

 This article provides an overview of the chemotypes that have been investigated or under investigation for the development of novel neuropathic pain treatments targeting the CB1 receptor.
This article provides an overview of the chemotypes that have been investigated or under investigation for the development of novel neuropathic pain treatments targeting the CB1 receptor.
Abstract
Neuropathic pain is caused by a lesion or dysfunction in the nervous system, and it may arise from illness, be drug-induced or caused by toxin exposure. Since the discovery of two G-protein-coupled cannabinoid receptors (CB1 and CB2) nearly three decades ago, there has been a rapid expansion in our understanding of cannabinoid pharmacology. This is currently one of the most active fields of neuropharmacology, and interest has emerged in developing cannabinoids and other small molecule modulators of CB1 and CB2 as therapeutics for neuropathic pain. This short review article provides an overview of the chemotypes currently under investigation for the development of novel neuropathic pain treatments targeting CB1 receptors.
Introduction
Neuropathic pain is a complex, chronic pain state caused directly by a lesion or disease affecting the somatosensory nervous system.1,2 Neuropathic pain may be spontaneous, such as a painful response to a non-painful stimulus (allodynia), or evoked as an exaggerated response to a painful stimulus (hyperalgesia).3 Diagnosis of neuropathic pain requires a history of injury to the nervous system (the peripheral nerve, dorsal root or dorsal root ganglion, or central nervous system) and is a common consequence of stroke, surgical or chemotherapeutic nerve trauma, and diabetic neuropathy.4
Neuropathic pain is estimated to affect one in every ten adults over the age of 30 in the United States,5 significantly impacts quality of life,6 is associated with a three-fold increase in direct healthcare costs,7 and contributes significantly to the $100 billion annual indirect costs attributed to chronic pain conditions due to absenteeism and decreased productivity.8,9
The current first-line treatments for neuropathic pain are tricyclic antidepressants (nortriptyline, desipramine)10 and anticonvulsants (gabapentin, pregabalin);11–13 however, many patients report incomplete relief as well as dose-limiting adverse effects of these drugs.14–16 Opioid drugs are a second-line treatment for neuropathic pain since they are ineffective in many patients and chronic opioid use is associated with adverse reactions, tolerance, and addiction.17,18 The current mini-review aims to provide an overview of the chemotypes currently under investigation for the development of novel neuropathic pain treatments targeting CB1 receptors.
The endocannabinoid system and neuropathic pain models
The endocannabinoid (eCB) system is ubiquitously expressed throughout the body and is responsible for the homeostatic control of many basic physiological processes. Modulation of the endocannabinoid system has been proposed as a promising platform for the treatment of nociceptive pain for over a decade.19–22 The eCB system is composed of two well-characterized G protein-coupled receptors (GPCRs), the cannabinoid type 1 and type 2 receptors (CB1 and CB2, respectively), endogenous signaling lipids such as anandamide (arachidonoylethanolamide, AEA, 1, Fig. 1)23 and 2-arachidonoylglycerol (2-AG, 2),24 and associated metabolic enzymes like fatty acid amide hydrolase (FAAH)25 and monoacylglycerol lipase (MAGL).26 Both FAAH and MAGL are key enzymes in the hydrolysis of the endocannabinoid 2-arachidonoylglycerol (2-AG). Due to their ability to regulate nociception, they are currently viewed as attractive drug targets for the treatment of pain. Activation of CB1 and CB2 receptors is known to reduce nociceptive signaling, and nociceptive processing is topically controlled by endocannabinoids, like AEA and 2-AG.27
Fig. 1. Selected endogenous cannabinoids.

