

Abstract

Protein footprinting coupled with mass spectrometry is being increasingly used for the study of protein interactions and conformations. The hydroxyl radical footprinting method, fast photochemical oxidation of proteins (FPOP), utilizes hydroxyl radicals to oxidatively modify solvent accessible amino acids. Here, we describe the further development of FPOP for protein structural analysis in vivo (IV-FPOP) with Caenorhabditis elegans. C. elegans, part of the nematode family, are used as model systems for many human diseases. The ability to perform structural studies in these worms would provide insight into the role of structure in disease pathogenesis. Many parameters were optimized for labeling within the worms including the microfluidic flow system and hydrogen peroxide concentration. IV-FPOP was able to modify several hundred proteins in various organs within the worms. The method successfully probed solvent accessibility similarily to in vitro FPOP, demonstrating its potential for use as a structural technique in a multiorgan system. The coupling of the method with mass spectrometry allows for amino-acid-residue-level structural information, a higher resolution than currently available in vivo methods.
Human diseases often involve dynamic interactions between molecular and cellular systems that influence pathogenesis. An understanding of these interactions is essential for the development of treatments for diseases. Studies, both in vitro and in cell cultures, have provided a wealth of information on protein conformations and interactions, but the interplay between the cellular systems of an in vivo system cannot be capitulated in these less complex systems. Cell-based studies are limited by the use of only one cell type in a monolayer culture. In some cases, substances that produce a response in a whole animal do not produce a response in isolated cells or tissues.1 The lack of whole animal physiology, interacting organ systems, and cell type variety limits the information that can be provided from monolayer cell culture. The use of three-dimensional cell culture aims to ease this issue by providing the ability to culture multiple cell types in a single dish. Still, these model systems do not demonstrate the effect of a multiorgan system on pathogenesis, highlighting the need to continue to use animals as models for human disease. Owing to limitations in currently available in vivo structural methods, the study of protein structure in animal models requires the development of new analytical tools.
Protein footprinting coupled with mass spectrometry is being increasingly used to study protein interactions and conformations. Footprinting methods utilize chemical labels to modify proteins, and these modifications are detected and quantified by mass spectrometry (MS). Methods such as hydrogen–deuterium exchange mass spectrometry (HDX-MS) have been instrumental in studying the higher order structure of proteins.2−4 However, because HDX-MS utilizes a reversible label, it would be difficult for this method to be used for in vivo studies that require time-consuming postlabeling sample processing. Another protein footprinting method, hydroxyl radical protein footprinting (HRPF), employs an irreversible label. HRPF utilizes hydroxyl radicals to oxidatively modify the side chains of solvent accessible amino acids.5 Solvent accessibility, which changes upon protein perturbation, is assessed by performing a differential experiment. HRPF techniques have been successfully used in vitro to identify protein–ligand interaction sites,6−9 protein–protein interactions sites,10,11 and regions of conformational change.12,13 The HRPF method, fast photochemical oxidation of proteins (FPOP), generates hydroxyl radicals via excimer laser (248 nm) photolysis of H2O2.14 Recently, the FPOP method has been extended for in-cell studies,15,16 demonstrating the flexibility of this method for studying complex systems. In-cell FPOP (IC-FPOP) takes into account the role macromolecular crowding plays in protein interactions and conformations.
Here, we report the extension of FPOP for in vivo protein structural analysis in C. elegans. C. elegans, a member of the nematode family, is widely used to study a variety of biological processes including gene regulation, aging, metabolism, cell signaling, and apoptosis.17 Genome sequencing of C. elegans has revealed that ∼80% of its proteins are conserved between worms and vertebrates.18 The neuronal signal pathways and cell biological principles are conserved between C. elegans and humans.19 This conservation contributes to the efficacy of using C. elegans as a model system for human disease. An important characteristic of C. elegans, which makes them especially suited for in vivo FPOP (IV-FPOP), is that they are transparent to the laser light, allowing it to penetrate the animal.20C. elegans have the ability to intake H2O2 by both passive and active diffusion through cuticle absorption and ingestion.21 A major advantage of using C. elegans as an animal model is the ease of growth and maintenance.
The ability of FPOP to oxidatively modify 19 of the 20 amino acids makes it advantageous for in vivo analysis.22,23 This signifies that multiple proteins, regardless of amino-acid sequence, can be oxidatively modified in the worms. In addition, the irreversible nature of the ligand means that the worms can be processed, including worm lysis, protein precipitation, and protein digestion, for MS analysis without the loss of the label. The current benchmark for in vivo analysis of proteins is fluorescence imaging, which relies on fusing the protein of interest to a fluorescent protein for experiments.24 In some cases, this can lead to experimental error, because the fluorescent protein can affect the function and subcellular localization of the protein of interest.25,26 An advantage of IV-FPOP is the protein of interest does not need to be tagged prior to analysis, so it is studied in its native conformation. In addition, fluorescence-based methods do not have the resolution to characterize conformational changes and interaction sites on the amino-acid level. FPOP coupled to high-resolution mass spectrometry is capable of providing data on the amino-acid-residue and subresidue level.10,27 Furthermore, MS-based proteomic methods have provided information on thousands of proteins in lysate samples, making IV-FPOP applicable for proteome-wide structural biology, where structural information can be obtained on hundreds of proteins across the proteome in a single experiment.28,29 In contrast, fluorescence imaging can only analyze one or two proteins in a single experiment.
