| 1 | 0059 | Week 1 | PM women | 9 | −66.22 | 9.45 | −50.66 | 4.33 | −29.98 | 5.70 | 101.41c | 17.48 |
| Week 4b | PM women | 8–9 | −67.59 | 7.06 | −63.59 | 4.08 | −30.31 | 12.14 | 103.83 | 16.12 | ||
| 014f , 11 | Week 1 | PM women | 8 | −46.97 | 21.96 | −58.33 | 13.48 | −25.11 | 24.34 | NA | ‐‐ | |
| Week 4 | PM women | 6–8 | −62.35 | 12.91 | −66.88 | 12.56 | −32.40 | 10.02 | NA | ‐‐ | ||
| 039 | Week 1 | PM women | 22 | −47.03 | 4.76 | −27.20 | 10.11 | −35.62 | 7.66 | 60.97 | 7.78 | |
| Week 4 | PM women | 22 | −49.05 | 6.29 | −22.14 | 11.91 | −25.06 | 8.74 | 53.64 | 7.75 | ||
| Week 12 | PM women | 22 | −45.87 | 6.16 | −40.27 | 6.85 | −30.36 | 7.56 | 76.78 | 10.94 | ||
| 059d , 19 | Week 1 | Healthy men (50–75 y) | 20–21 | −54.2 | 27.6 | −27.0e | 39.2 | −29.4 | 31.2 | 42.7 | 36.7 | |
| PM women | 10 | −58.6 | 26.6 | −44.7 | 40.7 | −14.6 | 16.3 | 44.3 | 43.5 | |||
| Week 4 | Healthy men (50–75 y) | 19–20 | −52.5 | 28.3 | −16.4 | 45.3 | −19.8 | 52.0 | 55.5 | 38.5 | ||
| PM women | 9 | −62.9 | 39.4 | −42.8 | 17.3 | −26.9 | 28.0 | 44.3 | 67.4 | |||
| 2/3 | 00424 | Week 1 | PM women | 67–70 | −65.06 | 2.87 | −55.88 | 2.85 | −28.63 | 2.60 | NA | ‐‐ |
| Week 4 | PM women | 68–69 | −73.21 | 2.45 | −63.82 | 2.24 | −33.87 | 3.14 | NA | ‐‐ | ||
| Week 12 | PM women | 61–62 | −69.63 | 3.49 | −62.13 | 2.46 | −34.61 | 3.83 | NA | ‐‐ | ||
| 022e, 30 | Week 1 | Osteoporotic men and PM women | 61–64 | −56.75 | (−61.88,‐50.93) | −49.39 | (−54.28,‐43.98) | −33.11 | (−38.73,‐26.98) | NA | ‐‐ | |
| Week 4 | Osteoporotic men and PM women | 61–64 | −74.07 | (−77.75, −69.78) | −62.19 | (−66.06, 57.88) | −46.50 | (−51.72,‐40.71) | NA | ‐‐ | ||
| Week 12 | Osteoporotic men and PM women | 61–64 | −69.25 | (−74.94, −62.28) | −60.24 | (−64.81,‐55.08) | −39.75 | (−46.00,‐32.77) | NA | ‐‐ | ||
| 05331 | Week 12 | Osteoporotic men | 128–134 | −65.56 | 1.56 | −46.53 | 3.95 | NA | ‐‐ | NA | ‐‐ | |
Range for n depends on the n per biomarker;
P005: Week 4 is actually Week 3;
P005: 1CTP is Week 1 Day 3 (not Week 1 Day 7 or 168 hours);
P059: variability is standard deviation, not standard error (SE). In addition, there was no Week 12‐time point;
P022: LS mean % change from baseline and 95% confidence intervals are presented; data are pooled as presented in CSR;
P014: variability is standard deviation, not SE.
sCTx = serum C‐telopeptides of type 1 collagen; uNTx/Cr = urinary aminoterminal crosslinked telopeptides of type 1 collagen to creatinine ratio; 1CTP = cross‐linked carboxy‐terminal telopeptide of type 1 collagen; uDPD/Cr = urinary deoxypyridinoline to creatinine ratio; PM = postmenopausal.
Figure 4.
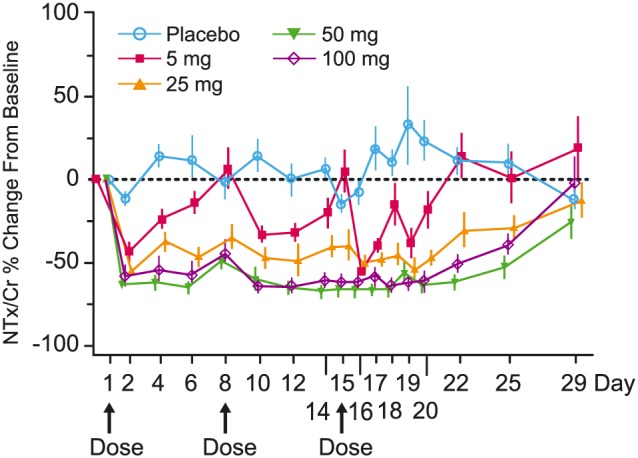
Percent change from baseline (geometric mean ± standard error) of urine aminoterminal crosslinked telopeptides of type 1 collagen to creatinine ratio (NTx/Cr) following administration of multiple once weekly oral doses of odanacatib in healthy postmenopausal women (n = 12 for placebo, n = 5 to 9 for 25 mg, n = 7 to 9 for 50 mg, n = 7 to 10 for 100 mg, n = 6 to 9 for 5 mg)9
In the Phase 2 global dose‐ranging study,24 sustained reductions in uNTx/Cr over time were demonstrated in PMW with osteoporosis at the 3 highest doses (10, 25 and 50 mg) with the magnitude of response generally increasing as the dose increased (referred to as the monotonic response). The reductions in resorption biomarkers were demonstrated out to 8 years of treatment with 50 mg weekly odanacatib in the P004 extension, confirming the durability of the treatment response.27, 32 At Month 24, treatment assignment was re‐randomized to allow assessment of patients previously treated with odanacatib for 2 years who were switched to placebo to further evaluate resolution‐of‐effect behaviour.26 This subset of patients demonstrated a transient increase in bone resorption markers (uNTx/Cr, sCTx) that returned to near baseline with a year off treatment (Figure 5, panel A). The 3‐mg group demonstrated increased bone turnover: resorption (uNTx/Cr) and formation (serum bone‐specific alkaline phosphatase [sBSAP], N‐terminal propeptide of type 1 procollagen sP1NP]) over placebo consistent with a nonmonotonic response in contrast with the monotonic reduction in bone turnover markers relative to placebo as observed at the higher doses (10, 25 and 50 mg).
