

Abstract
Transient, resolving inflammation plays a critical role in tissue repair and regeneration. In the context of joint disease, however, chronic inflammation following injury or with osteoarthritis can lead to irreversible articular cartilage degradation and joint pain. Developing tissue engineering strategies for the regeneration of articular cartilage remains challenging due to the harsh inflammatory environment of an injured or arthritic joint, which can promote degradation of engineered tissues as well as native articular cartilage. Here, we developed an artificial gene circuit for controlled, cell-based delivery of biologic drugs, based on a nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-responsive synthetic promoter. Using lentivirus-based gene therapy, we engineered murine induced pluripotent stem cells (iPSCs) capable of attenuating inflammation through controlled release of an anti-inflammatory drug, interleukin-1 receptor antagonist (IL-1Ra), subsequently inhibiting gene circuit activation in a self-regulating manner. Murine iPSCs were transduced with the synthetic gene circuit either in monolayer or through biomaterial-mediated transduction. Cells were maintained in monolayer or differentiated into cartilage constructs and stimulated with different doses of interleukin 1 alpha (IL-1α) to determine the ability of this synthetic NF-κB responsive system to inhibit inflammation and protect tissue-engineered constructs. In response to IL-1α, cells produced high levels of IL-1Ra, which inhibited inflammatory signaling and protected tissue-engineered cartilage from proteoglycan degradation. Our results show that the combination of gene therapy and tissue engineering can be used to successfully create iPSCs capable of producing biologic drugs in a controlled manner. This self-regulating system provides a tool for cell-based drug delivery as the basis for a novel therapeutic approach for a variety of diseases.
Impact Statement
We engineered a synthetic transcription system based on nuclear factor kappa-light-chain-enhancer of activated B cells signaling that can attenuate the effects of the inflammatory cytokine interleukin (IL)-1α in a self-regulating manner. This system responds in a time- and dose-dependent manner to rapidly produce therapeutic levels of IL-1 receptor antagonist (IL-1Ra). The use of lentiviral gene therapy allows this system to be utilized through different transduction methods and in different cell types for a variety of applications. Broadly, this approach may be applicable in developing autoregulated biologic systems for tissue engineering and drug delivery in a range of disease applications.
Keywords: synthetic biology, gene therapy, arthritis, regenerative medicine, stem cells
Introduction
Osteoarthritis (OA) is a debilitating joint disease that causes severe pain and loss of joint function, affecting over 32 million adults in the United States alone.1 OA is characterized by the degeneration of articular cartilage, the hyaline cartilage that covers the articulating surfaces of bones in diarthrodial joints. Because articular cartilage is avascular, aneural, and alymphatic, it lacks an intrinsic ability to repair.2 While there are many risk factors for OA—including injury, aging, metabolic disorders, obesity, and genetics—a common pathway for the pathogenesis and progression of joint degeneration and pain involves the proinflammatory activity of several cytokines, particularly the interleukin-1 (IL-1) family of cytokines, including IL-1α and IL-1β, and tumor necrosis factor-α (TNF-α).3 These cytokines primarily signal via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to induce extracellular matrix (ECM) degradation through the inhibition of anabolic activities and enhanced production of degradative enzymes and catabolic cytokines, particularly in articular chondrocytes.4,5 Furthermore, inflammation can significantly inhibit repair of cartilage and other joint tissues, as several studies have shown high sensitivity of stem cells and engineered cartilage to cytokines such as IL-1 and TNF.6–9
IL-1 receptor antagonist (IL-1Ra, anakinra), a competitive antagonist of IL-1, has been shown to alleviate symptoms of rheumatoid arthritis (RA) and post-traumatic OA.10–12 Furthermore, several gene therapy approaches to deliver IL-1Ra to the joints of patients with RA and OA are in progress or pending.5,13,14 However, despite the potential for success of anti-cytokine drugs and gene therapy approaches, there are currently no effective disease-modifying treatments to address both the symptoms and structural change of OA.14–16 Additionally, the inability of these approaches to accomplish a sustained delivery of biologic drugs in a dynamically and spatially controlled manner is an important limitation. Anti-cytokine therapies are often delivered at high doses, which may have significant off-target effects, including an increased susceptibility to infection and certain autoimmune diseases,17 as well as limited tissue regeneration and repair.18–20 Therefore, the long-term success of stem cell–based therapies for cartilage repair or OA may require engagement of intrinsic cellular abilities to regulate the inflammatory environment of the joint.
To address these limitations, tissue engineering and gene therapy approaches can be combined to create a cartilaginous tissue that is capable of replacing damaged tissue while delivering therapeutic drugs to diseased joints.21 Additionally, by using both gene delivery and synthetic biology, cells can be transduced ex vivo with expression vectors designed to produce a desired gene through specific inputs.22,23 Furthermore, the implantation of tissue-engineered constructs allows for localized delivery of biologic drugs to specific sites in the body.
The overall goal of this study was to create self-regulating (i.e., feedback-controlled) stem cells capable of attenuating inflammation in a prescribed manner for a controlled release of anti-inflammatory molecules. We engineered a synthetic transcriptional regulator system capable of producing a therapeutic drug, and packaged it into a lentiviral vector to allow for transduction into various cell types and through different transduction strategies. Specifically, we developed an NF-κB-inducible synthetic promoter that controls the release of a biologic drug, IL-1Ra, to maintain tissue homeostasis in response to the activation of NF-κB in a long-term and sustained manner (Fig. 1). We utilized this lentiviral vector to create murine induced pluripotent stem cells (iPSCs) capable of sensing and responding to inflammation. Additionally, we show the proof-of-concept for site-specific, scaffold-mediated delivery24,25 of this lentivirus to iPSCs. The transduced iPSCs were chondrogenically differentiated into articular cartilage tissue to determine the efficacy of this vector in protecting engineered tissue against cytokine-induced degradation. We hypothesized that this NF-κB-inducible biologic drug delivery system will allow for controlled, self-regulating production of anti-inflammatory molecules in direct response to dynamic changes in inflammatory stimuli. This type of cell-based approach could provide an effective method to treat OA and chronic inflammatory diseases while overcoming limitations of current drug delivery techniques.
FIG. 1.

Overview of synthetic promoter design and experimental approach. A synthetic promoter was designed with five NF-κB recognition motifs upstream of Il1rn (gene for IL-1Ra) to create an NF-κB-inducible promoter. An NF-κB NRE was inserted upstream of the promoter to reduce background signal. In a separate vector, the EF1α constitutive promoter was used to drive a continuous expression of IL-1Ra. iPSCs were transduced with lentivirus delivering an EF1α constitutive promoter that drives the expression of IL-1Ra, or an NF-κB-inducible promoter that drives the expression of IL-1Ra. In the presence of IL-1α, the synthetic promoter is activated and produces IL-1Ra, preventing IL-1α from binding to the IL-1 receptor and inhibiting the activation of inflammatory cascades within the cell. IL-1α, interleukin 1 alpha; IL-1Ra, interleukin-1 receptor antagonist; iPSCs, induced pluripotent stem cells; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NRE, negative regulatory element. Color images are available online.
