

Abstract
Background
Current diagnostic tests for hereditary spherocytosis (HS) focus on the detection of hemolysis or indirectly assessing defects of membrane protein, whereas direct methods to detect protein defects are complicated and difficult to implement. In the present study, we investigated the patterns of genetic variation associated with HS among patients clinically diagnosed with HS.
Methods
Multi-gene targeted sequencing of 43 genes (17 RBC membrane protein-encoding genes, 20 RBC enzyme-encoding genes, and six additional genes for the differential diagnosis) was performed using the Illumina HiSeq platform.
Results
Among 59 patients with HS, 50 (84.7%) had one or more significant variants in a RBC membrane protein-encoding genes. A total of 54 significant variants including 46 novel mutations were detected in six RBC membrane protein-encoding genes, with the highest number of variants found in SPTB (n = 28), and followed by ANK1 (n = 19), SLC4A1 (n = 3), SPTA1 (n = 2), EPB41 (n = 1), and EPB42 (n = 1). Concurrent mutations of genes encoding RBC enzymes (ALDOB, GAPDH, and GSR) were detected in three patients. UGT1A1 mutations were present in 24 patients (40.7%). Positive rate of osmotic fragility test was 86.8% among patients harboring HS-related gene mutations.
Conclusions
This constitutes the first large-scaled genetic study of Korean patients with HS. We demonstrated that multi-gene target sequencing is sensitive and feasible that can be used as a powerful tool for diagnosing HS. Considering the discrepancies of clinical and molecular diagnoses of HS, our findings suggest that molecular genetic analysis is required for accurate diagnosis of HS.
Electronic supplementary material
The online version of this article (10.1186/s13023-019-1070-0) contains supplementary material, which is available to authorized users.
Keywords: Hereditary spherocytosis, RBC membrane disorder, Molecular diagnosis
Background
Hereditary spherocytosis (HS) is the most common cause of hereditary hemolytic anemia (HHA) characterized by the presence of spherocytes in peripheral blood smear (PBS) [1, 2]. HS occurs in 1 in 2000 Caucasians, with less common frequency in Asians [1, 3, 4]. The crude incidence of HS in Korea was reported as 1 in every 5000 births [5]. Approximately 75% cases of HS are inherited as autosomal dominant (AD) mutations, whereas the remaining cases involve autosomal recessive (AR) or de-novo mutations [1].
HS is caused by a deficiency in or dysfunction of membrane proteins, including spectrin, ankyrin 1, band 3, and protein 4.2, associated with the RBC cytoskeleton [3, 4, 6]. Defective membrane proteins disrupt the vertical linkage between the RBC membrane cytoskeleton and the phospholipid bilayer, causing RBCs to lose its biconcave characteristics and become spherical in shape [3, 4, 6]. This abnormal RBC morphology leads to osmotically fragile cells that are selectively trapped and destroyed in the spleen [3, 4, 6]. A major clinical manifestation of HS is hemolytic anemia, which exhibits a wide range of clinical manifestations from asymptomatic to life-threatening anemia requiring regular RBC transfusions [1, 2]. Other clinical symptoms include splenomegaly, jaundice, and gallstones, depending on disease severity [1, 2].
We have been operating the Korean Hereditary Hemolytic Anemia Working Party (KHHAWP) of the Korean Society of Hematology for 7 years since 2010, which name has been changed to RBC Disorder Working Party since November 2016. From 2007 to 2011, 195 patients (121 males and 74 females) diagnosed with HHA from 25 institutions were registered [7]. The KHHAWP presented standard operating procedure (SOP) for the diagnosis of HHA (Fig. 1) [5], which is similar to ICSH (International Council for Standardization in Haematology) guideline [8] except for excluding acid glycerol lysis time test as a screening test. Instead of gel electrophoresis analysis of erythrocyte membranes, the KHHAWP adopted mass spectrometry method as a confirmatory test, which is performed in one central laboratory in Korea.
Fig. 1.
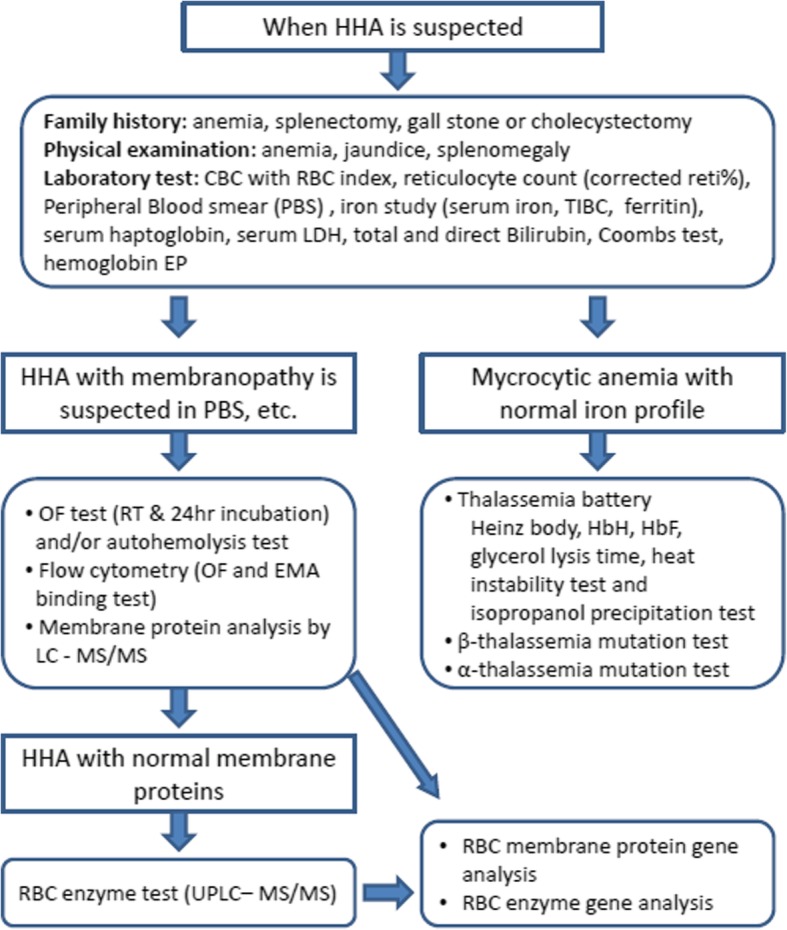
Standard operating procedure for the diagnosis of hereditary hemolytic anemia (HHA) by HHA Working Party of Korean Society of Hematology [5]
The diagnosis of HS is based upon a combination of positive family history, clinical features and presence of spherocytes in PBS, which are detectable in 97% of patients [9]. When the diagnosis of HS is equivocal, additional laboratory tests are recommended such as osmotic fragility test (OFT), autohemolysis test, flow cytometry [OFT and eosin-5-maleimide (EMA) binding test] for screening test, and protein analysis using gel electrophoresis or mass spectrometry can be additionally tested [10–16]. However, none of the current diagnostic test can detect all patients with HS.
Considering the limitations of existing diagnostic tests, development of a simple and direct method to measure RBC membrane protein abnormalities to confirm HS is required. Analysis of RBC membrane protein-encoding genes is expected that it can be used complementarily with the conventional confirmatory tests [1, 11]. Multi-gene target sequencing for RBC membrane protein-encoding genes is feasible and reliable diagnostic method to detect mutations in patients affected by various disorders of the RBC membrane. Particularly, gene testing is important in young children with congenital anemia, transfusion-dependent patients, and in families with variable clinical expression or complex inheritance patterns [17–19].