Here, we describe the IV-FPOP workflow, which includes the flow of worms, laser irradiation, and LC/MS/MS analysis (Figure 1), and detail the optimization of conditions to successfully perform FPOP in C. elegans. We demonstrate the ability to oxidatively modify proteins within multiple tissues of the worm, signifying that IV-FPOP would be efficacious in studying proteins in an animal model for human disease. IV-FPOP has the potential to fill a gap in knowledge in studying protein structure directly in animals.
Figure 1.

In vivo FPOP workflow. Worms are grown to the fourth larvae stage (L4) on nematode growth media plates. For IV-FPOP, worms are flowed through a 250 μm fused silica capillary in the presence of H2O2, and radicals are generated using a 248 nm wavelength excimer laser. Immediately after irradiation, excess H2O2 and radicals are quenched, worms are lysed, the protein extract is digested and prepared for mass spectrometry analysis, and the extent of FPOP modifications is calculated for proteins of interest.
Experimental Section
Nematode and Bacterial Culture
Nematodes were maintained using standard laboratory procedures at 20 °C on bacterial lawns containing either OP50 or NA22 E. coli on nematode growth medium (NGM) or 8P plates, respectively.30C. elegans strain BY205 (Pdat-1::GFP) were a kind gift from Dr. Richard Nass at Indiana University School of Medicine.
In Vivo FPOP
L4 larvae were harvested as described above. Each sample was prepared with approximatively 10 000 worms in 3 mL of M9 buffer. The flow system was adapted from a homemade flow system described by Konnerman et al.31,32 In this flow system, two fused silica capillaries (Polymicro Technologies) of 250 μm inner diameter (ID) and 360 μm outer diameter (OD) were connected to a third capillary of the same ID. The three capillaries are connected by a homemade tee made of FEP tubing (IDEX Heath & Science, 1/16″ OD × 0.02″ ID) with a dead volume of ∼3 nL. Two 5 mL syringes were each connected to a fused silica capillary and advanced by a syringe pump (KD Scientific, Legato Model 101). Worms were kept separated from H2O2 and mixed at the tee just prior to IV-FPOP, for approximately 1.5 s. Worms were mixed in the syringe using a VP710 tumble stirrer (V&P Scientific) with six stir discs (VP722fF, V&P Scientific) to prevent settling. The final H2O2 concentrations used were 20, 60, 100, 140, and 200 mM. The KrF excimer laser (GAM Laser Inc.) at a wavelength of 248 nm was set to pulse frequencies of 20 and 50 Hz, and the pump flow was set at 147.26 and 375.53 μL/min, respectively. The laser energy was 150 ± 2.32 mJ with no exclusion volume and a pulse width of 2.55 mm. Worms were collected in a 15 mL conical tube containing the quench buffer. DMSO (1%) was added to the quench buffer to inhibit methionine sulfoxide reductase. Labeling studies were performed in biological duplicates, each in technical triplicates with an equal number of controls (no laser irradiation, and no peroxide and no laser irradiation).
Protein Extraction and Digestion
Following IV-FPOP, the worm samples were pelleted by centrifugation at 2000 rpm, and the quench buffer was removed. Pellets were resuspended in lysis buffer (8 M urea, 0.5% SDS, 50 mM HEPES, 50 mM NaCl, 1 mM EDTA, 1 mM PMSF) and transferred to clean microcentrifuge tubes. Protein homogenate was sonicated for 10 s, followed by 60 s of incubation on ice. Small aliquots (2 μL) of homogenate were observed under a stereomicroscope (Olympus SSZ Model, 45× magnification) to check for homogenization. The sonication cycle was repeated until homogenization was complete. After homogenization, samples were centrifuged at 400 × g for 4 min at 4 °C, and the supernatant was transferred to a clean microcentrifuge tube. BenzNuclease (ACROBiosystems) was added to the lysate following the manufacturer’s protocol. The protein lysate was reduced with 10 mM dithiothreitol (DTT) for 45 min at 50 °C, cooled down at room temperature for 10 min, and then alkylated with 20 mM iodoacetamide (IAA) for 20 min in the dark at room temperature. Protein lysate was purified by an overnight acetone precipitation. The sample was resuspended in 25 mM Tris HCl pH 8 and quantified using a BCA assay (Thermo Scientific). An overnight trypsin (Thermo Scientific) digestion of 100 μg of protein was performed at a final ratio of 50:1 of protein/enzyme at 37 °C. The digestion was quenched using 5% formic acid. Peptides were dried by cold trap centrifugation and then resuspended with 0.1% formic acid at final concentration 10 μg/μL.
LC/MS/MS Analysis
Digested samples (50 μg) were loaded onto an M-Class C18 trap column (Waters) and washed for 10 min with 0.1% formic acid at 15 μL/min with an M-Class Acquity liquid chromatograph. Peptides were eluted and separated on a silica capillary column that was packed in-house with C18 reverse phase material (Aqua, 0.075 × 20 mm, 5 μm, 125 A, Phenomenex). The gradient was pumped at 300 nL/min for 120 min as follows: 0–1 min, 3% solvent B (acetonitrile, 0.1% formic acid); 2–90 min, 10–45% B; 100–105 min, 100% B; 106–120 min, 3% B. Eluted peptides were analyzed under positive ion mode nanoflow electrospray using an Orbitrap Lumos Fusion mass spectrometer (Thermo Scientific). The mass spectrometer was operated in data-dependent acquisition mode. MS1 spectra were acquired from over an m/z range of 375–1500 with 60 000 resolution, with a dynamic exclusion of 60 s. The AGC target was set at 5.5e5 with a maximum injection time of 50 ms and 5.0E4 intensity threshold. Ions with charge states of +1 and >6 were ejected. MS2 ions were subjected to high-energy collisional dissociation (HCD) (32% normalized collision energy) and detected in the orbitrap with 15 000 resolution and a 5.0e4 AGC target.