Anandamide is believed to serve as a natural pain modulator28 as several pertinent anandamide targets within the eCB system have been explored for pain treatment.29 Recent evidence from rodent models indicates that novel approaches targeting the eCB system might be beneficial in refractory neuropathic pain. However, consensus on whether single or multitarget modulators of the cannabinoid system represent the most effective therapeutic platforms for the treatment of neuropathic pain has not been reached.30,31
Translational efforts to date have focused on agonists of CB1 and CB2, as well as inhibitors of endocannabinoid metabolism.32 Ligands that activate central CB1 receptors via their orthosteric site produce psychoactive effects, limiting the broad utility of such drugs. Selective CB2 agonists have demonstrated efficacy in preclinical models of neuropathic pain, but despite extensive efforts by the pharmaceutical industry, no CB2 agonist has advanced to the market.33 Selective inhibitors of FAAH and MAGL, as well as dual inhibitors, have also shown promise in preclinical models, but have largely failed to meet end-points in clinical trials.34
Many rodent models of neuropathic pain were developed to evaluate the activity of new drug candidates wherein the efficacy of the eCB system's modulators was noted.35 All rodent models of neuropathic pain involve surgical manipulation of the peripheral nervous system to induce injury and inflammatory response. Although countless other models are available,36 the most commonly employed model for persistent neuropathic pain in rodents is partial sciatic nerve injury,37–39 which is caused by partial ligation of the sciatic nerve,40 chronic constriction injury (CCI),41 or L5 and L6 spinal nerve ligation (SNL).42 Rodent models of neuropathic pain aim to reproduce characteristics of human neuropathic pain, including mechanical allodynia and thermal hyperalgesia, and it is typically these features of pain that are assessed.43
The CB1 receptor and pain
The cannabinoid 1 (CB1) receptor is one of the most abundant G protein-coupled receptors (GPCRs) in the central nervous system, with key roles during neurotransmitter release and synaptic plasticity.44–46 CB1 has been cloned and encodes 473 amino acid protein.47 Like other class A G protein-coupled receptors (GPCRs), CB1R possesses six helical transmembrane domains, an extracellular N-terminus and an intracellular C-terminus. Upon ligand activation, CB1Rs may signal in three different spatiotemporal waves. The first wave, which is transient (<10 minutes) and initiated by heterotrimeric G proteins, is followed by a second wave (>5 minutes) that is mediated by β-arrestins.46–48 The third and final wave occurs at intracellular compartments and could be elicited by G proteins or β-arrestins. This complexity presents multiple challenges, including the correct classification of receptor ligands, the identification of the signaling pathways regulated by each wave, and the underlying molecular mechanisms and physiological impacts of these waves. It has been shown that upon small molecule agonist-induced activation, β-arrestins are recruited to the plasma membrane to initiate clathrin-mediated endocytosis (CME) and signaling. Compounds that activate CB1 were suggested as possible treatments for a wide range of medical disorders including pain, inflammation, glaucoma, nausea and emesis, neurodegenerative disorders, anxiety, and hypertension.
Like many GPCRs, the CB1 receptor is capable of ligand- and heterodimer-directed functional selectivity.49 The CB2 receptor is also known to possess diverse, ligand-directed signaling bias.50 There is also the physical and functional interplay between CB1 receptors and other CNS targets of relevance to pain, including D2 dopamine receptors, mu-, kappa-, and delta-opioid receptors (MORs, KORs, and DORs, respectively), the orexin-1 receptor, and the A2A adenosine receptor. It has been seen that following peripheral nerve lesion in rats, there is upregulation of a CB1–DOR heteromeric complex in the cortex of neuropathic animals, with concomitant changes in the activity of each protein.51
The development of CB1 knock-out (KO) mice has provided insights regarding the role of CB1 receptors in nociception. CB1 KO mice are hypoalgesic52 and demonstrate a reduced analgesic response to cannabinoids,53 however, CB1 KO mice still develop neuropathic pain,54,55 consistent with the complex pathophysiology of neuropathic pain. Spinal and supraspinal CB1 receptors are upregulated following nerve injury, and this may account for the efficacy of CB1 agonists in neuropathic pain.56 Indeed, CB1 receptors are upregulated in many rat models of neuropathic pain.57–59
Phytocannabinoids targeting CB1
The therapeutic activity of exogenous phytocannabinoids found in the cannabis plant supports the potential of the CB1 receptor as a relevant platform for the development of novel analgesics.60 The earliest indication that modulation of the CB1 receptor might be effective in the treatment of pain comes from the historical use of the cannabis plant. The recorded medicinal use of the cannabis plant (Cannabis sativa L.) for pain and other conditions dates back to approximately 2700 BCE.61 The total number of natural compounds identified in cannabis exceeds 480, of which the most important pharmacologically active members are the phenolic terpenoids collectively known as phytocannabinoids.62,63 The most abundant phytocannabinoids are Δ9-tetrahydrocannabinol (Δ9-THC, 3, Fig. 2)64 and cannabidiol (CBD, 4), both found in cannabis as their acid derivatives, each of which have found application in the treatment of neuropathic pain.
Fig. 2. Selected phytocannabinoids and analogs.

Phytocannabinoids interact non-selectively with many targets of the human endocannabinoid system beyond CB1 receptors. Many also interact with non-cannabinoid targets, some of which are relevant to neuropathic pain.65
The efficacy of medical cannabis itself in numerous pain conditions has been established and reviewed elsewhere.66,67 The utility of phytocannabinoids as lead structures for the development of novel analgesics targeting CB1 is confounded by their complex pharmacology.68 For example, Δ9-THC acts as a potent, low efficacy agonist of CB1 and CB2 receptors, while CBD is a negative allosteric modulator (NAM) of CB1,69 yet both compounds produce analgesic effects in rodent models. It is believed that non-cannabinoid targets like spinal α3 glycine receptors mediate the analgesic properties of CBD.70,71
Despite the therapeutic potential of phytocannabinoid-derived drugs, surprisingly few phytocannabinoids or phytocannabinoid analogues have reached the clinic. For example, oral Δ9-THC, known generically as dronabinol (Marinol®, Unimed), was approved by the FDA for nausea associated with chemotherapy in 1985. Nabiximols (Sativex®, GW Pharmaceuticals), first passed in the United Kingdom in 2010, is an oral spray formulation of Δ9-THC and CBD and is now available in numerous countries. Nabiximols is currently undergoing phase III trials in the US for the treatment of cancer pain. A synthetic analogue of Δ9-THC, nabilone (5), was approved by the FDA in 1985 and is sold as Cesamet® (Valeant Pharmaceuticals) for the treatment of refractory chemotherapy-induced nausea and vomiting (CINV).