In the present study, we investigated the genetic variation of RBC membrane protein-encoding genes using multi-gene target sequencing, comparing with clinical features. A total of 43 genes was included; 17 RBC membrane protein-encoding genes and 20 RBC enzyme-encoding genes, in context with six additional candidate genes for the purpose of differential diagnoses [thalassemia, congenital dyserythropoietic anemia (CDA), paroxysmal nocturnal hemoglobinuria (PNH), and Gilbert syndrome].
Methods
Patients
A total of 59 patients with HS including 31 males and 28 females with a median age of 7 years (range: 1–81 years), were registered between July 2013 and July 2014 from the pediatrics and internal medicine departments of 25 institutions in Korea. HS was diagnosed according to the SOP recommended by the KHHAWP of the Korean Society of Hematology (Fig. 1) [5].
Along with clinical data including age, sex, symptoms and family history, we collected the results of laboratory tests including CBC with RBC index, reticulocyte count, total and direct bilirubin concentration, lactate dehydrogenase (LDH), iron, total iron-binding capacity (TIBC), ferritin, PBS, and OFT by reviewing medical records (Table 1). Blood samples were collected from each patient after obtaining their written consent.
Table 1.
Clinical characteristics of patients with HS in Korea
| Characteristics | Total patients (n = 59) |
Patients with gene mutation (n = 50) |
Patients without gene mutation (n = 9) |
P value between group with mutation vs. without mutation |
|---|---|---|---|---|
| Sex, n (%) | 0.597 | |||
| Male | 31 (52.0) | 27 (54.0) | 4 (44.4) | |
| Female | 28 (48.0) | 23 (46.0) | 5 (55.6) | |
| Age (years) | 0.566 | |||
| Median | 7 | 7 | 8 | |
| Range | 1–81 | 1–81 | 2–17 | |
| Family history of HS, n (%) | 0.139 | |||
| Positive | 20 (33.9) | 16 (32.0) | 4 (44.4) | |
| Negative | 39 (66.1) | 34 (68.0) | 5 (55.6) | |
| Clinical symptoms, n (%) | ||||
| Splenomegaly | 38/59 (64.4) | 31/50 (62.0) | 7/9 (77.8) | 0.363 |
| Neontal jaundice | 28/54 (51.9) | 24/45 (53.3) | 4/9 (44.4) | 0.724 |
| Hepatomegaly | 9/53 (17.0) | 9/44 (20.5) | 1/9 (11.1) | 1.000 |
| Splenectomy | 13/58 (22.4) | 10/49 (20.4) | 3/9 (40.0) | 0.398 |
| Aplastic crisis | 14/56 (25.0) | 11/47 (23.4) | 3/9 (30.0) | 0.676 |
| Gallstones | 10/57 (17.5) | 9/48 (18.8) | 1/9 (33.3) | 1.000 |
| Hematologic parameters, mean | ||||
| Hemoglobin (g/dL) (range) | 8.4 (3.6–13.6) | 8.4 (3.6–13.6) | 8.3 (5.8–12.1) | 0.476 |
| MCV (fL) (range) | 80.9 (62.3–107.0) | 80.6 (62.3–107.0) | 85.3 (70.4–107.0) | 0.209 |
| MCHC (g/dL) (range) | 35.3 (30.8–38.2) | 35.2 (30.8–38.2) | 35.2 (31.5–37.9) | 0.279 |
| Markers of hemolysis, mean | ||||
| Reticulocyte count (%) (range) | 7.5 (0.5–24.8) | 7.4 (0.5–24.8) | 7.2 (3.4–13.3) | 0.461 |
| Total bilirubin (mg/dL) (range) | 4.1 (0.8–19.1) | 4.0 (0.8–19.1) | 4.3 (1.1–6.4) | 0.320 |
| Direct bilirubin (mg/dL) (range) | 0.7 (0.2–1.3) | 0.7 (0.3–1.3) | 0.6 (0.4–0.8) | 0.640 |
| LDH (IU/L) (range) | 508 (187–1557) | 522 (187–1557) | 448 (198–737) | 0.843 |
| Iron status parameters, mean | ||||
| Iron (μr/dL) (range) | 101 (26–245) | 98 (26–159) | 111 (51–245) | 0.198 |
| TIBC (μT/dL) (range) | 266 (108–486) | 269 (108–486) | 241 (195–274) | 0.769 |
| Ferritin (ng/mL) (range) | 342 (32–4671) | 360 (32–4671) | 339 (74–278) | 0.657 |
| Grading of peripheral spherocytes, n (%) | 0.622 | |||
| 0 | 5 (8.5) | 4 (8.0) | 1 (11.1) | |
| 1+ or slight (2–5%), | 18 (30.5) | 15 (30.0) | 3 (33.3) | |
| 2+ or moderate (6–15%), | 20 (33.9) | 16 (32.0) | 4 (44.4) | |
| 3+ or marked (> 16%) | 16 (27.1) | 15 (30.0) | 1 (11.1) | |
| Sex, n (%) | 0.597 | |||
| Male | 31 (52.0) | 27 (54.0) | 4 (44.4) | |
| Female | 28 (48.0) | 23 (46.0) | 5 (55.6) | |
| Severity, n (%) | 0.678 | |||
| Mild | 6 (10.2) | 5 (10.0) | 1 (11.1) | |
| Moderate | 27 (45.8) | 24 (48.0) | 3 (33.3) | |
| Severe | 26 (44.1) | 21 (42.0) | 5 (55.6) | |
| Osmotic fragility tests, n (%) | 0.614 | |||
| Positive | 41 (69.5) | 33 (66.0) | 8 (88.9) | |
| Negative | 6 (10.2) | 5 (10.0) | 1 (11.1) | |
| NA | 12 (20.3) | 12 (24.0) | 0 | |
Abbreviation: HS hereditary spherocytosis, NA not assessable
Targeted sequencing
To gain insight into the genetic variations, we performed targeted sequencing for 43 gene panel (Additional file 1: Table S1). gDNA shearing to generate the standard library and the hybridization step targeting only exonic regions were performed by Celemics Inc. (Seoul, Korea). The final quality was assessed using the Agilent 2200 TapeStation System (Santa Clara, CA, USA). We sequenced a total target length of 259-kb regions using the paired-end 150-bp rapid-run sequencing mode on an Illumina HiSeq 2500 platform. The mean sequencing depth for the targeted regions (259-kb) was 231-fold (n = 59). Because a matched control sample was not included in this study, we applied a stringent variant selection pipeline to prioritize the high-confidence set of somatic mutations.
Variant calling
The filtration process was performed as follows. Variants within non-exonic regions were removed. Variants that do not have enough depth were also filtered out to remove false positives. Common variants on 1000 genome projects with more than 5% of allele frequency were filtered out. CADD score shows predictive pathogenicity of variants. It considers diverse annotations from allelic diversity to functionality, in order to estimate pathogenic variants. In this study, CADD scores below 10 were cut-off for filtration. After these filters, in-house variants were also removed to make filtered variant lists. Validation of variant call was performed by target gene sequencing of involved genes.