Figure 5.
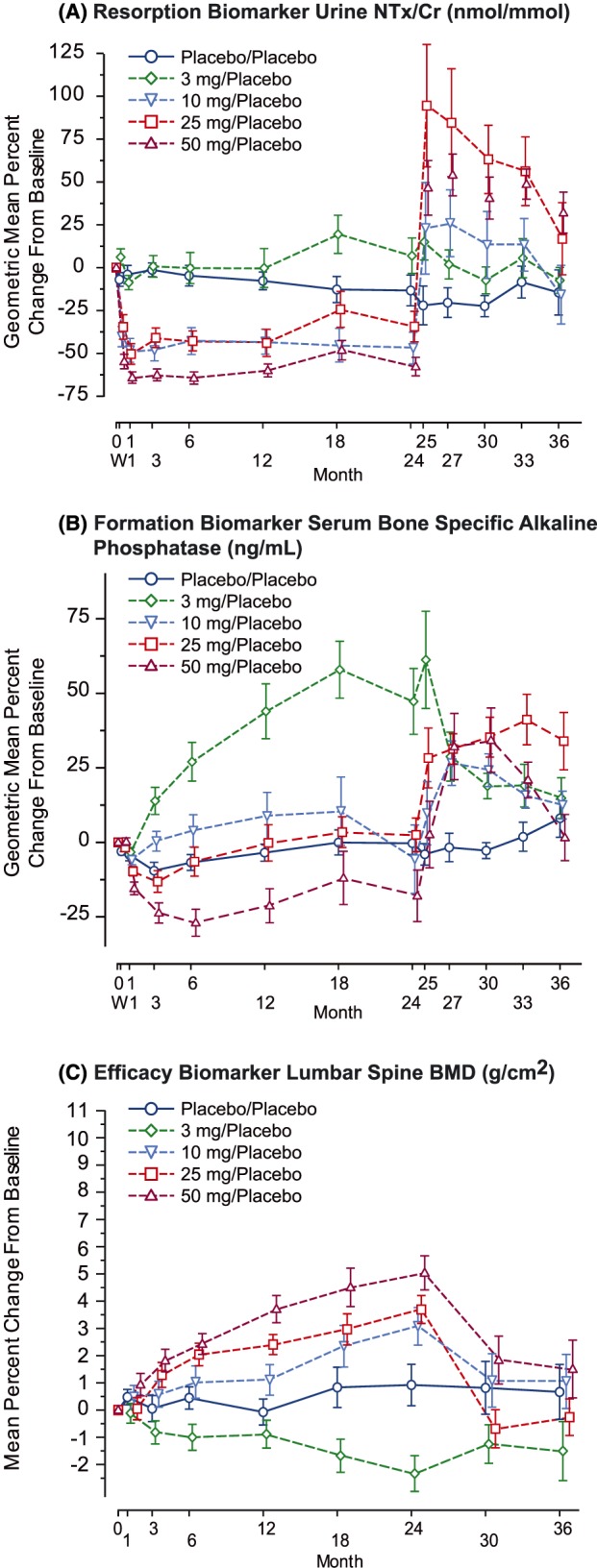
Longitudinal dose–response of key biomarkers over 3 years in the subset of phase 2 (P004) patients switched to placebo at 24 months (geometric mean ± standard error) per‐protocol population placebo.24, 26 A, Resorption biomarker urine aminoterminal crosslinked telopeptides of type 1 collagen to creatinine ratio (NTx/Cr; nmol/mmol). B, Formation biomarker serum bone specific alkaline phosphatase (ng/mL). C, Efficacy biomarker lumbar spine bone mineral density (BMD; g/cm2)
Serum bone formation markers—BSAP, P1NP and osteocalcin—were measured following administration of odanacatib, as these markers have been shown to correlate with increases in BMD for other classes of osteoporosis therapies.46 Since, during bone remodelling, there is tight coupling between bone resorption and formation, bone formation markers may be impacted indirectly several months following administration of anti‐resorptive therapies. For this reason, formation markers were not generally collected in short term Phase 1 studies, with the exception of the 12‐week mechanism study. Bone formation markers were initially modestly reduced in Phase 2 dose‐ranging studies (10, 25 and 50 mg)24, 30 to a lesser extent than observed with traditional antiresorptives (bisphosphonates, RANKL‐Ab; Figure 5, panel B).44, 45, 46 A notable increase in formation biomarkers (serum BSAP, serum P1NP) relative to placebo was observed with the 3‐mg dose in P004.24
BMD, as assessed by DXA at multiple anatomical sites, was included in the Phase 2 and 3 studies as a primary or secondary efficacy measure. The global dose‐range finding Phase 2 study showed a dose–response relationship for BMD at all 5 anatomical sites evaluated: lumbar spine (a primarily trabecular site), at the femoral neck, trochanter and total hip (mixed trabecular/cortical sites), and at the ⅓ radius (a primarily cortical site).24 Figure 5 (panel C) displays the mean percent change from baseline over time in lumbar spine BMD for the 3‐, 10‐, 25‐ and 50‐mg odanacatib weekly doses and for placebo over the first 24 months of treatment, a period during which all patients were treated based on their original treatment assignment. Lumbar spine BMD showed numerical gains over time in the 3 highest active doses (10 mg [3.39%] 25 mg [4.45%] and 50 mg [(5.67%] placebo corrected change from baseline at 24 months) with the magnitude of response increasing with increasing dose (monotonic response), while BMD remained relatively stable in the placebo group. The 3‐mg group showed lower BMD values than placebo (−0.84%), demonstrating a nonmonotonic response behaviour, which contrasts with the monotonic increases over placebo exhibited at the higher doses and which was considered a paradoxical finding given the large increase in bone formation biomarkers obtained at this dose. This general pattern of dose–response (monotonic increases in BMD response over the dose‐range of 10 to 50 mg with nonmonotonic reductions relative to placebo at 3 mg) was seen at the other 4 anatomical sites in P004.24 In the 1‐year Japanese dose‐range finding study,30 doses of 10‐, 25‐ and 50‐mg odanacatib weekly were compared to each other, and to placebo. Consistent with the dose–response pattern observed in global DR study, a monotonic dose–response relationship was seen for 50‐mg odanacatib at the lumbar spine, femoral neck, trochanter, and total hip compared to the 25‐ and 10‐mg doses and placebo. At all anatomical sites across both dose‐range finding studies, the BMD response at 50 mg was numerically superior to that observed at lower doses, including the 25‐mg dose.
