

Abstract
Nanopore sequencing offers a portable and affordable alternative to sequencing-by-synthesis methods but suffers from lower accuracy and cannot sequence ultrashort DNA. This puts applications such as molecular diagnostics based on the analysis of cell-free DNA or single-nucleotide variants (SNVs) out of reach. To overcome these limitations, we report a nanopore-based sequencing strategy in which short target sequences are first circularized and then amplified via rolling-circle amplification to produce long stretches of concatemeric repeats. After sequencing on the Oxford Nanopore Technologies MinION platform, the resulting repeat sequences can be aligned to produce a highly accurate consensus that reduces the high error-rate present in the individual repeats. Using this approach, we demonstrate for the first time the ability to obtain unbiased and accurate nanopore data for target DNA sequences <100 bp. Critically, this approach is sensitive enough to achieve SNV discrimination in mixtures of sequences and even enables quantitative detection of specific variants present at ratios of <10%. Our method is simple, cost-effective, and only requires well-established processes. It therefore expands the utility of nanopore sequencing for molecular diagnostics and other applications, especially in resource-limited settings.
Nanopore sequencing technology, most notably commercialized as the hand-held MinION instrument by Oxford Nanopore Technologies (ONT), has emerged as a powerful sequencing modality due to its low cost, portability, and capacity to sequence very long strands of DNA. These features have made nanopore sequencing indispensable for the assembly of genomes that were previously inaccessible to conventional short-read, sequencing-by-synthesis methods.1,2 Despite these positive attributes, nanopore sequencing is ill-suited for important applications, such as the profiling of microRNA (miRNA)3 or cell-free DNA,4 which require sequencing of ultrashort DNA (<100 bp). Specifically, it has been demonstrated that quality scores drop off dramatically for sequences shorter than 1 kb.5 To date, the shortest reported DNA sequence to be directly targeted and sequenced on a MinION was 434 bp.6,7 Current nanopore sequencing technologies also have higher error rates relative to the sequencing-by-synthesis chemistry employed in the widely used Illumina platforms.8 Because of this, conventional nanopore sequencing is incompatible with applications that require high accuracy, such as the detection and quantification of single-nucleotide variants (SNVs). To date, nanopore-based quantification of SNV abundance has only been achieved through high sequence coverage or by cosequencing on a high-accuracy short-read platform such as the Illumina MiSeq.9 Therefore, a novel methodology that enables sequencing of ultrashort DNA with low error-rates would greatly expand the utility of nanopore sequencing technology.
We describe a simple sample-preparation strategy that converts ultrashort DNA into long stretches of tandem repeats (concatemers) that can be sequenced on the MinION with sufficient accuracy to achieve reliable SNV detection. To the best of our knowledge, this represents the first successful nanopore sequencing of target DNA shorter than 100 bp, as well as the first successful resolution of SNVs from single reads on a nanopore sequencer. Our high-fidelity short reads method (HiFRe) entails the circularization of short DNA sequences, followed by the generation of concatemers via rolling-circle amplification (RCA). These concatemers can be readily sequenced on the MinION, with the tandem repeats providing highly accurate reads through in silico reconstruction of the target sequence. Previous work in this area has illustrated that circularizing and concatemerizing DNA is a robust approach to improve the fidelity of sequencing in general.10−12 Intramolecular-ligated nanopore consensus sequencing (INCseq),11 for example, uses blunt-end ligation to circularize double-stranded (ds) DNA of 600–800 bp followed by RCA to create linear DNA with tandem copies of the template molecule. INCseq improves the accuracy of MinION sequencing through computational alignment of the tandem repeats to reconstruct the original sequence. However, its reliance on blunt-end ligation requires the input DNA to be long enough to overcome the curvature-induced strain in double-stranded DNA to achieve circularization, preventing the method from being adapted to ultrashort reads.
HiFRe offers important advantages over this and other previously reported approaches.6,7,11,12 Most importantly, whereas existing methods for nanopore sequencing of small DNA are still limited to relatively long fragments of 500–1000 bp, HiFRe enables accurate targeted analysis of sequences <100 bp. As a demonstration, we have used HiFRe to sequence targeted 52 bp segments of DNA with high fidelity. Furthermore, we show that HiFRe enables SNV resolution from a single nanopore read, with the capacity to accurately quantify mixtures of sequences based on the discrimination of single-nucleotide differences at ratios as low as 10:90.
