

Abstract
Aims
Cetuximab associated with cisplatin and 5‐fluorouracil is used to treat patients with inoperable or metastatic head and neck squamous cell carcinomas (HNSCC) up until disease progression or unacceptable toxicities. To date, no biomarkers of efficacy are available to select patients who will benefit from treatment.
Methods
An ancillary pharmacokinetics (PK) exploration was performed in the context of a prospective study investigating circulating‐tumour cells vs progression‐free survival (PFS). Cetuximab plasma concentrations were analysed according to a population PK model. Individual exposure parameters were confronted with soluble epidermal growth factor receptor (sEGFR) concentrations, tumour response and PFS.
Results
PK data (28 patients, 203 observations) were best described by a two‐compartment model with linear elimination. Performance status (PS) significantly correlated to both cetuximab clearance and central volume of distribution with both parameters increasing by 33.3% (95% CI 1–65.6) for each 1‐point increase of PS compared to PS = 0. Univariate analysis showed that patients with higher trough cetuximab concentrations at Day 7 (Cmin,D7) had better tumour response (P = 0.03) and longer PFS (P = 0.035). However, multivariate analysis revealed that only PS and tumour size at baseline remained significantly associated with PFS. Levels of sEGFR increased during cetuximab treatment but were not associated with PFS in the multivariate analysis.
Conclusions
Our study prospectively indicates that PS is likely a confounding factor in the relationship between cetuximab PK and PFS, patients with a poor PS having lower cetuximab plasma exposure and lower PFS.
Keywords: cetuximab, concentration, efficacy, HNSCC, pharmacokinetics
1.
What is already known about this subject
Cetuximab is an anti‐EGFR monoclonal antibody that improves overall and progression‐free survival in metastatic and/or recurrent head and neck squamous cell carcinomas patients.
To date, cetuximab is given up until disease progression or unacceptable toxicities, and early biomarkers of efficacy are needed to select responder patients who would benefit from treatment.
Several previous studies have described a relationship between cetuximab plasma exposure and patients' outcome.
What this study adds
The present study confirms this PK‐PD relationship, but indicates that pejorative characteristics at baseline, such as performance status, are likely confounding factors associated with, on the one hand, greater cetuximab clearance and, on the other, poor outcome.
Monitoring of sEGFR showed that HNSCC patients have a higher baseline sEGFR concentration than healthy volunteers and display a significant sEGFR increase during cetuximab treatment.
2. INTRODUCTION
Head and neck squamous cell carcinomas (HNSCC) represent 90% of head and neck cancers1 and are widely spread malignancies with 150 000 new cases and 70 000 deaths per year in Europe.2 Inoperable recurrent and/or metastatic HNSCC are currently treated with combined cisplatin, fluorouracil (5‐FU), and cetuximab, an anti‐epidermal growth factor receptor monoclonal antibody. The addition of cetuximab to standard chemotherapy (EXTREME protocol) increased progression‐free survival (PFS) from 3.3 to 5.6 months and overall survival (OS) from 7.4 to 10.1 months.3 Whereas cisplatin/5‐FU is given every 3 weeks for six cycles, cetuximab is administered weekly until the disease progresses or unacceptable toxicities occur. The identification of early clinical or biological biomarkers of cetuximab efficacy could help to select patients who would benefit from treatment. Tumour EGFR expression is increased in HNSCC, and has been identified as a prognosis factor, but does not predict EGFR‐targeted therapy efficacy.4 Retrospective studies have reported an association between low phosphatase and tensin homolog expression and PFS and OS in patients under the EXTREME regimen,5 but they did not emerge as sufficiently effective to select patients. Recently, two independent studies have suggested that a high cetuximab plasma exposure may be associated with an improved clinical benefit. Indeed, a pharmacokinetic analysis of cetuximab concentrations from HNSCC patients revealed a link between global clearance (lower or higher than 0.747 L/day) and PFS (14.1 vs 11.6 months respectively, P = 0.0369) or OS (16.6 vs 6.3, P = 0.007).6 Recently, Becher et al.7 showed that residual cetuximab concentration was predictive of tumour response in 25 HNSCC patients.
The present study corresponds to an ancillary study of a multicentre clinical trial for which the main objective was to explore the predictive value of circulating tumour cells (CTCs) on PFS in recurrent and/or metastatic HNSCC patients. The objective of this research was to investigate cetuximab pharmacokinetics (PK) and its relationships with pharmacodynamic endpoints (plasma soluble EGFR, CTCs, tumour response, patient survival).
3. METHODS
3.1. Patients
The ancillary PK study was part of a prospective, non‐randomized, multicentre, open‐label study designed to explore the predictive value of CTCs on the PFS in recurrent and/or metastatic HNSCC patients treated by the EXTREME regimen that associates a platinum salt, fluorouracil and cetuximab as a first‐line of chemotherapy. This study was approved by the ethics committee Sud‐Méditerranée III (Number: 2012.03.02). Patients were eligible if they were over 18 years old with metastatic or recurrent histologically‐proven HNSCC with a WHO performance status ≤2 and a minimal life expectancy of two months after inclusion. Eighty patients (referred to as the “whole cohort”) were recruited from October 2012 to April 2014, with a maximum follow‐up of 3 years. Twenty‐eight patients participated in the pharmacokinetic study (PK cohort). The study was registered on ClinicalTrials.gov (NCT02119559).