Available clinical data support that patients continue to gain BMD during long term use of odanacatib. Results from the Phase 2 study (P004) extension indicate that continued gains in BMD out to 8 years are observed at many bone sites and that continued improvement in BMD relative to placebo is seen at nearly all bone sites.27, 29, 32 In LOFT, these BMD increases were associated with fracture risk reduction and increases in estimated bone strength.28, 29, 47 Corroborating evidence for the relationship between reduced bone resorption, increased BMD and increased bone strength was also demonstrated in preclinical rhesus studies48 and the utility of this animal model to allow for in vitro to in vivo translation has been confirmed.49
In contrast, data from patients who were switched to placebo following odanacatib treatment demonstrate that BMD rapidly (within ~1 year) returns to levels similar to baseline and placebo response (Figure 5, panel C).26
Existing antiresorptive agents reduce the number of functional osteoclasts, the principal bone resorbing cells responsible for bone resorption. Monitoring of osteoclast specific tartrate‐resistant acid phosphatase 5b (TRAP5b), an indicator of viable osteoclast cell number, confirmed that CatK inhibition with odanacatib did not affect osteoclasts cell number in the Phase 1 12‐week mechanism study (data on file, Aubrey Stoch, Merck & Co., Inc). There are several lines of evidence that longer term odanacatib treatment acts to increase osteoclast number as well as other osteoclast activity effects (including build‐up of CatK enzyme). In a preclinical model, in ovariectomized rhesus monkeys treated for 21 months, histomorphometric analysis indicate that both osteoclast number and osteoclast surface increased approximately 4‐fold by odanacatib treatment compared to vehicle.48 Similarly, in the bone biopsy substudy of LOFT,50 osteoclast surface area at Month 36 was approximately doubled in patients receiving 50 mg weekly odanacatib relative to patients receiving placebo in the histomorphometric analysis of transilial bone biopsies.51
Key mineral homeostasis parameters in the odanacatib programme included serum and urine calcium, serum phosphorus and serum parathyroid hormone (PTH). Serum calcium and phosphorus were routinely evaluated in clinical trials, with serum and urine calcium, serum phosphorus and serum PTH being intensively sampled in a dedicated Phase I mechanism‐of‐action study. Consistent with the mechanism of action, reductions in serum calcium and phosphorus have been observed with compensatory increases in PTH (data on file, Aubrey Stoch, Merck & Co., Inc).
1.3. PK/PD and dose–response
The PK/PD relationship between odanacatib concentrations and the bone resorption biomarkers was evaluated in Phase 18, 9, 11, 14, 19, 35 and as a component of a mechanistic PK/PD model of bone turnover applied to Phase 2 data.20, 22, 23 A pooled analysis including data from all applicable Phase 1 studies provides the most definitive assessment of Phase 1 PK/PD relationships for resorption biomarkers.35 Exposure–response between odanacatib plasma concentrations and the resorption biomarker response is well characterized by an instantaneous (no time delay between concentration and resulting response) sigmoid Emax relationship. The suitability of uNTx/Cr as an appropriate generalizable bone resorption biomarker for the assessment of a CatK inhibition by odanacatib was established by a cross‐study analysis of 3 biomarkers, uNTx/Cr, sCTx and uDPD/Cr in which a consistent exposure–response relationship was demonstrated (normalized to the biomarkers specific maximal response), including similarity of the EC50 and sigmoidicity or Hill(s) values, across all 3 biomarkers (Figure 6). Although maximal response (Imax, maximal reduction at high concentrations) varied from 39% to 70% response among these 3 biomarkers, this variation is not thought to reflect differences in drug effects on bone resorption but rather is likely to be due to differences in the relative contribution of nonbone and non‐CatK mediated bone sources of these biomarkers to baseline values, which would not be affected by CatK inhibition.
Figure 6.
