

Conspectus
Enzymes are the essential catalytic components of biology and adsorbing redox-active enzymes on electrode surfaces enables the direct probing of their function. Through standard electrochemical measurements, catalytic activity, reversibility and stability, potentials of redox-active cofactors, and interfacial electron transfer rates can be readily measured. Mechanistic investigations on the high electrocatalytic rates and selectivity of enzymes may yield inspiration for the design of synthetic molecular and heterogeneous electrocatalysts. Electrochemical investigations of enzymes also aid in our understanding of their activity within their biological environment and why they evolved in their present structure and function. However, the conventional array of electrochemical techniques (e.g., voltammetry and chronoamperometry) alone offers a limited picture of the enzyme–electrode interface.
How many enzymes are loaded onto an electrode? In which orientation(s) are they bound? What fraction is active, and are single or multilayers formed? Does this static picture change over time, applied voltage, or chemical environment? How does charge transfer through various intraprotein cofactors contribute to the overall performance and catalytic bias? What is the distribution of individual enzyme activities within an ensemble of active protein films? These are central questions for the understanding of the enzyme–electrode interface, and a multidisciplinary approach is required to deliver insightful answers.
Complementing standard electrochemical experiments with an orthogonal set of techniques has recently allowed to provide a more complete picture of enzyme–electrode systems. Within this framework, we first discuss a brief history of achievements and challenges in enzyme electrochemistry. We subsequently describe how the aforementioned challenges can be overcome by applying advanced electrochemical techniques, quartz-crystal microbalance measurements, and spectroscopic, namely, resonance Raman and infrared, analysis. For example, rotating ring disk electrochemistry permits the simultaneous determination of reaction kinetics and quantification of generated products. In addition, recording changes in frequency and dissipation in a quartz crystal microbalance allows to shed light into enzyme loading, relative orientation, clustering, and denaturation at the electrode surface. Resonance Raman spectroscopy yields information on ligation and redox state of enzyme cofactors, whereas infrared spectroscopy provides insights into active site states and the protein secondary and tertiary structure. The development of these emerging methods for the analysis of the enzyme–electrode interface is the primary focus of this Account. We also take a critical look at the remaining gaps in our understanding and challenges lying ahead toward attaining a complete mechanistic picture of the enzyme–electrode interface.
Introduction
The study of enzymes enhances our knowledge of catalysis and biology while leading to applications in medicine, sensing, energy, and more. To this end, the electrochemistry of protein-modified electrodes known as protein film electrochemistry (PFE) has served as a powerful tool for probing the thermodynamic and kinetic properties of redox-active proteins and enzymes since the second half of the 20th century.1−3 The topic is mature, and several excellent reviews have been published describing the information available from immobilized enzymes and routine PFE experiments.4−7 The unique insights provided by PFE are largely enabled by direct interfacial electron transfer between the electrode surface and the enzyme cofactors, including its active site. The catalytic rates often exceed those accessible by intermolecular electron transfer with traditional solution phase mediators (limited by their diffusion) as many enzymes display very fast intrinsic turnover rates. As a result, quantification of enzyme kinetics can be limited in solution assays and capturing the rapid active site reactivity by PFE can allow for unravelling of the active site’s intrinsic thermodynamic and kinetic properties.
PFE provides direct, real-time information on transient and steady-state catalytic behavior using a minuscule amount of electroactive enzyme (Figure 1a,b). The effect of changing the electrode potential, substrate or inhibitor concentration, pH value, and atmospheric composition can therefore be probed with relative ease within a single experiment. The readout from PFE experiments (typically voltammetry and chronoamperometry) is usually current, which can be readily analyzed as it directly relates to stoichiometric cofactor oxidation/reduction or catalytic rate. Furthermore, these conditions can exceed those available to traditional solution assays: the continuum of electrode potentials exceeds the limited reduction potential ranges of solution phase electron donors and acceptors, and the electrode potential can readily be modified in situ to gauge reaction kinetics. PFE has allowed a variety of potential-dependent effects to be observed such as catalytic bias,8 cooperative two-electron transfer,9 oxidation state dependent inhibitor binding,10 and anaerobic oxidative inactivation.11
Figure 1.
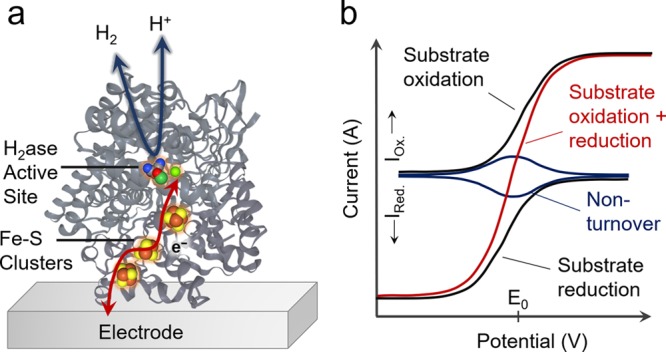
Protein film electrochemistry. (a) Schematic representation of the electron transfer between an electrode surface and the enzyme active site, which catalyzes interconversion between oxidized and reduced substrates (given example is a hydrogenase). (b) Simulated voltammetry showing the enzyme’s redox couple under non-turnover conditions and catalytic voltammetry under turnover conditions.
Theoretical descriptions of catalytic waveshapes12,13 have arisen with the development of PFE, highlighting both its strengths and weaknesses. The reproducible, facile, and precise measurements of PFE provide an ideal platform for modeling. A wealth of data can readily be generated as a function of electrode potential, potential scan rate, substrate concentration, and pH value to inform and test modeling approaches.
Despite its advantages, PFE does not provide a fully comprehensive picture of the enzyme–electrode interface. However, it can be coupled to a variety of complementary techniques to quantify aspects such as enzyme adsorption, product generation, and cofactor states and answer the questions posed in the conspectus. The application of these techniques is summarized in this Account.
Limitations of Protein Film Electrochemistry
First, reliable quantification of active enzyme surface coverage is often a challenge, which can lead to significant uncertainty in the turnover rates of immobilized enzymes. Some examples exist of non-catalytic turnover voltammetry (of inhibited enzymes or in the absence of substrate) allowing the quantification of electroactive enzymes on the electrode surface.9 However, non-turnover signals are not always possible to observe due to low enzyme surface coverage and significant capacitive currents (especially when employing structured electrode geometries). Potential-step voltammetry, Fourier-transform voltammetry, and other related techniques may aid in increasing sensitivity to this end.14,15 Product detection is challenging when using small enzyme quantities in PFE experiments and is therefore not routinely performed.
Although a theoretical model has been proposed,13 a comprehensive understanding of electron transfer through multiple electron transfer relays within an enzyme has yet to be completely elucidated experimentally. There is also a lack of information on the surface interactions, orientations, and structures of enzymes on electrode surfaces.16 The dispersion of enzymes resulting from different enzyme orientations and a distribution of distances of the enzyme’s surface electron relay to the electrode surface makes analytical interpretations challenging.17,18 Precious metals such as gold and graphite electrodes are most commonly used and best understood, but there is much scope for electrode design with alternative materials such as porous carbon-based systems and metal oxides.16,19,20 Within this context, a poor enzyme–electrode interface is often the reason when an electrochemical signal is not observed in PFE and new electrode materials and 3D architectures may improve the interactions.
Protein Film Photo-electrochemistry
PFE was initially applied to enzymes catalyzing “dark” reactions. Coupling illumination to the standard PFE apparatus, protein-film photo-electrochemistry (PF-PEC) applies the fundamental concepts and resulting insights of PFE to (i) photoactive enzymes wired to an electrode surface (Figure 2a,b),21 (ii) electroactive enzymes wired to dye-sensitized or semiconductor electrodes,22 or (iii) photoactive enzymes wired to sensitized or semiconductor electrodes.23 In the latter two cases, the semiconductor (or dye) provides energized charge carriers (electrons or holes) to drive the enzymatic reaction. In the case of photosystem II (PSII) as an example for the former case, electrons are extracted from water at the Mn4Ca active site and transferred through the electron transport chain to the electrode, resulting in anodic currents under illumination (Figure 2a–c).
Figure 2.