3.2. Treatment
Patients were treated with the EXTREME regimen. On Day 1 of each 3‐week cycle, patients received a 1‐h infusion of cisplatin (100 mg/m2) or carboplatin (in case of renal insufficiency, target area under the curve of 5 mg·min/mL) plus 1000 mg/m2 of fluorouracil per day for 4 days, with a maximum of six cycles. Cetuximab was administered on Day 1 at a dose of 400 mg/m2 as a 2‐h infusion, and every week at a dose of 250 mg/m2 as a 1‐h infusion. Cetuximab administrations were maintained weekly after the six cycles of chemotherapy up until disease progression or unacceptable adverse effects.
3.3. Cetuximab concentration measurements
Blood samples (5 mL) to determine cetuximab concentrations were collected 3 (Cmax,D0), 6, 24, 48, 72 and 168 hours (Cmin,D7) after the beginning of the first infusion. Blood samples were also taken at cycle 2 and 3, before (Cmin,D21, Cmin,D42), and 2 hours after the start of the infusion (Cmax,D21, Cmax,D42). Cetuximab concentrations were analysed with liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) according to a previously published method7 with two minor modifications. The first one consists of adding bovine serum albumin (BSA, Sigma‐Aldrich, St Louis, MO) to the suspension buffer composition. Indeed, dry eluted samples were suspended in 50 μL water/acetonitrile [95/ (5 + 0.22% BSA) v/v] containing 0.1% formic acid. After centrifugation at 20000g for 5 min, supernatants were transferred into an LC vial. Secondly, a gradient elution starting from 95–5% of mobile phase A (H2O/0.1% formic acid) and B (acetonitrile/0.1% formic acid), respectively, to 5–95% of mobile phase A and B respectively, was set for 10 minutes. Finally, quantifying cetuximab was carried out by means of LT3 peptide measurements only, with the lower limit of quantification (LLOQ) set at 4.8 mg/L.
3.4. Pharmacokinetic analysis
Cetuximab concentration–time data were analysed using a population PK approach and the nonlinear mixed effects software NONMEM® version 7.2 (Icon Development Solution, Ellicott City, MD) with a first‐order conditional estimation (FOCE) method. A log‐normal distribution of PK parameters and a proportional residual error model were assumed. Structural model selection was based on the decrease of the objective function value (OFV) and of the residual variability. Structural models with one or two compartments were tested. Combinations of different approaches were studied to properly describe the particular elimination of cetuximab: a linear elimination process with clearance (CL), with or without an additional saturable mechanism (Michaelis–Menten equation)8 or a saturated process with a zero‐order constant.6
Model validation was performed by inspecting standard goodness of fit plots. Visual predictive check (VPC) was carried out by simulating concentration profiles of 1000 patients based on the estimates of the final PK model parameters.
The influence of covariates was studied using an allometric equation for continuous variables ( ) and a linear equation for discrete covariates ( ), with θi being the value of the parameter for the ith patient, the typical value of the parameter, covi the value of the covariate for the ith patient, covmed the median value of the covariate in the population, and θcov the effect of the covariate. The addition of a covariate was considered significant if OFV drop was greater than 3.84 (χ2, α < 0.05). Tested covariates were age, body weight (BW), height (HT), body surface area (BSA), baseline albuminemia (ALB), WHO performance status (PS), tumour size at baseline and plasma soluble EGFR (sEGFR) concentration both at baseline and as a time‐varying covariate. Baseline tumour size was missing for several patients, and was thus only tested in the subpopulation for whom data was available.
To assess pharmacokinetic/pharmacodynamics relationships and survival analysis, individual PK and exposure parameters were obtained as the empirical Bayesian estimator (EBE) from the analysis without covariates. End of infusion concentrations (Cmax) at D0, D21 and D42, as well as trough concentrations (Cmin) at D7, D21 and D42, were also studied to assess pharmacokinetic‐pharmacodynamic relationships.
3.5. Soluble EGFR (sEGFR) concentration determination
Plasma sEGFR9 was explored in 66 HNSCC patients of the current study before the first administration of cetuximab (D0), on Day 7 and Day 21, as well as in 44 healthy volunteers. The concentrations were measured with a commercial Human EGFR/ErbB1 Quantikine® ELISA kit (R&D Systems Europe, Lille, France).
3.6. Circulating tumour cell (CTC) determination
Techniques used for CTC detection are described in Supplementary methods 1. After quantification of CTCs, six different dichotomic variables were generated:
Positive if presence of CTCs at Day 0, vs negative if not, with the CK19/EGFR‐EPISPOT technique.
Positive if increase of CTCs between D0 and D7 (or if the same values were quantified), vs negative if decrease (or if quantification was equal to zero for both), with the EGFR‐EPISPOT technique.
Positive if increase of CTCs between D0 and D7 (or if the same values were quantified), vs negative if decrease (or if quantification was equal to zero for both), with the CellSearch technique.
Positive if presence of CTCs at Day 7 with EPISPOT and/or CellSearch, vs negative if double‐absence.
Positive if presence of CTCs at Day 7 with EPISPOT and/or Cytometry, vs negative if double‐absence.
Positive if presence of CTCs at Day 7 with CellSearch and/or Cytometry, vs negative if double‐absence.
3.7. Clinical endpoints
Radiological examination, including scanner, magnetic resonance imaging or positron emission tomography depending on the tumour localization, were performed every 2 months during the first year of the study, then every 3 months during the following 2 years. Tumour lesions were measured in accordance with Response Evaluation Criteria in Solid Tumor (RECIST) version 1.1 standards. The sum of the longest diameters of measurable tumours was collected as an estimation of the tumour burden at inclusion and after 2 months.