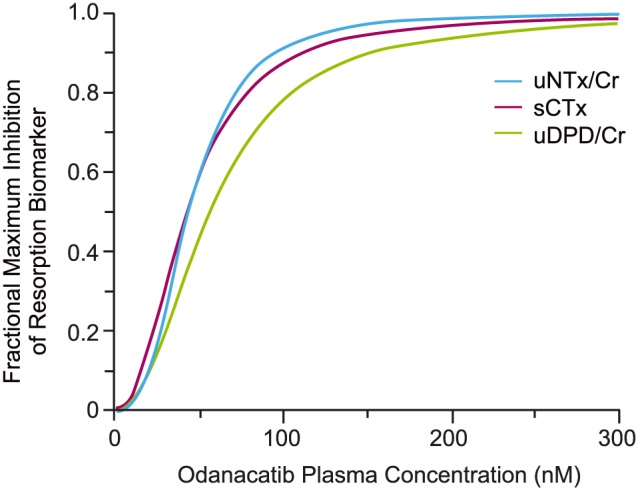
Consistency of the phase 1 concentration–response relationship for odanacatib across 3 resorption biomarkers in cross study analysis35
The odanacatib potency (IC50) estimated from the pooled Phase 1 model was 41.2 nM (5.41% standard error), which is consistent with estimates of the potency of odanacatib across individual study analyses, which ranged approximately 2‐fold, from a low of 28.6 nM to a high of 60.7 nM.35 This value is also similar to the typical value of 37.2 nM (9.41% standard error) estimated from pooled Phase 2 dose‐ranging data in women with osteoporosis.22, 23
Mechanistic PK/PD modelling was used to integrate the time course of odanacatib plasma concentrations, uNTx/Cr, and BMD data from the Phase 2 studies to enhance understanding of the exposure‐response pharmacology of odanacatib.20, 22, 23 The model structure was developed and qualified by testing its ability to well represent the key features from the Phase 2 studies, including: (i) sustained long term reduction of resorption biomarkers and increases in BMD on treatment at 50 mg; (ii) transiently increased resorption biomarkers over baseline and loss of BMD to near placebo levels within ~1 year following cessation of treatment; and (iii) the nonmonotonic behaviour at 3 mg. The PK/PD model structure incorporated 2 drug effects: resorption inhibition effect represents the direct, reversible inhibition effect of odanacatib on CatK; and osteoclast effects that probably reflect an amalgam of changes in osteoclast numbers, CatK enzyme levels, and/or precursor pools and feedback regulation set points. In the model, the modelled osteoclast number and level of CatK inhibition then informed the bone resorption rate which together with the formation rate (empirically modified by an effect of calcium and vitamin D supplements, but not odanacatib concentration) defined the change in BMD over time. The semi‐mechanistic model was simultaneously fit to the pooled uNTx and BMD data from the 2 Phase 2 studies. Separate models were estimated for BMD from 5 different bone sites (lumbar spine, femoral neck, trochanter, total hip and distal forearm) with consistent drug effect parameter values estimated for all sites such that variation in BMD response among bone sites was largely accounted for by differences in the underlying endogenous rates of bone formation and resorption at these sites. The model‐estimated concentration–response relationships for both drug effects based on the Phase 2 analysis are illustrated in Figure 7 with the most notable finding being a modest rightward shift in the concentration–response of the resorption inhibition effect relative to the osteoclast effects.
Figure 7.
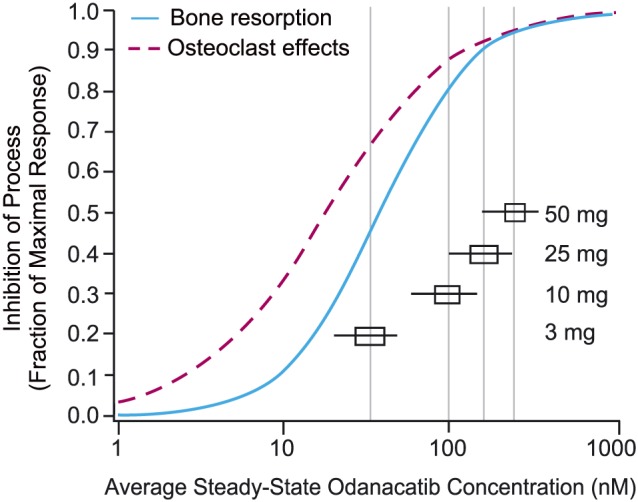
Model estimated fractional inhibition of bone resorption and osteoclast effects vs odanacatib concentration in postmenopausal women with osteoporosis; horizontal box (25th to 75th percentiles) and whisker (5th to 95th percentiles) plots depict the predicted distribution of average steady‐state concentrations for various weekly odanacatib doses22, 23
At exposures maintained over the dosing interval at 50 mg weekly, both drug effects approach maximal response, with the net effect being a strong inhibition of resorption effect as demonstrated by the sustained and substantial reductions in uNTx/Cr relative to placebo throughout the dosing interval and continual increases in BMD observed over multiple years of treatment. Doses <50 mg fall further down the exposure–response relationships identified through the model. These results in part supported the choice of 50 mg weekly for study in the Phase 3 trials.
The resolution of uNTx/Cr and BMD response after cessation of odanacatib treatment was characterized in the model by a build‐up of osteoclast numbers during therapy. Following cessation of treatment, the resorption inhibition effect washes out, in keeping with the declining odanacatib plasma concentrations, while the osteoclast effect washes out more slowly, resulting in transient elevations in resorption (uNTx/Cr) over baseline and the rapid loss of BMD that eventually returns to near‐placebo levels.