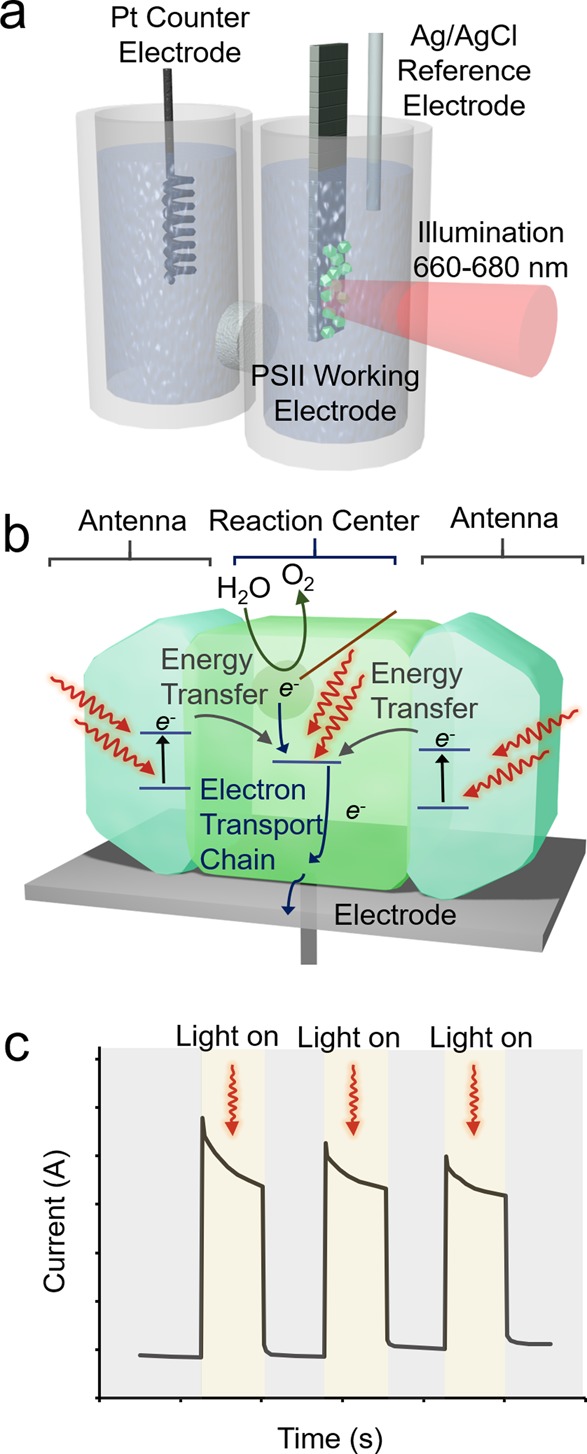
Protein film photo-electrochemistry. (a,b) PSII absorbs light to carry out water oxidation at its [Mn4Ca] active site and transfers electrons through the electron transfer chain. (c) Photoanodic currents of PSII-electrodes during chronoamperometry under representative dark–light cycles.
Uniquely, studying light-driven reactions in this context can serve as inspiration for artificial photosynthesis.24 Key considerations for the semiconductor are the energy levels, charge transfer kinetics, wavelength-dependent absorption coefficient, and surface biocompatibility. To avoid rate limitations from light irradiation, the energy of photons must be equal to or greater than the energy gap of the HOMO and LUMO within the photosensitizer (or band gap in a semiconductor), and the photon-driven electron flux should surpass the rate of the slowest step in the catalytic cycle. Transparent conductive oxide electrode substrates are typically employed to minimize parasitic light absorption.
In systems featuring multiple light-absorbing components such as the water oxidation enzyme PSII and a dye, dual-wavelength action spectra can be used to probe how well each component functions within the entire system.23 Electrode design considerations also become important to maximize light absorption, particularly in highly structured mesoporous and inverse opal electrodes.25 Therefore, chromophore-containing enzyme distribution and penetration into the depth of porous electrodes can be quantified with confocal fluorescence microscopy.26
Electrochemical Impedance Spectroscopy
While a wealth of information can be extracted from the standard array of voltammetric and chronoamperometric techniques in probing the enzyme–electrode interface, the data are limited to the net current flow. Extending the scope of electrochemical analysis is often needed to provide critical details regarding the deconvolution of charge transfer across various interfaces and through cofactors, precise kinetics of chemical catalysis, interfacial capacitance, and effects of substrate diffusion. Thus, electrochemical impedance spectroscopy (EIS) has been applied to enzymatic systems.27
EIS functions by applying an alternating current (AC) voltage with small perturbation (∼10 mV) at a given potential and recording the impedance. The resultant data is then modeled and fit to an equivalent circuit. Because of the complexity of the enzyme–electrode interface that often features several points of charge and mass transfer, multiple resistive, capacitive, and inductive elements may be present. Extracting quantitative information from EIS data that corresponds to physical processes is reliant on accurately capturing each of these elements in the construction of an equivalent circuit to fit the experimental data. Hence, the data interpretation is not completely unambiguous.
EIS can be used to detect formation of multienzyme assemblies through recording changes in electron transfer under catalytic turnover,28 analyzing reaction kinetics,29 and extracting enzymatic rate constants by plotting charge transfer resistance versus substrate concentration.30 Recently, an EIS study of a hydrogenase (H2ase) [enzyme catalyzing the reversible reduction of protons to H2] revealed that the charge transfer resistance through the FeS clusters is lower at voltages more negative than the reversible hydrogen electrode (RHE),31 which could be due to chemical effects or possible changes in orientation. Consequently, this was interpreted to result in a catalytic bias toward H2 evolution as opposed to H2 oxidation. Charge transfer resistance from chemical catalysis at the active site was found not to be limiting for oxidation currents as a similar H2ase natively lacking the FeS clusters (electrons transferred directly to the active site) did not feature a catalytic bias for H2 evolution and possessed a similar EIS-derived charge transfer resistance in each direction.
Rotating Ring Disk Electrochemistry
Product detection with PFE is notoriously difficult due to the use of tiny amounts of protein and the small amount of product being distributed in the bulk electrolyte solution. To this end, an instrumental approach applied to probe reaction mechanisms and products of immobilized enzymes is the use of a rotating ring disk electrode (RRDE). Here, two working electrodes are employed: an enzyme-loaded disk electrode and usually a Pt ring electrode. The apparatus is rotated to generate a well-defined hydrodynamic convective flow to first transfer reactants from the bulk solution to the central disk, and then, along with any products formed, to the outer ring electrode. By oxidizing or reducing substrates at the disk, one can gather insights about the occurring chemistry. Reaction products can be detected via their reduction or oxidation at the ring, providing insight into competing reaction mechanisms.
However, the ring is not selective for any one reaction and will perform all favorable reactions. Hence, the potential is set where only one possible reaction can be carried out. As an example of RRDE application, H2O2, originating from the reduction of O2 by glucose oxidase, was detected by selectively oxidizing H2O2 at the ring electrode.3 Recently, the RRDE technique was coupled with PF-PEC to study the oxygenic photoreactivity of PSII (Figure 3a,b).32 O2 was detected from the well-established light-driven water oxidation reaction by its reduction at the ring. Interestingly, products from reduction reactions of O2 at a more negative applied potential were found to be primarily H2O2, indicated by catalase-induced suppression of ring currents.
Figure 3.
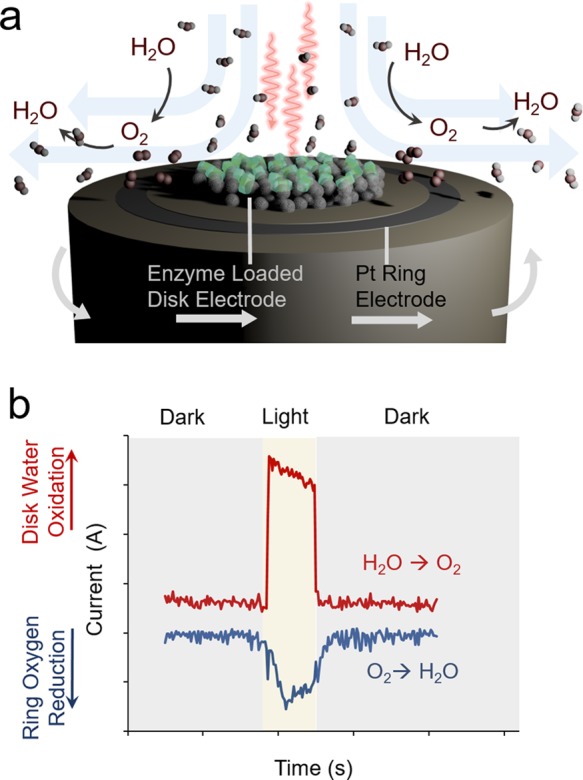
Rotating ring disk electrochemistry. (a) RRDE provides a controlled flux of substrates from the bulk solution to the disk and for the formed products to the ring electrode. (b) Illumination of PSII at a positive potential results in photoanodic disk currents from light-driven water oxidation and cathodic ring currents stemming from oxygen reduction.