Tumour response allowed classifying patients into the following categories according to the RECIST 1.1 guidelines: progressive disease (PD), stable disease (SD) or partial response (PR). Because of the small size of the cohort, patients were classified into two groups for efficacy evaluation: tumour response (SD + PR) vs progression (PD). PFS was defined as the time elapsed between inclusion and death or tumour progression according to the RECIST 1.1 guidelines.
3.8. Statistical analysis
The relationship between continuous variables was assessed by linear regression. Comparison of sEGFR plasma rates between healthy volunteers and cancer patients at D0, and differences in Cmin between patients with tumour response vs patients with progressive disease were tested using a t‐test, and a paired t‐test for longitudinal follow‐up of sEGFR concentrations in cancer patients. CTC variables were compared to PK and sEGFR variables by t‐test with a significance threshold of P < 0.05. PFS was analysed with the proportional hazards Cox method. A univariate analysis was first performed with PK variables (CL, AUCD0‐D7, Cmin,D7, Cmin,D21, Cmin,D42, Cmax,D0, Cmax,D21, Cmax,D42), sEGFR variables (sEGFRD0, sEGFRD7, sEGFRD21, and sEGFRD7/sEGFRD0, sEGFRD21/sEGFRD0, sEGFRD21/sEGFRD7 ratios), initial tumour size, WHO PS, ALB, age, BW, HT, and BSA. Secondly, significant variables (P < 0.2) were included in a multivariate Cox model, with a maximum of one variable for PK and sEGFR to avoid adding of non‐independent variables. A backward deletion procedure was used to exclude non‐significant covariates in the multivariate analysis (likelihood ratio test, P < 0.05, 1 degree of freedom). Kaplan–Meier analyses were performed with a log‐rank test. Analyses were performed using R® version 3.2.0 (R Foundation for Statistical Computing, Vienna, Austria).
3.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.10, 11
4. RESULTS
4.1. Patients
Overall, our analyses included up to 66 patients (the “whole cohort”), with PK data available for 28 individuals (referred to as the “PK cohort”). Patients' baseline characteristics are shown in Table 1.
Table 1.
Baseline characteristics of patients
| Whole cohort (n = 66) | PK cohort (n = 28) | |||
|---|---|---|---|---|
| Median (min–max) | n | Median (min–max) | n | |
| Age (year) | 61 (22–81) | 66 | 61.5 (44–77) | 28 |
| Body weight (kg) | 61.5 (39–94) | 66 | 62 (43–81.8) | 28 |
| Height (cm) | 171.5 (152–186) | 66 | 170 (152–180) | 28 |
| Body surface area (m2) | 1.70 (1.31–2.09) | 66 | 1.70 (1.38–1.92) | 28 |
| sEGFR (ng/mL) | 41.4 (23.9–67.6) | 65 | 39.2 (23.9–55.9) | 27 |
| Tumour size (mm) | 43.5 (11.1–170) | 46 | 48.5 (14–170) | 20 |
| Albuminemia (g/L)a | 33.4 (17.5–55.3) | 28 | ||
| CTCs | ||||
| Epispot | 0 (0–320) | 65 | 0 (0–320) | 27 |
| Cellsearch sEGFR− | 0 (0–6) | 57 | 0 (0–6) | 25 |
| Cellsearch sEGFR+ | 0 (0–47) | 57 | 0 (0–13) | 25 |
| Percentage | n | Percentage | n | |
|---|---|---|---|---|
| Sex | ||||
| F | 13.6% | 9 | 3.6% | 1 |
| M | 86.4% | 57 | 96.4% | 27 |
| Primary localization b | ||||
| Oral cavity | 25.8% | 17 | 14.3% | 4 |
| Oropharynx | 45.5% | 30 | 46.4% | 13 |
| Larynx | 9.1% | 6 | 14.3% | 4 |
| Hypopharynx | 16.7% | 11 | 21.4% | 6 |
| Unknown | 4.5% | 3 | 7.1% | 2 |
| WHO Performance status | ||||
| 0 | 24.2% | 16 | 46.4% | 13 |
| 1 | 50.0% | 33 | 35.7% | 10 |
| 2 | 16.7% | 11 | 14.3% | 4 |
| Unknown | 9.1% | 6 | 3.6% | 1 |
| Disease extent at inclusion | ||||
| Locoregional relapse | 34.8% | 23 | 25.0% | 7 |
| Metastatic relapse | 31.8% | 21 | 42.9% | 12 |
| Locoregional and metastatic relapse | 24.2% | 16 | 14.3% | 4 |
| Unknown | 9.1% | 6 | 17.9% | 5 |
Albuminaemia was only determined in the patients of the PK cohort.
One patient from the PK cohort had an oral cavity + oropharynx primary localization, and another patient had oropharynx + hypopharynx.
4.2. Pharmacokinetic analysis
Pharmacokinetic data were available for 28 patients. A total of 205 cetuximab concentrations were quantified, but only 203 were kept for the model set‐up, as two concentrations were excluded after visual inspection of individual PK profiles. One concentration (3.6 mg/L) was below the LLOQ but was considered as meaningful based on analytical consistency and therefore was kept for the PK analysis. Mean residual concentrations (% CV) were 36.6 (67%), 45.1 (90%) and 63.6 (61%) μg/mL on Days 7 (n = 23), 21 (n = 21) and 42 (n = 18), respectively. A two‐compartment model with linear elimination, parameterized with volumes of central (V1) and peripheral compartments (V2), an inter‐compartmental clearance (Q) and plasma clearance (CL), best described cetuximab concentration data. Adding a nonlinear elimination process was associated neither with an OFV decrease nor loss of residual variability, whether using a Michaelis–Menten equation, a zero‐order term or combinations of both with a linear elimination. Inter‐occasion variability on central volume greatly improved the goodness of fit (OFV decrease = 22.6) and lowered residual variability from 32.2% to 24.7%. Inter‐individual variability was estimated for all parameters except Q. The parameter estimates of the model without covariates are shown in Table 2. The relationship between CL (obtained with this PK model) and Cmin,D7 was well described by the linear equation CL = 1.591–0.300*ln (Cmin,D7) (CL in L/day, Cmin,D7 in μg/mL, P = 10−6, r 2 = 0.68) (see Supplementary Figure S1). Goodness of fit plots of this model are reported in Supplementary Figure S2. Visual predictive checks are reported in Supplementary Figure S3. The area under the curve after the first administration (AUCD0‐D7) was calculated by dividing the first dose by the CL. The cumulative area under the curve of cetuximab concentrations vs time (AUCcum) was calculated as the sum of the amount of administered cetuximab during the three cycles of the PK evaluation (ie, a maximum of nine administrations) divided by CL.