In the model the low dose nonmonotonic features of the dose–response derive from the small difference in potency (IC50) of odanacatib on the 2 drug effects—inhibition of resorption (Imax 71%, IC50 37.1 nM) and osteoclast effect (Imax 73%, IC50 17.9 nM)—based on the Phase 2 analysis underlying the rightward shift in the PK/PD curves.20, 22, 23 At very low doses associated with nonmonotonic response, average exposures over the dosing interval fall below the IC50 for resorption inhibition effect but above that for osteoclast effect, with a net effect of slightly enhanced resorption. Viewed across the dosing interval, this created at 3 mg a repeating pattern of reduction of uNTx/Cr early in the weekly dosing interval, but increased resorption (uNTx/Cr) over placebo in the 2nd half of the week as odanacatib plasma concentrations decline below levels associated with inhibition of resorption activity. The mechanistic PKPD modelling does not directly explain the paradoxical finding of increased bone resorption biomarkers at 3 mg as formation biomarker data was not used in the model development. However, one could speculate that increased osteoclast signalling in a partially uninhibited state at 3 mg could influence osteoblast activity through coupling without the expected increase in actual bone formation. The results from the Phase 2 modelling analysis and the Phase I mechanism study are consistent with alterations in osteoclast number and other potential related effects on osteoclast pharmacology (e.g., build‐up of CatK enzyme) with slower washout rates than the inhibition of resorption effect and slight shifts in drug potency on these 2 drug effects being the underlying mechanisms of the nonmonotonic response observed at 3 mg. In the Phase I mechanism study, serum PTH at 50 mg was elevated relative to placebo over the dosing interval at Week 12 consistent with inhibition of bone resorption (and thus reduced PTH release) at that dose; however, at 3 mg, PTH was not elevated relative to placebo, which is probably consistent with a lack of sustained inhibition of bone resorption.
The integrated characterization of the pharmacology of odanacatib effects on bone turnover provided by the PK/PD model was used throughout clinical development to: aid dose and regimen selection, inform understanding of covariates influences and clinical relevance; explore the potential for patient nonadherence to mimic the nonmonotonic response (bone loss); and predict the outcome of long‐term usage and resolution of effects following treatment discontinuation.
2. DISCUSSION AND CONCLUSIONS
This review highlights the clinical pharmacology of odanacatib as a representative CatK inhibitor. Two other CatK inhibitors progressed to date to clinical studies: balicatib (Phase 1 and 2 studies) and ONO‐5334 (Phase 1 and 2 studies). PD results from these programmes are generally consistent with the key findings from odanacatib, including: robust reversible inhibition of bone resorption demonstrated by biomarkers52, 53, 54, 55 and associated with BMD increases56, 57, 58, 59, 60; modest reductions of formation biomarkers (small relative to other osteoporosis therapies) at doses with near maximal resorption inhibition,56, 57, 59, 60 and increased formation biomarkers over baseline at subtherapeutic (very low) dose56, 57, 59, 60; transient acceleration of bone resorption on discontinuation of treatment of ≥3 months duration,56, 57, 59, 60 and osteoclast effects enhancing size, number and/or CatK enzyme levels.61 Of note, both balicatib and ONO‐5334 were dosed daily in Phase 2, which contrasts with the weekly dosing regimens studied in Phase 2 and 3 for odanacatib. The frequency of dosing required to maintain antiresorptive effects of CatK inhibitors depends on the pharmacokinetic half‐life, as CatK inhibition of resorption is freely reversible for all these compounds and declines consistent with the fall in drug concentration. This contrasts with bisphosphonates that exhibit a prolonged pharmacodynamic activity related to bone hydroxyapatite binding which offsets the effects limited PK exposures allowing for extended duration regimens—weekly or less frequent already in clinical use. Because the CatK inhibitors act as direct reversible inhibitors of an enzyme mediating the bone resorption process and odanacatib equilibrates rapidly between plasma and bone sites, the onset and offset of the antiresorption effects parallel the rise and fall of drug concentrations. These onset and offset rates are faster than observed with denosumab or bisphosphonates, which act indirectly on turnover of osteoclasts with slower inherent dynamics. In addition, for bisphosphonates, pharmacokinetic accumulation over time at the bone sites slows the dynamics of the direct inhibition of mineral bone resorption component of response.
In summary, the extensive clinical programme for odanacatib provides important insights into the clinical pharmacology of CatK inhibition and the potential role of CatK in metabolic bone disorders. Key findings include:
The odanacatib 50‐mg weekly dosing regimen evaluated in LOFT sustained reduction in resorption throughout treatment and, therefore, the trial results probably reflect near maximal effect possible with this mechanism.
CatK inhibition provides sustained reductions in resorption markers, increases in BMD across all bone sites (even non–weight‐bearing sites relative to placebo), and demonstrated fracture risk reduction
CatK inhibition is associated with a formation sparing phenotype; sustained resorption reduction is achieved unaccompanied by large reductions in bone formation. Increases in formation biomarkers (sBSAP, sP1NP, OC) at low dose (3 mg) was not associated with BMD increase (rather slight reductions in BMD), suggesting a lack of 1:1 correlation of formation markers with formation rate
CatK inhibition appears to increase osteoclast numbers as well as other osteoclast effects (including build‐up of CatK enzyme), which may have led to transient increases in resorption activity following treatment discontinuation and the potential for a nonmonotonic response at subtherapeutic doses.
2.1. Nomenclature of target and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,62 and are permanently achieved in the Concise Guide to PHARMACOLOGY 2017/2018.63
COMPETING INTERESTS
All authors are or were employed at Merck & Co., Inc., Kenilworth, NJ, USA.
ACKNOWLEDGEMENTS
The authors would like to thank all patients and investigators who participated in these studies. In addition, we would like to thank Boyd Scott, PhD (Merck & Co. Inc., USA) for his careful review of this manuscript. In addition, we would like to thank Jennifer Pawlowski, MS (Merck & Co. Inc., USA) for assistance in the submission of this manuscript.