Synthetic cannabinoids targeting CB1
Beyond phytocannabinoids, many synthetic cannabinoids were explored as potential analgesics in the 1980s, and several pharmaceutical companies had active cannabinoid analgesic research programs. These cannabinoid drug development programs produced some of the earliest CB1/CB2 and CB1-selective agonists still in use as pharmacological tool molecules, including WIN 55,212-2 (6, Fig. 2) from Sterling Winthrop and CP 55,940 (7) from Pfizer. Mechoulam and colleagues at Hebrew University also produced phytocannabinoid-inspired ligands intended to probe the cannabinoid function, such as HU-210 (8).72
Like Δ9-THC, these cannabinoids are typically orthosteric CB1 agonists, albeit more potent and efficacious than Δ9-THC. For example, in the chronic constriction injury (CCI) model of neuropathic pain in rats, mechanical allodynia and thermal hyperalgesia were strongly attenuated by Δ9-THC at doses of 3 mg kg–1 and 6 mg kg–1 (per os p.o.), respectively, whereas CP 55,940 produced similar effects at 0.05 mg kg–1 and 0.025 mg kg–1 (intraperitoneal i.p. injection), respectively.73 Both cannabinoids were more potent than traditional neuropathic pain treatments such as morphine (8 mg kg–1 and 16 mg kg–1 p.o., respectively) and gabapentin (50 mg kg–1 i.p. in both tests). CP 55,940 also attenuated tactile allodynia in the rat spinal nerve ligation (SNL) model of neuropathic pain, an effect that was reversed by a selective CB1 antagonist.74 The antinociceptive and motor effects of CP 55,940 observed in wild-type mice are absent in CB1 but not CB2 KO mice, indicating that CP 55,940 exerts its analgesic properties via orthosteric activation of CB1 receptors.75 CP 55,940, WIN 55,212-2 and HU-210 all produced complete reversal of mechanical hyperalgesia in the partial sciatic nerve ligation model of neuropathic pain.56,76–81
The psychoactivity and motor impairment of Δ9-THC and synthetic CB1 orthosteric agonists limit their broad application as an analgesic agent; therefore, CB1 agonists without central activity have been suggested as therapeutics.82 Several strategies for selective modulation of CB1 have been proposed, with focus on reducing adverse reactions and psychotropic activity.83,84 In this regard, the peripherally restricted CB1 agonists retain antinociceptive effects without apparent psychoactivity and are currently under development.85 Another alternative is the development of CB1 positive allosteric modulators (PAMs) or functionally selective CB1 ligands that induce biased signaling.86
Peripherally-restricted CB1 agonists
The use of centrally active CB1 agonists for neuropathic pain treatment is limited by centrally mediated side effects. The same is true for low efficacy agonists such as Δ9-THC and synthetic partial agonists with a similar pharmacological profile, like BAY 59-3074 (9).87 However, there is pharmacological evidence that both central and peripheral CB1 receptors mediate CB1 antihyperalgesia.76,88
Ajulemic acid (CT-3, 10), a synthetic analogue of Δ9-THC, is efficacious in neuropathic pain in rodents and humans and showed activity in a phase II trial.89,90 Curiously, ajulemic acid is an agonist at both CB1 and CB2 receptors,91 yet it does not produce psychotropic effects.89,90 The action mechanisms of ajulemic acid in neuropathic pain are not entirely understood,92 but its negligible adverse effect profile is believed to be due to reduced CNS penetration.91,93,94 Other potential peripherally restricted CB1 antagonists are under development for metabolic disorders and provide proof-of-principle for the development of CB1 agonists with reduced CNS liabilities.95,96 Although ajulemic acid has shown promise in clinical trials, phytocannabinoids are not ideal drug leads for peripherally-restricted CB1 agonists owing to their high lipophilicity and limited scope of structural modification.
The aminoalkylindole (AAI) class of CB1 agonists, typified by drugs like WIN 55,212-2, has served as a useful scaffold for the development of cannabinoid agonists with an array of physicochemical and functional properties. WIN 53,365 (11, Fig. 3) was selected as the lead for the generation of peripheral CB1 agonists by Novartis.97 The indole core was substituted with naphthalene based on previous bioequivalency data,98 and a 1,4-disubstitution pattern was selected to maintain optimal spatial orientation of key functional groups (12). Investigation of numerous pendant ether substituents was based on prior structure–activity relationships reported for other AAIs,99,100 with a pentyl ether found providing greater hCB1 binding (CRA13, 13, IC50 = 15 nM) than a butyl (14, IC50 = 48 nM) or hexyl (15, IC50 = 160 nM) ether.