Data Analysis
Data analysis was performed as previously described.33 Briefly, raw files were analyzed using Proteome Discover 2.2 software (Thermo Scientific) using Sequest version 1.4. All files were searched against the Uniprot C. elegans database containing 26 794 sequences. Sequest was searched with a fragment mass tolerance of 0.02 Da and a precursor mass tolerance of 10 ppm. All known hydroxyl radical side-chain modifications34,35 were searched as variable modifications. Carbamidomethylation of cysteine was specified as a fixed modification. The enzyme specificity was set to trypsin allowing for one missed cleavage. A target decoy database search was performed. Peptide identification was established at a greater than 95% confidence (medium) and 99% confidence (high) for both peptide- and residue-level analysis, respectively. The false discovery rate (FDR) was set at 1%. All data was exported to Excel and analyzed summarized using the PowerPivot add-in. The extent of oxidation per peptide or residue was determined according to the following equation
| 1 |
For peptide-level analysis, EIC area modified is the area of the peptide with a modified residue, and EIC area is the total area of the same peptide with and without the modified residue. For residue-level analysis, EIC area modified is the area of the residue with a modified residue, and EIC area is the total area of the same residue with and without the modified residue.
Label-free quantification was calculated by the summation of each protein associated peptide using the following equation
| 2 |
TSA is the total sample abundance area for identified proteins in laser irradiated samples in biological replicates 1 and 2.
Results and Discussion
Microfluidic System Design
To limit over oxidation, both in vitro and IC-FPOP utilize flow to transport the sample past the laser aperture, ensuring a single exposure to irradiation.14,16 However, the increased size of C. elegans heightens the possibility of clogging in the flow system. This is compounded by the tendency of C. elegans to curl their bodies for locomotion. To overcome these issues, a new microfluidic system, adapted from Konermann et al.,31 was used for in vivo labeling (Figure 2a). FEP tubing, with a dead volume of ∼3 nL, was used as a sleeve to connect two capillaries of equal inner (ID) and outer diameter (OD) to a third capillary of equal diameter (Figure S1). One capillary is used to flow H2O2, while the second is used to flow the C. elegans. The two streams are mixed at the sleeve junction and flowed together through the third capillary. By constructing the flow system without physical connectors, clogging is reduced. Keeping worms separate from peroxide until just prior to laser irradiation limits peroxide clearance by the various biological mechanisms utilized by different cell types.36
Figure 2.

Flow system adapted for in vivo labeling in C. elegans. (a) Schematic of IV-FPOP flow system. Worms are kept separated from H2O2 until just prior to labeling; the window for laser irradiation is in light blue. (b) Percent recovery of worms after the flow system for two biological replicates (BR) with fused silica of 250 (gray) and 150 (black) μm ID. Error bars are calculated from the standard deviation across technical triplicates.
Two capillary IDs, 150 and 250 μm, were tested to determine optimal size for worm recovery. The 150 μm size tubing was previously used for in vitro and parts of the IC-FPOP flow system. The 250 μm capillary size was chosen based on a commercially available flow-sorting system for C. elegans.37 Worms were weighed and then flowed through the system constructed with either 150 or 250 μm ID tubing as they would for an FPOP experiment but without laser irradiation. The worms were collected, dried, and weighed again to calculate percent recovery. A comparison of the two IDs indicates that the 250 μm capillary recovered 63–89% across two biological replicates (BR) (Figure 2b). This is ∼3-fold higher recovery than with the 150 μm ID capillary, where only 22–31% is recovered over two BR (Figure 2b). The larger diameter capillary allows for better unimpeded flow for the worms (Figure S1b,c). For subsequent IV-FPOP experiments, the 250 μm diameter capillaries were used.
Viability of C. elegans in the Presence of H2O2
For IV-FPOP to effectively monitor biologically relevant interactions and conformations, it is imperative to label proteins in live worms. One means to limit worm death is to perform IV-FPOP promptly after harvesting worms, as food source depletion could be lethal. Another is to limit the H2O2 exposure time prior to laser irradiation. Previous studies have demonstrated that the effects of H2O2 on worm viability are heavily dependent on incubation time. In a study testing the effects of time and peroxide concentration, continuous incubation with 1 or 5 mM H2O2 for 24 or 4 h, respectively, was lethal for the worms.38 However, Kumsta et al. demonstrated that after incubation with 6 or 10 mM H2O2 for 30 min, greater than 95% of worms survived, and worm life span was not altered.39 By using the flow system described above with a 250 μm ID and a flow rate of 375 or 147 μL/min, dependent on the laser frequency, worms are exposed to peroxide for only 1.5 or 3.8 s, respectively, prior to laser irradiation. Even with this short incubation time, it remains necessary to determine whether the peroxide is lethal to the worms, especially at the higher concentrations of H2O2 required for IV-FPOP.