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Stone JA, McCrea J, Witter R, Zajic S, Stoch SA. Clinical and translational pharmacology of the cathepsin K inhibitor odanacatib studied for osteoporosis. Br J Clin Pharmacol. 2019;85:1072–1083. 10.1111/bcp.13869
REFERENCES
- 1. Duong le T, Leung AT, Langdahl B. Cathepsin K inhibition: a new mechanism for the treatment of osteoporosis. Calcif Tissue Int. 2016;98:381‐397. [DOI] [PubMed] [Google Scholar]
- 2. Gauthier JY, Chauret N, Cromlish W, et al. The discovery of odanacatib (MK‐0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett. 2008;18(3):923‐928. [DOI] [PubMed] [Google Scholar]
- 3. Boonen S, Rosenberg E, Claessens F, Vanderschueren D, Papapoulos S. Inhibition of cathepsin K for treatment of osteoporosis. Curr Osteoporos Rep. 2012;10(1):73‐79. [DOI] [PubMed] [Google Scholar]
- 4. Delaisse JM, Andersen TL, Engsig MT, Henriksen K, Troen T, Blavier L. Matrix metalloproteinases (MMP) and cathepsin K contribute differently to osteoclastic activities. Microsc Res Tech. 2003;61(6):504‐513. [DOI] [PubMed] [Google Scholar]
- 5. Yasuda Y, Kaleta J, Bromme D. The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005;57(7):973‐993. [DOI] [PubMed] [Google Scholar]
- 6. Gelb BD, Shi GP, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science. 1996;273(5279):1236‐1238. [DOI] [PubMed] [Google Scholar]
- 7. Zajic S, Rossenu S, Hreniuk D, et al. The absolute bioavailability and effect of food on the pharmacokinetics of odanacatib: a stable‐label i.v./oral study in healthy postmenopausal women. Drug Metab Dispos. 2016;44(9):1450‐1458. [DOI] [PubMed] [Google Scholar]
- 8. Stoch SA, Zajic S, Stone JA, et al. Odanacatib, a selective cathepsin K inhibitor to treat osteoporosis: safety, tolerability, pharmacokinetics and pharmacodynamics—results from single oral dose studies in healthy volunteers. Br J Clin Pharmacol. 2013;75(5):1240‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stoch SA, Zajic S, Stone J, et al. Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double‐blind, randomized, placebo‐controlled phase I studies. Clin Pharmacol Ther. 2009;86(2):175‐182. [DOI] [PubMed] [Google Scholar]
- 10. Kassahun K, McIntosh I, Koeplinger K, et al. Disposition and metabolism of the cathepsin K inhibitor odanacatib in humans. Drug Metab Dispos. 2014;42(5):818‐827. [DOI] [PubMed] [Google Scholar]
- 11. Uemura N, Willmer JP, Nakamichi N, et al. Safety, pharmacokinetics and pharmacodynamics (bone turnover) of odanacatib: two double‐blind, randomized, placebo‐controlled phase I studies in Japanese men and postmenopausal women. Clin Pharmacol Ther. 2013;93:S88‐S88. [Google Scholar]
- 12. Chen X, Jiang J, Hu P, et al. Odanacatib pharmacokinetics comparison between Chinese and non‐Chinese postmenopausal women. Clin Pharm Drug Dev. 2018;7(7):744‐750. [DOI] [PubMed] [Google Scholar]
- 13. Zajic S, Trucksis M, Garrett G, et al. Evaluation of odanacatib in subjects with hepatic insufficiency. J Bone Miner Res. 2013;28(Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=c1ad774e-d2ab-41d5-8073-080dd15ef61e. Accessed March 4, 2019. [Google Scholar]
- 14. Stoch A, Liu CC, Zajic S, et al. The pharmacokinetics of odanacatib 50 mg are not affected by severe renal insufficiency. J Bone Miner Res. 2013;28(Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=906aead6-0d77-4c42-b49e-c4fdd0810387. Accessed March 4, 2019. [Google Scholar]
- 15. Stoch SA, Ballard J, Gibson C, et al. Coadministration of rifampin significantly reduces odanacatib concentrations in healthy subjects. J Clin Pharmacol. 2017;57(1):110‐117. [DOI] [PubMed] [Google Scholar]
- 16. Marcantonio EE, Ballard J, Gibson CR, et al. Prednisone has no effect on the pharmacokinetics of CYP3A4 metabolized drugs ‐ midazolam and Odanacatib. J Clin Pharmacol. 2014;54(11):1280‐1289. [DOI] [PubMed] [Google Scholar]
- 17. Stoch SA, Witter R, Hreniuk D, et al. Absence of a clinically relevant drug‐drug interaction between odanacatib and digoxin after concomitant administration. Int J Clin Pharm Th. 2013;51(08):688‐692. [DOI] [PubMed] [Google Scholar]
- 18. Stoch SA, Witter R, Hrenuik D, et al. Odanacatib does not influence the single dose pharmacokinetics and pharmacodynamics of warfarin. J Popul Ther Clin Pharmacol. 2013;20(3):e312‐e320. [PubMed] [Google Scholar]
- 19. Anderson MS, Gendrano IN, Liu C, et al. Odanacatib, a selective cathepsin K inhibitor, demonstrates comparable pharmacodynamics and pharmacokinetics in older men and postmenopausal women. J Clin Endocr Metab. 2014;99(2):552‐560. [DOI] [PubMed] [Google Scholar]
- 20. Stone J, Jaworowicz D, Zajic S, Humphrey R, Santora A, Stoch A. Lack of clinically important gender differences in the pharmacokinetics and exposure‐response relationship of Odanacatib. J Bone Miner Res. 2014;29(Suppl 1):S193‐S193. [Google Scholar]
- 21. Uchida S, Shiraki M, Fukunaga M, et al. The results of a double‐blind, randomized, phase 2 dose‐finding study of odanacatib, a potent cathepsin‐K inhibitor, in Japanese patients with osteoporosis with a model‐based pharmacokinetic (PK) analysis. J Bone Miner Res. 2012;27(Suppl 1). Available at http://www.asbmr.org/Meetings/AnnualMeeting/AbstractDetail.aspx?aid=42973cf1-376a-4fc7-9629-d489950434de. Accessed March 4, 2019. [Google Scholar]