Materials and Methods
Overall strategy
The overall strategy for this work was to create a synthetic transcription system that is activated by inflammatory cytokines and, when incorporated into a lentiviral vector, can be readily delivered to different types of cells for applications in cell therapy or tissue engineering. Cells transduced by the lentiviral vectors were programmed to express anti-inflammatory biologic drugs downstream of the synthetic promoter, providing a negative feedback system that blocks the action of an inflammatory cytokine (Fig. 1). Here, we specifically tested the ability of our synthetic transcription system to protect engineered cartilage from its intrinsic response to inflammatory cytokines through three different methods: in monolayer, in tissue-engineered cartilage, and with biomaterial-mediated delivery of vectors.
Vector design
A synthetic NF-κB-inducible promoter was designed to incorporate multiple NF-κB response elements that drive a target gene of interest.26 Briefly, a synthetic promoter was developed containing five consensus sequences approximating the NF-κB canonical recognition motif based on genes that are upregulated by inflammatory challenge: InfB1, Il6, Mcp1, Adamts5, and Cxcl10.26,27 Murine Il1rn or firefly luciferase from the pGL3 basic plasmid (Promega) was cloned downstream of this synthetic promoter; a TATA box derived from the minimal CMV promoter was cloned between the synthetic promoter and downstream target genes; and an NF-κB-negative regulatory element (NRE—5′-AATTCCTCTGA-3′)28 was cloned upstream of the promoter to reduce background signal (Fig. 1). This engineered NRE-IL1Ra cassette results in transgene expression when the promoter is activated in response to NF-κB-based inflammatory stimuli, resulting in a “self-regulating” system. A constitutive control vector was also tested using murine Il1rn cloned into the lentiviral transfer vector (No. 12250; Addgene) downstream of the EF1α promoter sequence29 (EF1α-IL1Ra) using Gibson Assembly.30 A nuclear-targeted green fluorescent protein (GFP)31 (No. 11680; Addgene) was cloned into a constitutive, lentiviral vector32 (No. 11645; Addgene) and was used as a transduction control (GFP).
Lentivirus production
HEK293T cells were co-transfected with an expression transfer vector, second-generation packaging plasmid psPAX2 (No. 12260; Addgene), and an envelope plasmid pMD2.G (No. 12259; Addgene) by calcium phosphate precipitation to make vesicular stomatitis virus glycoprotein pseudotyped lentivirus.33 The expression transfer vectors include the NRE-IL1Ra, NRE-Luc, EF1α-IL1Ra, and GFP plasmids. The lentivirus was stored at −80°C until further use. The functional titer of each virus group was determined via quantitative real-time polymerase chain reaction to determine the number of lentiviral DNA copies integrated into the genome of transduced HeLa cells.33
Cell culture and differentiation
Murine iPSCs, generated from tail fibroblasts from adult C57BL/6 mice and validated for pluripotency as described by Diekman et al.,34,35 were maintained on mitomycin C-treated mouse embryonic fibroblasts (Millipore). To track inflammatory activity in cells, an iPSC reporter cell line was created via the CRISPR-Cas9 genome editing system to incorporate firefly luciferase downstream of the Ccl2 locus (Ccl2-Luc).26 In this cell line, proinflammatory signaling activates Ccl2 promoter activity, resulting in the transcription of luciferase.36
Unedited and Ccl2-Luc cells were then differentiated toward a mesenchymal state using a high-density micromass culture. Differentiation medium contained Dulbecco's modified Eagle's medium high glucose (DMEM-HG); 1% culture medium supplement containing recombinant human insulin, human transferrin, and sodium selenite (ITS+); minimum essential medium (MEM) nonessential amino acids; 55 μM 2-mercaptoethanol; 24 ng/mL gentamicin; 50 μg/mL l-ascorbic acid; and 40 μg/mL l-proline. On days 3–5, this medium was supplemented with 100 nM dexamethasone and 50 ng/mL bone morphogenetic protein 4 (BMP-4; R&D Systems).35 After 15 days of culture, the micromasses were dissociated with pronase and collagenase type II and the predifferentiated iPSCs (PDiPSCs) were plated on gelatin-coated dishes in expansion medium containing DMEM-HG, 10% fetal bovine serum, 1% ITS+, MEM nonessential amino acids, 55 μM 2-mercaptoethanol, 1% penicillin/streptomycin, 50 μg/mL l-ascorbic acid, 40 μg/mL l-proline, and 4 ng/mL of basic fibroblast growth factor (bFGF; R&D Systems). These cells were then expanded, transduced, and either used for monolayer experiments or in pellet cultures to produce engineered cartilage to evaluate the ability of these cells to protect against inflammation.
Lentiviral transduction and culture of PDiPSCs
For initial characterization, PDiPSCs were transduced with the NRE-Luc virus. For all other monolayer experiments, passage 4 PDiPSCs and Ccl2-Luc cells were transduced with NRE-IL1Ra and EF1α-IL1Ra virus, and non-transduced (NT) cells were used as control.
In pellet experiments, PDiPSCs were transduced at passage 1 with NRE-IL1Ra, EF1α-IL1Ra, or GFP virus. Passage 1 cells were trypsinized, and pellet cultures were created by centrifuging 250k cells at 200 g for 5 min. The pellets were cultured in chondrogenic media containing DMEM-HG, 1% ITS+, MEM nonessential amino acids, 55 μM 2-mercaptoethanol, 1% penicillin-streptomycin, 50 μg/mL l-ascorbic acid, 40 μg/mL l-proline, 100 nM dexamethasone, and 10 ng/mL transforming growth factor-β3 (TGF-β3) for 21 days.35
For all monolayer and pellet studies, transduction media consisted of expansion medium supplemented with 4 μg/mL polybrene (Sigma-Aldrich) and the desired number of viral particles to achieve a multiplicity of infection = 3. Transduction media were exchanged with expansion medium after 24 h of transduction.
Biomaterial-mediated delivery
Biomaterial-mediated lentiviral transduction was tested using a model system based on poly(ɛ-caprolactone) (PCL; molecular weight 70,000–90,000; Sigma-Aldrich), a scaffold material commonly used for cartilage tissue engineering.37,38 PCL was dissolved in glacial acetic acid at a 10% wt/vol ratio. Tissue culture-treated plates were coated with the dissolved PCL, and the acid was evaporated overnight. The acid was quenched with 1 N NaOH, washed with phosphate buffered saline (PBS), and sterilized with an ethanol gradient. Plates were then incubated in 0.002% poly-l-lysine (PLL) solution (Sigma-Aldrich). PLL was aspirated, and two groups of lentivirus (NRE-IL1Ra and EF1α-IL1Ra) or PBS for NT controls were added to the plates and allowed to incubate for 4 h at 37°C.24,37 After virus immobilization, viral supernatant was aspirated, wells were washed with PBS, and passage 4 PDiPSCs and Ccl2-Luc control cells were plated in the wells. After an additional 3 days of culture, the ability of these cells to respond to and attenuate inflammation was evaluated through a luminescence activity assay, protein production, and gene expression analysis.