Fig. 3. Structures and activities of selected ligands reported during the discovery of CRA13.

CRA13 (also known as SAB-378), i.e., naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone, was advanced as a development candidate and found to possess potent, orally bioavailable CB1/CB2 agonist activity with restricted CNS penetration.101 CRA13 produced up to 90% hyperalgesia reversal when administered orally at 3 mg kg–1 in a rat neuropathic model of mechanical hyperalgesia with rapid onset and long duration of action and without apparent CNS-mediated side effects. The antihyperalgesic effects of CRA13 were inhibited by a CB1-selective inverse agonist/antagonist, rimonabant, but not by a CB2-selective antagonist, SR 144528 (need structure). In rats, CRA13 only produced central side effects at doses 170-fold higher than those required for antihyperalgesic activity. The human pharmacokinetic data for CRA13 were reported, and adverse effects were observed at high doses similar to those reported for Δ9-THC, which are likely due to CNS penetration at high plasma concentrations.102
AstraZeneca has explored many peripherally-restricted CB1 agonists for their efficacy in rodent models of neuropathic pain, resulting in clinical candidates AZ11713908 (17, Fig. 4)103 and AZD1940 (18),104 arising from the extensive exploration of the 5-sulfonamide benzimidazole scaffold.
Fig. 4. Peripherally-restricted CB1 agonists under development by AstraZeneca.

AZ11713908 demonstrated high affinity for CB1 and CB2 receptors derived from mouse, rat, and human sources (hCB1 pIC50 8.4, hCB2 pIC50 9.0), but little selectivity between subtypes.103 This drug showed potent, efficacious agonist activity at hCB1 in the GTPγS assay (pEC50 8.0, Emax = 115%) compared to WIN 55,212. It was noted that compared to WIN 55,212, which showed a 2.2- and 4.2-fold increase in the brain compared to plasma in rat and mouse, respectively, AZ11713908 showed just 7% and 5% brain uptake compared to plasma in the same species. In the rat spinal nerve ligation (SNL) model of neuropathic pain, systemic administration of 2.5 μmol kg–1 AZ11713908 reduced mechanical allodynia with 100% efficacy, whereas 8 μmol kg–1 WIN 55,212 was required to achieve the same effect.
AZD1940 is a high affinity hCB1/hCB2 agonist with pKi values of 7.93 and 9.06 at each cannabinoid receptor, respectively.104 AZD1940 has a brain–plasma partition coefficient of 0.04 in rats, comparable to that of AZ11713908. Low brain uptake was confirmed using positron emission tomography (PET) imaging of its carbon-11-labeled isotopologue ([11C] AZD1940).105 Despite its promising preclinical profile, AZD1940 showed limited analgesic efficacy against capsaicin-induced pain and hyperalgesia in healthy humans and mild-to-moderate CNS and gastrointestinal adverse effects.106 AZD1940 also failed to reduce post-operative pain after surgical molar removal and produced central cannabinoid side-effects.107 Although AZD1940 reached phase II trials, it was discontinued in 2009.107
By scaffold hopping from a benzimidazole class of cannabinoid ligands,108 researchers at AstraZeneca identified hit 19 (Fig. 5) as the first of a new γ-carboline class of cannabinoids.10919 demonstrated moderate affinity for both CB receptors (hCB1Ki = 143 nM, hCB2Ki = 14 nM) and functioned suitably as an agonist (rCB1 EC50 = 1440 nM, Emax = 47%). Additionally, 18 had reasonable aqueous solubility (>3 mg mL–1 at pH 7.4), unlike many classes of cannabinoids.
Fig. 5. Effect of the terminal carboline amine substituent on CB1 agonist activity and PSA.

Starting from hit 19, with a moderate brain-to-plasma ratio of 0.74, AstraZeneca aimed to increase the polar surface area (PSA) based on the finding that marketed non-CNS drugs have higher PSA values than CNS drugs (mean PSA of 56 and 40, respectively).110 A range of alicyclic, heterocyclic, aromatic, and heteroaromatic groups were explored at the terminal carboline nitrogen, with all leading to increased hCB1 potency and comparable efficacy, except for various pyridines. The cyclopentyl (20, EC50 = 33 nM, Emax = 65%) and 4-tetahydropyranyl (21, EC50 = 60 nM, Emax = 57%) analogues showed the greatest improvement in potency without compromising metabolic stability (HLM clearance of 93 and 42 μl min–1 mg–1, respectively) or decreasing the PSA (21 and 30, respectively) (Fig. 5).