The fluorescent dye propidium iodide (PI) was used to ascertain the viability of C. elegans in the presence of H2O2. PI intercalates the DNA of dead worms, while viable worms expel the dye.40 Several different peroxide concentrations, from 20 to 200 mM, were tested. Worms were incubated with peroxide for 30 s, a 20-fold longer time than actual IV-FPOP exposure. After incubation, 20 mM N′-dimethylthiourea (DMTU) and 20 mM N-tert-butyl-α-phenylnitrone (PBN) were used to quench H2O2 and OH radicals, respectively. Fluorescence imaging reveals some minor worm death (Figure 3a); yet, even at high peroxide concentrations, the worm death is significantly lower than that with incubation with methanol (Figure 3), which is known to kill C. elegans.40 Comparison of the bright field image of worms to the fluorescence image after incubation indicates that for all concentrations, the proportion of dead worms is very low (Figure 3a). The percent viability of H2O2 treated worms is similar to that of the positive control of untreated worms (<2% loss of viability), even at concentrations as high as 200 mM (Figure 3b). There is not a statistically significant difference between the various concentrations, indicating at this short time frame, concentration-dependence is minimal. The negligent worm death in all concentrations shows that IV-FPOP is probing live worms even at high H2O2 concentrations.
Figure 3.

C. elegans viability in the presence of H2O2. (a) Bright field (black and white) and fluorescent (red) images of C. elegans (∼1000) at various H2O2 concentrations after 30 s of incubation. Fluorescent images show dead worms stained with PI. (b) Percent viability of worms using nine different H2O2 concentrations for two biological replicates. Negative control worms are in the presence of 50% methanol, and positive control worms have no H2O2 added. Error bars are calculated from the standard deviation across technical triplicates.
In Vivo FPOP Oxidatively Modifies Proteins within Different Organs and Tissues
Various concentrations of H2O2 were tested to determine the best concentration for labeling proteins in worms. To calculate the number of modified proteins, background oxidation was taken into account (Figure S2). Modified proteins were identified at all H2O2 concentrations with 200 mM proving the highest number with 199 proteins modified in BR2 (Figure S3). To improve the number of modified proteins, we increased the laser frequency from 20 to 50 Hz to provide multiple exposures across the body of the worms. Under these conditions and using 200 mM H2O2, a total of 545 proteins across two BR were oxidatively modified, approximately 3-fold greater than the 191 proteins modified across two BR at a laser frequency of 20 Hz (Figure 4a). An increased number of modified proteins were observed in BR2. This was attributed to a higher abundance of expressed proteins in BR2, leading to an increased number of modifications (Figure S4a,b). Also, there were only 36 proteins commonly modified in both BR. This discrepancy could be attributed to gene variability between BR samples, which has previously been linked to protein abundances.41
Figure 4.
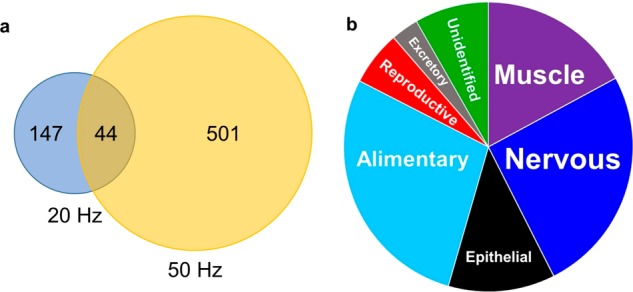
IV-FPOP oxidatively modifies proteins within C. elegans. (a) Venn diagram of modified proteins at 20 and 50 Hz laser frequencies. (b) Pie chart of oxidatively modified proteins within different body systems.
The potential of IV-FPOP lies in its possible use as a general strategy to simultaneously study a wide variety of proteins in C. elegans. This would allow the method to probe the role of protein interactions and conformations in disease states within the relevant tissue or organ. For this, IV-FPOP must demonstrate the ability to modify proteins in all body systems within the worm. To this end, we wanted to analyze the location of the modified proteins. Each of the 545 proteins modified at 50 Hz was matched to its primary gene through UniProt and rematched to its corresponding organ and tissue using the online Gene Search tool by The Genome BC C. elegans Gene Expression Consortium.42 All the various body systems within the worm are represented by a modified protein (Figure 4b and Table S1). The majority are found in the nervous, alimentary, and muscle systems; although proteins from the epithelial, reproductive, and excretory systems are also represented (Figure 4b). Because C. elegans can ingest H2O2, it is understandable that the majority of the modified proteins are found in the alimentary system. A large number are also found in the nervous system. C. elegans can also uptake H2O2 through the skin, and once absorbed, neurons are the initial body system the peroxide will encounter. The presence of oxidatively modified proteins in various body systems demonstrates that IV-FPOP would be useful in the study of proteins within various organs and tissues.