Inflammatory challenge
To evaluate the response of the synthetic promoter to inflammatory cytokines, unedited PDiPSCs transduced with NRE-Luc were challenged with 1 ng/mL IL-1α or 20 ng/mL TNF and evaluated at 72 h after challenge.
To determine the sensitivity and kinetics of the synthetic promoter, all groups of Ccl2-Luc and unedited PDiPSCs (NT, NRE-IL1Ra, and EF1α-IL1Ra) were evaluated at 0, 4, 12, 24, and 72 h post supplementation of media with 0.05, 0.1, 0.5, or 1 ng/mL IL-1α and removal of bFGF. Control cells were cultured in the absence of IL-1α.
For characterizing the pellets' response to inflammation, after 21 days of chondrogenic culture, pellets were challenged with 0.5 or 1 ng/mL IL-1α and removal of TGF-β and dexamethasone for 72 h. Control pellets were cultured in the absence of IL-1α.
Inflammation activity assay
Luciferase activity from NRE-Luc transduced cells and Ccl2-Luc cells in all monolayer experiments was measured using the BrightGlo Luminescence kit (Promega) and a Cytation5 plate reader (Biotek). Luciferase activity is reported as a fold change of IL-1α-stimulated cells over control cells cultured without IL-1α (n = 4–6).
Gene expression
Unedited PDiPSCs from monolayer studies and pellets were harvested for quantitative, reverse transcription polymerase chain reaction (qRT-PCR) after inflammatory challenge (n = 4). Monolayer cells were rinsed in PBS, lysed in Buffer RL (total RNA purification; Norgen Biotek), and stored at −80°C until RNA isolation. Pellets of engineered cartilage tissue were rinsed in PBS and stored at −80°C until RNA isolation. Pellets were homogenized using a miniature bead beater. RNA isolation was carried out following the manufacturer's protocol (Norgen Biotek). Reverse transcription was performed using Superscript VILO complementary DNA (cDNA) master mix (Invitrogen). qRT-PCR was performed using Fast SyBR Green master mix (Applied Biosystems) following the manufacturer's protocol. Primer pairs (Supplementary Table S1) were synthesized by Integrated DNA Technologies, Inc. Fold changes were calculated using the ΔΔCT method and are shown relative to the 0 h no cytokine, NT control samples for monolayer experiments, or GFP pellets without cytokines. For samples with no amplification, CT threshold was set to the cycle limit.
Enzyme-linked immunosorbent assays
Culture media were collected from all unedited PDiPSC monolayer and pellet samples after inflammation challenge and stored at −20°C. IL-1Ra concentration was measured with DuoSet enzyme-linked immunosorbent assay (ELISA) specific to mouse IL-1Ra/IL-1F3 (n = 4; R&D Systems). Each sample was measured in technical duplicates. Absorbance was measured at 450 and 540 nm.
Biochemical analysis of pellet cultures
After 72 h of inflammatory challenge, pellets were washed with PBS and stored at −20°C until processing. Pellets were digested overnight in 125 μg/mL papain at 65°C for biochemical analysis. DNA content was measured with PicoGreen assay (Thermo Fisher), and total sulfated glycosaminoglycan (sGAG) content was measured using a 1,9-dimethylmethylene blue assay at 525 nm wavelength39 (n = 4).
Histological processing of pellet culture
After 72 h of cytokine challenge, pellets were washed with PBS and fixed in 10% neutral buffered formalin for 24 h, paraffin-embedded, and sectioned at 8 μm thickness. Slides were stained for Safranin-O/hematoxylin/fast green using a standard protocol.40
Statistical analysis
Statistical analysis was performed with the JMP Pro software package. Luminescence data and biomaterial-mediated delivery qRT-PCR data were analyzed using analysis of variance (ANOVA) with Dunnett's post hoc test using NT as the control (α = 0.05). A two-way ANOVA with Tukey's HSD post hoc test was used to analyze all ELISA data, pellet biochemistry, and pellet qRT-PCR data (α = 0.05).
Results
Responsiveness of the NF-κB synthetic promoter to IL-1α or TNF
PDiPSCs receiving the NRE-Luc vector responded to 1 ng/mL IL-1α and 20 ng/mL TNF with a 10.17 ± 0.54- and 8.40 ± 0.32-fold increase in luminescence after 72-h cytokine stimulation, respectively (Fig. 2A).
FIG. 2.

Cell response to cytokine stimulation in monolayer to determine the responsiveness of NRE-IL1Ra vector. (A) NRE-Luc cells were stimulated with 20 ng/mL TNF-α or 1 ng/mL IL-1α, and luminescence was measured after 72 h. Bars represent mean RLU ± SEM (n = 4). (B) Fold change of NF-κB activity measured by luminescence signal from Ccl2-Luc cells. Bars represent the mean fold change in RLU ± SEM (n = 6). Asterisks represent significance (p < 0.05) compared with NT control. (C) NT and NRE-IL1Ra cells were treated with IL-1α, and an ELISA was performed on samples to determine IL-1Ra protein production. Values represent mean ± SEM (n = 4). Groups not sharing same letter are statistically significant (p < 0.05). ELISA, enzyme-linked immunosorbent assay; NT, non-transduced; RLU, relative luminescence unit; SEM, standard error of the mean; TNF-α, tumor necrosis factor-α. Color images are available online.
Self-regulating attenuation of inflammation by iPSCs
Attenuation of inflammatory signaling was tested in Ccl2-Luc reporter cells that produce luciferase in response to inflammatory stimuli. After transduction, cells transduced with either the NRE-IL1Ra or EF1α-IL1Ra lentivirus and NT control cells were treated with IL-1α at a range of doses from physiologic (0.1 ng/mL) to supraphysiologic (1 ng/mL)41 and compared with cells without cytokine. In all doses of IL-1α, by 4 h there was significantly less luminescence output in both NRE-IL1Ra and EF1α-IL1Ra groups compared with NT control. This trend was sustained up to 24 h in all doses in the NRE-IL1Ra groups and up to 72 h in all doses except 0.05 ng/mL of IL-1α (Fig. 2B) (p < 0.01). This decrease in luminescence shows that there was attenuation of IL-1 signaling with the NRE-IL1Ra group and that this system is responsive at a range of cytokine concentrations.