Simulation of the effect of mutated genes on protein structure
To predict how gene mutation affect protein structure, we visualized three-dimensional (3-D) spatial protein structure following acquisition of their structural information (http://www.proteinmodelportal.org) (Additional file 1: Table S2). We used PyMOL (http://www.pymol.org) to visualize 3-D representations of the protein, modified protein structures based on genetic mutation profiles from next-generation sequencing (NGS) results.
Statistical analyses
Stata/SE (v.14; StataCorp, College Station, TX, USA) was used for data analyses. Statistical differences in terms of continuous clinical characteristic variables were estimated by two sample t test. The significance of differences in categorical variables between groups was determined by the Pearson χ2 test or Fisher’s exact test. The level of significance was set at P < 0.05.
Results
Clinical characteristics
Among 59 patients with HS, 20 (33.9%) had a family history of HS, whereas symptoms of splenomegaly, neonatal jaundice, and hepatomegaly were exhibited in 38 of 59 (64.4%), 28 of 54 (51.9%), and 10 of 59 (16.7%) patients, respectively. Mean values for laboratory tests were as follows: hemoglobin concentration 8.4 g/dL (3.6–13.6 g/dL); corpuscular volume 80.9 fL (62.3–107.0 fL); corpuscular hemoglobin concentration 35.3 g/dL (30.8–38.2 g/dL); reticulocyte count indicating hemolysis 7.5% (0.5–24.8%); total bilirubin/direct bilirubin 4.1/0.7 mg/dL (0.8–19.1/0.2–1.3 mg/dL); LDH 508 IU/L (187–1557 IU/L); parameters representing iron profile, including iron 101 μg/dL (26–245 μg/dL), TIBC 266 μg/dL (108–486 μg/dL); and ferritin concentration, 342 ng/mL (32–4671 ng/mL). PBS was rated for spherocytes on a four-point scale [20] from 0, 1+ or slight (2–5%), 2+ or moderate (6–15%), and 3+ or marked (> 16%) and the number of smears returning 0, 1+ or slight, 2+ or moderate and 3+ or marked were 5 (8.5%), 18 (30.5%), 20 (33.9%), and 16 (27.1%) patients, respectively. According to HS-severity criteria [11], severe, moderate, and mild cases were 26 (44.1%), 27 (45.8%), and 6 (10.2%) patients, respectively (Table 1).
Variants profile of RBC membrane protein-encoding genes
Among 17 RBC membrane protein-encoding genes examined, significant disease-related mutations were observed in six: SPTB (spectrin, beta), ANK1 (ankyrin 1), SLC4A1 (solute carrier family 4, member 1), SPTA1 (spectrin, alpha 1), EPB41 (erythrocyte membrane protein band 4.1), and EPB42 (erythrocyte membrane protein band 4.2) (Fig. 2). A total of 54 significant mutations were observed, of which eight were previously reported as pathogenic in patients with HS and 46 variants were novel mutations (Additional file 1: Table S3). The highest number of mutations were found in SPTB (n = 28), and followed by ANK1 (n = 19), SLC4A1 (n = 3), SPTA1 (n = 2), EPB41 (n = 1), and EPB42 (n = 1). According to the American College of Medical Genetics and Genomics guidelines [21], 12 were pathogenic mutations (including eight previously reported variants), 29 were likely pathogenic mutations, and 13 were classified as having uncertain significance. All the variants have been confirmed by Sanger sequencing using 35 primer sets (Additional file 1: Table S4).
Fig. 2.

Characteristics of significant variants for RBC membrane protein-encoding genes; SPTB, ANK1, SLC4A1, SPTA1, EPB41, EPB42. Abbreviations: SPTB, spectrin, beta; ANK1, ankyrin 1; SLC4A1, solute carrier family 4, member 1; SPTA1, spectrin, alpha 1; EPB41, erythrocyte membrane protein band 4.1; EPB42, erythrocyte membrane protein band 4.2
Variant characteristics in patients with HS
Among 59 patients with HS, 50 (84.7%) had at least one mutation in a RBC membrane protein-encoding gene (Fig. 3). Twenty eight patients carried mutations in the SPTB gene, and 20 patients had mutations in the ANK1 gene. Forty patients (67.8%) carried a single mutation, and 10 patients (16.9%) carried two mutations. Among 40 patients with a single mutation, the most frequently mutated genes were SPTB and ANK1, which were mutated in 21 and 17 patients, respectively. The SCL4A1 mutation was found in two patients. Among the 10 patients harboring two mutations, one carried two mutations in a single gene (ANK1), and three patients carried mutations in both SPTB and SPTA1. Combinations of mutations in SPTB and ANK1, SPTB and EPB41, and SPTB and EPB42 were detected in one patient each. In addition, combination with RBC enzyme-encoding gene mutations were found in three patients [SLC4A1 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), ANK1 and GSR (glutathione reductase), SPTB and ALDOB (aldolase B)] (Additional file 1: Table S5).
Fig. 3.

Number of patients with RBC membrane protein-encoding gene mutations. Abbreviations: SPTB, spectrin, beta; SPTA1, spectrin, alpha 1; EPB41, erythrocyte membrane protein band 4.1; EPB42, erythrocyte membrane protein band 4.2; ALDOB, aldolase B; ANK1, ankyrin 1; GSR, glutathione reductase; SLC4A1, solute carrier family 4, member 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase
Nine patients carried no mutation on the RBC membrane protein- or enzyme-encoding genes. Coexisting mutations of UGT1A1 (UDP glycosyltransferase 1 family, polypeptide A1) gene were detected in 24 of 59 HS patients (40.7%), with UGT1A1 mutations combined with other gene mutations in 20 patients and without other gene mutation in four patients (Table 2, Additional file 1: Table S6). Total bilirubin level or presence of neonatal jaundice did not differ significantly from those without UGT1A1 mutations.
Table 2.