Microelectrode-Based Techniques
Scanning electrochemical current microscopy (SECM), electrochemical scanning tunneling microscopy (EC-STM), and related techniques have been useful in analyzing activities of enzyme assemblies and even single enzymes. Deconvoluting single enzyme activities from the ensemble average is important in elucidating the catalytic activity of enzymes in discrete functional states, orientations or different levels of integrity. With SECM/EC-STM, the enzyme-loaded substrate is the primary working electrode, while a cantilever or fine tip functions as a secondary working electrode, both connected to a bipotentiostat (Figure 4a,b).33 The secondary electrode acts to reduce or oxidize any reaction products formed in its vicinity or to image surface-bound enzymes. In the context of PFE, EC-STM has been used to measure potential-dependent turnover frequencies of H2ase by correlating EC-STM-derived enzyme-surface coverage with voltammetry.34 On a self-assembled monolayer (SAM)-functionalized Au surface, a TOF of ∼1000 s–1 was observed and was extrapolated to 21 000 if the enzyme were in direct contact with the Au without the interfacial SAM spacer.34
Figure 4.

Scanning electrochemical current microscopy. (a) SECM configuration with the secondary working electrode detecting locally produced reaction products. (b) Principle to study activities of individual enzymes or clusters of enzymes by SECM (H2ase shown as example).
In SECM, a microelectrode directly oxidizes or reduces the product of an enzymatic reaction. A large challenge yet to be overcome is extrapolating this down to a single enzyme level, though it has already been achieved with single nanoparticle catalysts.35 For example, an SECM-measured H2 oxidation current will drop when the tip is positioned over an inactive enzyme or bare substrate. Conversely, the current will significantly increase when the tip is positioned over an active enzyme due to the increased H2 supply (Figure 4b). Alternatively, single-enzyme activity can be potentially quantified through temporary contact (and consequently, catalytic current generated) from a diffusing enzyme to a microelectrode.35,36 As with most miniaturized techniques, detecting low signals is a challenge and impedes unambiguous quantification of single enzyme activity.
Quartz Crystal Microbalance with Dissipation
Quartz crystal microbalance with dissipation (QCM-D) is an invaluable tool toward understanding enzyme binding on electrode surfaces and can be operated in parallel to electrochemical analysis. Briefly, the QCM-D instrument operates by measuring the resonance frequency of a piezoelectric quartz chip upon the application of a voltage. The frequency is typically proportional to the mass on the electrode in accordance with the Sauerbrey equation (assuming a rigid thin film).37 Enzyme adsorption onto a coated quartz electrode is readily measured in a flow cell, illustrated in Figure 5a, from a frequency decrease upon the introduction of enzymes into the flowing solution. This was used for quantifying enhanced enzyme accommodating capacities of planar, high surface area mesoporous, and inverse opal metal–oxide electrode architectures for H2ase and formate dehydrogenase (FDH) [enzyme catalyzing the oxidation of formate to CO2].22,38 If the enzyme specific activity is known or can be electrochemically determined, comparing the enzyme loading as measured with QCM-D to the enzyme activity can elucidate the proportion of active enzymes oriented for direct electron transfer to or from the electrode.39
Figure 5.

Quartz crystal microbalance with dissipation. (a) Typical setup of a QCM-D flow cell. Complementary spectroscopic and electrochemical analysis can be simultaneously coupled in the system. (b) Quantity of adsorbed enzyme and rigidity of binding is measured through changes in frequency and dissipation, respectively. (c) QCM-D measurements provide information about coverage as well as multilayer formation.
Furthermore, the dissipation, a measure of how quickly the quartz oscillation decays following perturbation, reflects both the mass and its viscoelasticity (rigidity) on the electrode surface (Figure 5b). Cross-comparing changes in frequency and dissipation can indicate the rigidity of enzyme binding, enzyme-assembly formation, enzyme desorption, denaturation, reorientation, and conformational changes (Figure 5c).40 A notable work deconvoluted mass loss (changes in frequency) and non-desorptive structural changes (changes in dissipation) to electrocatalytic performance decays of O2-reducing bilirubin oxidase.41 It was concluded that the primary mechanism of activity loss in the system was likely due to effects such as enzyme dehydration or denaturation as opposed to enzyme desorption. There remains some uncertainty in interpreting changes in dissipation and directly linking to the enzyme at a molecular level.
To expand the scope of QCM-D, the use of a transparent window integrated into a QCM cell enables simultaneous photochemical and spectroscopic measurements to be carried out. First applied to understand electrocatalysts,42 probing the activity of adsorbed photosystems is now an intriguing possibility. Moreover, coupling spectroscopy with QCM-D can reveal how the redox state of cofactors changes as a function of enzyme conformation, loading density, assembly formation, and catalytic activity.
UV–Vis Absorption Spectroscopy
Many metalloenzymes possess chromophoric active sites (or other cofactors) that absorb in the visible spectrum. UV–vis spectroscopy has therefore emerged as a simple method to characterize the active sites for determining enzyme loading via the Lambert–Beer relationship.43,44 Heme-containing proteins immobilized on transparent, conductive, and 3D-structured metal oxide electrodes have been studied using UV–vis spectro-electrochemistry in transmission mode (Figure 6a). The redox activity of microperoxidase immobilized in a mesoporous ITO electrode was monitored to optimize enzyme loading,45 and time-resolved UV–vis spectro-electrochemistry showed that the reduction of the Fe occurred within 50 ms, supporting good electronic contact with the enzyme. The isolated electron conduit decaheme protein MtrC was also adsorbed on mesoporous ITO electrodes and monitored using UV–vis spectro-electrochemistry to determine the redox potentials of the heme cofactors by recording changes in their oxidation state-dependent absorption.46,47
Figure 6.
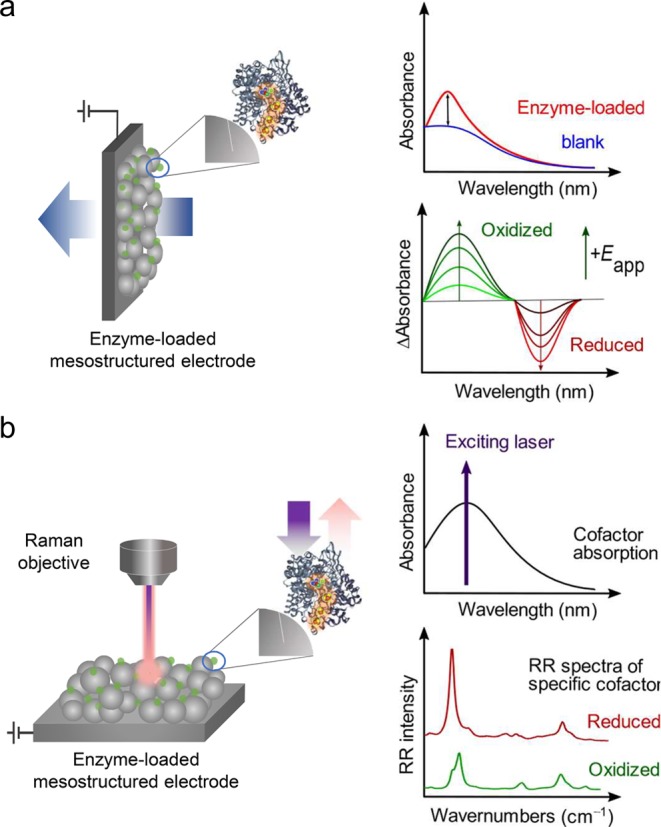
Spectro-electrochemistry for investigation of immobilized enzymes. (a) UV–vis spectro-electrochemistry measured in transmission mode. The enzyme is loaded on transparent 3D-nanostructured metal oxide electrodes. (b) Resonance Raman spectro-electrochemistry measured in confocal mode. The exciting laser line is tuned to match the cofactor absorption band yielding selectively strongly enhanced resonance Raman spectra of the cofactor unit. For both techniques, applying potentials allows for monitoring changes in redox states of the enzyme cofactor(s).