To evaluate the ability of these vectors to produce therapeutic levels of IL-1Ra, unedited PDiPSCs were lentivirally transduced with the NRE-IL1Ra or EF1α-IL1Ra vectors, and IL-1Ra protein production was measured. The transduced cells and NT control cells were administered with IL-1α and compared with cells without cytokine. Culture media were collected at 0, 4, 12, 24, and 72 h to measure the production of IL-1Ra in response to an inflammatory challenge. NRE-IL1Ra groups had an increase in IL-1Ra production over time and exhibited higher IL-1Ra production with increased doses of IL-1α. There was a significant increase in IL-1Ra production in the NRE-IL1Ra groups challenged with IL-1α compared with the NRE-IL1Ra cells without cytokine at 24 and 72 h (p < 0.0001) with 140.64 ± 8.78 and 163.41 ± 16.04 ng/mL IL-1Ra produced with 0.5 and 1 ng/mL IL-1α stimulation at 72 h, respectively. Additionally, there was an increase in IL-1Ra in NRE-IL1Ra groups compared with the NT and EF1α-IL1Ra groups (Fig. 2C and Supplementary Fig. S1) (p < 0.0001). These results taken together show that this iPSC-based NRE-IL1Ra system responds to an inflammation challenge by producing increased levels of the biologic drug.
Transduced NRE-IL1Ra iPSCs form tissue-engineered cartilage that is protected from IL-1α
Following lentiviral delivery to iPSCs and 21 days of chondrogenic differentiation, pellets were administered 0.5 or 1 ng/mL IL-1α for 72 h and were compared with pellets that did not receive cytokine. GFP control pellets showed rich Safranin-O staining for sGAG, one of the primary components of cartilage ECM. Pellets treated with 0.5 ng/mL or 1 ng/mL IL-1α for 72 h displayed reduced Safranin-O staining (Fig. 3A). Pellets made from NRE-IL1Ra PDiPSCs produced 72.90 ± 2.42 and 92.24 ± 2.31 ng/mL IL-1Ra in response to 0.5 and 1 ng/mL IL-1α, respectively, which was significantly higher than both GFP and EF1α-IL1Ra pellets (p < 0.0001). EF1α-IL1Ra pellets produced significantly higher levels of IL-1Ra compared with GFP control pellets (Fig. 3B) (p < 0.001). NRE-IL1Ra pellets showed protection against IL-1α-mediated matrix degradation, as indicated by robust Safranin-O staining and quantitative biochemical analysis. NRE-IL1Ra pellets that received IL-1α had significantly higher amounts of sGAG when normalized to the concentration of DNA in each pellet (normalized to pellets that did not receive IL-1α) compared with EF1α-IL1Ra and GFP control pellets (Fig. 3C) (p = 0.0322 and 0.0005, respectively). Despite the increased levels of IL-1Ra production, the EF1α-IL1Ra pellets showed little or no inhibition of IL-1α-mediated sGAG loss, as indicated by the loss of Safranin-O staining and sGAG/DNA content (Fig. 3C).
FIG. 3.

Assessment of engineered cartilage to determine if NRE-IL1Ra protects tissues from cytokine stimulation. (A) Safranin-O/fast green/hematoxylin-stained tissue sections of engineered cartilage (scale bar = 200 μm). (B) IL-1Ra production collected from media samples after IL-1α stimulation (n = 4). Groups not sharing same letter are statistically significant (p < 0.05). (C) Percent conserved sGAG/DNA in pellets after the addition of IL-1α (n = 4). Groups not sharing same letter are statistically significant (p < 0.05). (D) Relative gene expression compared with 0 h control measured by qRT-PCR (n = 4). Hash represents significance (p < 0.05). Primer sequences are available in Supplementary Table S1. qRT-PCR, quantitative, reverse transcription polymerase chain reaction; sGAG, sulfated glycosaminoglycan. Color images are available online.
Gene expression analysis of the pellets showed a significantly decreased expression of inflammation-related genes, Ccl2 and Il6, in pellets engineered with the NRE-IL1Ra vector compared with GFP control (p = 0.0164 and 0.0004, respectively) and EF1α-IL1Ra (p = 0.0054 and 0.0008, respectively) pellets. Furthermore, NRE-IL1Ra pellets had significantly higher levels of expression of cartilage matrix-related genes, Col2a1 and Acan, compared with GFP control (p = 0.0088 and 0.0090, respectively) and EF1α-IL1Ra pellets (Fig. 3D) (p = 0.0044 and 0.041, respectively).
Biomaterial-mediated lentiviral delivery shows self-regulating production of IL-1Ra and attenuation of inflammation
Ccl2-Luc reporter cells were seeded on a PCL film and were transduced through biomaterial-mediated delivery of either NRE-IL1Ra or EF1α-IL1Ra vectors. Cell were also seeded on PCL without virus as a NT control. Cells were then administered with IL-1α. For all doses of IL-1α, the NRE-IL1Ra group had significantly less luminescence output compared with NT controls by 24 h (p < 0.001) (Fig. 4A). At 72 h post stimulation, NRE-IL1Ra and EF1α-IL1Ra cells had decreased luminescent output compared with NT controls in all doses, showing attenuation of inflammation (p < 0.0001).
FIG. 4.