Table 2.
Population estimates of the PK parameters obtained with the final model
| Parameters | Estimate | RSE (%) | Shrinkage (%) |
|---|---|---|---|
| Population parameters | |||
| V1 (L) | 4.4 | 10.5 | |
| V2 (L) | 8.1 | 32.9 | |
| Q (mL/h) | 27.7 | 15.7 | |
| CL (mL/h) | 19.4 | 15.6 | |
| Inter‐individual variability (CV%) | |||
| V1 | 43.0 | 18.0 | 14.1 |
| V2 | 132 | 16.1 | 38.9 |
| CL | 75.8 | 13.8 | 18.7 |
| Inter‐occasion variability (CV%) | |||
| V1 | 25.9 | 28.7 | |
| Residual variability (CV%) | |||
| Proportional error | 24.7 | 12.3 | 21.0 |
The study of covariates revealed that PS was the only significant covariate, with the same coefficient applied both to clearance (CL) and volume of central compartment (V1) (+33.3% per point of PS score, 95% CI 1–65.6%). This covariate decreased OFV by 10.0, and inter‐individual variability of CL and V1 by 10% (from 75.8% to 65.8%) and 4.1% (from 43% to 38.9%), respectively.
4.3. sEGFR
A description of sEGFR plasma concentrations is provided in Figure 1. Mean sEGFR blood concentrations were significantly higher in healthy volunteers than in cancer patients (56.6 vs 41.5 ng/mL, P < 2.2 × 10−16). The longitudinal follow‐up performed for patients treated with cetuximab revealed a significant increase of sEGFR levels from D0 to D7 (P < 2.2 × 10−16, n = 56), D0 to D21 (P < 2.2 × 10−16, n = 54) and D7 to 21 (P = 8.4 × 10−6, n = 48), with a mean rise of +67, +89 and +15%, respectively. There was no link between the size of the tumour at baseline and the sEGFR amount at Day 0 (P = 0.246, n = 46). At Day 7, there was a significant relationship between sEGFR increase and tumour size at baseline: the larger the tumour, the greater the increase at Day 7 (P = 0.0106, n = 39).
Figure 1.
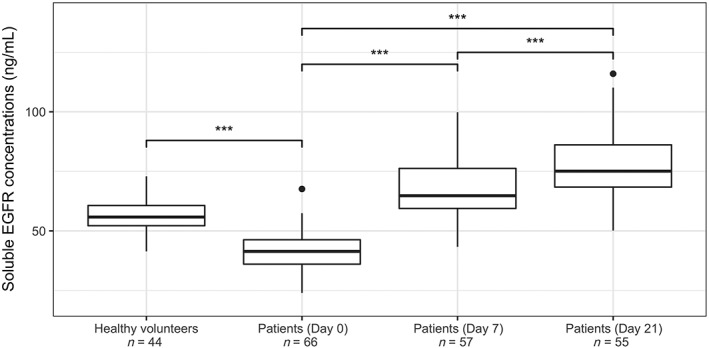
Soluble EGFR concentrations (ng/mL) in healthy volunteers and patients at Day 0, 7 and 21. ***: P < 0.001
5. PHARMACOKINETICS/PHARMACODYNAMICS RELATIONSHIPS
5.1. PK vs tumour response
Tumour response was available in 16 patients from the PK cohort. Patients with tumour response had a significantly higher Cmin,D7 (Figure 2A, 42.6 vs 18.8 μg/mL, P = 0.03) and Cmin,D21 (47.4 vs 19.0 μg/mL, P = 0.04) compared to patients with tumour progression. The same trend was observed with AUCD0‐D7 (P = 0.09) and AUCcum (P = 0.10) but not with Cmax. However, as a consequence of the aforementioned effect of PS on CL, Cmin,D7 (Figure 2B) and Cmin,D21 were higher in patients having a PS score equal to 0, compared to patients scoring 1 or 2 (P = 0.03 and 0.01, respectively, n = 23), suggesting that patients with tumour response are those with a favourable PS score.
Figure 2.
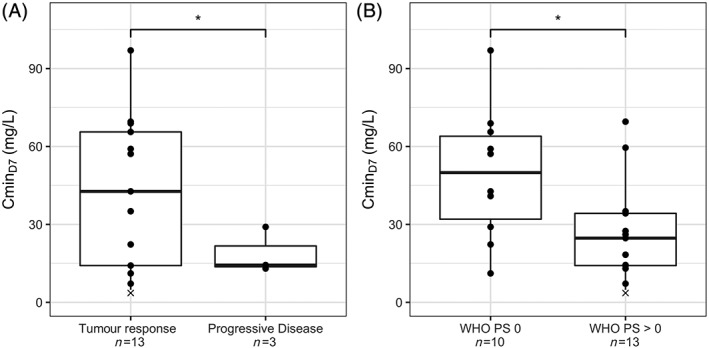
Cmin,D7 vs tumour response (A), or vs WHO PS (B). *: P < 0.05; ×: this concentration is below LLOQ (4.8 µg/mL)
5.2. PK vs CTCs
In the 28 patients of the PK cohort, there was no association between PK (Cmin, Cmax or AUCs) and CTC variation during treatment.