- 22. Zajic S, Stone JA, Jaworowicz D, et al. Semi‐mechanistic PK/PD model of the effect of odanacatib, a cathepsin K inhibitor, on bone turnover to characterize lumbar spine Bone mineral density in two phase II studies of postmenopausal women. J Bone Miner Res. 2010;25(Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=58f9bb62-f97e-47bf-aaa6-95afdf475eed. Accessed March 4, 2019. [Google Scholar]
- 23. Zajic S, Stone JA, Jaworowicz D, et al. Semi‐mechanistic PK/PD model of the effect of odanacatib, a cathepsin k inhibitor, on bone turnover to characterize lumbar spine and distal forearm bone mineral density in a phase iib study of postmenopausal women. Athens, Greece: PAGE; 2011. [Google Scholar]
- 24. Bone HG, McClung MR, Roux C, et al. Odanacatib, a cathepsin‐K inhibitor for osteoporosis: a two‐year study in postmenopausal women with low bone density. J Bone Miner Res. 2010;25(5):937‐947. [DOI] [PubMed] [Google Scholar]
- 25. Bonnick S, De Villiers T, Odio A, et al. Effects of odanacatib on BMD and safety in the treatment of osteoporosis in postmenopausal women previously treated with alendronate: a randomized placebo‐controlled trial. J Clin Endocrinol Metab. 2013;98(1):4727‐4735. [DOI] [PubMed] [Google Scholar]
- 26. Eisman JA, Bone HG, Hosking DJ, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: three‐year continued therapy and resolution of effect. J Bone Miner Res. 2011;26(2):242‐251. [DOI] [PubMed] [Google Scholar]
- 27. Langdahl B, Binkley N, Bone H, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res. 2012;27(1):2251‐2258. [DOI] [PubMed] [Google Scholar]
- 28. McClung MR, Langdahl B, Papapoulos S, et al. Odanacatib antifracture efficacy and safety in postmenopausal women with osteoporosis: results from the phase iii Long‐term Odanacatib Fracture Trial (Loft). Osteoporosis Int. 2015;26:S35‐S36. [Google Scholar]
- 29. McClung MR, Langdahl B, Papapoulos S, et al. Odanacatib efficacy and safety in postmenopausal women with osteoporosis: 5‐year data from the extension of the phase 3 Long‐term Odanacatib Fracture Trial (LOFT). Menopause. 2016;23:1369‐1369. [Google Scholar]
- 30. Nakamura T, Shiraki M, Fukunaga M, et al. Effect of the cathepsin K inhibitor odanacatib administered once weekly on bone mineral density in Japanese patients with osteoporosis—a double‐blind, randomized, dose‐finding study. Osteoporos Int. 2014;25(1):367‐376. [DOI] [PubMed] [Google Scholar]
- 31. Orwoll E, Adami S, Binkley N, et al. Randomized controlled trial to assess the safety and efficacy of Odanacatib in the treatment of men with osteoporosis. Arthritis Rheumatol. 2014;66:S408‐S409. [DOI] [PubMed] [Google Scholar]
- 32. Rizzoli R, Benhamou CL, Halse J, et al. Continuous treatment with odanacatib for up to 8 years in postmenopausal women with low bone mineral density: a phase 2 study. Osteoporos Int. 2016;27(6):2099‐2107. [DOI] [PubMed] [Google Scholar]
- 33. Mullard A. Merck & Co. drops osteoporosis drug odanacatib. Nat Rev Drug Discov. 2016;15:669. [DOI] [PubMed] [Google Scholar]
- 34. Kassahun K, Black WC, Nicoll‐Griffith D, et al. Pharmacokinetics and metabolism in rats, dogs, and monkeys of the cathepsin k inhibitor odanacatib: demethylation of a methylsulfonyl moiety as a major metabolic pathway. Drug Metab Dispos. 2011;39(6):1079‐1087. [DOI] [PubMed] [Google Scholar]
- 35. Zajic S, Hreniuk D, Witter R, Panebianco D, Stone JA, Stoch A. Odanacatib, a cathepsin‐K inhibitor, has similar clinical concentration‐response relationships for urinary N‐terminal telopeptide (uNTx) and deoxypyridinoline (uDPD). J Bone Miner Res. 2012;27(Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=77d8f4e2-f354-469c-8b35-5e535a286ac5. Accessed March 4, 2019. [Google Scholar]
- 36. Miller PD. Chronic kidney disease and the skeleton. Bone Res. 2014;2(1):14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cremers S, Garnero P. Biochemical markers of bone turnover in the clinical development of drugs for osteoporosis and metastatic bone disease—potential uses and pitfalls. Drugs. 2006;66(16):2031‐2058. [DOI] [PubMed] [Google Scholar]
- 38. Garnero P, Darte C, Delmas PD. A model to monitor the efficacy of alendronate treatment in women with osteoporosis using a biochemical marker of bone turnover. Bone. 1999;24(6):603‐609. [DOI] [PubMed] [Google Scholar]
- 39. Garnero P, Shih WCJ, Gineyts E, Karpf DB, Delmas PD. Comparison of new biochemical markers of bone turnover in late postmenopausal osteoporotic women in response to alendronate treatment. J Clin Endocr Metab. 1994;79(6):1693‐1700. [DOI] [PubMed] [Google Scholar]
- 40. Seibel MJ, Baylink DJ, Farley JR, et al. Basic science and clinical utility of biochemical markers of bone turnover—a congress report. Exp Clin Endocr Diab. 1997;105:125‐133. [DOI] [PubMed] [Google Scholar]
- 41. Tesch G, Amur S, Schousboe JT, Siegel JN, Lesko LJ, Bai JP. Successes achieved and challenges ahead in translating biomarkers into clinical applications. AAPS J. 2010;12(3):243‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bauer DC, Black DM, Bouxsein ML, et al. Treatment‐related changes in bone turnover and fracture risk reduction in clinical trials of anti‐resorptive drugs: a meta‐regression. J Bone Miner Res. 2018;33(4):634‐642. [DOI] [PubMed] [Google Scholar]