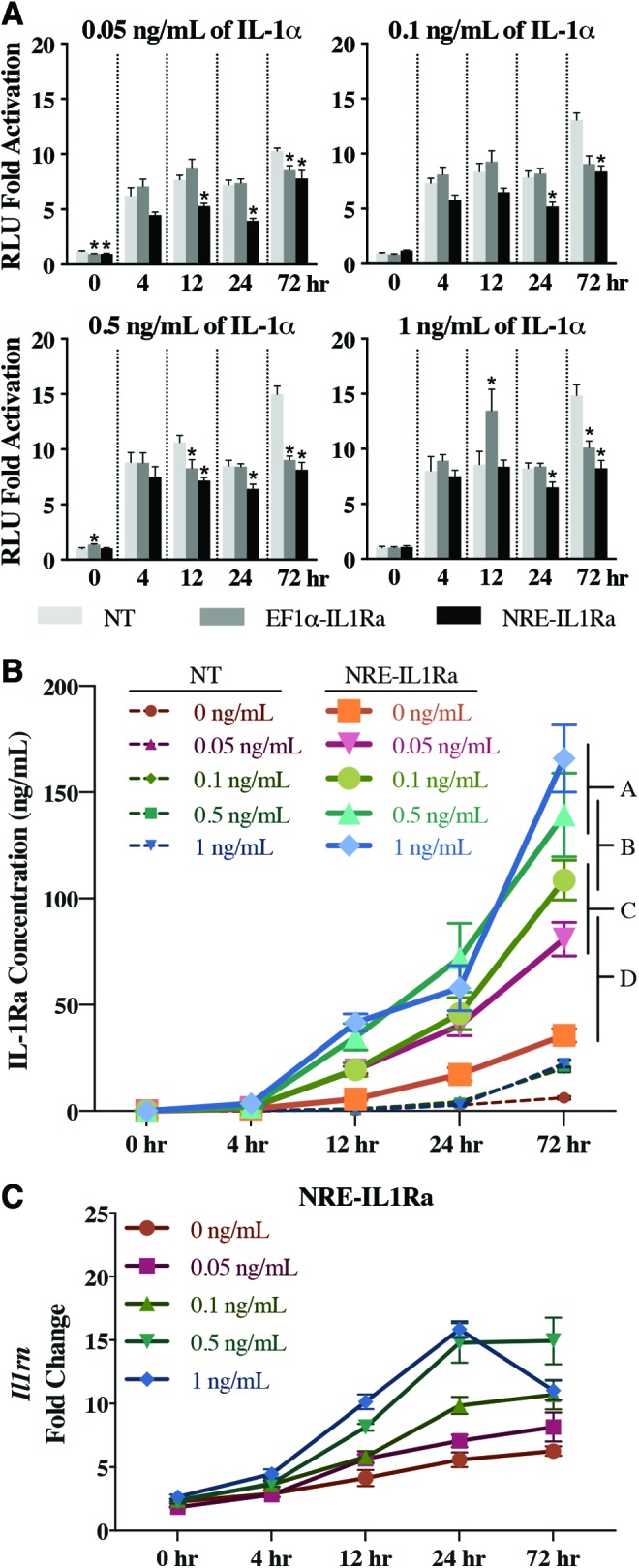
Evaluation of cells transduced via biomaterial-mediated lentiviral delivery. (A) Fold change of NF-κB activity measured by luminescence signal from Ccl2-Luc cells. Bars represent the mean fold change in RLU ± SEM of cells treated with IL-1α compared with controls cultured with no cytokine (n = 6). Asterisks represent significance (p < 0.05) compared with NT control. (B) NT and NRE-IL1Ra cells were treated with IL-1α, and an ELISA was performed on samples to determine IL-1Ra protein production. Values represent mean ± SEM (n = 4). Groups not sharing same letter are statistically significant (p < 0.05). (C) Il1rn gene expression. Fold changes were determined relative to 0 h NT cells without cytokine. Error bars represent means of fold change ± SEM (n = 6). Primer sequences are available in Supplementary Table S1. Color images are available online.
Culture media were collected at 0, 4, 12, 24, and 72 h to measure IL-1Ra production from cells transduced via biomaterial-mediated transduction. The NRE-IL1Ra group exhibited a time- and dose-dependent response to IL-1Ra production. IL-1Ra protein production was significantly increased at higher doses of IL-1α, with 139.31 ± 19.6 and 165.91 ± 15.83 ng/mL IL-1Ra being produced when stimulated with 0.5 and 1 ng/mL IL-1α, respectively, at 72 h (p < 0.001). Additionally, IL-1Ra production was increased in the NRE-IL1Ra group compared with NT and EF1α-IL1Ra groups (Fig. 4B and Supplementary Fig. S2) (p < 0.0001). There was an increase in Il1rn gene expression in all cells that received the NRE-IL1Ra vector at 24 h (Fig. 4C), and in EF1α-IL1Ra cells at 0, 12, and 24 h (Supplementary Fig. S3C). By 72 h, NT and NRE-IL1Ra groups were not significantly different from each other, indicating that a decreased inflammatory signaling led to an autoregulated decrease in Il1rn gene expression due to IL-1Ra-mediated inhibition of inflammation and subsequent gene circuit activation.
Gene expression analysis showed a dose-dependent increase in inflammation-related genes, Ccl2 and Il6, as early as 4 h following the delivery of inflammatory cytokines to all groups of cells, which persisted through 72 h. However, the biomaterial-mediated delivery of the self-regulating NRE-IL1Ra compared with NT controls significantly decreased the expression of Ccl2 starting at 4 h and was maintained through 72 h (Fig. 5A) (p < 0.01). Ccl2 was also decreased in EF1α-IL1Ra cells compared with NT controls at 24 and 72 h (Supplementary Fig. S3A). Il6 was significantly decreased in NRE-IL1Ra cells from 4 h through 24 h (Fig. 5B) (p < 0.0001) and was significantly decreased in EF1α-IL1Ra cells at 24 h (Supplementary Fig. S3B). Together, these results show that this system responds to inflammatory stimulus and suggests that the therapeutic levels produced could inhibit inflammation. Additionally, these findings show a successful proof-of-concept experiment for the delivery of either our transduced cells for implantation into the joint6,24,42 or of our gene therapy vectors for transduction of endogenous cells in vivo.
FIG. 5.

Gene expression of cells transduced through biomaterial-mediated delivery and challenged with IL-1α. Fold changes were determined relative to 0 h NT cells without cytokine. Error bars represent means of fold change ± SEM (n = 6). Asterisks represent significance relative to NT control group (p < 0.05). (A) Ccl2 gene expression. (B) Il6 gene expression. Color images are available online.
Discussion
A significant challenge in the field of regenerative medicine has been the ability to tissue-engineer cartilage that is capable of withstanding the harsh inflammatory environment of an injured or arthritic joint. We developed an iPSC-based lentiviral system in which cells sense and respond to inflammatory stimuli by producing anti-inflammatory mediators. Using a combination of regenerative medicine, synthetic biology, and gene therapy, we developed self-regulating iPSCs that are capable of forming engineered cartilage for the replacement of diseased tissue and mitigating the inflammatory effects of IL-1α.35 Additionally, the versatility of both the iPSCs and the lentiviral system allows for translation to various cell types or tissues. Lastly, by leveraging the flexibility of lentiviral transduction in tissue engineering applications, we demonstrated proof-of-concept of a targeted delivery method allowing for spatial control of therapy via biomaterial-mediated lentiviral transduction.