5.3. PK vs soluble EGFR
We observed a significant positive relationship between CL and sEGFR increase from Day 0 to Day 7 (P = 0.0341, n = 23) or to Day 21 (P = 1.43 × 10−5, n = 21): patients with high clearance values (and therefore the lowest AUC) had the highest increase of sEGFR (Supplementary Figure S4). This relationship was found with AUCD0‐D7 vs sEGFRD7/sEGFRD0 (P = 0.047, n = 23) and vs sEGFRD21/sEGFRD0 (P = 0.004, n = 21), but not with Cmin,D7 (P = 0.228, n = 20 and P = 0.0617, n = 17, respectively) or Cmin,D21 (P = 0.476, n = 19 and P = 0.135, n = 18, respectively).
5.4. PFS analysis
The median PFS in the PK cohort (n = 27, survival data unavailable for one patient) was 175 days (95% CI: 123–227). Among the significant variables in univariate analysis (Table 3), four were eligible for multivariate analysis: PS, Cmin,D7, initial tumour size and sEGFRD0.
Table 3.
Univariate and multivariate analysis of PFS
| Univariate analysis | ||||
|---|---|---|---|---|
| Group | Variable (n) | Hazard ratio | CI 95% | P‐value |
| PK | CL (n = 27) | 69.3 | 1.24e−08–3.88e+11 | 0.711 |
| AUCD0‐D7 (n = 27) | 1 | 1–1 | 0.127 | |
| Cmin,D7 (n = 23) | 0.981 | 0.964–0.999 | 0.035 | |
| Cmin,D21 (n = 21) | 0.99 | 0.979–1 | 0.075 | |
| Cmin,D42 (n = 18) | 0.996 | 0.982–1.01 | 0.563 | |
| Cmax,D0 (n = 27) | 0.994 | 0.988–1 | 0.081 | |
| Cmax,D21 (n = 20) | 0.997 | 0.993–1 | 0.218 | |
| Cmax,D42 (n = 19) | 0.997 | 0.994–1 | 0.178 | |
| sEGFR | sEGFRD0 (n = 27) | 1.05 | 0.983–1.12 | 0.151 |
| sEGFRD7 (n = 23) | 1.03 | 0.989–1.06 | 0.180 | |
| sEGFRD21 (n = 21) | 1.01 | 0.989–1.04 | 0.301 | |
| sEGFRD7/sEGFRD0 (n = 23) | 1.2 | 0.409–3.52 | 0.740 | |
| sEGFRD21/sEGFRD0 (n = 21) | 0.867 | 0.411–1.83 | 0.708 | |
| sEGFRD21/sEGFRD7 (n = 19) | 0.909 | 0.19–4.35 | 0.905 | |
| Age (n = 27) | 0.999 | 0.947–1.05 | 0.984 | |
| BSA (n = 27) | 0.989 | 0.0781–12.5 | 0.993 | |
| BW (n = 27) | 0.994 | 0.955–1.03 | 0.752 | |
| HT (n = 27) | 1.02 | 0.974–1.08 | 0.353 | |
| Initial tumour size (n = 20) | 1.01 | 0.998–1.02 | 0.149 | |
| WHO PS (n = 27) | 3.42 | 1.68–6.95 | 0.0007 | |
| ALB (n = 27) | 0.997 | 0.955–1.04 | 0.899 | |
| Multivariate analysis (n = 19) | ||||
|---|---|---|---|---|
| WHO PS | 4.67 | 1.833–11.91 | 0.0012 | |
| Initial tumour size | 1.01 | 1.001–1.02 | 0.039 | |
Figure 3 shows the Kaplan–Meier analysis for each of these four variables. PFS was higher in patients with PS = 0 vs PS ≥ 1 (P = 0.011, n = 27), and in patients with PS ≤ 1 vs PS = 2 (P < 10−4, n = 27). By stratifying patients according to their Cmin,D7 with the median as a cut‐off value (ie, 29 μg/mL), median PFS was 194 days in patients with a high Cmin,D7, vs 106 days in patients with a low Cmin,D7 (P = 0.0503, n = 23).
Figure 3.
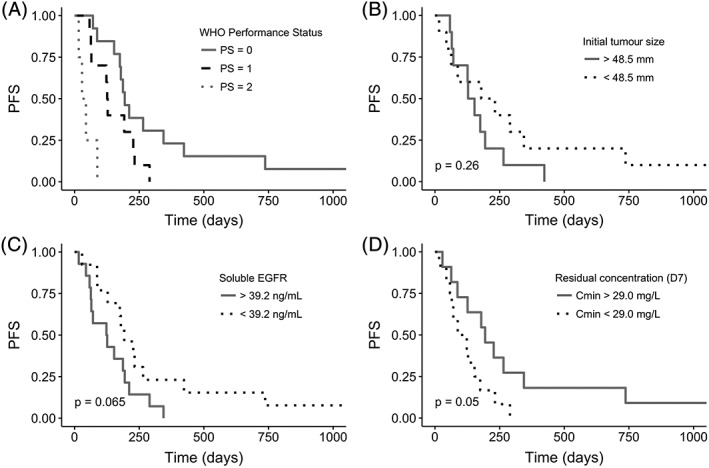
Progression‐free survival (PFS) as function of WHO PS (A), initial tumour size (B), sEGFR (C), and Cmin,D7 (D).