IV-FPOP Probes Solvent Accessibility in C. elegans
FPOP has been shown to report on protein solvent accessibility in vitro14,43 and in cell.44 To determine if IV-FPOP can do so within a multiorgan system, we looked at the peptide-level oxidation pattern of the myosin chaperone protein UNC-45. Although only two modified peptides were observed for UNC-45, it was chosen for further study, because a crystal structure is available for the C. elegans form of this protein alone (PDB ID 5MZU(45)) and in complex with a heat shock protein 90 (Hsp90) peptide (PDB ID 4I2Z46). Only multiresidue modified peptides were observed (Figure 5b), rendering the contribution of the different residues to the overall extent of modification indistinguishable. For UNC-45, peptides 669–680 and 698–706 were oxidatively modified. In 669–680, MS/MS analysis indicates that residues E675, E677, and D678 were modified each with a mass shift of −43.99 (Figure 5a,b), corresponding to a loss of CO2, a modification unique to carboxylic acids.35,47 Glu and Asp have low reactivity with hydroxyl radicals (2.3 × 108 and 7.5 × 107 M–1 s–1, respectively48), in contrast to other higher reactive residues such as Tyr, Phe, and Leu (1.3 × 1010, 6.9 × 109, and 1.7 × 109 M–1 s–1, respectively48) located within the same peptide. Although these residues have higher reactivity, they are not modified in peptide 669–680 by IV-FPOP. The calculated residue-level solvent accessible surface area (SASA) reveals that E675, E677, and D678 have a higher SASA than the highly reactive residues, with E677 and D678 being over 90% accessible (Table 1). The less reactive residues are preferentially modified in good correlation with their increased solvent accessibility, indicating that IV-FPOP is assessing solvent accessibility and could be used to detect solvent accessible changes similar to in vitro FPOP. This trend is not as straightforward in peptide 698–706, where only low-reactive amino acids, including Ala (7.7 × 107)48 and Lys (3.5 × 108),48 are present. It also includes a Ser and two Gly, residues seldom modified in a hydroxyl radical footprinting experiment.49 In this peptide, two carboxylic acids, E702 and E698, are modified with a mass shift of −43.99 (Figure 5b). E702 is highly solvent accessible with a 92.2% SASA, whereas E698 is also exposed with 45% SASA (Table 1).
Figure 5.

Correlating IV-FPOP modification to solvent accessibility. (a) Myosin chaperon protein UNC-45 (gray) (PDB ID 4I2Z(46)) highlighting two modified peptides identified by LC/MS/MS analysis, 669–680 and 698–706 (green, left inset). UNC-45 is bound to the Hsp90 peptide fragment (blue). Oxidatively modified residues within this fragment are shown in sticks (red), and UNC-45 is rendered as a surface (right inset). (b]) Tandem MS spectra of UNC-45 peptide 669–680 (top) and 698–706 (bottom) showing b- and y-ions for the loss of CO2, an FPOP modification. (c) The calculated ln(PF) for the Hsp90 oxidatively modified residues, R697, M698, E699, and E700. (d) Tandem MS spectra for R697, M698, E699, and E700 showing a +16 FPOP modification.
Table 1. Oxidatively Modified Peptides and Residues.
| protein | peptide | residue | SASA |
|---|---|---|---|
| UNC-45 | 669–680 | S669 | 0 |
| L670 | 0.02 | ||
| L671 | 0.13 | ||
| A672 | 0.23 | ||
| F673 | 0 | ||
| A674 | 0.01 | ||
| E675a | 0.49 | ||
| Y676 | 0.17 | ||
| E677a | 0.58 | ||
| D678a | 0.80 | ||
| L679 | 0.03 | ||
| R680 | 0.07 | ||
| 698–706 | E698a | 0.45 | |
| A699 | 0 | ||
| S700 | 0.46 | ||
| G701 | 0.95 | ||
| E702a | 0.59 | ||
| G703 | 0 | ||
| K704 | 0.47 | ||
| I705 | 0.55 | ||
| K706 | 0.21 | ||
| Hsp90 | 695–702 | A695 | 0.9 |
| S696 | 0.58 | ||
| R697a | 0.48 | ||
| M698a | 0.03 | ||
| E699a | 0.42 | ||
| E700a | 0.52 | ||
| V701 | 0 | ||
| D702 | 0.58 |
Oxidatively modified residues with identified MS/MS scans.
To further correlate IV-FPOP to solvent accessibility, the residue-level extent of modification for Hsp90 was calculated using eq 1. High-quality MS/MS data allowed single residue modification analysis (Figure 5c,d). The modification of four residues within the UNC-45-binding peptide on Hsp90, R697, M698, E699, and E700 was confirmed by MS/MS analysis (Figure 5d). To normalize for variations in reactivity with hydroxyl radicals, a protection factor (ln(PF)) was calculated from the extent of FPOP modification.33,50 Higher ln(PF) values are indicative of buried regions, whereas lower ln(PF) values indicate more accessible regions.50 A comparison of the ln(PF) indicates that E699 and E700 are more exposed than R697, whereas M698 is the most buried of the residues (Figure 5c). The Hsp90-bound UNC-45 structure shows M698 interacting with UNC-45 more extensively than R697, E699, and E700 (Figure 5a, inset). Calculations confirm that M698 has the lowest solvent accessibility of the four residues, whereas R697, E699, and E700 have similar SASA values (Table 1). In another example from the same data set, the peptide-level analysis of actin was performed. Actin has been previously studied by in vitro HRPF51 as well as by IC-FPOP.44 The IV-FPOP labeling of actin was compared to the in vitro and in-cell data. For IV-FPOP, four peptides in actin were oxidatively modified (Figure 6a). When the ln(PF) values of these peptides are compared to actin modification data from IC-FPOP and in vitro HRFP, the level of oxidation correlates well. The strong correlation of the data is interesting considering that in vivo actin can bind multiple proteins and can exist in a monomeric or polymeric state. This data provides direct evidence that IV-FPOP can probe solvent accessibility similar to in vitro studies. Both in vitro HRPF and IC-FPOP have a higher number of modified peptides than IV-FPOP (Figure 6b). This may be due to a lower number of modifications by IV-FPOP or a lack of identification by LC/MS/MS. An increase in hydrogen peroxide concentration may increase the number of modifications, and the use of fractionation or two-dimensional chromatography may increase the identification of oxidatively modified peptides
Figure 6.