Using lentivirus-based gene therapy, we engineered iPSCs with the NRE-IL1Ra vector, creating a system that can dynamically respond to and attenuate NF-κB signaling. Cells receiving the NRE-IL1Ra vector responded rapidly to IL-1α as all groups had reduced inflammatory signaling by 4 h after stimulation. Importantly, NRE-IL1Ra cells responded to both physiologic and supraphysiologic doses of IL-1α, and this response was sustained throughout 72 h for all doses, except 0.05 ng/mL IL-1α. In addition, all doses of IL-1α activated the synthetic promoter to produce therapeutic levels of IL-1Ra by 24 and 72 h and in a dose-dependent manner. This dose response provides a controlled production of the therapeutic drug in response to different levels of inflammation. Cells that received the EF1α-IL1Ra vector produced IL-1Ra, however, at a significantly lower amount than the NRE-IL1Ra vector. This could be due to the low production of the transgene from the constitutive promoter, which has previously been reported in lentiviral systems with constitutive reporters.43
An important advance of this work is the application of our lentiviral system in iPSCs. iPSCs are attractive for tissue engineering and regenerative medicine because they can be expanded, patient-matched, and differentiated into a variety of different cell types to treat multiple tissues, overcoming the limitations of other common cell sources.35,44 Here, we show that the NRE-IL1Ra vector can be delivered to PDiPSCs, and these cells can form cartilaginous tissues, and that in the presence of 0.05 or 1 ng/mL IL-1α, engineered cartilage pellets produced high levels of IL-1Ra. This indicates the potential use of this system for the repair of diseased articular cartilage and mitigation of inflammation through a soluble release of IL-1Ra into the joint. We observed less IL-1Ra production from tissue-engineered cartilage than cells in monolayer. This difference can be attributed to the different differentiation states of the cells, as well as their accessibility to the culture media. In pellets, cells are embedded within a dense ECM, which may bind or hinder the transport of IL-1Ra and/or IL-1α.45 Despite these transport limitations, NRE-IL1Ra pellets were protected from IL-1α-mediated degradation, as evidenced by rich Safranin-O staining and significantly higher levels of sGAG/DNA. Furthermore, NRE-IL1Ra pellets had decreased expression of inflammation-related genes, while cartilage matrix-related genes were sustained, showing the attenuation of inflammatory pathways and protection of the matrix.
In previous approaches, gene therapy for the treatment of OA has been performed with plasmid DNA,46 retrovirus,47 lentivirus,48–50 and most commonly non-integrating viral vectors such as adeno-associated virus.37,51–54 Lentiviral delivery is advantageous for this system due to its ability to stably integrate into the genome of dividing and non-dividing cells for long-term gene expression, its larger packaging capacity, low immunogenicity, and low cytotoxicity.55,56 The vector used in this study is self-inactivating and, therefore, replication-defective, overcoming safety concerns previously associated with viral gene therapy.57–59 Additionally, our group and others have shown that viral vectors can be immobilized to biomaterial surfaces or scaffolds for the delivery of therapeutic vectors to cells.6,24,25,42,60 Our study showed that biomaterial-mediated delivery of NRE-IL1Ra from PCL provided efficient transduction and effectively decreased inflammatory signaling. Specifically, biomaterial-mediated transduction of NRE-IL1Ra significantly decreased the expression of inflammation-related genes, Ccl2 and Il6, and stimulated high levels of IL-1Ra production in a dose-dependent manner. These results indicate that this approach provides an effective method for delivering therapeutic vectors and can be applied for broader tissue engineering applications. This strategy could address limitations of existing gene therapy approaches such as the loss of therapeutic transgene expression over time when using non-integrating delivery methods, lack of spatial control of transduction when using systemic injections of vectors, and lack of controllable or inducible production of transgenes when using constitutive expression vectors.
Previously, investigators have developed various systems for inflammation-inducible expression of pro-regenerative or anti-inflammatory transgenes. Rachakonda et al. created a self-limiting promoter construct that was based on truncated promoter sequences of cyclooxygenase-2 upstream of IL-4 to express IL-4 only in the presence of inflammation.61 Others have also created expression systems based on NF-κB binding sequences for luciferase reporting vectors62,63 or inducible systems driving the expression of anti-inflammatory mediators in adeno-associated viral vectors.64,65 Previous work in our lab utilized CRISPR-Cas9 technology to genome-engineer stem cells capable of using the endogenous systems within the cells to sense inflammation and produce therapeutic transgenes.26 While this system has specific advantages in terms of the precision of CRISPR-based gene editing, by packaging our NRE-IL1Ra system into a lentiviral expression cassette, we expand the vector's applicability to transduce different cell types and tissues, such as mesenchymal stem cells, adipose-derived stem cells, or primary cells such as articular chondrocytes or synovial cells, all of which are commonly used in tissue engineering or targeted for gene therapy but are more challenging to edit with CRISPR-Cas9. The sensitivity of the synthetic promoter can be tuned dependent on the sequence, number of tandem repeats, and neighboring regulatory elements, as well as through gene editing66 or epigenetic modification67 of the cells' receptors. Additionally, other inflammatory cytokines, such as TNF, or intracellular signaling components can be targeted for specific or broad inhibition of inflammation.
Our synthetic lentiviral system can create self-regulating cells capable of sensing and responding to inflammation with a therapeutic level of biologic drug. The continued development of designer circuits through gene switches,68–70 microRNA classifiers,71,72 and synthetic transcription systems73–75 provides a toolkit to engineer more complex circuits with specialized control. This work adds to our long-term goals of developing a molecular toolbox76,77 of biological building blocks for novel synthetic applications in mammalian cells for ameliorating chronic diseases.
The autoregulatory capabilities of this system allow applications for regenerative medicine or for use as a preventative approach to inflammatory disease. Applying this methodology not only allows for protection against inflammation to aid in cartilage repair, but also provides protection of any tissue-engineered constructs inserted to help repair an osteoarthritic joint or biomaterial-mediated lentiviral transduction of any infiltrating cells through localized transduction. The customization aspect of this system and its functionality in monolayer, an engineered tissue replacement, and through scaffold-mediated delivery gives innovative opportunities for effective treatments that are applicable in a variety of diseases.
Supplementary Material
Acknowledgments
The authors thank Dr. Charles Gersbach and Dr. Caitlin Guenther for important discussions and assistance in the early stages of this work. This study was supported, in part, by NIH grants AR50245, AR48852, AG15768, AR48182, AG46927, AR073752, OD10707, AR060719, AR057235, AR073752, the AO Foundation, the Arthritis Foundation, and the Nancy Taylor Foundation for Chronic Diseases, the Philip and Sima Needleman Fellowship for Regenerative Medicine from the Washington University Center of Regenerative Medicine to L.P. and A.K.R., a National Science Foundation Graduate Research Fellowship to A.K.R., and NIH Fellowship F32EB024391 to J.M.B.
Authors' Contributions
L.P., A.K.R., J.M.B., and F.G. designed the study, L.P., A.K.R., and J.M.B. performed the experiments and data analysis, L.P., A.K.R., and F.G wrote the article.
Disclosure Statement
F.G. is a paid employee of Cytex Therapeutic, Inc. For the remaining authors, no competing financial interests exist.