Experimental Section
Reagents
Unless noted otherwise, all DNA sequences were purchased from Integrated DNA Technologies and all reagents were purchased from New England Biolabs. Initial DNA concentrations were measured and normalized to 100 μM via A260 measurement on a Nanodrop 2000 (Thermo Scientific).
Probe Design
Molecular inversion probes (MIPs) were carefully designed to minimize secondary structure in the hybridization regions using a combination of the NUPACK13 and mfold(14) DNA folding applications. We established a threshold of >90% conversion from linear to circular DNA after five cycles of annealing, extension, and ligation. As described in Calculation S1 of the Supporting Information (SI), this means that any secondary structures in the MIP’s anchor sites must have a ΔG > −0.33 kcal/mol. This was an important consideration for the MIP design as a whole as well as for the barcodes listed in Table S1.
Preamplification and Generation of dsDNA Input Templates
dsDNA was generated from the ssDNA mixtures listed in Table S2. Standard PCR reactions were prepared as follows: 18 μL ssDNA input, 60 μL 2× GoTaq Master Mix (Promega), 12 μL 10 μM forward primer (FP), 12 μL 10 μM reverse primer (RP), and 18 μL nuclease-free water (Ambion). Reactions were carried out by thermocycling with an initial denaturation at 95 °C for 3 min and 20 cycles of 95 °C for 10 s, 55 °C for 30 s, and 72 °C for 30 s on a vapo.protect Mastercycler Pro thermocycler (Eppendorf). After cleanup with a MinElute PCR Purification Kit (Qiagen) via the manufacturer’s protocol, amplified dsDNA product was normalized to 100 nM in nuclease-free water based on Qubit reading. The resultant amplified dsDNA simulates a typical input to the HiFRe workflow (Figure S1, top).
Circularization Reaction
Optimal MIP annealing temperature was determined to be 60.4 °C based on maximum yield after circularization with a gradient of annealing temperatures ranging from 58 to 70 °C on a CFX96 Real-Time qPCR System (Bio-Rad). For all of these experiments, 15 nM target dsDNA was circularized with 20 nM MIP. Circularization reactions (10 μL total volume) were prepared on ice containing 1.5 μL 100 nM target, 2 μL 100 nM MIP, 5 μL 2X Phusion Master Mix (New England Biolabs), 0.5 μL ampligase (10 U/μL, Lucigen), and 1 μL 10X ampligase buffer (Lucigen). Circularization was achieved under the following conditions: 95 °C for 3 min followed by 5 cycles of 95 °C for 30s, 60.4 °C for 1 min, and 37 °C for 2 min. After circularization, linear DNA was degraded by adding 0.45 μL of exonuclease I (20 U/μL) and 0.45 μL of exonuclease III (100 U/μL). The reaction was incubated at 37 °C for 90 min followed by heat inactivation at 65 °C for 20 min. Exonuclease inactivation was determined to be sufficiently efficient based on the lack of exonuclease activity observed in samples left at 37 °C for 2 days, thus no cleanup step was performed prior to the RCA reaction.
Rolling Circle Amplification
The RCA reaction was performed under the following conditions: 2 μL nuclease-treated circularization product, 1 μL φ29 (10 U/μL), 1 μL 10 μM RCA FP, 4 μL dNTP mix (2 mM each, Novagen), 2 μL 10× φ29 buffer, 7.8 μL nuclease-free water, and 0.2 μL 100X BSA. Reactions were carried out with 12 h incubation at 30 °C, 10 min at 60 °C, and a hold at 4 °C. We also tested 2-, 4-, and 12-h RCA reaction times, and observed no difference in median product length, which indicates that the protocol could be shortened substantially with further optimization of the RCA protocol.
We then amplified the single-stranded RCA product to dsDNA with standard PCR using the following reaction conditions: 9 μL RCA product, 15 μL 2× GoTaq Master Mix (Promega), 1.5 μL 10 μM FP, 1.5 μL 10 μM RP, and 3 μL nuclease-free water. The reaction was initiated with a 2 min hot start (95 °C) followed by 5–8 cycles of 95 °C for 10 s, 58 °C for 30 s, and 72 °C for 10 min, with a final extension at 72 °C for 2 min and a hold at 4 °C. The number of cycles was determined by a pilot PCR with 4–10 cycles of amplification, where we chose the minimum cycle number that produced a total DNA concentration of >50 ng/μL with no observed laddering. The resultant dsDNA was purified using a MinElute PCR Purification Kit (Qiagen) following the manufacturer’s protocol.