Gene mutations, laboratory tests and clinical characteristics
| Patient ID | Membrane gene mutation | Other mutation | OFT | PB spherocytes | Splenectomy | Family history of HS | Severity of HS | Additional tests with positive results |
|---|---|---|---|---|---|---|---|---|
| 1 | SPTB, EPB41 | UGT1A1 | NA | ♦ | ▲▲▲ | SDS-PAGE (Spectrin) | ||
| 2 | ANK1 | + | ♦♦♦ | (HA, father) | ▲▲ | |||
| 3 | SPTB | NA | ♦♦ | AD | ▲▲▲ | Flow cytometrya | ||
| 4 | SPTB | UGT1A1 | + | ♦♦ | ▲▲ | |||
| 5 | + | ♦♦ | ● | AD | ▲▲▲ | SDS-PAGE (Spectrin) | ||
| 6 | ANK1 | – | ♦♦ | ● | ▲▲▲ | SDS-PAGE (Spectrin) | ||
| 7 | SPTB | + | ♦ | ▲▲▲ | ||||
| 8 | SPTB, SPTA1 | + | ♦ | AD | ▲▲▲ | |||
| 9 | SPTB, SPTA1 | NA | ♦ | AD | ▲▲▲ | Flow cytometrya | ||
| 10 | + | ♦♦ | ● | (HA, mother) | ▲▲▲ | |||
| 11c | SPTB | UGT1A1 | NA | ♦♦♦ | ▲▲▲ | |||
| 12 | ANK1 | + | ♦♦ | ● | ▲▲▲ | |||
| 13 | SPTB | UGT1A1 | + | ♦ | ▲▲ | |||
| 14 | + | ♦ | ● | AD | ▲▲▲ | |||
| 15 | ANK1 b | NA | ♦♦ | AD | ▲▲ | SDS-PAGE (Spectrin) | ||
| 16 | ANK1 b | UGT1A1 | NA | ♦♦♦ | AD | ▲▲▲ | ||
| 17 | UGT1A1 | + | ♦♦♦ | ▲▲ | ||||
| 18 | ANK1 | NA | ♦♦♦ | AD | ▲▲▲ | |||
| 19 | ANK1 | UGT1A1, UGT1A1 | + | ♦♦ | ▲▲ | |||
| 20 | SPTB, SPTA1 | UGT1A1 | – | ♦♦ | AD | ▲▲▲ | ||
| 21c | SLC4A1 b | UGT1A1 | NA | – | (HA, sibling) | ▲▲ | ||
| 22 | UGT1A1 | + | ♦ | AD | ▲▲▲ | |||
| 23 | SPTB | + | ♦♦♦ | AD | ▲▲▲ | |||
| 24 | UGT1A1 | + | ♦♦ | (HA, mother) | ▲▲▲ | |||
| 25c | ANK1 b | NA | ♦♦♦ | ▲▲ | ||||
| 26 | ANK1 | + | ♦♦♦ | ● | AD | ▲▲ | ||
| 27 | ANK1 | + | ♦ | ▲▲▲ | ||||
| 28 | SPTB | + | ♦♦ | ● | AD | ▲▲ | ||
| 29 | ANK1 b | GSR | + | ♦♦ | ▲▲ | |||
| 30 | SPTB | ALDOB | + | ♦♦♦ | ▲▲ | |||
| 31c | SPTB | – | NA | ♦♦♦ | ▲▲▲ | |||
| 32 | SLC4A1 b | UGT1A1, UGT1A1 | + | ♦♦♦ | ▲▲ | |||
| 33 | SPTB | UGT1A1 | + | ♦♦ | ● | ▲▲▲ | ||
| 34 | SPTB | – | – | ♦ | AD | ▲▲ | ||
| 35 | SPTB b , EPB42 | UGT1A1 | + | – | ▲▲ | Autohemolysis | ||
| 36 | SPTB | + | ♦♦ | ● | ▲▲ | |||
| 37 | ANK1 | + | ♦♦ | ● | ▲▲ | |||
| 38 | UGT1A1 | + | ♦ | ▲ | SDS-PAGE (Spectrin) | |||
| 39c | ANK1 | UGT1A1 | – | ♦ | ▲ | |||
| 40 | SPTB | + | ♦♦ | ● | ▲▲▲ | |||
| 41 | ANK1 | + | ♦♦♦ | ▲ | ||||
| 42 | ANK1 | + | ♦♦ | ▲▲▲ | ||||
| 43c | ANK1, ANK1 | UGT1A1 | NA | ♦ | ▲▲ | |||
| 44 | SPTB,ANK1 | UGT1A1 | + | ♦ | ▲▲ | |||
| 45 | ANK1 | UGT1A1 | + | ♦♦♦ | ▲▲ | |||
| 46 | SPTB | UGT1A1 | + | ♦♦♦ | ▲▲ | |||
| 47 | SPTB | + | ♦ | (HA, sibling) | ▲▲▲ | |||
| 48c | SPTB b | NA | ♦ | ▲▲▲ | ||||
| 49c | SPTB | UGT1A1 | + | – | ▲▲ | |||
| 50 | ANK1 | + | – | AD | ▲▲ | |||
| 51 | SPTB | UGT1A1 | + | ♦ | AD | ▲▲ | ||
| 52 | + | ♦♦ | ▲▲ | |||||
| 53 | ANK1 | + | ♦ | ▲ | ||||
| 54 | – | – | ● | AD | ▲▲▲ | |||
| 55 | SPTB | – | ♦♦♦ | AD | ▲ | |||
| 56 | SLC4A1 | UGT1A1, GAPDH | + | ♦ | ▲ | |||
| 57 | SPTB | + | ♦♦♦ | AD | ▲▲▲ | |||
| 58 | SPTB | UGT1A1 | + | ♦♦ | AD | ▲▲ | ||
| 59 | SPTB | + | ♦ | ▲▲▲ |
aFlow cytometry (OFT and EMA binding test), bPreviously reported variants (see Additional file 1: Table S3), cEight patients who did not meet the diagnostic criteria of HS without genetic testing
PB spherocytes [20] ♦, 1+; ♦♦, 2+; ♦♦♦, 3+, Severity of HS [8] ▲, mild; ▲▲, moderate; ▲▲▲, severe
Abbreviations: AD autosomal dominant, ALDOB aldolase B, ANK1 ankyrin 1, EPB41 erythrocyte membrane protein band 4.1, EPB42 erythrocyte membrane protein band 4.2, GAPDH glyceraldehyde-3-phosphate dehydrogenase, GSR glutathione reductase, HA hemolytic anemia, SLC4A1 solute carrier family 4, member 1, SPTA1 spectrin, alpha 1, SPTB spectrin, beta, UGT1A1, UDP glycosyltransferase 1 family, polypeptide A1, OFT osmotic fragility test, NA not assessable
Genotype and phenotype correlations in patients with HS
Comparisons of laboratory findings and clinical characteristics showed no significant differences in hematologic parameters, hemolysis markers, iron status parameters, sex, family history of HS, number of splenectomized patients, and disease severity according to the gene mutation type and number of mutation or presence of UGT1A1 mutation (Table 1, Additional file 1: Table S6).
Among 59 patients with HS, nine patients (15.3%) without mutation associated with RBC membrane protein-encoding genes showed similar baseline characteristics in most aspects as compared with those with mutations (Table 1). Median age of patients without mutation was 8 years, and the proportion of family history, clinical symptoms, grading of peripheral spherocytes, and OFT results did not differ significantly from those with mutation.
Intercorrelations between gene mutations and laboratory findings: OFT, the presence of spherocytes in PBS, and gene mutations
The results of genetic test were matched with routine diagnostic tests for HS including OFT and the presence of spherocytes in PBS (Table 3, Fig. 4). Among 59 patients with clinical HS, results of NaCl induced OFT (room temperature and/or 24 h incubated) was available in 47 patients and 41 of them (87.2%) showed positive results (Additional file 1: Figure S2). Thirty three of 47 patients (70.2%) showed positivity in both OFT and gene test, while one patients (2.1%) showed negative results in both OFT and gene test. In six out of 47 patients (12.7%) with negative OFT, five carried mutations in RBC membrane protein-encoding genes. Among 38 patients harboring HS-related gene mutations, 33 showed positive OFT (86.8%).
Table 3.
Comparison of OFT, PBS and gene test results in patients with HS
| RBC membrane protein-encoding genes | |||
|---|---|---|---|
| No. of patients with mutation (%) | No. of patients without mutation (%) | ||
|
OFT (n = 47) |
Positive | 33 (70.2) | 8 (17.0) |
| Negative | 5 (10.6) | 1 (2.1) | |
|
PBS (n = 59) |
Positive | 46 (78.0) | 8 (13.6) |
| Negative | 4 (6.8) | 1 (1.7) | |
Abbreviation: OFT osmotic fragility test, PBS peripheral blood cell smear
Fig. 4.