Supplementary Material
References
- 1. Cisternas M.G., Murphy L., Sacks J.J., Solomon D.H., Pasta D.J., and Helmick C.G. Alternative methods for defining osteoarthritis and the impact on estimating prevalence in a US population-based survey. Arthritis Care Res (Hoboken) 68, 574, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abramson S.B., and Attur M. Developments in the scientific understanding of osteoarthritis. Arthritis Res Ther 11, 227, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kapoor M., Martel-Pelletier J., Lajeunesse D., Pelletier J.P., and Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol 7, 33, 2011 [DOI] [PubMed] [Google Scholar]
- 4. Rigoglou S., and Papavassiliou A.G. The NF-kappaB signalling pathway in osteoarthritis. Int J Biochem Cell Biol 45, 2580, 2013 [DOI] [PubMed] [Google Scholar]
- 5. Wehling P., Reinecke J., Baltzer A.W.A., et al. Clinical responses to gene therapy in joints of two subjects with rheuamtoid arthritis. Hum Gene Ther 20, 97, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moutos F.T., Glass K.A., Compton S.A., et al. . Anatomically shaped tissue-engineered cartilage with tunable and inducible anticytokine delivery for biological joint resurfacing. Proc Natl Acad Sci U S A 113, E4513, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ousema P.H., Moutos F.T., Estes B.T., et al. . The inhibition by interleukin 1 of MSC chondrogenesis and the development of biomechanical properties in biomimetic 3D woven PCL scaffolds. Biomaterials 33, 8967, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wehling N., Palmer G.D., Pilapil C., et al. . Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum 60, 801, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McNulty A.L., Moutos F.T., Weinberg J.B., and Guilak F. Enhanced integrative repair of the porcine meniscus in vitro by inhibition of interleukin-1 or tumor necrosis factor alpha. Arthritis Rheum 56, 3033, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Choy E.H., Kavanaugh A.F., and Jones S.A. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol 9, 154, 2013 [DOI] [PubMed] [Google Scholar]
- 11. Furman B.D., Mangiapani D.S., Zeitler E., et al. . Targeting pro-inflammatory cytokines following joint injury: acute intra-articular inhibition of interleukin-1 following knee injury prevents post-traumatic arthritis. Arthritis Res Ther 16, R134, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chevalier X., Goupille P., Beaulieu A.D., et al. . Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum 61, 344, 2009 [DOI] [PubMed] [Google Scholar]
- 13. Evans C.H., Robbins P.D., Ghivizzani S.C., et al. . Gene transfer to human joints: progress toward a gene therapy of arthritis. Proc Natl Acad Sci U S A 102, 8698, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evans C.H., Ghivizzani S.C., and Robbins P.D. Gene delivery to joints by intra-articular injection. Hum Gene Ther 29, 2, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matthews G.L., and Hunter D.J. Emerging drugs for osteoarthritis. Expert Opin Emerg Drugs 16, 479, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goldring M.B., and Berenbaum F. Emerging targets in osteoarthritis therapy. Curr Opin Pharmacol 22, 51, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramos-Casals M., Brito-Zeron P., Soto M.J., Cuadrado M.J., and Khamashta M.A. Autoimmune diseases induced by TNF-targeted therapies. Best Pract Res Clin Rheumatol 22, 847, 2008 [DOI] [PubMed] [Google Scholar]
- 18. Gopinath S.D., and Rando T.A. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell 7, 590, 2008 [DOI] [PubMed] [Google Scholar]
- 19. Kimmerling K.A., Furman B.D., Mangiapani D.S., et al. . Sustained intra-articular delivery of IL-1Ra from a thermally-responsive elastin-like polypeptide as a therapy for post-traumatic arthritis. Eur Cells Mater 29, 124, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mozzetta C., Minetti G., and Puri P.L. Regenerative pharmacology in the treatment of genetic diseases: the paradigm of muscular dystrophy. Int J Biochem Cell Biol 41, 701, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cucchiarini M., and Madry H. Biomaterial-guided delivery of gene vectors for targeted articular cartilage repair. Nat Rev Rheumatol 15, 18, 2019 [DOI] [PubMed] [Google Scholar]
- 22. Madry H., Orth P., and Cucchiarini M. Gene therapy for cartilage repair. Cartilage 2, 201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Auslander S., and Fussenegger M. Engineering gene circuits for mammalian cell-based applications. Cold Spring Harb Perspect Biol 8, a023895, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brunger J.M., Huynh N.P., Guenther C.M., et al. . Scaffold-mediated lentiviral transduction for functional tissue engineering of cartilage. Proc Natl Acad Sci U S A 111, E798, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gersbach C.A., Coyer S.R., Le Doux J.M., and Garcia A.J. Biomaterial-mediated retroviral gene transfer using self-assembled monolayers. Biomaterials 28, 5121, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brunger J.M., Zutshi A., Willard V.P., Gersbach C.A., and Guilak F. Genome engineering of stem cells for autonomously regulated, closed-loop delivery of biologic drugs. Stem Cell Reports 8, 1202, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hou C.H., Huang J., and Qian R.L. Identification of alpha NF-κB site in the negative regulatory element (e-NRAII) of human e-globin gene and its binding protein NF-κB p50 in the nuclei of K562 cells. Cell Res 12, 79, 2002 [DOI] [PubMed] [Google Scholar]
- 28. Nourbakhsh M., Hoffmann K., and Hauser H. Interferon-β promoters contain a DNA element that acts as a position-independent silencer on the NF-. EMBO J 12, 451, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wiznerowicz M., Szulc J., and Trono D. Tuning silence: conditional systems for RNA interference. Nat Methods 3, 682, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, and Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Kanda T., Sullivan K.F., and Wahl G.M. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol 8, 377, 1998 [DOI] [PubMed] [Google Scholar]
- 32. Szulc J., Wiznerowicz M., Sauvain M.O., Trono D., and Aebischer P. A versatile tool for conditional gene expression and knockdown. Nat Methods 3, 109, 2006 [DOI] [PubMed] [Google Scholar]
- 33. Salmon P., and Trono D. Production and titration of lentiviral vectors. Curr Protoc Hum Genet Chapter 12, Unit 12, 10, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Carey B.W., Markoulaki S., Hanna J., et al. . Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci U S A 106, 157, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Diekman B.O., Christoforou N., Willard V.P., et al. . Cartilage tissue engineering using differentiated and purified induced pluripotent stem cells. Proc Natl Acad Sci U S A 109, 19172, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hao S., and Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10, 281, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu W.W., Elkasabi Y., Chen H.Y., et al. . The use of reactive polymer coatings to facilitate gene delivery from poly (epsilon-caprolactone) scaffolds. Biomaterials 30, 5785, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moutos F.T., and Guilak F. Functional properties of cell-seeded three-dimensionally woven poly(epsilon-caprolactone) scaffolds for cartilage tissue engineering. Tissue Eng Part A 16, 1291, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farndale R.W., Buttle D.J., and Barrett A.J. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta 883, 173, 1986 [DOI] [PubMed] [Google Scholar]