Sample Pooling
In order to test multiple conditions on a single flow cell, we incorporated barcodes into the N12 region of the MIPs to enable multiplexing. Samples were prepared with varying input ratios of target molecules (Table S2). Post-RCA dsDNA products were pooled together before sequencing based on the measured concentration (HS dsDNA, Qubit) normalized by the median length reported by the 4200 Tape Station (Agilent). The median length of amplified RCA product was 6,600 bp.
Nanopore Sequencing and Data Analysis
The pooled library was prepared for sequencing via ONT’s Ligation Sequencing 1D Kit, and then sequenced on a R9.4 flow cell for 24 h according to the manufacturer’s protocol. We obtained 769,934 raw reads. Initial base-calling was done through ONT’s MinKNOW software. We avoided use of more sophisticated nanopore alignment tools, as we wanted to test the worst-case scenario raw input, with as little data processing as possible in between the nanopore run and HiFRe analysis. Custom MATLAB scripts (available at https://github.com/btotherad77/hifre) were developed to parse and analyze the raw nanopore reads. Individual repeats were identified as regions that mapped significantly to the FP sequence. The alignment threshold was determined as the average Smith-Waterman local alignment15 of random sequences to the FP plus five standard deviations (p < 3 × 10–7). We observed 200,532 raw reads with 4 or more identifiable repeats. The individual repeats were collected and aligned to each other using a progressive multialignment algorithm (multialign, MATLAB). The consensus sequence was then determined base-wise with a winner-take-all strategy and any resulting gaps were removed. The individual repeats and the consensus sequence were then compared back to the original template in order to assess the accuracy before and after alignment, respectively.
Gel Protocol
Gels were run using a PowerPac Basic (Bio-Rad) and imaged on a Molecular Imager Gel Doc XR+ (Bio-Rad). For amplified input DNA and post-RCA dsDNA, native gels were run with 5 μL per well of a 1:5:1 mixture of sample/water/BlueJuice loading dye (Thermo Fisher Scientific) in a 10% TBE, 1 mm, 12-well gel (Thermo Fisher Scientific) for 45 min at 150 V. For circularized ssDNA and post-RCA ssDNA, denaturing gels were run with 6 μL per well of a 1:1 mixture of sample and Gel Loading Buffer II with formamide (Ambion) in a 10% TBE-Urea, 1 mm, 12-well gel (Thermo Fisher Scientific) for 1 h at 200 V. Sample-dye mixtures were denatured at 95 °C for 7 min prior to loading into the gel.
Results and Discussion
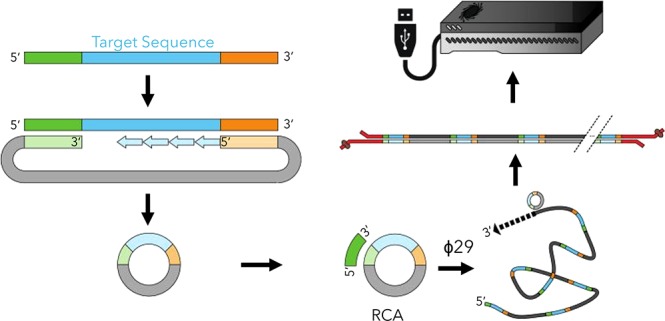
HiFRe enables sequencing analysis of ultrashort DNA targets through a straightforward procedure of circularization and amplification (Figure 1a). First, we employ molecular inversion probes (MIPs)16,17 to copy a target DNA sequence into a circular single-stranded (ss) DNA construct. For this reaction, we employ the Phusion polymerase, which lacks 5′ → 3′ exonuclease activity, thereby ensuring that extension halts at the 5′ end of the second hybridization site, where ligation is to occur. We took care to design MIPs that are predicted to exhibit minimal secondary structure—especially in the anchor sites—to ensure highly efficient circularization. We established a threshold for secondary structure energetics based on the ΔGfolding for which the conversion from linear to circular DNA is >90% after five temperature cycles. In order to achieve efficient circularization, we estimate that any secondary structure involving an anchor site must have ΔGfolding ≥ −0.33kcal/mol (Calculation S1). Prior works that did not consider this aspect of MIP design used 99 cycles of melting, annealing, extension, and ligation to achieve circularization,17 whereas we are consistently able to obtain complete conversion to circular product in just five cycles. After ligation, we incubate the reaction with a mixture of exonucleases I and III to degrade any remaining linear single- or double-stranded DNA while leaving circular DNA intact. We note that this combination of circularization and degradation results in excellent specificity, allowing us to efficiently isolate and purify target amplicons even in a large background of nonspecific byproducts of PCR amplification (Figure S1).