P‐values correspond to log‐rank tests. No P‐value was provided in Figure 3A because the log‐rank test must be performed between two groups, however the PFS was higher in patients with PS = 0 vs PS ≥ 1 (P = 0.011, n = 27), or in patients with PS ≤ 1 vs PS = 2 (P < 10−4, n = 27)
For multivariate analysis, backward deletion led to a final model with only PS (HR 4.672, 95% CI: 1.833–11.91, P = 0.00124) and initial tumour size (HR 1.010, 95% CI: 1.001–1.020, P = 0.039) as variables associated with PFS. As initial tumour size was missing for several patients, this multivariate analysis could be performed on only 19 patients: a second multivariate analysis was performed with PS, Cmin,D7, and sEGFRD0 in order to increase the sample size (n = 23) and ensure the robustness of our findings. After the backward exclusion, only the PS score remained as a significant covariate (HR 3.372, 95% CI: 1.59–7.151, P = 0.00153).
6. DISCUSSION
To the best of our knowledge, this is the first study to assess the impact of cetuximab exposure on PFS in a multivariate analysis including other factors in a prospective cohort of HNSCC patients.
A population pharmacokinetics model was used to analyse the multiple concentration data gathered over consecutive cycles for each patient. As for many monoclonal antibodies, cetuximab PK was expected to be driven by the antigen mass due to a target‐mediated drug disposition phenomenon (TMDD).12, 13 The nonlinear PK of cetuximab have previously been described with a Michaelis–Menten equation instead of a linear elimination,8 or, more recently, with a combination of a zero‐ and a first‐order elimination.6, 14 Here, the elimination was parametrized with a single first‐order clearance, as analysis of our data by adding a nonlinear elimination did not result in a better model. This discrepancy with previous studies might be explained by the fact that we had a limited number of low plasma concentrations (maximum three trough samples per individual) compared to the previously reported models.6, 14 Moreover, we observed only one trough concentration (out of 84 values) below 4.8 mg/L (LLOQ) contrary to the PK study of Azzopardi et al.,14 where several concentrations were below LLOQ, especially in patients for whom a weekly administration was delayed. These low detectable values might therefore have helped the authors to describe the nonlinear elimination in their PK model.
Although the global fit of the model is satisfying, regarding the low residual variability and the goodness of fit plots, the absence of nonlinear pharmacokinetics may be responsible for the incapacity to fit some of the lowest concentrations (Supplementary Figure S2).
Performance status was the only covariate of the final model. Pejorative status was associated with a greater clearance and a higher central compartment volume (+33% for each increase of 1 point of WHO PS score from PS = 0 to PS = 2). Pharmacokinetics driven by the disease status is commonly described with monoclonal antibodies, along with tumour size, disease type, inflammatory status (ie, CRP, leucocyte count, albuminaemia) or performance status mainly found as covariates that impact clearance or volume of distribution (or both) of monoclonal antibodies.12, 15 Baseline tumour size was not found to influence the PK of cetuximab, although it was tested in a subpopulation of the PK cohort (n = 20). Soluble EGFR also failed the covariate screening, but its relevance as a marker of antigen mass is controversial, due to its imprecise physiopathology16 and the differences in terms of concentrations with cetuximab (units in ng/mL for sEGFR vs μg/mL for cetuximab but with a similar molecular weight; 110 kDa vs 152 kDa respectively). The inclusion of inter‐occasion variability on V1 improved the fit of the data but did not reflect a systematic variation. Although time‐varying pharmacokinetics have been described for some therapeutic antibodies,17 there is no mechanistic hypothesis to explain this IOV on V1. Moreover, the value of this IOV (25.9%) remains limited compared to the inter‐individual variability (43.0%) of this parameter.
The PK model allowed us to generate CL values and AUCs for each patient. As expected, Cmin,D7 appeared well correlated with cetuximab clearance: interestingly, the log‐linear regression between Cmin,D7 and CL led to an equation that is very close to that observed by Azzopardi et al.14 between Cmin,D14 (μg/mL) and CL (L/h).
Our study corroborates previous work that associated cetuximab pharmacokinetics and efficacy. We found that patients with higher Cmin,D7 had better tumour response (Figure 2), such as seen in a preliminary phase I escalating single‐dose study, although it was carried out in patients with different types of carcinomas and outside the context of the EXTREME regimen.18 In a cohort of 25 HNSCC patients, Becher et al. obtained similar results between tumour response and late trough concentrations, and suggested a threshold of 33.8 mg/L to select responders and non‐responders.7 Univariate Cox analysis showed that Cmin,D7 was associated with PFS (HR 0.981, 95% CI: 0.964–0.999, P = 0.035), similarly to previous studies associating global clearance (a time‐independent criterion inversely proportional to cetuximab exposure) and Cmin,D14 to PFS in colorectal cancer,14 or global clearance to PFS and OS in HNSCC cancer.6
Nevertheless, Cmin,D7 influence was no longer significant in the multivariate Cox analysis, and only PS and baseline tumour size (to a lesser extent) remained associated with PFS. Therefore, these latter patient characteristics are likely to be confounding factors associated with lower cetuximab plasma exposure and worsened PFS.
Besides clinical response, we aimed to investigate the effect of cetuximab exposure on supposed surrogate biomarkers such as CTCs. Unfortunately, no relationships between CTCs and cetuximab exposure were identified but the small number of patients in the PK cohort with positive CTCs detection may have hampered this analysis.