While retaining the cyclopentyl and 4-tetrahydropyranyl groups of 20 and 21, an array of polar substituents was introduced at the carboline central nitrogen. All alkyl substituents (methyl, ethyl, and propyl) were tolerated with minimal effects on CB1 agonist potency, efficacy, or PSA. Compared to propyl analogue 22 (EC50 = 103 nM, Emax = 72%, PSA 30), introduction of a carbonyl spacer as in 23 increased the PSA to 48 with little effect on potency (EC50 = 117 nM), but the efficacy was dramatically reduced (Emax = 17%). The PSA was further increased by replacing the carbonyl spacer with a sulfone (24, PSA 67), with little impact on potency and reinstatement of efficacy (Emax = 100%). Truncation of the propyl group of 24 to an ethyl group (25; EC50 = 49 nM, Emax = 120%, PSA 67) furnished a potent CB1 agonist with a suitably high PSA, whereas further truncation to a methylsulfone reduced the potency 10-fold (26; EC50 = 473 nM). Attempts to further increase the PSA by introduction of a sulfonamide (e.g., 27; PSA 83) completely eliminated the activity (Fig. 6).
Fig. 6. Effect of the central carboline amine substituent on CB1 agonist activity and PSA.

Compound 25 was selected as a potent, efficacious agonist at hCB1 (EC50 = 49 nM, Emax = 120%) and rCB1 (EC50 = 85 nM, Emax = 156%), with no appreciable activity at more than 50 other CNS targets, including pain targets. The brain–plasma partition coefficient for 25 was the same as for AZ11713908 (0.07), and it showed a promising pharmacokinetic profile as well as good aqueous solubility. In rats, carboline 25 showed reasonable oral bioavailability (16%) and low CYP inhibition but moderate hERG activity. In the rat carrageenan inflammatory pain model, 25 showed potent, dose-dependent reversal of thermal hyperalgesia with mild hypoactivity observed only at the highest dose (1.4 mg kg–1 subcutaneous, s.c.). No further reports of the translation of 25 have appeared.
Researchers at Merck have also employed the strategy of reducing CNS penetration by increasing the PSA.111 Using MONIKA, a web tool developed internally with Organon, Merck optimized parameters related to both oral bioavailability and CNS permeability (used as exclusion criteria), to further optimize a previously developed indole cannabinoid with desirable properties (28, Fig. 7). Compound 28 hydrochloride was a moderately potent CB1 agonist (pEC50 = 7.4) with good oral bioavailability in the rat. Previous SAR studies had indicated that both oxadiazole and thiadiazoles were tolerated, but 29 showed inferior potency and PSA compared to lead 30. Introducing a sulfone in place of the tetrahydropyran ether bridge, as in 29, increased the PSA but reduced the potency. Introduction of polar groups as pendant substituents of the oxadiazole ring was found to increase the potency and PSA (31 and 32).
Fig. 7. Peripherally-restricted indole-3-heterocycle CB1 agonists.

Compound 32 is a moderately potent agonist of CB1 (pKi 7.7, pEC50 7.1) with negligible selectivity over CB2 (pKi 7.7). LBP1 had a suitably low brain-to-plasma ratio in mouse (0.16) and good aqueous solubility (89 mg L–1) and oral bioavailability (45%). 32 showed antihyperalgesic effects in the rat Chung Hagreaves model and antiallodynic effects in the rat Chung Von Frey model, while the CB1 antagonist rimonabant blocked these effects. As with other centrally penetrant CB1 agonists, WIN 55,212-2 produced robust catalepsy in rats, whereas 32 induced no effects in the rat catalepsy model up to a maximum oral dose of 160 μmol kg–1. Compound 32 has progressed to phase I clinical trials, but no further details have been reported.
Inspired by WIN 55,225 (or JWH-200) and to utilize the benefits of CBs to ameliorate neuropathic pain, a study with a series of 3-alkylated indenes was recently reported, where peripherally restricted cannabinoids were designed and tested for selectivity by introduction of charge to CB1. Screening of compounds at various doses and drug administration routes identified 4-{2-[-(1E)-1[(4-propylnaphthalen-1-yl)methylidene]-1H-inden-3-yl]ethyl}morpholine (PrNMI, 33) as the most promising compound (Fig. 8).112
Fig. 8. Peripherally restricted CB1 agonists with efficacy in rodent models of neuropathic pain.