Figure 1.

Sequencing ultrashort reads on the MinION. (a) (1) Molecular inversion probes (MIPs) anneal adjacent to the target sequence (blue) at anchor site 1 (AS1, orange) and anchor site 2 (AS2, green). Phusion polymerase copies the target sequence into the MIP; the lack of 5′ → 3′ exonuclease activity ensures that extension halts when the polymerase reaches AS2. (2) Ampligase ligates the extended template to the phosphorylated 5′ end of the MIP, generating circular ssDNA. Linear ss- or dsDNA fragments are degraded by a combination of exonuclease I and exonuclease III. (3) The circular DNA is subjected to RCA to generate tandem repeats of the original target, yielding ultralong, concatemerized ssDNA. (4) The RCA product is converted to dsDNA with Taq polymerase and subjected to ONT library preparation. (5) Sequencing reads are collected from a new MinION R9.4 flow-cell run for 24 h. (b) The raw sequences are compiled and analyzed. The identified repeats have poor accuracy in isolation, but since the sequencing errors vary across repeats, they can be aligned together to produce a high-fidelity consensus sequence.
We then initiate RCA18 on the circular product with the strand-displacing polymerase φ29, which generates long single-stranded concatemers (>1000 kb) of the complement to the circular template. Shorter template circles with commensurately smaller target sequence inserts are generally advantageous here, as they can generate more repeats within a fixed RCA length, which in turn positively impacts the accuracy of the resulting computational alignment. There are certain limitations, including the fact that the ring strain in very small DNA circles can cause φ29 to dissociate prematurely.19 However, since the MIP itself is 102 nt, HiFRe circumvents this limitation and can target inserts as short as a single nucleotide (Figure 2, lane 2). The ssDNA generated by the RCA reaction is incompatible with the dsDNA required by the MinION library preparation protocol. We therefore implement a brief secondary amplification with Taq polymerase to generate the required starting material of ∼1 μg dsDNA. The presence of multiple primer sequences within the concatamer creates opportunities for laddering and shortening in the amplification product pool. We exploit the 5′ → 3′ exonuclease capacity of Taq polymerase to overcome this problem: any sequences that are undesirably primed from an internal primer-binding site are likely to be degraded by the exonucleolytic activity of an upstream Taq enzyme, greatly favoring the synthesis of full-length products. Finally, the dsDNA products are prepared for sequencing via ONT’s standard 1D amplicon by ligation kit and sequenced on a R9.4 flow cell. The tandem repeats identified from the raw reads are then used to computationally reconstruct the original sequence (Figure 1b).
Figure 2.

Circularization can be performed on target sequences as short as a single nucleotide with reaction efficiency that is independent of target sequence length. After five rounds of temperature cycling and subsequent exonuclease treatment, we achieve consistently efficient circularization for target sequences ranging in length from 1 to 120 nt (lanes 2–8). In this denaturing gel, lane 1 contains a mixture of all the linear ssDNA target sequences. The lengths listed are the lengths of the target region; the full lengths in lane 1 have additional flanking 28- and 23-nt anchor sites, and the full lengths in lanes 2–8 have an additional 102 nt from the MIP. Lane 9 illustrates that no circular DNA is produced in the absence of the target sequence.
HiFRe Enables Accurate MinION Sequencing of Ultrashort DNA Sequences (<100 bp)
To demonstrate the generalizability of this approach, we first performed our circularization procedure on a variety of sequences with target lengths ranging from 1 to 120 bp. We found that all of the tested sequences were targeted and circularized with high efficiency, using the same 102 bp MIP design for each target. The observation that high circularization efficiency is preserved independent of the tested target length (Figure 2) illustrates the generalizability of this approach over a range of ultrashort sequence lengths.