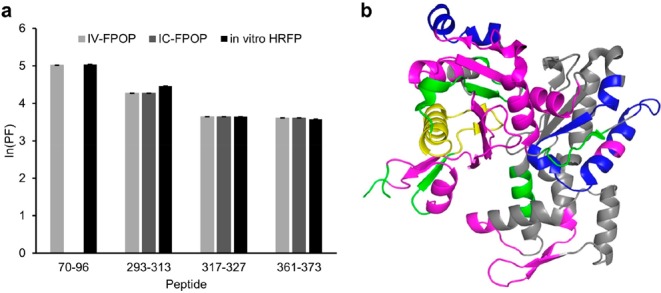
Comparison of actin modification. Oxidation of actin correlates with solvent accessibility through IV-FPOP. (a) ln(PF) of peptides modified by IV-FPOP, IC-FPOP (adapted from Espino et al.44), and in vitro HRPF (adapted from Guan et al.51). (b) Peptides oxidatively modified with in vitro HRPF, IC-FPOP, and IV-FPOP (blue), IV-FPOP and in vitro HRFP only (yellow), in vitro HRFP and IC-FPOP only (green), and HRFP only (magenta).
Conclusions
We report the development of a new method to study protein structure directly in an animal model for human disease. By optimizing conditions for performing FPOP in C. elegans, the method can be used to study the role that changes in protein interactions and conformational states play in disease pathogenesis. Variations in H2O2 concentration and laser frequency improve the number of proteins that can be modified by the method. The ability of IV-FPOP to modify proteins in the different body systems within the worm makes it useful as a general strategy that can used to study a wide variety of proteins regardless of their organ location. The correlation between the extent of FPOP modification with solvent accessible surface area and in vitro HRPF studies indicates the ability of the method to successfully probe solvent accessibility within the worm. This demonstrates the future possibility of using the method to study a protein system in vivo by comparing the labeling pattern in multiple states including perturbations such as ligand-binding, mutations, growth conditions, signaling, and the physiological state of the organism. Information on the amino-acid-residue level can be obtained from the method, which is higher resolution than other in vivo structural methods. Ongoing development of the method to increase the number of modified proteins and the number of modifications per protein will further increase its utility as a method for in vivo structural biology. Taken together, this data demonstrates the potential of the method in being used to study interactions and conformations in an animal model for human disease.
Acknowledgments
The authors would like to thank Daniel Deredge for his help in editing the manuscript. This work was supported by a grant from the NIH 1R01 GM 127595-01 and start-up funds from the University of Maryland, Baltimore.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b00244.
Supplementary methods; Figure S1: IV-FPOP flow system; Figure S2: Background oxidation in the presence and absence of hydrogen peroxide; Figure S3: Comparison of the total number of oxidatively modified proteins through IV-FPOP between biological replicates 1 and 2 in hydrogen peroxide; Figure S4a: Venn diagrams of oxidatively modified proteins in the presence of 200 mM hydrogen peroxide at 50 Hz; Figure S4b: Protein abundance differences between biological replicates 1 and 2 for 200 mM hydrogen peroxide at 50 Hz (PDF)
Author Contributions
L.M.J. devised the project, J.A.E. performed the experiments, and L.M.J. and J.A.E. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Murphy H. C. The Use of Whole Animals Versus Isolated Organs or Cell Culture in Research. Transactions of the Nebraska Academy of Sciences and Affiliated Societies 1991, 105–108. [Google Scholar]
- Huang R. Y.; Krystek S. R. Jr.; Felix N.; Graziano R. F.; Srinivasan M.; Pashine A.; Chen G. Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction. mAbs 2018, 10 (1), 95–103. 10.1080/19420862.2017.1393595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Y.; O’Neil S. R.; Lipovsek D.; Chen G. Conformational Assessment of Adnectin and Adnectin-Drug Conjugate by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29 (7), 1524–1531. 10.1007/s13361-018-1966-2. [DOI] [PubMed] [Google Scholar]
- Weis D. D., Ed. Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications; Wiley, 2016. [Google Scholar]
- Chea E. E.; Jones L. M. Analyzing the structure of macromolecules in their native cellular environment using hydroxyl radical footprinting. Analyst 2018, 143 (4), 798–807. 10.1039/C7AN01323J. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Gau B. C.; Jones L. M.; Vidavsky I.; Gross M. L. Fast photochemical oxidation of proteins for comparing structures of protein-ligand complexes: the calmodulin-peptide model system. Anal. Chem. 2011, 83 (1), 311–8. 10.1021/ac102426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson M.; Wang L.; van Gils N.; Stephens B. S.; Zhang P.; Schall T. J.; Yang S.; Abagyan R.; Chance M. R.; Kufareva I.; Handel T. M. Structural basis of ligand interaction with atypical chemokine receptor 3. Nat. Commun. 2017, 8, 14135. 10.1038/ncomms14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Wei H.; Krystek S. R. Jr.; Bond D.; Brender T. M.; Cohen D.; Feiner J.; Hamacher N.; Harshman J.; Huang R. Y.; Julien S. H.; Lin Z.; Moore K.; Mueller L.; Noriega C.; Sejwal P.; Sheppard P.; Stevens B.; Chen G.; Tymiak A. A.; Gross M. L.; Schneeweis L. A. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal. Chem. 2017, 89 (4), 2250–2258. 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. S.; Chen G.; Mo J.; Huang R. Y. C.; Deyanova E. G.; Beno B. R.; O’Neil S. R.; Tymiak A. A.; Gross M. L. Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Anal. Chem. 2017, 89 (14), 7742–7749. 