Confocal Resonance Raman Spectroscopy
Resonance Raman (RR) spectroscopy has been utilized to investigate structure–function relationships of cofactors of various enzymes.48 The strength of RR spectroscopy is its ability to selectively provide signals from the chromophore unit of interest by tuning the exciting laser line to match the chromophore’s absorption. RR spectroscopy therefore largely neglects the otherwise interfering protein matrix and can provide signal enhancements of up to several orders of magnitude. In contrast to UV–vis spectroscopy, it can inherently reveal a higher depth of information by probing molecule specific vibrational modes. Optimal conditions for RR spectroscopy involve a combination of a strong electronic transition in the enzyme and a high electrode surface area maximizing sample concentration within the focused laser spot (Figure 6b). For example, RR spectro-electrochemistry was used to study adsorption and redox behavior of cytochrome c (cytc) on mesoporous ITO electrodes.49 From the unique RR signals of the ferric and ferrous cytc species, the apparent redox potential of the immobilized enzyme was calculated. Furthermore, RR spectro-electrochemical studies on MtrC adsorbed on mesoporous ITO electrodes revealed crucial insights into the origin of its recently discovered peroxidase activity.46 A transition from a six-coordinated to a five-coordinated heme in the ferrous state in some hemes was observed. These heme groups exhibited a potentially weaker ligation that allowed for effective H2O2 binding and catalysis. In all, this technique helps to provide a molecular basis for the activity measured by PFE.
Infrared Absorption Spectroscopy
Fourier-transform infrared (FTIR) absorption spectroscopy is a powerful tool to study the enzymes’ secondary and tertiary structures, as well as their active sites.50 The IR vibrational spectrum contains a wealth of information about structure and environment of amino acid side chains, as well as protein conformation and the polypeptide backbone. To enable in situ IR study of enzymes adsorbed on electrodes, the method is usually carried out in attenuated total reflection (ATR) mode.51 The electrode is thereby deposited on an IR active waveguide typically made of Si or Ge, and the IR light is coupled in from below (Figure 7a). At the interface, the IR beam is reflected, giving rise to an evanescent wave (penetrating approximately 500 nm, depending on the apparatus, into the electrode structure) that probes surface-bound enzymes. This configuration offers the advantage of avoiding the interfering total absorption of water and readily enables electrochemical control by coating the waveguide with a conductive layer. Moreover, operation in difference mode allows for detection of very small changes at the protein backbone, amino acid side chains, or cofactors (Figure 7b,c).52
Figure 7.
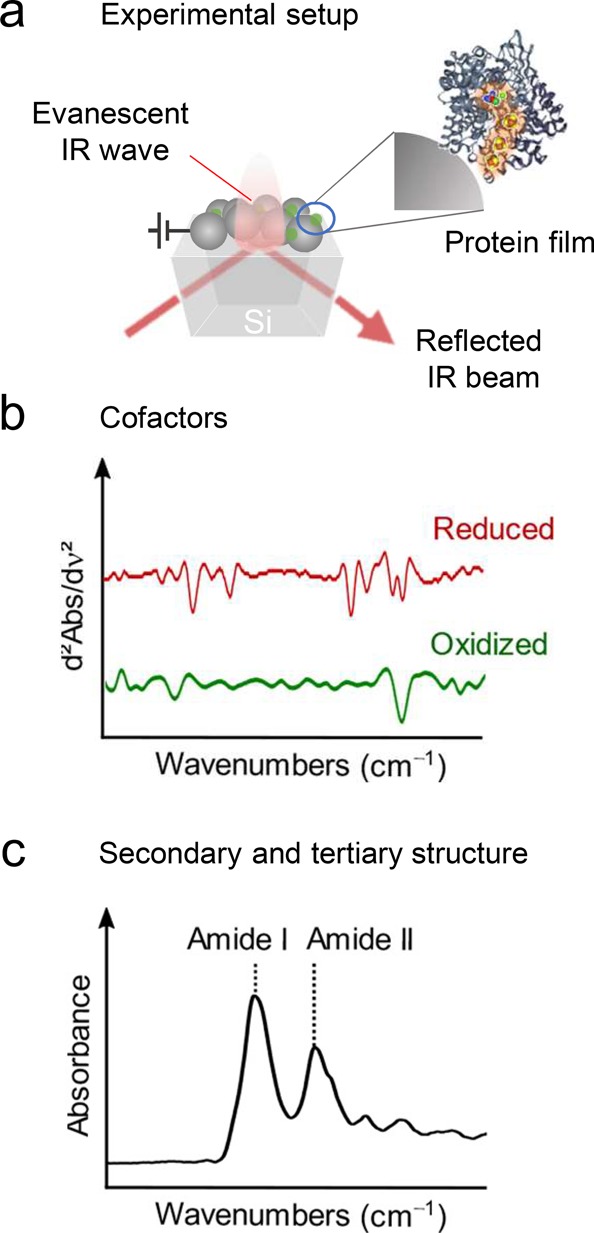
Fourier transform infrared absorption spectro-electrochemistry in attenuated total reflection mode. (a) Enzymes are loaded onto a 3D-nanostructured electrode deposited on an IR waveguide (typically a Si or Ge prism). The IR beam is reflected at the interface giving rise to an evanescent IR wave. (b) IR signatures of cofactors can be obtained and are usually plotted in second derivative mode due to intrinsically low signal intensities. (c) The highly intense amide bands in the region from around 1550 to 1650 cm–1 contains information on the secondary and tertiary structure of the immobilized enzyme.
ATR-FTIR spectro-electrochemistry has been used to study immobilized [NiFe] H2ases on a 3D-structured electrode consisting of highly conductive carbon particles embedded in a phosphate buffered Nafion network.53 The electrode was designed to chemically resemble graphite electrodes that are commonly employed in PFE to enable a direct comparison of voltammetric and spectroscopic data. The employed experimental configuration enabled IR detection of the NiFe active site. The observed unique IR bands from 1900 to 2110 cm–1 reflect the CN– and CO ligands attached to the Fe metal atom that are sensitive to electron density changes at the metal center(s). Studies using a [NiFe] H2ase also demonstrated electrochemically induced oxidation state changes at the active site of the enzyme in the immobilized state.54 In another line of investigation, the spectroscopic signatures of the active site of an oxygen-sensitive [NiFe] H2ase were monitored to demonstrate the protection from damage at high potentials conferred to the enzyme by a surrounding viologen-modified redox hydrogel.55 The electrode modification with redox-active hydrogels enabled the recording of the measurement in transmission mode in an optically transparent thin layer electrochemical cell, which is usually employed in solution studies.
Beyond active sites, the strongest features in ATR-FTIR spectra of enzymes are typically the amide bands (e.g., amide I at around 1650 cm–1 and amide II at around 1550 cm–1) that arise from the amide bonds in the polypeptide backbone and sensitively encode the enzyme’s secondary structure.50 In this respect, a maintained amide I and amide II pattern in the ATR-FTIR spectra supports a largely conserved enzyme structure upon adsorption.56 The intensity of the amide bands has been used to study the immobilization and infiltration process of H2ase, PSII, and FDH throughout a 3D-structured metal oxide electrode.22,26,38 By following the H2ase’s amide band intensities during the incubation process, it was found that hierarchical inverse opal TiO2 electrodes allowed for full penetration of enzymes into the macrostructure, whereas mesoporous TiO2 led to an accumulation of proteins only at the top layers. ATR-FTIR spectroscopic studies on FDH immobilized on a planar TiO2 surface gave insights into the nature of the binding between enzyme and metal oxide surface, revealing that the interaction is stronger than purely electrostatic.38
Another relevant technique is polarization modulated infrared reflection absorption spectroscopy (PMIRRAS), which is well-suited for studying particularly the enzyme orientation on electrode surfaces. The polarized IR light is detected in reflection from the top on mirror metal surfaces thus impeding in situ applications of this method, that is, in aqueous conditions under potential control. Nevertheless, important information was derived using this highly surface-sensitive technique. For example, the orientations of a laccase57,58 and a [NiFe] H2ase56 on SAM-coated Au electrodes could be studied via the analysis of the amide I/II intensity ratio when adsorbed on differently charged SAMs.58 The enzyme orientation could then be directly linked to the overall activity of the biomodified electrode. In addition, surface plasmon resonance spectroscopy and ellipsometry have been shown to complement PMIRRAS for determination of enzyme loading and biofilm thickness.58
Surface-Enhanced Vibrational Spectroscopy
Arising from locally elevated electromagnetic fields at the surfaces of certain metals, the high sensitivity of surface-enhanced (resonance) Raman (SE(R)R) (Figure 8a) and surface-enhanced IR absorption (SEIRA) (Figure 8b) spectroscopy make these techniques ideal for studying enzyme–electrode interactions. However, the requirement of surface-enhanced activity of the supporting substrate largely limits the application of SE(R)R and SEIRA spectroscopy to nanostructured Ag and Au electrodes.59 Due to the fast drop of the enhancement effect with distance from the electrode, SE(R)R and SEIRA spectroscopy effectively probe only the first few nanometers at the interface, rendering this technique exceptionally surface-sensitive.