- 40. Estes B.T., Diekman B.O., Gimble J.M., and Guilak F. Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nat Protoc 5, 1294, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McNulty A.L., Rothfusz N.E., Leddy H.A., and Guilak F. Synovial fluid concentrations and relative potency of interleukin-1 alpha and beta in cartilage and meniscus degradation. J Orthop Res 31, 1039, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glass K.A., Link J.M., Brunger J.M., Moutos F.T., Gersbach C.A., and Guilak F. Tissue-engineered cartilage with inducible and tunable immunomodulatory properties. Biomaterials 35, 5921, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Norrman K., Fischer Y., Bonnamy B., Wolfhagen Sand F., Ravassard P., and Semb H. Quantitative comparison of constitutive promoters in human ES cells. PLoS One 5, e12413, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Willard V.P., Diekman B.O., Sanchez-Adams J., Christoforou N., Leong K.W., and Guilak F. Use of cartilage derived from murine induced pluripotent stem cells for osteoarthritis drug screening. Arthritis Rheumatol 66, 3062, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fetter N.L., Leddy H.A., Guilak F., and Nunley J.A. Composition and transport properties of human ankle and knee cartilage. J Orthop Res 24, 211, 2006 [DOI] [PubMed] [Google Scholar]
- 46. Shea L.D., Smiley E., Bonadio J., and Mooney D.J. DNA delivery from polymer matrices for tissue engineering. Nat Biotechnol 17, 551, 1999 [DOI] [PubMed] [Google Scholar]
- 47. Phillips J.E., Burns K.L., Le Doux J.M., Guldberg R.E., and Garcia A.J. Engineering graded tissue interfaces. Proc Natl Acad Sci U S A 105, 12170, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yin G., Liu W., An P., et al. . Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther 5, 547, 2002 [DOI] [PubMed] [Google Scholar]
- 49. Gouze E., Pawliuk R., Pilapil C., et al. . In vivo gene delivery to synovium by lentiviral vectors. Mol Ther 5, 397, 2002 [DOI] [PubMed] [Google Scholar]
- 50. Kato K., Miyake K., Igarashi T., Yoshino S., and Shimada T. Human immunodeficiency virus vector-mediated intra-articular expression of angiostatin inhibits progression of collagen-induced arthritis in mice. Rheumatol Int 25, 522, 2004 [DOI] [PubMed] [Google Scholar]
- 51. Basile P., Dadali T., Jacobson J., et al. . Freeze-dried tendon allografts as tissue-engineering scaffolds for Gdf5 gene delivery. Mol Ther 16, 466, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu W.-W., Lang M.W., and Krebsbach P.H. Development of adenovirus immobilization strategies forin situgene therapy. J Gene Med 10, 1102, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hu W.W., Wang Z., Hollister S.J., and Krebsbach P.H. Localized viral vector delivery to enhance in situ regenerative gene therapy. Gene Ther 14, 891, 2007 [DOI] [PubMed] [Google Scholar]
- 54. Hu W.-W., Lang M.W., and Krebsbach P.H. Digoxigenin modification of adenovirus to spatially control gene delivery from chitosan surfaces. J Control Release 135, 250, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wiznerowicz M., and Trono D. Harnessing HIV for therapy, basic research and biotechnology. Trends Biotechnol 23, 42, 2005 [DOI] [PubMed] [Google Scholar]
- 56. Kumar M., Keller B., Makalou N., and Sutton R. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther 12, 1893, 2001 [DOI] [PubMed] [Google Scholar]
- 57. Zufferey R., Dull T., Mandel R.J., et al. . Self-inactivatig lentivirus vector for seafe and efficient in vivo gene delivery. J Virol 72, 9873, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schambach A., and Baum C. Clinical application of lentiviral vectors—concepts and practice. Curr Gene Ther 8, 474, 2008 [DOI] [PubMed] [Google Scholar]
- 59. Milone M.C., and O'Doherty U. Clinical use of lentiviral vectors. Leukemia 32, 1529, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Valonen P.K., Moutos F.T., Kusanagi A., et al. . In vitro generation of mechanically functional cartilage grafts based on adult human stem cells and 3D-woven poly(epsilon-caprolactone) scaffolds. Biomaterials 31, 2193, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rachakonda P.S., Rai M.F., and Schmidt M.F. Application of inflammation-responsive promoter for an in vitro arthritis model. Arthritis Rheum 58, 2088, 2008 [DOI] [PubMed] [Google Scholar]
- 62. Badr C.E., Niers J.M., Tjon-Kon-Fat L.-A., Noske D.P., Wurdinger T., and Tannous B.A. Real-time monitoring of nuclear factor κB activity in cultured cells and in animal models. Mol Imaging 8, 278, 2009 [PMC free article] [PubMed] [Google Scholar]
- 63. van de Loo F.A., de Hooge A.S., Smeets R.L., et al. . An inflammation-inducible adenoviral expression system for local treatment of the arthritic joint. Gene Ther 11, 581, 2004 [DOI] [PubMed] [Google Scholar]
- 64. Khoury M., Adriaansen J., Vervoordeldonk M.J.B.M., et al. Inflammation-inducible anti-TNF gene expression mediated by intra-articular injection of serotype 5 adeno-associated virus reduces arthritis. J Gene Med 9, 596, 2007 [DOI] [PubMed] [Google Scholar]
- 65. Adriaansen J., Khoury M., de Cortie C.J., et al. . Reduction of arthritis following intra-articular administration of an adeno-associated virus serotype 5 expressing a disease-inducible TNF-blocking agent. Ann Rheum Dis 66, 1143, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brunger J.M., Zutshi A., Willard V.P., Gersbach C.A., and Guilak F. CRISPR/Cas9 editing of murine induced pluripotent stem cells for engineering inflammation-resistant tissues. Arthritis Rheumatol 69, 1111, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Farhang N., Brunger J.M., Stover J.D., et al. . CRISPR-based epigenome editing of cytokine receptors for the promotion of cell survival and tissue deposition in inflammatory environments. Tissue Eng Part A 23, 738, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gossen M., and Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A 89, 5547, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Greber D., El-Baba M.D., and Fussenegger M. Intronically encoded siRNAs improve dynamic range of mammalian gene regulation systems and toggle switch. Nucleic Acids Res 36, e101, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kramer B.P., Viretta A.U., Daoud-El-Baba M., Aubel D., Weber W., and Fussenegger M. An engineered epigenetic transgene switch in mammalian cells. Nat Biotechnol 22, 867, 2004 [DOI] [PubMed] [Google Scholar]
- 71. Wroblewska L., Kitada T., Endo K., et al. . Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat Biotechnol 33, 839, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xie Z., Wroblewska L., Prochazka L., Weiss R., and Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science 333, 1307, 2011 [DOI] [PubMed] [Google Scholar]
- 73. Chen Y.Y., Jensen M.C., and Smolke C.D. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc Natl Acad Sci U S A 107, 8531, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Perez-Pinera P., Ousterout D.G., Brunger J.M., et al. . Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 10, 239, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qi L.S., Larson M.H., Gilbert L.A., et al. . Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Boyle P.M., and Silver P.A. Harnessing nature's toolbox: regulatory elements for synthetic biology. J R Soc Interface 6 Suppl 4, S535, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Auslander S., and Fussenegger M. From gene switches to mammalian designer cells: present and future prospects. Trends Biotechnol 31, 155, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.