A diagram showing the number of patients with positive results of gene mutation, osmotic fragility test, and peripheral blood (PB) spherocytes in 58 of 59 patients with HS. One of 59 patients who had anemia and family history of HS showed negative result on all three tests
Spherocytes in PBS were present in 54 of 59 patients (91.5%). Among five patients without spherocytes in PBS, four carried mutations in RBC membrane protein-encoding genes (Additional file 1: Table S7). One of 59 patients who had anemia and family history of HS showed negative results on all three tests.
Discussion
Using multi-gene target sequencing, 50 of 59 patients (84.7%) of clinically diagnosed HS proved to be molecular HS and three patients harbored coexisting gene mutations of RBC enzymes (ALDOB, GAPDH, and GSR) in this study. Mutations of six kinds of RBC membrane protein-encoding genes (total 54 variants) were detected in order of SPTB, ANK1, SLC4A1, SPTA1, EPB41, and EPB42.
To find whether there is an ethnic difference in HS related variants, we reviewed the literatures on the reports of HS related mutations in comparison with the results of the present study, although the methods are different among reported mutations of HS. Table 4 shows summary of comparison among previous reports by NGS [22–24]. With regards to the frequency of mutated gene, the SPTA1 mutation was the most common followed by the SPTB mutation in the reports from the United States [22, 23]. Meanwhile, a study in Netherland revealed that the ANK1 mutation was the most common mutation followed by the SPTA1 mutation [24]. In the present study, SPTB mutations was the most common mutation, followed by ANK1 mutations. Particularly noteworthy, SPTA1 mutations was rarely detected, compared to that of the United States. Briefly, mutation frequency by NGS study in Korean was different from those of Caucasian. Korean patients with HS showed higher frequency of ANK1 mutation. Consistent with our study, another study in Korea reported that 25 patients with HS carried one heterozygous mutation of ANK1 (n = 13) or SPTB (n = 12) but none carried mutations in SPTA1, SLC4A1, or EPB42 by Sanger sequencing [25]. Previous molecular testing demonstrated that mutations in the ANK1, SPTB, SLC4A1, SPTA1, and EPB42 genes account for 60, 10, 15, 10, and 5% cases of HS, respectively, in the United States and Europe [26, 27].
Table 4.
NGS results of RBC membrane protein-encoding genes in patients with HS
| RBC membrane-encoding gene | USA 1 [22] | USA 2 [23] | Netherlands [24] | Korea (this study) |
|---|---|---|---|---|
| No. of patients with mutation (%) | 10/20a (50.0) | 16 /19b (84.2) | 52 /66 (78.9) | 50/59 (84.7) |
| No. of total mutations | 13 | 21 | 73 | 57 |
| No. of different variants | 11 | 15 | 53 | 54 |
| ANK1 | 1 | 3 | 14 | 19 |
| SPTA | 6 | 5 | 25 | 2 |
| SPTB | 4 | 4 | 8 | 28 |
| SCL4A1 | 0 | 3 | 4 | 3 |
| EBP41 | NA | 0 | 1 | 1 |
| EBP42 | NA | 0 | 1 | 1 |
aincluding 2 patients suspected having hereditary elliptocytosis
bincluding patients with diagnosed as HHA
Abbreviation; NA not assessable
Ethnic differences in RBC membrane protein defects were also reported in previous studies according to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analyses (Table 5) [9, 16, 28–32]. A Korean study in 2000 [28] reported that protein 4.2 defects were detected at a higher frequency than those of band 3 in the United States and Europe. That study also reported that most defects were found in ankyrin 1 according to SDS-PAGE analysis, whereas most mutations were detected in the SPTB followed by ANK1, according to our NGS results. Additionally, protein defects were not observed was nine out of 27 patients (33.3%) [28]. Meanwhile, single defects in band 3 and spectrin constitute the primary variants reported in Italy [9, 16], and a combined defect in spectrin/ankyrin is frequently detected in patients in the United States and Spain [6, 29, 30]. Regarding to the incidence of HS, an incidence of Japan is highest among Asian countries, and the defect in the 4.2 protein in Japan is more frequent as compared to the United States and Europe [31, 32]. Those different profiles of HS among countries might be due to complexity associated with SDS-PAGE methods and lack of objectiveness in the interpretation of the results. The interpretation of SDS-PAGE is based on the comparison with normal healthy control. For that reason, the standardization is not possible and the comparison of SDS-PAGE results cannot give a meaningful conclusion. By contrast, nucleotide sequence analysis gives us straightforward results, and the interpretation of results is objective.
Table 5.
Literature review on SDS-PAGE results of RBC membrane protein abnormalities in patients with HS (%)
| RBC membrane protein | Italy2[16] (n = 87) |
Italy1[9] (n = 300) |
USA2[6] (n = 55) |
USA1[29] (n = 166) |
Spain[30] (n = 62) |
Japan2*[31] (n = 60) |
Japan1[32] (n = 47) |
Korea[28] (n = 27) |
|---|---|---|---|---|---|---|---|---|
| Band 3 | 23 (26) | 158 (53) | 10 (18) | 38 (23) | 0 | (20) | 15 (32) | 3 (11) |
| Spectrin only | 36 (41) | 98 (33) | 7 (13) | 0 | 19 (31) | 0 | 8 (15) | 2 (7) |
| Ankyrin only | 0 | 13 (4)† | 0 | 0 | 4(6) | (7) | 1 (2) | 8 (30) |
| Spectrin/ankyrin | 16 (18) | 6 (11) | 100 (60) | 34 (55) | 0 | 1 (2) | 1 (4) | |
| Other combination | – | – | – | – | – | – | 15 (34) | – |
| 4.2 protein | 6 (7) | 2 (1) | 0 | 3 (2) | 0 | (45) | 3 (6) | 4 (15) |
| Undetected | 6 (7) | 29 (10) | 32 (58) | 25 (15) | 5 (8) | (28) | 4 (9) | 9 (33) |
*Only % without the number of the patients was presented in this study
†Including both Ankyrin only and Spectrin/ankyrin
Inherited pattern of HS differs depending on the gene. In most HS patients, inheritance is AD and each of HS patients has a unique mutation [11]. However, SPTA1 or EPB42 mutation is inherited with AR pattern. Rarely, double dominant HS due to defects in SLC4A1 or SPTB are reported [33], which results in fetal death or severe transfusion-dependent hemolytic anemia presenting in the neonatal period. SPTB and SPTA1 mutations can be AD or de novo, whereas ANK1mutation can be AD, AR, or de novo. SLC4A1 mutation is AD and EPB42 is AR. Inherited pattern is not clearly revealed in EPB41. Of note, all the significant variants in RBC membrane protein-encoding genes are heterozygous. Hence, mutations of genes inherited in AR pattern such as EPB41 and EPB42 gene possibly cannot be a direct cause of HS, requiring additional mutation to cause hemolytic phenotype. In the present study, two patients harboring EPB41 and EPB42 mutations also carried another mutation in the SPTB gene (EPB41 and SPTB, EPB42 and SPTB in each patient).