Figure 8.
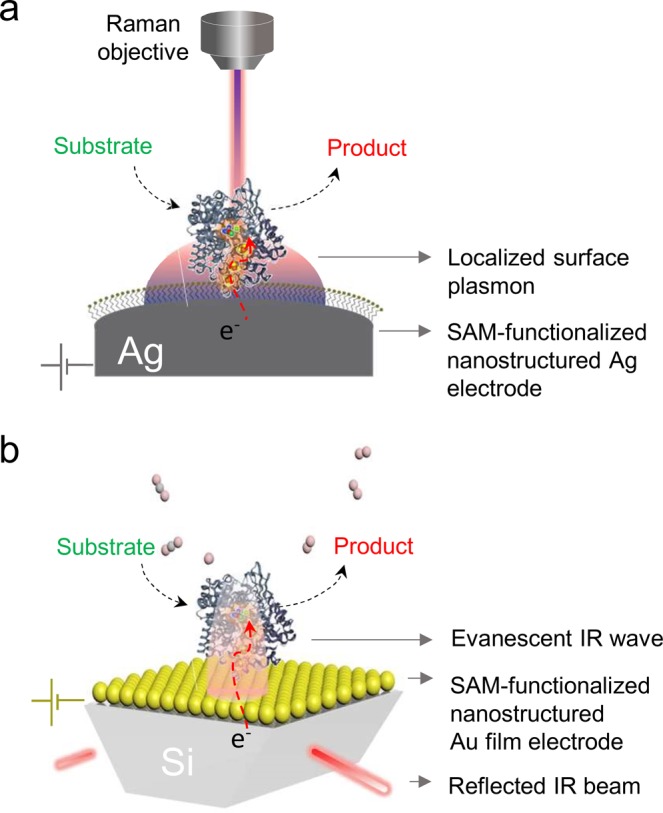
Surface-enhanced vibrational spectroscopy. (a) Surface-enhanced (resonance) Raman and (b) IR absorption spectro-electrochemistry on SAM-coated Ag and Au electrodes, respectively.
SERR spectroscopy has been extensively employed to investigate a range of immobilized proteins, with heme-containing proteins being the most studied.59 For measurements, a laser line matching the electronic transition of the heme and concomitantly exciting the surface plasmons of a nanostructured Ag support is utilized. Stationary and time-resolved potential-controlled SERR spectroscopy have been applied to study the interfacial electron transfer of cytc,60 the communication between the heme cofactors in cytochrome c oxidase,61 the heme cofactor of immobilized cytochrome P450,62 the interaction between cytochrome cd1 nitrite reductase and its physiological partner cytochrome c552 on an electrode surface,63 and the active site of a heme peroxidase64 at biomimetically coated rough Ag electrodes. SERR spectroscopic investigations of the non-heme DNA-repairing enzyme endonuclease III revealed redox activity of the [4Fe–4S] cluster also in the absence of DNA-binding.65 Also, electron transfer from the electrode to a membrane-bound [NiFe] H2ase heterotrimer was demonstrated to predominantly occur through the enzyme’s HoxGK subunit rather than the terminal HoxZ subunit on the basis of the SERR-derived reduction rate of the heme b cofactors in HoxZ.66 Important to these studies is that SERR spectroscopy allows for selectively monitoring of protein cofactors even at very low surface coverages.
In the absence of a resonance Raman effect, SER spectroscopy is also useful. For example, SER studies on a laccase immobilized on Au nanoparticle decorated electrodes revealed that the enzyme binds via the T2/T3 Cu site instead of the T1 Cu site.67 Moreover, increasing SER bands assigned to COO– and CH2 groups indicated also a change in orientation of the enzyme with increasing positive potentials. Beyond nanostructured Au and Ag electrodes, surface-enhancement can be exploited from metal oxides, which are typically more biocompatible than metals. Recently, periodic metal oxide nanostructures gave rise to strongly increased Raman signals.68 Particularly, the enhancement enabled detection of a cytochrome b unit, which was found to maintain its secondary structure and redox properties upon binding to a TiO2 nanotube electrode surface.
Analogous to ATR-FTIR spectroscopy, SEIRA spectroscopy has been used to study an immobilized H2ase on SAM-coated Au electrodes (Figure 8b).69 The results indicated that the enzyme conserved its structure upon adsorption and was in excellent electronic contact to the electrode. Moreover, a recent SEIRA study supported by molecular dynamics simulations and electrocatalytic measurements showed that the immobilization complex of a [NiFe] H2ase and the coated electrode can control the enzyme’s electrocatalytic performance.70 In some of the occupied orientations, which varied depending on the charge of the electrode coating, no direct electronic contact with the electrode was possible and the use of redox mediators was required. Moreover, the orientation of a nitric oxide reductase on a SAM-coated Au electrode has been determined by SEIRA spectroscopy.71 By exploiting the high affinity of carbon monoxide to FeII and following the potential-dependent intensity of the ν(CO) modes, the formal potentials of the binuclear Fe reaction center could be determined. Aside from probing orientations, SEIRA spectroscopy has been applied to monitor formation of a hybrid complex of PSI and a H2ase utilized toward photochemical H2 generation.72
Concluding Remarks
PFE has been established as a powerful tool in enzymology, but complementary electrochemical techniques are required to more completely understand the enzyme–material interface and catalytic performance of immobilized enzymes. Quantifying enzyme adsorption, inter- and intraprotein electron transfer, activity and orientation distributions, and product generation are significant challenges. To this end, we discussed several emerging methods that facilitate these investigations and provide new insights into the aforementioned parameters to answer the questions posed in the conspectus. The combination of this suite of techniques allows probing the enzyme–electrode interface in depth and is instrumental in accelerating the development of bioelectrochemical systems such as enzymatic sensors, biofuel cells, and semiartificial photosynthetic devices, as well as enhancing our fundamental knowledge of biology.
Acknowledgments
N.K. was supported by a Royal Society Newton International Fellowship, NF160054. E.R., W.E.R., N.H., and J.Z.Z. acknowledge the European Research Council (ERC) Consolidator Grant “MatEnSAP” (682833). K.H.L. has been funded by a Marie Sklodowska-Curie Individual Fellowship (GAN 701192-VSHER).
Biographies
Nikolay Kornienko obtained his B.S. at the University of Pittsburgh (with Prof. Sanford Asher from 2010 to 2011) and his Ph.D. at the University of California, Berkeley (with Prof. Peidong Yang from 2011 to 2016) studying materials chemistry and electrocatalysis. He then carried out a Royal Society Newton Fellowship at the University of Cambridge with Prof. Erwin Reisner (2016 to 2018) investigating electroactive enzymes and microorganisms. He has recently been appointed as an Assistant Professor at the University of Montreal. His laboratory’s research is centered on developing supramolecular bioinspired photo- and electrocatalysts through rational design and establishing operando spectroscopic techniques to probe catalyst dynamics in the course of reaction.
Khoa H. Ly obtained his diploma in chemistry (2002–2007) and his Ph.D. (2008–2012) under the supervision of Prof. Peter Hildebrandt from the Technische Universität Berlin. He subsequently carried out postdoctoral research at the Technische Universität Berlin with Prof. Inez Weidinger (2013–2015) and as a Marie Skłodowska-Curie Fellow with Prof. Erwin Reisner at the University of Cambridge (2016–2018). Since April 2018, he has been working at the Chair of Electrochemistry at the Technische Universität Dresden and is entrusted with the establishment of a junior research group as part of the Open Topic Postdoc Program. His research focuses on chemical energy conversion using molecular and heterogeneous catalysis. In particular, he develops vibrational spectro-electrochemical approaches that allow tailor-made in operando investigation of the catalytic systems to derive relevant structure–activity relationships.
William E. Robinson obtained a Master of Chemistry degree from the University of Sheffield (2008–2012) before completing a Ph.D. at the University of Cambridge as part of the NanoDTC Doctoral Training Centre with Prof. Erwin Reisner (2012–2017). He continued for another year in the Reisner group as an ERC research associate (2017–2018) before moving to his current postdoctoral position in the Physical Organic Chemistry Group at Radboud University Nijmegen. His research interests revolve around understanding how networks of abiotic chemical reactions can model or mimic biological processes using chemical kinetics to provide insight into their behavior.
Nina Heidary received her diploma and Ph.D. from the Technische Universität Berlin in the group of Prof. Peter Hildebrandt and Prof. Anna Fischer. Her research centered on developing infrared spectroscopy geared towards investigating dynamics at hydrogenase–electrode interfaces. She next carried out postdoctoral work under the supervision of Prof. Erwin Reisner studying in situ mechanisms of biological catalysts for energy conversion. At the University of Montreal, Dr. Heidary focuses on applying vibrational spectro-electrochemical methods to probe metal–organic framework electrocatalysts.