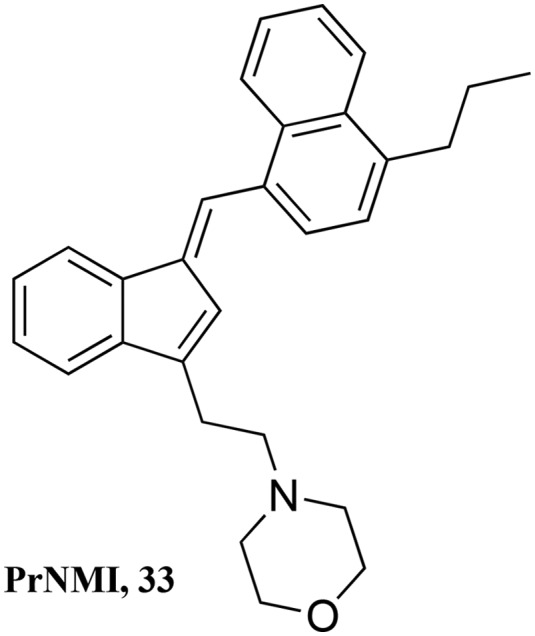
This CB1 agonist showed repeated suppression of neuropathy symptoms with a lack of side effects mediated by activation of central CB1 receptors. The potency, peripheral selectivity, in vivo efficacy, and absence of CNS side effects of the peripherally restricted cannabinoids suggest promising further development of such compounds for viable treatment for neuropathic pain states.
Positive allosteric modulators (PAMs) of CB1
The therapeutic utility of allosteric interactions with GPCRs is an emerging area of rational drug design enabled by advances in structural and computational biology.113–115 As with many class A GPCRs, CB1 contains an allosteric site that can be targeted by small molecules.116–119 Several endogenous allosteric modulators of CB1 were recently identified, including pregnenolone (34, Fig. 9), lipoxin A4 (35)120 and the dodecapeptide pepcan-12. The off-target activity of these ligands for CB1 allosteric modulation limits their utility as lead structures for the rational design of CB1 PAMs.
Fig. 9. Selected endogenous and exogenous allosteric modulators of CB1.

The first identified CB1 AM was the indole-2-carboxamide Org27569 (35). Org27569 enhances the CB1 binding of selective cannabinoid ligands, yet acts as an insurmountable antagonist in several biochemical assays.121 The activity of Org27569 was recently explored in vivo, and it is unclear if Org27569 functions as a positive or negative allosteric modulator of CB1 in rodents.122–124 Nguyen et al. found that the modification of 1H-indole-2-carboxamides compounds related to Org27569 (35), i.e. Org27759 (36), and Org29647 (37), showed modulation potency of this series at the CB1 receptor which was enhanced by the presence of a diethylamino group at the 4-position of the phenyl ring, a chloro or fluoro group at the C5 position and short alkyl groups at the C3 position on the indole ring.125 The most active compound (37) had an IC50 value of 79 nM which is ∼2.5- and 10-fold more potent than the parent compounds 35 and 36, respectively. These compounds appeared to be negative allosteric modulators at the CB1 receptor.125 Org27569 has served as a useful scaffold for the development of CB1 NAMs intended to probe the CB1 structure and function,126–130 and several structurally distinct CB1 NAMs have also been identified, including PSNCBAM-1 (38).131,132
Although CB1 NAMs may have utility in obesity and other conditions, fewer CB1 PAMs are known. By enhancing the signaling of endogenous CB1 agonists, CB1 PAMs may represent a relatively safe and efficacious treatment option for neuropathic pain. ZCZ-011 (39) was recently shown to enhance CB1 signaling and reverse nociceptive behavior in neuropathic and inflammatory pain models, without apparent psychoactive effects in mice.133,134 GAT-211 (40) is a CB1 PAM but has not been explored in vivo. GAT-211 was also resolved into its (+)- and (–)-enantiomers (GAT-228, 41, and GAT-229, 42), revealing a stereochemical basis for differences in signaling bias.135 Because the endocannabinoid system can affect dopamine neurotransmission and cause hypolocomotion,136 the anomalous pharmacology of the dopamine transporter (DAT) inhibitor JHW007 (43) led to the discovery of RTI-371 (44) as the first tropane with PAM activity at CB1 receptors (Fig. 10).137
Fig. 10. Selected PAM modulator of CB1.

The potential of biased CB1 ligands for pain
The cannabinoid CB1 receptor has been implicated in the treatment of drug addiction, pain, appetite disorders, and other CNS related diseases.138–141 For example, Huntington disease (HD) is a neurodegenerative disorder in which there is a decrease in the levels of type 1 cannabinoid receptor (CB1) mRNA and protein in the medium spiny projection neurons of the caudate and putamen.142–145 A comparative analysis of six cannabinoids tested for signaling bias in in vitro models of medium spiny projection neurons expressing wild-type (STHdhQ7/Q7) or mutant huntingtin protein (STHdhQ111/Q111) showed that Gαi/o- and Gβγ-selective CB1 ligands are probably the most therapeutically useful cannabinoids in the treatment of HD. However, highly potent synthetic cannabinoids, such as WIN, could produce unwanted psychoactive effects and their chronic use would probably result in receptor desensitization or downregulation.146,147 When administered directly, endocannabinoids, which enhance Gαi/o- and Gβγ-dependent signaling in the STHdh cell culture system, are rapidly metabolized in vivo and consequently have limited efficacy.148,149 The inhibitor of endocannabinoid catabolism URB597 has demonstrated limited efficacy at improving motor control deficits in R6/2 HD mice,150,151 but additional studies are needed to understand how elevated endocannabinoid levels affects the signs and symptoms of HD in vivo (Fig. 11).