Soluble EGFR was also measured in our study and analysed as a surrogate or a PD endpoint. HNSCC patients had lower mean plasma concentrations than healthy volunteers (41.5 vs 56.6 ng/mL). Such a difference was previously found by Lemos‐González et al. in head and neck cancer patients, however with different ranges of concentrations (21.2 vs 35.9 ng/mL) despite an identical ELISA method (R&D Systems, Minneapolis, MN).19 A longitudinal follow‐up of sEGFR plasma concentrations was also carried out, and an overall increase was observed from inclusion, to Day 7 and Day 21 (Figure 1). Previously, only Argiris et al. had studied this biomarker at different times, in 20 HNSCC patients with a cetuximab + bevacizumab co‐medication (no additional chemotherapy).20 They showed a 59% increase of sEGFR after 21 days of treatment compared to baseline, whereas we observed an 89% increase. The binding of a soluble antigen to an immunoglobulin—with a longer half‐life due to its Fc portion—creates a circulating antibody–antigen complex with larger residence time compared to that of the free soluble antigen. This phenomenon is often considered to explain the apparent accumulation of antigen over time.21 Surprisingly, we found that the higher percentages of increase in sEGFR were related to higher CL and lower AUC0–7. The reason for this correlation to the PK of cetuximab is poorly understood. One could hypothesize that the higher clearance could reflect a larger antigen mass that would release more sEGFR, as the sEGFR increase at D7 was higher in patients with larger tumours. However, sEGFR is a 110 kDa isoform of membrane EGFR, and is produced by alternative mRNA splicing9 or by EGFR ectodomain shedding.22 The impact of cancer or anticancer drugs on the mechanism of sEGFR production remains unclear,16 but sEGFR concentrations seem lower in cancer patients than in healthy volunteers, whatever the malignancy: non‐squamous cell lung cancer,19, 23, 24 breast cancer25 or epithelial ovarian cancer.9, 26 One study reported higher sEGFR in gastric cancer patients vs healthy controls.27 Overall our data showed that sEGFR may be related to HNSCC cancer detection, but cannot be used as a surrogate marker of cetuximab efficacy.
To conclude, the residual concentration of cetuximab at Day 7 (Cmin,D7) was found to be associated with better outcome in our study, both in terms of tumour response and survival. Because low cetuximab concentration was associated with worsened PS, we cannot conclude that a low exposure rather than the patients' state is responsible for shorter PFS. The small size of our cohort does not allow us to study the impact of PK within each PS group, but this question should be addressed in larger studies.
Indeed, we cannot definitively exclude that inter‐patient PK variability of monoclonal antibodies (and cetuximab in particular) may contribute (independently of disease status) to efficacy. Moreover, regarding the impressive bibliography describing the impact of tumoral disease on the pharmacokinetics of monoclonal antibodies (ie, lower plasma mAb concentrations in patients with progressive disease) prospective studies showing benefits of dose increase in patients with low concentration are needed before considering any implementation of TDM of these compounds.
COMPETING INTERESTS
F.L.L., C.A.‐P., T.L., B.C.A., R.G., L.D., D.C., J.‐P.D., B.L., M.A., K.A., M.M., F.B., F.P., E.C. and F.T. have no competing interests to declare. J.G. has served on advisory boards for AstraZeneca, Bristol‐Myers Squibb, Innate Pharma, Merck KGaA and Nanobiotix, and has received grants for research from GSK, Bristol‐Myers Squibb, Chugai and Merck KGaA.
CONTRIBUTORS
C.A.‐P., R.G, D.C., E.C. and F.T. were involved in conception and design of the study. C.A.‐P., T.L., B.C.A., R.G., L.D., J.G., D.C., J.‐P.D., B.L., M.A, K.A, M.M. and F.B. were involved in acquisition of the data. F.L.L, C.A.‐P., M.M., F.P. and E.C. were involved in analysis and interpretation of the data. F.L.L., F.T. and E.C. drafted the manuscript and all other authors critically revised the manuscript. All authors gave final approval of the manuscript to be published. R.G. was the principal investigator.
Supporting information
DATA S1 Supporting information
DATA S2 Supporting information
ACKNOWLEDGEMENTS
This work was supported by the PAIR VADS 2011 programme funded by the French “Institut National du Cancer” (INCa), “Fondation ARC pour la Recherche sur le Cancer” and the “Ligue contre le Cancer” foundation. This study was approved by the ethics committee Sud‐Méditerranée III (Number: 2012.03.02), and was performed in accordance with the Declaration of Helsinki. It was registered on ClinicalTrials.gov (NCT02119559). Every patient gave written informed consent.
Le Louedec F, Alix‐Panabières C, Lafont T, et al. Cetuximab pharmacokinetic/pharmacodynamics relationships in advanced head and neck carcinoma patients. Br J Clin Pharmacol. 2019;85:1357–1366. 10.1111/bcp.13907
The authors confirm that the Principal Investigator for this paper is Renaud Garrel and that he had direct clinical responsibility for patients.