Jenny Z. Zhang obtained her M.Sc. and Ph.D. at the University of Sydney in bioinorganic chemistry, following which she joined Professor Erwin Reisner as a Marie Curie postdoctoral Fellow at the University of Cambridge. She is currently a BBSRC David Phillips Fellow at the same institution, where she focuses on the use of photoelectrochemistry to study photosynthetic protein films and biofilms. In particular, she is interested in developing photoelectrochemical platforms to rewire biocatalysts for power and fuel generation.
Erwin Reisner received his education at the University of Vienna (Diploma in 2002, Ph.D. with Prof. Bernhard K. Keppler in 2005 and Habilitation in 2010). He worked as an Erwin Schrödinger postdoctoral Fellow at the Massachusetts Institute of Technology with Prof. Stephen J. Lippard (2005–2007), followed by further postdoctoral research at the University of Oxford with Prof. Fraser A. Armstrong (2008–2009). He joined the University of Cambridge as a University Lecturer in the Department of Chemistry and as a Fellow of St. John’s College in 2010. He was appointed to Reader in 2015, and to his current position as Professor of Energy and Sustainability in 2017. His laboratory explores chemical biology, synthetic chemistry, materials science, and engineering relevant to the development of solar-driven chemistry for the sustainable synthesis of fuels and chemicals.
Author Contributions
⊥ N.K. and K.H.L. contributed equally.
The authors declare no competing financial interest.
References
- Eddowes M. J.; Hill H. A. O. Novel method for the investigation of the electrochemistry of metalloproteins: cytochrome c. J. Chem. Soc., Chem. Commun. 1977, 771–772. 10.1039/c3977000771b. [DOI] [Google Scholar]
- Guilbault G. G.; Lubrano G. J. An enzyme electrode for the amperometric determination of glucose. Anal. Chim. Acta 1973, 64, 439–455. 10.1016/S0003-2670(01)82476-4. [DOI] [PubMed] [Google Scholar]
- Shu F. R.; Wilson G. S. Rotating ring-disk enzyme electrode for surface catalysis studies. Anal. Chem. 1976, 48, 1679–1686. 10.1021/ac50006a014. [DOI] [PubMed] [Google Scholar]
- Armstrong F. A.; Hirst J. Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14049–14054. 10.1073/pnas.1103697108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léger C.; Bertrand P. Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. 10.1021/cr0680742. [DOI] [PubMed] [Google Scholar]
- Vincent K. A.; Parkin A.; Armstrong F. A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. 10.1021/cr050191u. [DOI] [PubMed] [Google Scholar]
- Grunwald P. Immobilized Biocatalysts. Catalysts 2018, 8, 386. 10.3390/catal8090386. [DOI] [Google Scholar]
- Adamson H.; Robinson M.; Wright J. J.; Flanagan L. A.; Walton J.; Elton D.; Gavaghan D. J.; Bond A. M.; Roessler M. M.; Parkin A. Retuning the catalytic bias and overpotential of a [NiFe]-hydrogenase via a single amino acid exchange at the electron entry/exit site. J. Am. Chem. Soc. 2017, 139, 10677–10686. 10.1021/jacs.7b03611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoke K. R.; Cobb N.; Armstrong F. A.; Hille R. Electrochemical studies of arsenite oxidase: an unusual example of a highly cooperative two-electron molybdenum center. Biochemistry 2004, 43, 1667–1674. 10.1021/bi0357154. [DOI] [PubMed] [Google Scholar]
- Robinson W. E.; Bassegoda A.; Reisner E.; Hirst J. Oxidation-state-dependent binding properties of the active site in a Mo-containing formate dehydrogenase. J. Am. Chem. Soc. 2017, 139, 9927–9936. 10.1021/jacs.7b03958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourmond V.; Infossi P.; Giudici-Orticoni M.-T.; Bertrand P.; Léger C. “Two-step” chronoamperometric method for studying the anaerobic inactivation of an oxygen tolerant NiFe hydrogenase. J. Am. Chem. Soc. 2010, 132, 4848–4857. 10.1021/ja910685j. [DOI] [PubMed] [Google Scholar]
- Fourmond V.; Baffert C.; Sybirna K.; Lautier T.; Abou Hamdan A.; Dementin S.; Soucaille P.; Meynial-Salles I.; Bottin H.; Léger C. Steady-state catalytic wave-shapes for 2-electron reversible electrocatalysts and enzymes. J. Am. Chem. Soc. 2013, 135, 3926–3938. 10.1021/ja311607s. [DOI] [PubMed] [Google Scholar]
- Hexter S. V.; Esterle T. F.; Armstrong F. A. A unified model for surface electrocatalysis based on observations with enzymes. Phys. Chem. Chem. Phys. 2014, 16, 11822–11833. 10.1039/c3cp55230f. [DOI] [PubMed] [Google Scholar]
- Guo S.-X.; Bond A. M.; Zhang J. Fourier transformed large amplitude alternating current voltammetry: principles and applications. Rev. Polarogr. 2015, 61, 21–32. 10.5189/revpolarography.61.21. [DOI] [Google Scholar]
- Mazurenko I.; Monsalve K.; Infossi P.; Giudici-Orticoni M.-T.; Topin F.; Mano N.; Lojou E. Impact of substrate diffusion and enzyme distribution in 3D-porous electrodes: a combined electrochemical and modelling study of a thermostable H2/O2 enzymatic fuel cell. Energy Environ. Sci. 2017, 10, 1966–1982. 10.1039/C7EE01830D. [DOI] [Google Scholar]
- Jenner L. P.; Butt J. N. Electrochemistry of surface-confined enzymes: Inspiration, insight and opportunity for sustainable biotechnology. Curr. Opin. Electrochem. 2018, 8, 81–88. 10.1016/j.coelec.2018.03.021. [DOI] [Google Scholar]
- Léger C.; Jones A. K.; Albracht S. P. J.; Armstrong F. A. Effect of a dispersion of interfacial electron transfer rates on steady state catalytic electron transport in [NiFe]-hydrogenase and other enzymes. J. Phys. Chem. B 2002, 106, 13058–13063. 10.1021/jp0265687. [DOI] [Google Scholar]
- Kranich A.; Ly H. K.; Hildebrandt P.; Murgida D. H. Direct observation of the gating step in protein electron transfer: electric-field-controlled protein dynamics. J. Am. Chem. Soc. 2008, 130, 9844–9848. 10.1021/ja8016895. [DOI] [PubMed] [Google Scholar]
- Wen D.; Eychmüller A. Enzymatic biofuel cells on porous nanostructures. Small 2016, 12, 4649–4661. 10.1002/smll.201600906. [DOI] [PubMed] [Google Scholar]
- Sarma A. K.; Vatsyayan P.; Goswami P.; Minteer S. D. Recent advances in material science for developing enzyme electrodes. Biosens. Bioelectron. 2009, 24, 2313–2322. 10.1016/j.bios.2008.09.026. [DOI] [PubMed] [Google Scholar]
- Kato M.; Zhang J. Z.; Paul N.; Reisner E. Protein film photoelectrochemistry of the water oxidation enzyme photosystem II. Chem. Soc. Rev. 2014, 43, 6485–6497. 10.1039/C4CS00031E. [DOI] [PubMed] [Google Scholar]
- Nam D. H.; Zhang J. Z.; Andrei V.; Kornienko N.; Heidary N.; Wagner A.; Nakanishi K.; Sokol K. P.; Slater B.; Zebger I.; Hofmann S.; Fontecilla-Camps J. C.; Park C. B.; Reisner E. Solar Water Splitting with a Hydrogenase Integrated in Photoelectrochemical Tandem Cells. Angew. Chem., Int. Ed. 2018, 57, 10595–10599. 10.1002/anie.201805027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol K. P.; Robinson W. E.; Warnan J.; Kornienko N.; Nowaczyk M. M.; Ruff A.; Zhang J. Z.; Reisner E. Bias-free photoelectrochemical water splitting with photosystem II on a dye-sensitized photoanode wired to hydrogenase. Nat. Energy 2018, 3, 944–951. 10.1038/s41560-018-0232-y. [DOI] [Google Scholar]