Interestingly, concurrent mutations of genes encoding RBC enzymes (ALDOB, GAPDH, and GSR) were detected along with heterozygous mutations of RBC membrane protein-encoding genes in three patients. Further analysis of enzyme activities in these patients is necessary for validation. Of the 59 patients with HS examined in this study, 24 (40.7%) had significant UGT1A1 variants. It was reported that a polymorphism of UGT1A1 gene promoter homozygous insertion of TA pairs (genotype UGT1A1*28/*28) might results in a decrease in bilirubin glucuronidation activity, leading to hyperbilirubinemia and late complication of patients with HS, such as development gallstones [34, 35]. In contrast, there are debates on the late impact of genotype of UGT1A1 [36]. However, a polymorphism of UGT1A1 gene promoter was not included in this study. Based on the results of the present study showing high frequency of UGT1A1 variant with low enzymatic activity, we infer that genotyping of UGT1A1 polymorphism might help to predict the development of gallstones in HS.
The laboratory diagnosis of HS routinely relies on the presence of spherocytes in PBS, OFT, and more recently EMA binding test [10, 11, 37, 38]. Yet, there is no single test that can confirm HS. We have matched the results of genetic test with those of routine diagnostic tests (Table 3). Among 50 patients harboring mutations of encoding RBC membrane protein, 86.8% showed positive OFT, while 70.2% of clinical HS showed positive OFT. On the contrary, eight patients (17.0%) with positive OFT result revealed no mutation of membrane genes, and five (10.6%) with negative OFT proved to harbor membrane gene mutation. Regarding to spherocytes, four of 50 patients (8%) harboring membrane gene mutation did not show spherocytes in PBS. We retrospectively reviewed PBS to determine the presence of spherocytes in those four patients who did not show spherocytes in PBS but with RBC membrane protein-encoding gene mutations. However, we could not detect additional spherocytes. Conclusively, OFT and spherocytes in PBS can be used in conjunction with genetic test for the -diagnosis of HS, giving higher sensitivity and specificity.
With regards to the genotype-phenotype relationship, we could not find any correlation between the genetic test results and clinical characteristics including disease severity, mean hemoglobin concentrations, splenomegaly, gallstones, aplastic crisis and bilirubin levels according to mutations of four genes (SPTB, ANK1, SPTA1, and SLC4A1), except EPB41 and EPB42, which were found in only one patient each, However, one study reported that anemia was most severe in HS patients with mutations on the ANK1 spectrin-binding domain and splenectomy was more frequently performed in patients with ANK1 mutations than in those with SPTB mutations [25]. In addition, the other reported that hemoglobin concentration was slightly lower in patients with spectrin deficiency than with band 3 deficiency [39].
Other NGS study on RBC membrane diseases reported similar results (86.3%, 44 of 51 patients) [24]. This finding suggested a close correlation between clinical diagnosis and gene mutations. In the present study, molecular test could detect additional HS which could be missed without molecular test (Fig. 4). Furthermore, molecular test would be an effective method for neonates or transfused individuals, since the result of OFT and spherocytes in PBS can be unreliable, especially when the patients are transfused [11]. Collectively, our results suggest that mutation analyses will complement with other conventional tests for accurate diagnosis of HS. We consider the molecular test needs to be integrated to the diagnostic criteria of HS.
The limitation of this study is that we did not perform the analysis on RBC membrane protein as a validation. Instead, we simulated 3-D spatial structure of protein encoding mutated genes, predicting the effects of gene mutations in silico. Although exact changes in protein structure cannot be predicted based on 3-D spatial structure, large-scale modification of the protein due to frame shift or nonsense mutations can be visualized and subsequent functional changes can be expected from structure analysis. Further family study or functional studies using knockout mice needs to be conducted to validate the significance of variants. Another limitation is that we could not match the results of EMA binding test with genetic results, since our study was done retrospectively. Nine patients who did not harbor gene mutation of RBC membrane protein (Additional file 1: Table S8), satisfied the diagnostic criteria of HS suggested in the guideline [11]. Though they satisfied those criteria, there are two possibilities that they have other forms of hemolytic anemia or other membrane gene mutations that is not included in our multi-gene panel (e.g. channel defects such as KCNN4 as found in hereditary stomatocytosis) [40].
When we target the most frequent mutations only, composition of gene panel with genes over 10% frequency (SPTB and ANK1) will cover 94% (47 of 50 patients) of the diagnosis of HS. This could provide a cheaper and more convenient method than current strategies for diagnosis of HS. Regarding to the diagnostic guidelines suggested by international working parties, we suggest that genetic test should be conducted at least in patients without clues of laboratory tests in spite of clinically suspected HS.
Conclusions
This constitutes the first large-scaled genetic study of Korean patients with HS. We detected 54 significant HS-related mutations, including 46 novel mutations in RBC membrane protein-encoding genes. We demonstrated that multi-gene target sequencing is sensitive and feasible that can be used as a powerful tool for diagnosing HS. Considering the discrepancies between clinical and molecular diagnoses, use of molecular genetics analysis provides an effective method for improving the accuracy of HS diagnosis.
Additional file
Figure S1. Significant variants diagrams for UGT1A1 gene. Figure S2. Results of NaCl induced OFT. Table S1. Multi-gene panel for targeted sequencing. Table S2. List of protein simulation templates. Table S3. List of significant variants detected in RBC membrane protein-encoding genes. Table S4. Primer sets for all significant variants in RBC membrane protein-encoding genes. Table S5. List of significant variants detected in RBC enzyme-encoding genes among patients with HS. Table S6. List of UGT1A1 gene variants in patients with HS in Korea. Table S7. Clinical characteristics of patients with HS without peripheral blood spherocytes. Table S8. Patients without RBC membrane-encoding gene mutation. (DOCX 114 kb)
Acknowledgments
The authors thank the participating patients and their families. We also thank Dr. YM Park and the Division of Statistics at the Medical Research Collaborating Center, Seoul National University Bundang Hospital for assistance with statistical analysis.
Funding
Support was provided by: the National Research Foundation of Korea (NRF) grant funded by the Korea government(MSIT) (NRF-2017R1A2A1A17069780) http://www.nrf.re.kr/.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AD
Autosomal dominant
- ALDOB
Aldolase B
- ANK1
Ankyrin 1
- AR
Autosomal recessive
- CDA
Congenital dyserythropoietic anemia
- EMA
Eosin-5-maleimide
- EPB42
Erythrocyte membrane protein band 4.2
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- GSR
Glutathione reductase
- HHA
Hereditary hemolytic anemia
- HS
Hereditary spherocytosis
- ICSH
International Council for Standardization in Haematology
- IRB
Institutional Review Board
- KHHAWP
The Korean Hereditary Hemolytic Anemia Working Party
- LDH
Lactate dehydrogenase
- NA
Not assessable
- NGS
Next-generation sequencing
- OFT
Osmotic fragility test
- PBS
Peripheral blood smear
- PNH
Paroxysmal nocturnal hemoglobinuria
- SLC4A1
Solute carrier family 4, member 1
- SNP
Single nucleotide polymorphism
- SOP
Standard operating procedure
- SPTA1
Spectrin, alpha 1
- SPTB
Spectrin, beta
- TIBC
Total iron-binding capacity
Authors’ contributions
HLJ, JHK and DSL designed the study. HSC, QC, HYS, HJK, HK, SJK, IK, JAK, HK, KDP, KBP, MP, SKP, ESP, JAP, JEP, JKP, HJB, JHS, YJS, HSA, KHY, HSY, KSL, KCL, MJL, SAL, JML, JHL, JAL, JWL, YWW, YTL, HWC, EJC, HLJ and DSL collected study samples and data. QC, JAK, KOI, SNP, YP, JHK, and DSL processed blood samples, performed mutation analysis and analyzed the study data. CHS, QC, and DSL wrote the manuscript. HLJ, JHK and DSL provided final review of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board (IRB) of each participating institution (Seoul National University Hospital IRB No. 1308–006-507).