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. M.; Sperry J. B.; Carroll J. A.; Gross M. L. Fast Photochemical Oxidation of Proteins for Epitope Mapping. Anal. Chem. 2011, 83 (20), 7657–7661. 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Grant O. C.; Ito K.; Wallace A.; Wang S.; Zhao P.; Wells L.; Lu S.; Woods R. J.; Sharp J. S. Structural Analysis of the Glycosylated Intact HIV-1 gp120-b12 Antibody Complex Using Hydroxyl Radical Protein Footprinting. Biochemistry 2017, 56 (7), 957–970. 10.1021/acs.biochem.6b00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poor T. A.; Jones L. M.; Sood A.; Leser G. P.; Plasencia M. D.; Rempel D. L.; Jardetzky T. S.; Woods R. J.; Gross M. L.; Lamb R. A. Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E2596. 10.1073/pnas.1408983111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks B. B.; Konermann L. Time-dependent changes in side-chain solvent accessibility during cytochrome c folding probed by pulsed oxidative labeling and mass spectrometry. J. Mol. Biol. 2010, 398 (2), 362–73. 10.1016/j.jmb.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Hambly D. M.; Gross M. L. Laser Flash Photolysis of Hydrogen Peroxide to Oxidize Protein Solvent-Accessible Residues on the Microsecond Timescale. J. Am. Soc. Mass Spectrom. 2005, 16 (12), 2057–2063. 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Espino J. A.; Mali V. S.; Jones L. M. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal. Chem. 2015, 87 (15), 7971–8. 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- Rinas A.; Mali V. S.; Espino J. A.; Jones L. M. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal. Chem. 2016, 88 (20), 10052–10058. 10.1021/acs.analchem.6b02357. [DOI] [PubMed] [Google Scholar]
- Riddle D. L.; Blumenthal T.; Meyer B. J.; Priess J. R.. Introduction to C. elegans. In C. elegans II, 2nd ed.; Riddle D. L.; Blumenthal T.; Meyer B. J.; Priess J. R., Eds.; Cold Spring Harbor Laboratory Press, 1997. [PubMed] [Google Scholar]
- Lai C. H.; Chou C. Y.; Ch’ang L. Y.; Liu C. S.; Lin W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10 (5), 703–13. 10.1101/gr.10.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussbaum-Krammer C. I.; Morimoto R. I. Caenorhabditis elegans as a model system for studying non-cell-autonomous mechanisms in protein-misfolding diseases. Dis. Models & Mech. 2014, 7 (1), 31–9. 10.1242/dmm.013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C. I.; Calkins J.; Hartman P. S.; Rupert C. S. UV photobiology of the nematode Caenorhabditis elegans: action spectra, absence of photoreactivation and effects of caffeine. Photochem. Photobiol. 1987, 46 (4), 483–8. 10.1111/j.1751-1097.1987.tb04799.x. [DOI] [PubMed] [Google Scholar]
- Miranda-Vizuete A.; Veal E. A. Caenorhabditis elegans as a model for understanding ROS function in physiology and disease. Redox Biol. 2017, 11, 708–714. 10.1016/j.redox.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie B.; Sood A.; Woods R. J.; Sharp J. S. Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection. Sci. Rep. 2017, 7, 4552. 10.1038/s41598-017-04689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Moniz H.; Wang S.; Ramiah A.; Zhang F.; Moremen K. W.; Linhardt R. J.; Sharp J. S. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. J. Biol. Chem. 2015, 290 (17), 10729–40. 10.1074/jbc.M115.648410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans B. N.; Adams S. R.; Ellisman M. H.; Tsien R. Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312 (5771), 217–24. 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Jensen E. C. Use of fluorescent probes: their effect on cell biology and limitations. Anat. Rec. 2012, 295 (12), 2031–6. 10.1002/ar.22602. [DOI] [PubMed] [Google Scholar]
- Tavare J. M.; Fletcher L. M.; Welsh G. I. Using green fluorescent protein to study intracellular signalling. J. Endocrinol. 2001, 170 (2), 297–306. 10.1677/joe.0.1700297. [DOI] [PubMed] [Google Scholar]
- Cornwell O.; Radford S. E.; Ashcroft A. E.; Ault J. R. Comparing Hydrogen Deuterium Exchange and Fast Photochemical Oxidation of Proteins: a Structural Characterisation of Wild-Type and DeltaN6 beta2-Microglobulin. J. Am. Soc. Mass Spectrom. 2018, 29 (12), 2413–2426. 10.1007/s13361-018-2067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur U.; Meng H.; Lui F.; Ma R.; Ogburn R. N.; Johnson J. H. R.; Fitzgerald M. C.; Jones L. M. Proteome-Wide Structural Biology: An Emerging Field for the Structural Analysis of Proteins on the Proteomic Scale. J. Proteome Res. 2018, 17 (11), 3614–3627. 10.1021/acs.jproteome.8b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur U.; Johnson D. T.; Chea E. E.; Deredge D.; Espino J. A.; Jones L. M. Evolution of Structural Biology Through the Lens of Mass Spectrometry. Anal. Chem. 2019, 91, 142. 10.1021/acs.analchem.8b05014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974, 77 (1), 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L.; Collings B. A.; Douglas D. J. Cytochrome c Folding Kinetics Studied by Time-Resolved Electrospray Ionization Mass Spectrometry. Biochemistry 1997, 36 (18), 5554–5559. 10.1021/bi970046d. [DOI] [PubMed] [Google Scholar]
- Douglas D. J.; Konermann L.; Collings B.. Method and apparatus for determining the rates of reactions in liquids by mass spectrometry. U.S. Patent 6054709, 2000.