- Kornienko N.; Zhang J. Z.; Sakimoto K. K.; Yang P.; Reisner E. Interfacing nature’s catalytic machinery with synthetic materials for semi-artificial photosynthesis. Nat. Nanotechnol. 2018, 13, 890–899. 10.1038/s41565-018-0251-7. [DOI] [PubMed] [Google Scholar]
- Mersch D.; Lee C.-Y.; Zhang J. Z.; Brinkert K.; Fontecilla-Camps J. C.; Rutherford A. W.; Reisner E. Wiring of photosystem II to hydrogenase for photoelectrochemical water splitting. J. Am. Chem. Soc. 2015, 137, 8541–8549. 10.1021/jacs.5b03737. [DOI] [PubMed] [Google Scholar]
- Fang X.; Sokol K. P.; Heidary N.; Kandiel T. A.; Zhang J. Z.; Reisner E. Structure-activity relationships of hierarchical three-dimensional electrodes with photosystem II for semi-artificial photosynthesis. Nano Lett. 2019, 19, 1844–1850. 10.1021/acs.nanolett.8b04935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E.; Willner I. Probing biomolecular interactions at conductive and semiconductive surfaces by impedance spectroscopy: routes to impedimetric immunosensors, DNA-sensors, and enzyme biosensors. Electroanalysis 2003, 15, 913–947. 10.1002/elan.200390114. [DOI] [Google Scholar]
- Kharitonov A. B.; Alfonta L.; Katz E.; Willner I. Probing of bioaffinity interactions at interfaces using impedance spectroscopy and chronopotentiometry. J. Electroanal. Chem. 2000, 487, 133–141. 10.1016/S0022-0728(00)00178-9. [DOI] [Google Scholar]
- Vidaković-Koch T.; Mittal V. K.; Do T. Q. N.; Varničić M.; Sundmacher K. Application of electrochemical impedance spectroscopy for studying of enzyme kinetics. Electrochim. Acta 2013, 110, 94–104. 10.1016/j.electacta.2013.03.026. [DOI] [Google Scholar]
- Zayats M.; Katz E.; Willner I. Electrical contacting of flavoenzymes and NAD(P)+-dependent enzymes by reconstitution and affinity interactions on phenylboronic acid monolayers associated with Au-electrodes. J. Am. Chem. Soc. 2002, 124, 14724–14735. 10.1021/ja027919y. [DOI] [PubMed] [Google Scholar]
- Pandey K.; Islam S. T.; Happe T.; Armstrong F. A. Frequency and potential dependence of reversible electrocatalytic hydrogen interconversion by [FeFe]-hydrogenases. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3843–3848. 10.1073/pnas.1619961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornienko N.; Zhang J. Z.; Sokol K. P.; Lamaison S.; Fantuzzi A.; van Grondelle R.; Rutherford A. W.; Reisner E. Oxygenic Photoreactivity in Photosystem II Studied by Rotating Ring Disk Electrochemistry. J. Am. Chem. Soc. 2018, 140, 17923–17931. 10.1021/jacs.8b08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard A. J.; Fan F.-R. F.; Kwak J.; Lev O. Scanning Electrochemical Microscroscopy. Introduction and principles. Anal. Chem. 1989, 61, 132–138. 10.1021/ac00177a011. [DOI] [Google Scholar]
- Madden C.; Vaughn M. D.; Díez-Pérez I.; Brown K. A.; King P. W.; Gust D.; Moore A. L.; Moore T. A. Catalytic turnover of [FeFe]-hydrogenase based on single-molecule imaging. J. Am. Chem. Soc. 2012, 134, 1577–1582. 10.1021/ja207461t. [DOI] [PubMed] [Google Scholar]
- Mirkin M. V.; Sun T.; Yu Y.; Zhou M. Electrochemistry at one nanoparticle. Acc. Chem. Res. 2016, 49, 2328–2335. 10.1021/acs.accounts.6b00294. [DOI] [PubMed] [Google Scholar]
- Lin C.; Kätelhön E.; Sepunaru L.; Compton R. G. Understanding single enzyme activity via the nano-impact technique. Chem. Sci. 2017, 8, 6423–6432. 10.1039/C7SC02084H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbrey G. Verwendung von Schwingquarzen zur Wägung dünner Schichten und zur Mikrowägung. Eur. Phys. J. A 1959, 155, 206–222. 10.1007/BF01337937. [DOI] [Google Scholar]
- Miller M.; Robinson W. E.; Oliveira R. A.; Heidary N.; Kornienko N.; Warnan J.; Pereira I. A. C.; Reisner E. Interfacing formate dehydrogenase with metal oxides for reversible electrocatalysis and solar-driven reduction of carbon dioxide. Angew. Chem., Int. Ed. 2019, 58, 4601–4605. 10.1002/anie.201814419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan D. G. G.; Marritt S. J.; Kemp G. L.; Gordon-Brown P.; Butt J. N.; Jeuken L. J. C. The impact of enzyme orientation and electrode topology on the catalytic activity of adsorbed redox enzymes. Electrochim. Acta 2013, 110, 79–85. 10.1016/j.electacta.2013.01.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.; Blanford C. F. Electrochemical Quartz Crystal Microbalance with Dissipation Monitoring: A Technique to Optimize Enzyme Use in Bioelectrocatalysis. ChemCatChem 2014, 6, 921–929. 10.1002/cctc.201300900. [DOI] [Google Scholar]
- Singh K.; McArdle T.; Sullivan P. R.; Blanford C. F. Sources of activity loss in the fuel cell enzyme bilirubin oxidase. Energy Environ. Sci. 2013, 6, 2460–2464. 10.1039/c3ee00043e. [DOI] [Google Scholar]
- Kornienko N.; Heidary N.; Cibin G.; Reisner E. Catalysis by design: development of a bifunctional water splitting catalyst through an operando measurement directed optimization cycle. Chem. Sci. 2018, 9, 5322–5333. 10.1039/C8SC01415A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topoglidis E.; Cass A. E. G.; O’Regan B.; Durrant J. R. Immobilisation and bioelectrochemistry of proteins on nanoporous TiO2 and ZnO films. J. Electroanal. Chem. 2001, 517, 20–27. 10.1016/S0022-0728(01)00673-8. [DOI] [Google Scholar]
- Kato M.; Cardona T.; Rutherford A. W.; Reisner E. Covalent immobilization of oriented photosystem II on a nanostructured electrode for solar water oxidation. J. Am. Chem. Soc. 2013, 135, 10610–10613. 10.1021/ja404699h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault C.; Harris K. D.; Brett M. J.; Balland V.; Limoges B. Time-resolved UV-visible spectroelectrochemistry using transparent 3D-mesoporous nanocrystalline ITO electrodes. Chem. Commun. 2011, 47, 1863–1865. 10.1039/C0CC04154H. [DOI] [PubMed] [Google Scholar]
- Reuillard B.; Ly K. H.; Hildebrandt P.; Jeuken L. J. C.; Butt J. N.; Reisner E. High performance reduction of H2O2 with an electron transport decaheme cytochrome on a porous ITO electrode. J. Am. Chem. Soc. 2017, 139, 3324–3327. 10.1021/jacs.6b12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-Y.; Reuillard B.; Sokol K. P.; Laftsoglou T.; Lockwood C. W. J.; Rowe S. F.; Hwang E. T.; Fontecilla-Camps J. C.; Jeuken L. J. C.; Butt J. N.; Reisner E. A decahaem cytochrome as an electron conduit in protein-enzyme redox processes. Chem. Commun. 2016, 52, 7390–7393. 10.1039/C6CC02721K. [DOI] [PubMed] [Google Scholar]
- Horch M.; Schoknecht J.; Mroginski M. A.; Lenz O.; Hildebrandt P.; Zebger I. Resonance raman spectroscopy on [NiFe] Hydrogenase provides structural insights into catalytic intermediates and reactions. J. Am. Chem. Soc. 2014, 136, 9870–9873. 10.1021/ja505119q. [DOI] [PubMed] [Google Scholar]
- Frasca S.; von Graberg T.; Feng J. J.; Thomas A.; Smarsly B. M.; Weidinger I. M.; Scheller F. W.; Hildebrandt P.; Wollenberger U. Mesoporous indium tin oxide as a novel platform for bioelectronics. ChemCatChem 2010, 2, 839–845. 10.1002/cctc.201000047. [DOI] [Google Scholar]