In order to further explore the performance and capabilities of our HiFRe method, we subjected a 52 bp target sequence (Table 1) to the entire workflow, including sequencing and data analysis. We circularized our target sequence with a 102 nt MIP, yielding an RCA product with a repeat length of 154 bases. To account for potential sequence-induced biases that could arise from using a single test sequence, we incorporated two randomized N10 regions at the ends of the 52 bp target sequence, adjacent to the anchor sites. Notably, the rest of the workflow can be adapted to any other assay that also results in a circular DNA output, such as MiR-ID20 or cPLA.21
Table 1. DNA Sequences Used in This Worka.

Sequences in blue represent the target sequences, orange and green represent universal anchor sites 1 & 2, respectively, and grey represents the MIP scaffold sequence. Bases in red indicate the three SNVs that were introduced into Target #2. The barcode in the MIP sequences is a 12 nt region that allows for multiplexing in a single nanopore run. Barcode sequences are listed in Table S1.
The resulting consensus reads achieved excellent accuracy despite the short length of the original template. In general, short reads are less tolerant of errors than long sequences in terms of sequence identity. For example, an E. coli genome sequence with a 20% error rate will be accurately identified as E. coli since there are still millions of accurately sequenced bases and the sequence space is very sparse. Alternatively, a 20-bp miRNA with a 20% error rate has a high probability of being misidentified as a completely different miRNA, as the sequence space is far more dense.22 Therefore, we developed a computational algorithm to process the data from HiFRe, identify tandem repeats in the raw base-called reads, and reconstruct the original sequence. The source code for this algorithm is publicly available on GitHub (see Methods).
A representative alignment illustrates the utility of using individual tandem repeats to reconstruct an accurate consensus sequence (Figure 3a). In order to recover the individual repeats, the algorithm scans the raw base-called reads for regions that map to the RCA initiator. These individual repeats are compared to the initial target sequence in order to define the average unmapped accuracy (Figure 3b, gray). Using a progressive multiple alignment algorithm, we then generate a consensus sequence from the ensemble of consecutive repeats. The consensus sequence is compared to the original target sequence to define the postalignment accuracy (Figure 3b, red). We observed marked improvement in the alignment scores after consensus sequence generation, with the median alignment score increasing from 0.65 to 0.87 after alignment. It should be noted that we applied the algorithm to any read that had more than three identifiable repeats, regardless of the quality score or pass/fail designation reported by the MinKNOW software—in stark contrast to previous methods that only consider high-quality, passing reads.11
Figure 3.

Improving read accuracy through repeat-based consensus. (a) Representative consensus sequence generation. A single, base-called read is split into its individual repeats. These repeats are aligned with each other to generate a consensus sequence via a winner-take-all base-calling strategy. Gaps are removed and the consensus sequence is then compared back to the original sequence to assess the postalignment accuracy. (b) Histogram of alignment scores before (gray) and after (red) consensus sequence generation. The “before” alignment score is an average over the alignment scores of all the repeats found within a single raw read. Data includes all reads with more than three identified repeats, regardless of the quality score or pass/fail designation of the MinION software.
As anticipated, we found that the improvement in alignment is a strong function of the number of tandem repeats used to generate the consensus sequence (Figure 4). Contrary to previous reports that show a plateau in accuracy at 15 repeats,11 we found that we could achieve significant increases in accuracy with up to 29 repeats. The average normalized Smith-Waterman alignment score increased from 0.74 ± 0.13 to 0.95 ± 0.04 going from 4 to 29 aligned repeats, as opposed to the average alignment score of 0.57 ± 0.14 for unaligned repeats. Additional gains may be possible beyond 29 repeats, but the read depth at these lengths was too low to determine the statistical significance of any further increases in accuracy. Improvements to the RCA protocol23 could enable the production of higher fractions of longer concatemers, and therefore yield an even more pronounced increase in overall accuracy. Additionally, we observed that the standard deviation of the alignment score also decreases with greater numbers of repeats (Figure 4), yielding alignments that are both more accurate and more precise.
Figure 4.

Increased accuracy from alignment of tandem repeats. Plots show normalized Smith-Waterman alignment scores as a function of the number of repeats before (gray) and after (red) alignment. Before consensus sequence generation, alignment score exhibits no dependence on repeat count. Since each “before point” represents an average over all repeats in that read, the observed narrowing arises solely because the increased number of repeats decreases the standard deviation of the average alignment score. After the consensus sequence is generated, the alignment accuracy exhibits a strong dependence on the number of repeats used.