- Rinas A.; Espino J. A.; Jones L. M. An efficient quantitation strategy for hydroxyl radical-mediated protein footprinting using Proteome Discoverer. Anal. Bioanal. Chem. 2016, 408 (11), 3021–3031. 10.1007/s00216-016-9369-3. [DOI] [PubMed] [Google Scholar]
- Xu G.; Chance M. R. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem. Rev. 2007, 107 (8), 3514–3543. 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- Gau B. C.; Chen H.; Zhang Y.; Gross M. L. Sulfate radical anion as a new reagent for fast photochemical oxidation of proteins. Anal. Chem. 2010, 82 (18), 7821–7. 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner B. A.; Witmer J. R.; van ’t Erve T. J.; Buettner G. R. An Assay for the Rate of Removal of Extracellular Hydrogen Peroxide by Cells. Redox Biol. 2013, 1 (1), 210–217. 10.1016/j.redox.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulak R. Techniques for Analysis, Sorting, and Dispensing of C. elegans on the COPAS Flow-Sorting System. C. elegans: Methods and Applications 2006, 351, 275–286. 10.1385/1-59745-151-7:275. [DOI] [PubMed] [Google Scholar]
- Jansen W. T.; Bolm M.; Balling R.; Chhatwal G. S.; Schnabel R. Hydrogen peroxide-mediated killing of Caenorhabditis elegans by Streptococcus pyogenes. Infect. Immun. 2002, 70 (9), 5202–7. 10.1128/IAI.70.9.5202-5207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumsta C.; Thamsen M.; Jakob U. Effects of Oxidative Stress on Behavior, Physiology, and the Redox Thiol Proteome of Caenorhabditis elegans. Antioxid. Redox Signaling 2011, 14 (6), 1023–1037. 10.1089/ars.2010.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira S. R.; Mendes T. A. O.; Bueno L. L.; de Araújo J. V.; Bartholomeu D. C.; Fujiwara R. T. A New Methodology for Evaluation of Nematode Viability. BioMed Res. Int. 2015, 2015, 879263. 10.1155/2015/879263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K. D.; Roschitzki B.; Snoek L. B.; Grossmann J.; Zheng X.; Elvin M.; Kamkina P.; Schrimpf S. P.; Poulin G. B.; Kammenga J. E.; Hengartner M. O. Natural Genetic Variation Influences Protein Abundances in C. elegans Developmental Signalling Pathways. PLoS One 2016, 11 (3), e0149418 10.1371/journal.pone.0149418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt-Newbury R.; Viveiros R.; Johnsen R.; Mah A.; Anastas D.; Fang L.; Halfnight E.; Lee D.; Lin J.; Lorch A.; McKay S.; Okada H. M.; Pan J.; Schulz A. K.; Tu D.; Wong K.; Zhao Z.; Alexeyenko A.; Burglin T.; Sonnhammer E.; Schnabel R.; Jones S. J.; Marra M. A.; Baillie D. L.; Moerman D. G. High-Throughput In Vivo Analysis of Gene Expression in Caenorhabditis elegans. PLoS Biol. 2007, 5 (9), e237 10.1371/journal.pbio.0050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau B. C.; Chen J.; Gross M. L. Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: data processing and application to barstar. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834 (6), 1230–8. 10.1016/j.bbapap.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino J. A.; Mali V. S.; Jones L. M. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal. Chem. 2015, 87 (15), 7971–7978. 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- Hellerschmied D.; Roessler M.; Lehner A.; Gazda L.; Stejskal K.; Imre R.; Mechtler K.; Dammermann A.; Clausen T. UFD-2 is an adaptor-assisted E3 ligase targeting unfolded proteins. Nat. Commun. 2018, 9 (1), 484. 10.1038/s41467-018-02924-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda L.; Pokrzywa W.; Hellerschmied D.; Lowe T.; Forne I.; Mueller-Planitz F.; Hoppe T.; Clausen T. The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell 2013, 152 (1–2), 183–95. 10.1016/j.cell.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. M.; Sperry J.; Carroll J.; Gross M. L. Fast Photochemical Oxidation of Proteins (FPOP) for Epitope Mapping. Anal. Chem. 2011, 83 (20), 7657–7661. 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Chance M. R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107 (8), 3514–43. 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- Xu G.; Chance M. R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 2005, 77 (14), 4549–55. 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- Huang W.; Ravikumar K. M.; Chance M. R.; Yang S. Quantitative Mapping of Protein Structure by Hydroxyl Radical Footprinting-Mediated Structural Mass Spectrometry: A Protection Factor Analysis. Biophys. J. 2015, 108 (1), 107–115. 10.1016/j.bpj.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J.-Q.; Almo S. C.; Reisler E.; Chance M. R. Structural Reorganization of Proteins Revealed by Radiolysis and Mass Spectrometry: G-Actin Solution Structure Is Divalent Cation Dependent. Biochemistry 2003, 42 (41), 11992–12000. 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.