Consent for publication
As details on individuals reported within the manuscript are entirely unidentifiable, consent for publication in OJRD was not requested from parents.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hyoung Soo Choi, Email: choihs1786@gmail.com.
Qute Choi, Email: moname@hanmail.net.
Jung-Ah Kim, Email: jungah0928@gmail.com.
Kyong Ok Im, Email: greenteaok@hanmail.net.
Si Nae Park, Email: brook0910@gmail.com.
Yoomi Park, Email: yoomip89@gmail.com.
Hee Young Shin, Email: hyshin@snu.ac.kr.
Hyoung Jin Kang, Email: kanghj@snu.ac.kr.
Hoon Kook, Email: hoonkook@chonnam.ac.kr.
Seon Young Kim, Email: ksuny55@gmail.com.
Soo-Jeong Kim, Email: alvin97@yuhs.ac.kr.
Inho Kim, Email: kim_dajung@hanmail.net.
Ji Yoon Kim, Email: phojyk@gmail.com.
Hawk Kim, Email: kimhawkmd@gmail.com.
Kyung Duk Park, Email: kd4kid@snu.ac.kr.
Kyung Bae Park, Email: kbpark@schmc.ac.kr.
Meerim Park, Email: ming2a@hanmail.net.
Sang Kyu Park, Email: sang@uuh.ulsan.kr.
Eun Sil Park, Email: espark@gnu.ac.kr.
Jeong-A Park, Email: jeonga95@hanmail.net.
Jun Eun Park, Email: pedpje@ajou.ac.kr.
Ji Kyoung Park, Email: pjk4285@hanmail.net.
Hee Jo Baek, Email: swan93@naver.com.
Jeong Ho Seo, Email: duketm@naver.com.
Ye Jee Shim, Email: dapdapgirl@hanmail.net.
Hyo Seop Ahn, Email: hsahn@snu.ac.kr.
Keon Hee Yoo, Email: hema2170@skku.edu.
Hoi Soo Yoon, Email: snoopyi@hanmail.net.
Young-Woong Won, Email: dr.wonyw@gmail.com.
Kun Soo Lee, Email: kslee@knu.ac.kr.
Kwang Chul Lee, Email: kwcl5609@korea.ac.kr.
Mee Jeong Lee, Email: lmjped@dankook.ac.kr.
Sun Ah. Lee, Email: mozarteffect@hanmail.net
Jun Ah Lee, Email: junahlee@kcch.re.kr.
Jae Min Lee, Email: mopic@hanmail.net.
Jae Hee Lee, Email: pedjhl@gmail.com.
Ji Won Lee, Email: agnesjw@hanmail.net.
Young Tak Lim, Email: limyt@pusan.ac.kr.
Hyun Joo Jung, Email: free1109@ajou.ac.kr.
Hee Won Chueh, Email: caaf80@empas.com.
Eun Jin Choi, Email: ejchoi2@cu.ac.kr.
Hye Lim Jung, Email: hl.jung@samsung.com.
Ju Han Kim, Phone: 82-2-2072-4081, Email: juhan@snu.ac.kr.
Dong Soon Lee, Phone: 82-2-2072-3986, Email: soonlee@snu.ac.kr.
The Hereditary Hemolytic Anemia Working Party of the Korean Society of Hematology, Email: hematology@kams.or.kr.
References
- 1.Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426. doi: 10.1016/S0140-6736(08)61588-3. [DOI] [PubMed] [Google Scholar]
- 2.Iolascon A, Del Giudice EM, Perrotta S, Alloisio N, Morlé L, Delaunay J. Hereditary spherocytosis: from clinical to molecular defects. Haematologica. 1998;83(3):240–257. [PubMed] [Google Scholar]
- 3.Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27(4):167–178. doi: 10.1016/j.blre.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Barcellini W, Bianchi P, Fermo E, Imperiali FG, Marcello AP, Vercellati C, et al. Hereditary red cell membrane defects: diagnostic and clinical aspects. Blood Transfus. 2011;9(3):274–277. doi: 10.2450/2011.0086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung HL. A new paradigm in the diagnosis of hereditary hemolytic anemia. Blood Res. 2013;48(4):237–239. doi: 10.5045/br.2013.48.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cynober T, Mohandas N, Tchernia G. Red cell abnormalities in hereditary spherocytosis: relevance to diagnosis and understanding of the variable expression of clinical severity. J Lab Clin Med. 1996;128(3):259–269. doi: 10.1016/S0022-2143(96)90027-X. [DOI] [PubMed] [Google Scholar]
- 7.Park ES, Jung HL, Kim HJ, Park SS, Bae SH, Shin HY, et al. Hereditary hemolytic anemia in Korea from 2007 to 2011: a study by the Korean hereditary hemolytic Anemia working Party of the Korean Society of hematology. Blood Res. 2013;48(3):211–216. doi: 10.5045/br.2013.48.3.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King MJ, Garcon L, Hoyer JD, Iolascon A, Picard V, Stewart G, et al. ICSH guidelines for the laboratory diagnosis of nonimmune hereditary red cell membrane disorders. Int J Lab Hematol. 2015;37(3):304–325. doi: 10.1111/ijlh.12335. [DOI] [PubMed] [Google Scholar]
- 9.Mariani M, Barcellini W, Vercellati C, Marcello AP, Fermo E, Pedotti P, et al. Clinical and hematologic features of 300 patients affected by hereditary spherocytosis grouped according to the type of the membrane protein defect. Haematologica. 2008;93(9):1310–1317. doi: 10.3324/haematol.12546. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi P, Fermo E, Vercellati C, Marcello AP, Porretti L, Cortelezzi A, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97(4):516–523. doi: 10.3324/haematol.2011.052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis–2011 update. Br J Haematol. 2012;156(1):37–49. doi: 10.1111/j.1365-2141.2011.08921.x. [DOI] [PubMed] [Google Scholar]
- 12.Park SH, Park CJ, Lee BR, Cho YU, Jang S, Kim N, et al. Comparison study of the eosin-5′-maleimide binding test, flow cytometric osmotic fragility test, and cryohemolysis test in the diagnosis of hereditary spherocytosis. Am J Clin Pathol. 2014;142(4):474–484. doi: 10.1309/AJCPO7V4OGXLIIPP. [DOI] [PubMed] [Google Scholar]
- 13.Streichman S, Gescheidt Y. Cryohemolysis for the detection of hereditary spherocytosis: correlation studies with osmotic fragility and autohemolysis. Am J Hematol. 1998;58(3):206–212. doi: 10.1002/(SICI)1096-8652(199807)58:3<206::AID-AJH8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 14.Shim YJ, Won DI. Flow cytometric osmotic fragility testing does reflect the clinical severity of hereditary spherocytosis. Cytometry B Clin Cytom. 2014;86(6):436–443. doi: 10.1002/cytob.21143. [DOI] [PubMed] [Google Scholar]
- 15.Kar R, Mishra P, Pati HP. Evaluation of eosin-5-maleimide flow cytometric test in diagnosis of hereditary spherocytosis. Int J Lab Hematol. 2010;32(1p2):8–16. doi: 10.1111/j.1751-553X.2008.01098.x. [DOI] [PubMed] [Google Scholar]
- 16.Miraglia del Giudice E, Iolascon A, Pinto L, Nobili B, Perrotta S. Erythrocyte membrane protein alterations underlying clinical heterogeneity in hereditary spherocytosis. Br J Haematol. 1994;88(1):52–55. doi: 10.1111/j.1365-2141.1994.tb04976.x. [DOI] [PubMed] [Google Scholar]
- 17.Del Orbe Barreto R, Arrizabalaga B, De la Hoz AB, García-Orad Á, Tejada MI, Garcia-Ruiz JC, et al. Detection of new pathogenic mutations in patients with congenital haemolytic anaemia using next-generation sequencing. Int J Lab Hematol. 2016;38(6):629–638. doi: 10.1111/ijlh.12551. [DOI] [PubMed] [Google Scholar]
- 18.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 19.Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7(2):111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 20.Constantino BT. Reporting and grading of abnormal red blood cell morphology. Int J Lab Hematol. 2015;37(1):1–7. doi: 10.1111/ijlh.12215. [DOI] [PubMed] [Google Scholar]
- 21.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hui P, Shultz V, Krauthammer M, Holford M, Chen S, Silva T, et al. Yale Blood Cell Disease Reference Laboratory Programs: Rapid Mutation Scanning by Next Generation Sequencing. Available from: https://medicine.yale.edu/pathology/programs/moleculardiagnostics/YBDRL/Morrow_2012_YBCDRL_Program_265397_33208_v1.pdf. Accessed 24 Dec 2016.