HiFRe Enables Quantitative Discrimination of Sequence Variants
Our method is also quantitative, enabling ratiometric quantification of different target sequences. This is useful for many sequencing-based assays, such as transcriptome analysis,24 miRNA profiling,25,26 and cell-free DNA detection.4 Most uses of nanopore sequencing to date have generally not explored molecular counting, except for quantifying chromosome copy number for aneuploidy detection.6 RNA-seq can be performed with the nanopore system,27,28 but this application is still in the nascent stages of commercial development. We have found that HiFRe can accurately quantify input ratios of two target sequences ranging from 0 to 90% with an R2 of 0.993 (Figure 5a), yielding a limit of detection29 of 3.3 ± 2.1%. This strong linear relationship between input and output abundances illustrates the power of HiFRe for quantifying relative abundances of multiple sequences.
Figure 5.

Quantitative analysis with HiFRe. a) Counting relative molecular abundance for two sequences present in mixtures at different ratios. A linear fit to y = mx + b yielded m = 0.97 ± 0.04 and b = 0.02 ± 0.02 with R2 = 0.993. This strong linear relationship results in a limit of detection of 3.3 ± 2.1%. b) Discrimination and quantitation of SNVs in short DNA sequences. Two sequences differing by three SNVs were mixed together in different ratios, and the plot shows the output ratios recovered after HiFRe analysis. Green bars represent the fraction of the original sequence and yellow bars show the sequence with three SNVs. In both panels, the error bars represent the standard deviation for the mean of two multiplexed sequencing runs.
This discriminatory power is important, as many clinical applications of DNA sequencing involve resolving the relative abundance of sequences with single-nucleotide differences—for example, the detection of cancerous mutations in cell-free DNA4 or the analysis of closely related miRNA families.25,26 Until recently, SNV resolution on nanopore-based platforms has required either extremely high read depth or cosequencing with another sequencing method. HiFRe now enables the resolution of SNVs from single reads within a standard MinION experimental workflow. To demonstrate this, we mixed together two 52 bp test sequences that were identical apart from three nucleotides (Table 1). Using HiFRe, we were able to accurately resolve the representation of the two sequences for all three SNVs at a variety of different input ratios (Figure 5b). Although we tested three different SNVs (G → C, A → T, C → G) to account for potential biases in nanopore base-calling errors,30 we observed no impact on sequence discrimination from these individual SNVs. We note that some SNVs not tested in this work are particularly challenging to detect via nanopore. For example, A[G/C/T]A versus AAA would be difficult to quantify due to the nanopore platform’s propensity to introduce deletions when reading homopolymers of As. However, future iterations of the HiFRe algorithm could be designed to account for these biases.
Conclusions
HiFRe offers a simple and straightforward strategy for the accurate nanopore sequencing of ultrashort sequences (<100 bp). This method deploys RCA on a MIP-circularized template to generate long concatemers that can readily be sequenced on the MinION platform. Each raw read contains numerous repeats of the original target sequence that can be computationally aligned to generate a highly accurate consensus sequence. Using a 52 bp target sequence, we demonstrate over multiple experiments that the accuracy of this method is sufficiently high to enable relative molecular counting and SNV resolution, resulting in accurate ratiometric quantification of DNA sequences that are present at input ratios below 10%. With the added ability to analyze short targets, nanopore sequencing can be used not only for genomic analysis but also as a readout for the many bioassays that feature the production of short DNA oligonucleotides as an output, such as PLA,31 PLAYR,32 and AbSeq.33 Ultimately, assays that previously required a fully equipped laboratory and a desktop sequencer could potentially be performed at a fraction of the cost in a portable format, greatly expanding their utility and accessibility, especially in resource-limited settings.
Acknowledgments
This work was financially supported by the Chan-Zuckerberg Biohub and the Bill and Melinda Gates Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b00856.