- Barth A.; Zscherp C. What vibrations tell about proteins. Q. Rev. Biophys. 2002, 35, 369–430. 10.1017/S0033583502003815. [DOI] [PubMed] [Google Scholar]
- Hoarau M.; Badieyan S.; Marsh E. N. G. Immobilized enzymes: understanding enzyme-surface interactions at the molecular level. Org. Biomol. Chem. 2017, 15, 9539–9551. 10.1039/C7OB01880K. [DOI] [PubMed] [Google Scholar]
- Glassford S. E.; Byrne B.; Kazarian S. G. Recent applications of ATR FTIR spectroscopy and imaging to proteins. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 2849–2858. 10.1016/j.bbapap.2013.07.015. [DOI] [PubMed] [Google Scholar]
- Healy A. J.; Reeve H. A.; Vincent K. A. Development of an infrared spectroscopic approach for studying metalloenzyme active site chemistry under direct electrochemical control. Faraday Discuss. 2011, 148, 345–357. 10.1039/C004274A. [DOI] [PubMed] [Google Scholar]
- Healy A. J.; Ash P. A.; Lenz O.; Vincent K. A. Attenuated total reflectance infrared spectroelectrochemistry at a carbon particle electrode; unmediated redox control of a [NiFe]-hydrogenase solution. Phys. Chem. Chem. Phys. 2013, 15, 7055–7059. 10.1039/c3cp00119a. [DOI] [PubMed] [Google Scholar]
- Plumeré N.; Rüdiger O.; Oughli A. A.; Williams R.; Vivekananthan J.; Pöller S.; Schuhmann W.; Lubitz W. A redox hydrogel protects hydrogenase from high-potential deactivation and oxygen damage. Nat. Chem. 2014, 6, 822–827. 10.1038/nchem.2022. [DOI] [PubMed] [Google Scholar]
- Ciaccafava A.; Infossi P.; Ilbert M.; Guiral M.; Lecomte S.; Giudici-Orticoni M. T.; Lojou E. Electrochemistry, AFM, and PM-IRRA spectroscopy of immobilized hydrogenase: role of a hydrophobic helix in enzyme orientation for efficient H2 oxidation. Angew. Chem., Int. Ed. 2012, 51, 953–956. 10.1002/anie.201107053. [DOI] [PubMed] [Google Scholar]
- Olejnik P.; Palys B.; Kowalczyk A.; Nowicka A. M. Orientation of Laccase on Charged Surfaces. Mediatorless Oxygen Reduction on Amino- and Carboxyl-Ended Ethylphenyl Groups. J. Phys. Chem. C 2012, 116, 25911–25918. 10.1021/jp3098654. [DOI] [Google Scholar]
- Hitaishi V. P.; Mazurenko I.; Harb M.; Clément R.; Taris M.; Castano S.; Duché D.; Lecomte S.; Ilbert M.; de Poulpiquet A.; Lojou E. Electrostatic-Driven Activity, Loading, Dynamics, and Stability of a Redox Enzyme on Functionalized-Gold Electrodes for Bioelectrocatalysis. ACS Catal. 2018, 8, 12004–12014. 10.1021/acscatal.8b03443. [DOI] [Google Scholar]
- Khoa Ly H.; Sezer M.; Wisitruangsakul N.; Feng J. J.; Kranich A.; Millo D.; Weidinger I. M.; Zebger I.; Murgida D. H.; Hildebrandt P. Surface-enhanced vibrational spectroscopy for probing transient interactions of proteins with biomimetic interfaces: electric field effects on structure, dynamics and function of cytochrome c. FEBS J. 2011, 278, 1382–1390. 10.1111/j.1742-4658.2011.08064.x. [DOI] [PubMed] [Google Scholar]
- Khoa Ly H.; Wisitruangsakul N.; Sezer M.; Feng J.-J.; Kranich A.; Weidinger I. M.; Zebger I.; Murgida D. H.; Hildebrandt P. Electric-field effects on the interfacial electron transfer and protein dynamics of cytochrome c. J. Electroanal. Chem. 2011, 660, 367–376. 10.1016/j.jelechem.2010.12.020. [DOI] [Google Scholar]
- Sezer M.; Kielb P.; Kuhlmann U.; Mohrmann H.; Schulz C.; Heinrich D.; Schlesinger R.; Heberle J.; Weidinger I. M. Surface enhanced resonance Raman spectroscopy reveals potential induced redox and conformational changes of cytochrome c oxidase on electrodes. J. Phys. Chem. B 2015, 119, 9586–9591. 10.1021/acs.jpcb.5b03206. [DOI] [PubMed] [Google Scholar]
- Bonifacio A.; Millo D.; Keizers P. H. J.; Boegschoten R.; Commandeur J. N. M.; Vermeulen N. P. E.; Gooijer C.; van der Zwan G. Active-site structure, binding and redox activity of the heme-thiolate enzyme CYP2D6 immobilized on coated Ag electrodes: a surface-enhanced resonance Raman scattering study. JBIC, J. Biol. Inorg. Chem. 2008, 13, 85–96. 10.1007/s00775-007-0303-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira C. M.; Quintas P. O.; Moura I.; Moura J. J. G.; Hildebrandt P.; Almeida M. G.; Todorovic S. SERR Spectroelectrochemical Study of Cytochrome cd1 Nitrite Reductase Co-Immobilized with Physiological Redox Partner Cytochrome c552 on Biocompatible Metal Electrodes. PLoS One 2015, 10, e0129940 10.1371/journal.pone.0129940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezer M.; Genebra T.; Mendes S.; Martins L. O.; Todorovic S. A DyP-type peroxidase at a bio-compatible interface: structural and mechanistic insights. Soft Matter 2012, 8, 10314–10321. 10.1039/c2sm26310f. [DOI] [Google Scholar]
- Moe E.; Sezer M.; Hildebrandt P.; Todorovic S. Surface enhanced vibrational spectroscopic evidence for an alternative DNA-independent redox activation of endonuclease III. Chem. Commun. 2015, 51, 3255–3257. 10.1039/C4CC09498K. [DOI] [PubMed] [Google Scholar]
- Sezer M.; Frielingsdorf S.; Millo D.; Heidary N.; Utesch T.; Mroginski M.-A.; Friedrich B.; Hildebrandt P.; Zebger I.; Weidinger I. M. Role of the HoxZ Subunit in the Electron Transfer Pathway of the Membrane-Bound [NiFe]-Hydrogenase from Ralstonia eutropha Immobilized on Electrodes. J. Phys. Chem. B 2011, 115, 10368–10374. 10.1021/jp204665r. [DOI] [PubMed] [Google Scholar]
- Dagys M.; Laurynėnas A.; Ratautas D.; Kulys J.; Vidžiu̅naitė R.; Talaikis M.; Niaura G.; Marcinkevičenė L.; Meškys R.; Shleev S. Oxygen electroreduction catalysed by laccase wired to gold nanoparticles via the trinuclear copper cluster. Energy Environ. Sci. 2017, 10, 498–502. 10.1039/C6EE02232D. [DOI] [Google Scholar]
- Öner I. H.; Querebillo C. J.; David C.; Gernert U.; Walter C.; Driess M.; Leimkühler S.; Ly K. H.; Weidinger I. M. High electromagnetic field enhancement of TiO2 nanotube electrodes. Angew. Chem., Int. Ed. 2018, 57, 7225–7229. 10.1002/anie.201802597. [DOI] [PubMed] [Google Scholar]
- Millo D.; Hildebrandt P.; Pandelia M. E.; Lubitz W.; Zebger I. SEIRA spectroscopy of the electrochemical activation of an immobilized [NiFe] hydrogenase under turnover and non-turnover conditions. Angew. Chem., Int. Ed. 2011, 50, 2632–2634. 10.1002/anie.201006646. [DOI] [PubMed] [Google Scholar]
- Heidary N.; Utesch T.; Zerball M.; Horch M.; Millo D.; Fritsch J.; Lenz O.; von Klitzing R.; Hildebrandt P.; Fischer A.; Mroginski M.; Zebger I. Orientation-controlled electrocatalytic efficiency of an adsorbed oxygen-tolerant hydrogenase. PLoS One 2015, 10, e0143101 10.1371/journal.pone.0143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M.; Nakagawa S.; Tosha T.; Shiro Y.; Masuda Y.; Nakata K.; Yagi I. Surface-Enhanced Infrared Absorption Spectroscopy of Bacterial Nitric Oxide Reductase under Electrochemical Control Using a Vibrational Probe of Carbon Monoxide. J. Phys. Chem. Lett. 2018, 9, 5196–5200. 10.1021/acs.jpclett.8b02581. [DOI] [PubMed] [Google Scholar]
- Krassen H.; Schwarze A.; Friedrich B.; Ataka K.; Lenz O.; Heberle J. Photosynthetic Hydrogen Production by a Hybrid Complex of Photosystem I and [NiFe]-Hydrogenase. ACS Nano 2009, 3, 4055–4061. 10.1021/nn900748j. [DOI] [PubMed] [Google Scholar]