ClinicalTrials.gov: NCT02119559 (CIRCUTEC)
REFERENCES
- 1. Haines GK. Pathology of head and neck cancer: Epithelial and related tumors In: Radosevich JA, ed. Head and Neck Cancer: Current Perspectives, Advances and Challenges. New York: Springer; 2013:257‐287. [Google Scholar]
- 2. Ferlay J, Steliarova‐Foucher E, Lortet‐Tieulent J, et al. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374‐1403. [DOI] [PubMed] [Google Scholar]
- 3. Vermorken JB, Mesia R, Rivera F, et al. Platinum‐based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116‐1127. [DOI] [PubMed] [Google Scholar]
- 4. Licitra L, Mesia R, Rivera F, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first‐line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol. 2011;22(5):1078‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. da Costa AABA, D'Almeida Costa F, Ribeiro AR, et al. Low PTEN expression is associated with worse overall survival in head and neck squamous cell carcinoma patients treated with chemotherapy and cetuximab. Int J Clin Oncol. 2015;20(2):282‐289. [DOI] [PubMed] [Google Scholar]
- 6. Pointreau Y, Azzopardi N, Ternant D, Calais G, Paintaud G. Cetuximab pharmacokinetics influences overall survival in patients with head and neck cancer. Ther Drug Monit. 2016;38(5):567‐572. [DOI] [PubMed] [Google Scholar]
- 7. Becher F, Ciccolini J, Imbs D‐C, et al. A simple and rapid LC‐MS/MS method for therapeutic drug monitoring of cetuximab: a GPCO‐UNICANCER proof of concept study in head‐and‐neck cancer patients. Sci Rep. 2017;7(1):2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dirks NL, Nolting A, Kovar A, Meibohm B. Population pharmacokinetics of cetuximab in patients with squamous cell carcinoma of the head and neck. J Clin Pharmacol. 2008;48(3):267‐278. [DOI] [PubMed] [Google Scholar]
- 9. Baron AT, Cora EM, Lafky JM, et al. Soluble epidermal growth factor receptor (sEGFR/sErbB1) as a potential risk, screening, and diagnostic serum biomarker of epithelial ovarian cancer. Cancer Epidemiol Prev Biomark. 2003;12(2):103‐113. [PubMed] [Google Scholar]
- 10. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol. 2017;174(Suppl 1):S225‐S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ternant D, Azzopardi N, Raoul W, Bejan‐Angoulvant T, Paintaud G. Influence of antigen mass on the pharmacokinetics of therapeutic antibodies in humans. Clin Pharmacokinet. 2019;58(2):169‐187. [DOI] [PubMed] [Google Scholar]
- 13. Levy G. Pharmacologic target‐mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248‐252. [DOI] [PubMed] [Google Scholar]
- 14. Azzopardi N, Lecomte T, Ternant D, et al. Cetuximab pharmacokinetics influences progression‐free survival of metastatic colorectal cancer patients. Clin Cancer Res. 2011;17(19):6329‐6337. [DOI] [PubMed] [Google Scholar]
- 15. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maramotti S, Paci M, Manzotti G, et al. Soluble epidermal growth factor receptors (sEGFRs) in cancer: Biological aspects and clinical relevance. Int J Mol Sci. 2016;17(4):593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu C, Yu J, Li H, et al. Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101(5):657‐666. [DOI] [PubMed] [Google Scholar]
- 18. Fracasso PM, Burris H, Arquette MA, et al. A phase 1 escalating single‐dose and weekly fixed‐dose study of cetuximab: pharmacokinetic and pharmacodynamic rationale for dosing. Clin Cancer Res. 2007;13(3):986‐993. [DOI] [PubMed] [Google Scholar]
- 19. Lemos‐González Y, Rodríguez‐Berrocal FJ, Cordero OJ, Gómez C, Páez de la Cadena M. Alteration of the serum levels of the epidermal growth factor receptor and its ligands in patients with non‐small cell lung cancer and head and neck carcinoma. Br J Cancer. 2007;96(10):1569‐1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Argiris A, Kotsakis AP, Hoang T, et al. Cetuximab and bevacizumab: Preclinical data and phase II trial in recurrent or metastatic squamous cell carcinoma of the head and neck. Ann Oncol. 2013;24(1):220‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Igawa T, Haraya K, Hattori K. Sweeping antibody as a novel therapeutic antibody modality capable of eliminating soluble antigens from circulation. Immunol Rev. 2016;270(1):132‐151. [DOI] [PubMed] [Google Scholar]
- 22. Wilken JA, Perez‐Torres M, Nieves‐Alicea R, et al. Shedding of soluble epidermal growth factor receptor (sEGFR) is mediated by a metalloprotease/fibronectin/integrin axis and inhibited by cetuximab. Biochemistry. 2013;52(26):4531‐4540. [DOI] [PubMed] [Google Scholar]
- 23. Jantus‐Lewintre E, Sirera R, Cabrera A, et al. Analysis of the prognostic value of soluble epidermal growth factor receptor plasma concentration in advanced non‐small‐cell lung cancer patients. Clin Lung Cancer. 2011;12(5):320‐327. [DOI] [PubMed] [Google Scholar]
- 24. Lococo F, Paci M, Rapicetta C, et al. Preliminary evidence on the diagnostic and molecular role of circulating soluble EGFR in non‐small cell lung cancer. Int J Mol Sci. 2015;16(8):19612‐19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Asgeirsson KS, Agrawal A, Allen C, et al. Serum epidermal growth factor receptor and HER2 expression in primary and metastatic breast cancer patients. Breast Cancer Res. 2007;9(6):R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baron AT, Boardman CH, Lafky JM, et al. Soluble epidermal growth factor receptor (SEG‐FR) and cancer antigen 125 (CA125) as screening and diagnostic tests for epithelial ovarian cancer. Cancer Epidemiol Prev Biomark. 2005;14(2):306‐318. [DOI] [PubMed] [Google Scholar]
- 27. Oh M‐J, Choi J‐H, Kim IH, et al. Detection of epidermal growth factor receptor in the serum of patients with cervical carcinoma. Clin Cancer Res. 2000;6(12):4760‐4763. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1 Supporting information
DATA S2 Supporting information