- 23.Agarwal AM, Nussenzveig RH, Reading NS, Patel JL, Sangle N, Salama ME, et al. Clinical utility of next-generation sequencing in the diagnosis of hereditary haemolytic anaemias. Br J Haematol. 2016;174(5):806–814. doi: 10.1111/bjh.14131. [DOI] [PubMed] [Google Scholar]
- 24.van Wijk R. Next generation sequencing in diagnosis and research on rare anemias. 20th Congress of the European Hematology Association Vienna, Austria, June 11–14, 2015. Oral Presentation, Not published.
- 25.Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. 2016;90(1):69–78. doi: 10.1111/cge.12749. [DOI] [PubMed] [Google Scholar]
- 26.Eber S, Lux SE. Hereditary spherocytosis–defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41(2):118–141. doi: 10.1053/j.seminhematol.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 27.An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008;141(3):367–375. doi: 10.1111/j.1365-2141.2008.07091.x. [DOI] [PubMed] [Google Scholar]
- 28.Lee YK, Cho HI, Park SS, Lee YJ, Ra E, Chang YH, et al. Abnormalities of erythrocyte membrane proteins in Korean patients with hereditary spherocytosis. J Korean Med Sci. 2000;15(3):284–288. doi: 10.3346/jkms.2000.15.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarolim P, Murray JL, Rubin HL, Taylor WM, Prchal JT, Ballas SK, et al. Characterization of 13 novel band 3 gene defects in hereditary spherocytosis with band 3 deficiency. Blood. 1996;88(11):4366–4374. [PubMed] [Google Scholar]
- 30.Ricard MP, Gilsanz F, Millan I. Erythroid membrane protein defects in hereditary spherocytosis. A study of 62 Spanish cases. Haematologica. 2000;85(9):994–995. [PubMed] [Google Scholar]
- 31.Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. 2000;71(2):118–135. [PubMed] [Google Scholar]
- 32.Inoue T, Kanzaki A, Yawata A, Wada H, Okamoto N, Takahashi M, et al. Uniquely higher incidence of isolated or combined deficiency of band 3 and/or band 4.2 as the pathogenesis of autosomal dominantly inherited hereditary spherocytosis in the Japanese population. Int J Hematol. 1994;60(4):227–238. [PubMed] [Google Scholar]
- 33.Iolascon A, Perrotta S, Stewart GW. Red blood cell membrane defects. Rev Clin Exp Hematol. 2003;7(1):22–56. [PubMed] [Google Scholar]
- 34.Chen Z, Su D, Ai L, Jiang X, Wu C, Xu Q, et al. UGT1A1 sequence variants associated with risk of adult hyperbilirubinemia: a quantitative analysis. Gene. 2014;552(1):32–38. doi: 10.1016/j.gene.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Takeuchi K, Kobayashi Y, Tamaki S, Ishihara T, Maruo Y, Araki J, et al. Genetic polymorphisms of bilirubin uridine diphosphate-glucuronosyltransferase gene in Japanese patients with Crigler-Najjar syndrome or Gilbert syndrome as well as in healthy Japanese subjects. J Gastroenterol Hepatol. 2004;19(9):1023–1028. doi: 10.1111/j.1440-1746.2004.03370.x. [DOI] [PubMed] [Google Scholar]
- 36.Warang P, Devendra R, D’Silva S, Chiddarwar A, Kedar P, Ghosh K, et al. Do UGT1A1 and HMOX1 gene promoter polymorphisms increase the risk of hyperbilirubinemia and gallstones in patients with hereditary spherocytosis? Ann Hematol. 2015;94(1):169–171. doi: 10.1007/s00277-014-2123-z. [DOI] [PubMed] [Google Scholar]
- 37.Farias MG. Advances in laboratory diagnosis of hereditary spherocytosis. Clin Chem Lab Med. 2017;55(7):944–948. doi: 10.1515/cclm-2016-0738. [DOI] [PubMed] [Google Scholar]
- 38.Arora RD, Dass J, Maydeo S, Arya V, Radhakrishnan N, Sachdeva A, et al. Flow cytometric osmotic fragility test and eosin-5′-maleimide dye-binding tests are better than conventional osmotic fragility tests for the diagnosis of hereditary spherocytosis. Int J Hematol. 2018;40(3):335–342. doi: 10.1111/ijlh.12794. [DOI] [PubMed] [Google Scholar]
- 39.Iolascon A, Avvisati RA. Genotype/phenotype correlation in hereditary spherocytosis. Haematologica. 2008;93(9):1283–1288. doi: 10.3324/haematol.13344. [DOI] [PubMed] [Google Scholar]
- 40.Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101(11):1284–1294. doi: 10.3324/haematol.2016.142463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Significant variants diagrams for UGT1A1 gene. Figure S2. Results of NaCl induced OFT. Table S1. Multi-gene panel for targeted sequencing. Table S2. List of protein simulation templates. Table S3. List of significant variants detected in RBC membrane protein-encoding genes. Table S4. Primer sets for all significant variants in RBC membrane protein-encoding genes. Table S5. List of significant variants detected in RBC enzyme-encoding genes among patients with HS. Table S6. List of UGT1A1 gene variants in patients with HS in Korea. Table S7. Clinical characteristics of patients with HS without peripheral blood spherocytes. Table S8. Patients without RBC membrane-encoding gene mutation. (DOCX 114 kb)
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.