Determination of threshold thermodynamics for MIP design; gel electrophoresis before and after circularization demonstrates high specificity of our reaction conditions; sequences of barcodes used for multiplexing; and list of experimental conditions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Jain M.; Koren S.; Miga K. H.; Quick J.; Rand A. C.; Sasani T. A.; Tyson J. R.; Beggs A. D.; Dilthey A. T.; Fiddes I. T.; et al. Nanopore Sequencing and Assembly of a Human Genome with Ultra-Long Reads. Nat. Biotechnol. 2018, 36 (4), 338–345. 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael T. P.; Jupe F.; Bemm F.; Motley S. T.; Sandoval J. P.; Lanz C.; Loudet O.; Weigel D.; Ecker J. R. High Contiguity Arabidopsis Thaliana Genome Assembly with a Single Nanopore Flow Cell. Nat. Commun. 2018, 9 (1), 1–8. 10.1038/s41467-018-03016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. S.; Parkin R. K.; Kroh E. M.; Fritz B. R.; Wyman S. K.; Pogosova-Agadjanyan E. L.; Peterson A.; Noteboom J.; Briant K. C. O.; Allen A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (30), 10513–10518. 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen E.; Nordentoft I.; Vang S.; Birkenkamp-Demtröder K.; Jensen J. B.; Agerbæk M.; Pedersen J. S.; Dyrskjøt L. Optimized Targeted Sequencing of Cell-Free Plasma DNA from Bladder Cancer Patients. Sci. Rep. 2018, 8 (1), 1–11. 10.1038/s41598-018-20282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R.; Sinha A.; Bird S. W.; Jayamohan H.; Edwards H. S.; Schoeniger J. S.; Patel K. D.; Branda S. S.; Bartsch M. S. Systematic and Stochastic Influences on the Performance of the MinION Nanopore Sequencer across a Range of Nucleotide Bias. Sci. Rep. 2018, 8 (1), 1–13. 10.1038/s41598-018-21484-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.; Williams Z. Rapid Short-Read Sequencing and Aneuploidy Detection Using MinION Nanopore Technology. Genetics 2016, 202 (1), 37–44. 10.1534/genetics.115.182311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.; Weiss Z. R.; Williams Z. Rapid Multiplex Small DNA Sequencing on the MinION Nanopore Sequencing Platform. G3: Genes, Genomes, Genet. 2018, 8 (5), 1649–1657. 10.1534/g3.118.200087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H.; Giordano F.; Ning Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics Bioinf. 2016, 14 (5), 265–279. 10.1016/j.gpb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis S.; Gansemans Y.; Deleye L.; Deforce D.; Van Nieuwerburgh F. Forensic SNP Genotyping Using Nanopore MinION Sequencing. Sci. Rep. 2017, 7, 1–5. 10.1038/srep41759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J.; Fehr A.; Gray J.; Luong K.; Lyle J.; Otto G.; Peluso P.; Rank D.; Baybayan P.; Bettman B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323 (5910), 133–138. 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Li C.; Chng K. R.; Boey E. J. H.; Ng A. H. Q.; Wilm A.; Nagarajan N. INC-Seq: Accurate Single Molecule Reads Using Nanopore Sequencing. GigaScience 2016, 5 (1), 34. 10.1186/s13742-016-0140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volden R.; Palmer T.; Byrne A.; Cole C.; Schmitz R. J.; Green R. E.; Vollmers C. Improving Nanopore Read Accuracy with the R2C2Method Enables the Sequencing of Highly Multiplexed Full-Length Single-Cell CDNA. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (39), 9726–9731. 10.1073/pnas.1806447115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. NUPACK: Analysis and Design of Nucleic Acid Systems. J. Comput. Chem. 2011, 32 (1), 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction. Nucleic Acids Res. 2003, 31 (13), 3406–3415. 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. F.; Waterman M. S. Comparison of Biosequences. Adv. Appl. Math. 1981, 2 (4), 482–489. 10.1016/0196-8858(81)90046-4. [DOI] [Google Scholar]
- Hardenbol P.; Banér J.; Jain M.; Nilsson M.; Namsaraev E. A.; Karlin-Neumann G. A.; Fakhrai-Rad H.; Ronaghi M.; Willis T. D.; Landegren U.; et al. Multiplexed Genotyping with Sequence-Tagged Molecular Inversion Probes. Nat. Biotechnol. 2003, 21 (6), 673–678. 10.1038/nbt821. [DOI] [PubMed] [Google Scholar]