Fig. 11. CB1 ligands for the treatment of Huntington disease.

Cannabinoid agonists displayed distinct biased signaling profiles at CB1.152 Therefore, the clinical application of cannabinoid ligands has been hampered owing to their adverse on-target effects. Ligand-biased signaling from, and allosteric modulation of, CB1 offer pharmacological approaches that may enable the development of improved CB1 drugs, through modulation of only therapeutically desirable CB1 signaling pathways. There is growing evidence that CB1Rs are subject to ligand-biased signaling and allosterism.153 Quantification of ligand-biased signaling and allosteric modulation at CB1 using cyclic adenosine monophosphate (cAMP)154 signaling assay showed that cannabinoid agonists displayed distinct biased signaling of CB1R. For instance, whereas 2-arachidonylglycerol and WIN 55,212-2 (6) [(R)-(1)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-napthalenylmethanone] showed little preference for inhibition of cAMP and phosphorylation of extracellular signal-regulated kinase 1/2 (pERK1/2), N-arachidonoylethanolamine (anandamide), methanandamide, CP55940 (7) [2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxy propyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol] and HU-210 (8) [11-hydroxy-d-THC-dimethylheptyl] were biased toward cAMP inhibition. The small-molecule allosteric modulator Org27569 [5-chloro-3-ethyl-1H-indole-2-carboxylic acid[2-(4-piperidin-1-yl-phenyl)ethyl]amide] displayed biased allosteric effects by blocking cAMP inhibition mediated by all cannabinoid ligands tested, at the same time having little or no effect on ERK1/2 phosphorylation mediated by a subset of these ligands. Org27569 also displayed negative binding cooperativity with [H] SR141716, i.e. [5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide]; however, it had minimal effects on binding of cannabinoid agonists. Furthermore, we highlight the need to validate the reported allosteric effects of the endogenous ligands lipoxin A4 and pregnenolone at CBRs. Pregnenolone but not lipoxin A4 displaced [H] SR141716A, but there was no functional interaction between either of these ligands and cannabinoid agonists. This study demonstrates an approach to validating and quantifying ligand-biased signaling and allosteric modulation at CBRs, revealing ligand-biased “fingerprints” that may ultimately allow the development of improved CBR-targeted therapies.
Evidence suggests that GPCRs can adopt multiple conformations and these might explain biased signaling—the phenomenon where different drugs binding to the same orthosteric site on the receptor can cause activation of different signaling pathways, such as β-arrestin signaling.155 The structural dynamic study of allosteric inactivation of CB1R showed that a previously unidentified structure is induced in the marijuana receptor CB1 by an unusual allosteric ligand that blocks G-protein signaling but increases agonist binding and elicits biased signaling, which suggests that a common structural state may exist for β-arrestin biased signaling, one that can also be attained by allosteric ligand binding.156 Together all these studies constitute a comprehensive description of signaling from CB1 and suggest modulation of receptor endocytic trafficking as a therapeutic approach.157 Therefore, to effectively match cannabinoids with therapeutic goals, these compounds must be screened for their signaling bias.
Overall, the CB1 receptor plays an important role in diverse processes such as pain, cognition, metabolism, etc. However, the psychoactive side effects of CB1 activation in the brain have limited the use of CB1 ligands as drugs. The endocannabinoid system is ubiquitously expressed throughout the body and is responsible for the homeostatic control of many basic physiological processes. Thus, a great opportunity exists for the development of cannabinoid-based drugs for a wide range of therapeutic applications. When new strategies are developed to mitigate the side effects, tremendous potential exists for future development of efficacious drugs targeting CB1 for a variety of disease states.
Abbreviations
- AAI
Aminoalkylindole
- CBD
Cannabidiol
- CCI
Chronic constriction injury
- GPCR
G protein-coupled receptor
- SNL
Spinal nerve ligation
- THC
Tetrahydrocannabinol
Conflicts of interest
There are no conflicts to declare.