- 43. Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48(5):677‐692. [DOI] [PubMed] [Google Scholar]
- 44. Hosking D, Adami S, Felsenberg D, et al. Comparison of change in bone resorption and bone mineral density with once‐weekly alendronate and daily risedronate: a randomised, placebo‐controlled study. Curr Med Res Opin. 2003;19(5):383‐394. [DOI] [PubMed] [Google Scholar]
- 45. Rosen CJ, Hochberg MC, Bonnick SL, et al. Treatment with once‐weekly alendronate 70 mg compared with once‐weekly risedronate 35 mg in women with postmenopausal osteoporosis: a randomized double‐blind study. J Bone Miner Res. 2005;20(1):141‐151. [DOI] [PubMed] [Google Scholar]
- 46. Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther. 2008;12(3):157‐170. [DOI] [PubMed] [Google Scholar]
- 47. Brixen K, Chapurlat R, Cheung AM, et al. Bone density, turnover, and estimated strength in postmenopausal women treated with odanacatib: a randomized trial. J Clin Endocrinol Metab. 2013;98(2):571‐580. [DOI] [PubMed] [Google Scholar]
- 48. Masarachia PJ, Pennypacker BL, Pickarski M, et al. Odanacatib reduces bone turnover and increases bone mass in the lumbar spine of skeletally mature ovariectomized rhesus monkeys. J Bone Miner Res. 2012;27(1):509‐523. [DOI] [PubMed] [Google Scholar]
- 49. Ma B, Luo B, Euler DH, et al. Applicability of in vitro‐in vivo translation of cathepsin K inhibition from animal species to human with the use of free‐drug hypothesis. Naunyn Schmiedebergs Arch Pharmacol. 2017;390(4):435‐441. [DOI] [PubMed] [Google Scholar]
- 50. Bone HG, Dempster DW, Eisman JA, et al. Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the long‐term Odanacatib fracture Trial. Osteoporos Int. 2015;26(2):699‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Recker R, Dempster D, Langdahl B, et al. Effects of odanacatib on transilial cortical remodeling/modeling and microarchitecture in postmenopausal women with osteoporosis: 5‐year data from the extension of the phase 3 Long‐term Odanacatib Fracture Trial (LOFT). J Bone Miner Res. 2016;31(Suppl 1). Available at http://www.asbmr.org/education/AbstractDetail?aid=58e6b175-c780-49ad-adfa-cb84267c0852. Accessed March 5, 2019. [DOI] [PubMed] [Google Scholar]
- 52. Eastell R, Dijk DJ, Small M, et al. Morning vs evening dosing of the cathepsin K inhibitor ONO‐5334: effects on bone resorption in postmenopausal women in a randomized, phase 1 trial. Osteoporos Int. 2016;27(1):309‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanaka M, Hashimoto Y, Hasegawa C. An oral cathepsin K inhibitor ONO‐5334 inhibits N‐terminal and C‐terminal collagen crosslinks in serum and urine at similar plasma concentrations in postmenopausal women. Bone. 2015;81:178‐185. [DOI] [PubMed] [Google Scholar]
- 54. Tanaka M, Hashimoto Y, Hasegawa C, Deacon S, Eastell R. Antiresorptive effect of a cathepsin K inhibitor ONO‐5334 and its relationship to BMD increase in a phase II trial for postmenopausal osteoporosis. BMC Musculoskelet Disord. 2017;18(1):267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hasegawa C, Ohno T, Umemura T, et al. Population pharmacokinetic and pharmacodynamic modeling of different formulations of ONO‐5334, cathepsin K inhibitor, in Caucasian and Japanese postmenopausal females. J Clin Pharmacol. 2014;54(1):23‐34. [DOI] [PubMed] [Google Scholar]
- 56. Adami S, Supronik J, Hala T, et al. Effect of one year treatment with the cathepsin‐K inhibitor, balicatib, on bone mineral density (BMD) in postmenopausal women with osteopenia/osteoporosis. J Bone Miner Density. 2006;21(Suppl 1):1085. [Google Scholar]
- 57. Collins WCJ, Roy SK, Pillai G, Woodworth TG. Safety and pharmacodynamics of ascending oral doses of the cathepsin K inhibitor, balicatib, in postmenopausal women. J Bone Miner Res. 2006;21(Suppl 1):S115‐S115. [Google Scholar]
- 58. Eastell R, Nagase S, Small M, et al. Effect of the cathepsin K inhibitor ONO‐5334 on biochemical markers of bone turnover in the treatment of postmenopausal osteopenia or osteoporosis: 2‐year results from the OCEAN atudy. J Bone Miner Res. 2011;26(Suppl 1). [Google Scholar]
- 59. Itabashi A, Kurata N, Sugita Y, Kawai Y. Balicatib, a novel cathepsin‐K inhibitor, increases serum intact PTH beyond diurnal patterns following 14‐daily administration in Japanese postmenopausal women. J Bone Miner Res. 2006;21(Suppl 1):S24‐S24. [Google Scholar]
- 60. Papanastasiou P, Ortmann CE, Olson M, Vigneron A, Trechsel U. Effect of three month treatment with the cathepsin‐k inhibitor, balicatib, on biochemical markers of bone turnover in postmenopausal women: evidence for uncoupling of bone resorption and bone formation. J Bone Miner Res. 2006;21(Suppl 1):S59‐S59. [Google Scholar]
- 61. Chappard D, Libouban H, Mindeholm L, Basle MF, Legrand E, Audran M. The cathepsin K inhibitor AAE581 induces morphological changes in osteoclasts of treated patients. Microsc Res Tech. 2011;73:726‐732. [DOI] [PubMed] [Google Scholar]
- 62. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alexander SP, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Supp 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]