- Akhras M. S.; Unemo M.; Thiyagarajan S.; Nyrén P.; Davis R. W.; Fire A. Z.; Pourmand N. Connector Inversion Probe Technology: A Powerful One-Primer Multiplex DAN Amplification System for Numerous Scientific Application. PLoS One 2007, 2 (9), 1–6. 10.1371/journal.pone.0000915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsen M. G.; Kool E. T. The Discovery of Rolling Circle Amplification and Rolling Circle Transcription. Acc. Chem. Res. 2016, 49 (11), 2540–2550. 10.1021/acs.accounts.6b00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A.; Xu S. Q. Rolling Replication of Short DNA Circles. Proc. Natl. Acad. Sci. U. S. A. 1995, 92 (10), 4641–4645. 10.1073/pnas.92.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Johnston B. H.; Kazakov S. A. MiR-ID: A Novel, Circularization-Based Platform for Detection of MicroRNAs. RNA 2011, 17 (2), 365–380. 10.1261/rna.2490111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili R.; Horecka J.; Swartz J. R.; Davis R. W.; Persson H. H. J. Streamlined Circular Proximity Ligation Assay Provides High Stringency and Compatibility with Low-Affinity Antibodies. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (5), E925–E933. 10.1073/pnas.1718283115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt E. M.; Kool E. T. Amplified MicroRNA Detection by Templated Chemistry. Nucleic Acids Res. 2012, 40 (9), e65–e65. 10.1093/nar/gkr1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducani C.; Bernardinelli G.; Högberg B. Rolling Circle Replication Requires Single-Stranded DNA Binding Protein to Avoid Termination and Production of Double-Stranded DNA. Nucleic Acids Res. 2014, 42 (16), 10596–10604. 10.1093/nar/gku737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino J. H.; Cook P.; Miller K. S. Accurate and Statistically Verified Quantification of Relative MRNA Abundances Using SYBR Green I and Real-Time RT-PCR. J. Immunol. Methods 2003, 283 (1–2), 291–306. 10.1016/S0022-1759(03)00103-0. [DOI] [PubMed] [Google Scholar]
- Sethi P.; Lukiw W. J. Micro-RNA Abundance and Stability in Human Brain: Specific Alterations in Alzheimer’s Disease Temporal Lobe Neocortex. Neurosci. Lett. 2009, 459 (2), 100–104. 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Denzler R.; Agarwal V.; Stefano J.; Bartel D. P.; Stoffel M. Assessing the CeRNA Hypothesis with Quantitative Measurements of MiRNA and Target Abundance. Mol. Cell 2014, 54, 766–776. 10.1016/j.molcel.2014.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne A.; Beaudin A. E.; Olsen H. E.; Jain M.; Cole C.; Palmer T.; DuBois R. M.; Forsberg E. C.; Akeson M.; Vollmers C. Nanopore Long-Read RNAseq Reveals Widespread Transcriptional Variation among the Surface Receptors of Individual B Cells. Nat. Commun. 2017, 8 (May), 16027. 10.1038/ncomms16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S. Native RNA-Sequencing Throws Its Hat into the Transcriptomics Ring. Trends Biochem. Sci. 2018, 43 (4), 225–227. 10.1016/j.tibs.2018.02.007. [DOI] [PubMed] [Google Scholar]
- Long G. L.; Winefordner J. D. Limit of Detection: A Closer Look at the IUPAC Definition. Anal. Chem. 1983, 55 (7), 712A–724A. 10.1021/ac00258a001. [DOI] [Google Scholar]
- Rang F. J.; Kloosterman W. P.; De Ridder J. From Squiggle to Basepair: Computational Approaches for Improving Nanopore Sequencing Read Accuracy. Genome Biol. 2018, 19 (1), 1–11. 10.1186/s13059-018-1462-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebai T.; Kamali-Moghaddam M.; Landegren U. Parallel Protein Detection by Solid-Phase Proximity Ligation Assay with Real-Time PCR or Sequencing. Curr. Protoc. Mol. Biol. 2015, 1–25. 10.1002/0471142727.mb2010s109. [DOI] [PubMed] [Google Scholar]
- Frei A. P.; Bava F.-A.; Zunder E. R.; Hsieh E. W. Y.; Chen S.-Y.; Nolan G. P.; Gherardini P. F. Highly Multiplexed Simultaneous Detection of RNAs and Proteins in Single Cells. Nat. Methods 2016, 13 (3), 269–275. 10.1038/nmeth.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi P.; Kim S. C.; Haliburton J. R.; Gartner Z. J.; Abate A. R. Abseq: Ultrahigh-Throughput Single Cell Protein Profiling with Droplet Microfluidic Barcoding. Sci. Rep. 2017, 7 (March), 44447. 10.1038/srep44447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.