References
- Treede R.-D., Jensen T. S., Campbell J. N., Cruccu G., Dostrovsky J. O., Griffin J. W., Hansson P., Hughes R., Nurmikko T., Serra J. Neurology. 2007;70:1630–1635. doi: 10.1212/01.wnl.0000282763.29778.59. [DOI] [PubMed] [Google Scholar]
- Jensen T. S., Baron R., Haanpaa M., Kalso E., Loeser J. D., Rice A. S., Treede R.-D. Pain. 2011;152:2204–2205. doi: 10.1016/j.pain.2011.06.017. [DOI] [PubMed] [Google Scholar]
- Treede R.-D., Rief W., Barke A., Aziz Q., Bennett M. I., Benoliel R., Cohen M., Evers S., Finnerup N. B., First M. B., Giamberardino M. A., Kaasa S., Kosek E., Lavand'homme P., Nicholas M., Perrot S., Scholz J., Schug S., Smith B. H., Svensson P., Vlaeyen J. W., Wang S.-J. Pain. 2015;156:1003–1007. doi: 10.1097/j.pain.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf C. J., Mannion R. J. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Yawn B. P., Wollan P. C., Weingarten T. N., Watson J. C., Hooten W. M., Melton L. J. Pain medicine. 2009;10:586–593. doi: 10.1111/j.1526-4637.2009.00588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen M. P., Chodroff M. J., Dworkin R. H. Neurology. 2007;68:1178–1182. doi: 10.1212/01.wnl.0000259085.61898.9e. [DOI] [PubMed] [Google Scholar]
- Berger A., Dukes E. M., Oster G. J. Pain. 2004;5:143–149. doi: 10.1016/j.jpain.2003.12.004. [DOI] [PubMed] [Google Scholar]
- McCarberg B. H., Billington R. Am. J. Manag. Care. 2006;12:S263–S268. [PubMed] [Google Scholar]
- Gaskin D. J., Richard P. J. Pain. 2012;13:715–724. doi: 10.1016/j.jpain.2012.03.009. [DOI] [PubMed] [Google Scholar]
- McQuay H. J., Tramér M., Nye B. A., Carroll D., Wiffen P. J., Moore R. A. Pain. 1996;68:217–227. doi: 10.1016/s0304-3959(96)03140-5. [DOI] [PubMed] [Google Scholar]
- Bennett M. I., Simpson K. H. Palliat. Med. 2004;18:5–11. doi: 10.1191/0269216304pm845ra. [DOI] [PubMed] [Google Scholar]
- Guay D. R. Am. J. Geriatr. Pharmacother. 2005;3:274–287. [PubMed] [Google Scholar]
- Gilron I., Bailey J. M., Tu D., Holden R. R., Weaver D. F., Houlden R. L. N. Engl. J. Med. 2005;352:1324–1334. doi: 10.1056/NEJMoa042580. [DOI] [PubMed] [Google Scholar]
- Kingery W. S. Pain. 1997;73:123–139. doi: 10.1016/S0304-3959(97)00049-3. [DOI] [PubMed] [Google Scholar]
- Lynch M. E., Watson C. P. N. Pain. Res. Manag. 2006;11:11–38. doi: 10.1155/2006/642568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk D. C., Wilson H. D., Cahana A. Lancet. 2011;377:2226–2235. doi: 10.1016/S0140-6736(11)60402-9. [DOI] [PubMed] [Google Scholar]
- Dworkin R. H., O'Connor A. B., Backonja M., Farrar J. T., Finnerup N. B., Jensen T. S., Kalso E. A., Loeser J. D., Miaskowski C., Nurmikko T. J., Portenoy R. K., Rice A. S. C., Stacey B. R., Treede R. D., Turk D. C., Wallace M. S. Pain. 2007;132:237–251. doi: 10.1016/j.pain.2007.08.033. [DOI] [PubMed] [Google Scholar]
- Finnerup N. B., Sindrup S. H., Jensen T. S. Pain. 2010;150:573–581. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. Prog. Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Summers T., Hanten B., Peterson W., Burrell B. Sci. Rep. 2017;7:1–9. doi: 10.1038/s41598-017-06114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerandzic V., Mrozkova P., Adamek P., Spicarova D., Nagy I., Palecek J. Br. J. Pharmacol. 2018;175:2322–2336. doi: 10.1111/bph.13849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storozhuk M. V., Zholos A. V. Curr. Neuropharmacol. 2018;16:137–150. doi: 10.2174/1570159X15666170424120802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W. A., Hanus L., Breuer A., Pertwee R. G., Stevenson L. A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Stella N., Schweitzer P., Piomelli D. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Deutsch D. G., Chin S. A. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- Makara J. K., Mor M., Fegley D., SzabóI S. I., Kathuria S., Astarita G., Duranti A., Tontini A., Tarzia G., Rivara S., Freund T. F., Piomelli D. Nat. Neurosci. 2005;8(9):1139–1141. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- Starowicz K., Malek N. and Przewlocka B., Wiley Interdisciplinary Reviews: Membrane Transport and Signaling, 2013, vol. 2, pp. 121–132. [Google Scholar]
- Walker J. M., Huang S. M., Strangman N. M., Tsou K., Sañudo-Peña M. C. Proc. Natl. Acad. Sci. U. S. A. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. P. Expert Opin. Invest. Drugs. 2014;23:1123–1140. doi: 10.1517/13543784.2014.918603. [DOI] [PubMed] [Google Scholar]
- Starowicz K., Przewlocka B. Philos. Trans. R. Soc., B. 2012;367:3286–3299. doi: 10.1098/rstb.2011.0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starowicz K., Di Marzo V. Eur. J. Pharmacol. 2013;716:41–53. doi: 10.1016/j.ejphar.2013.01.075. [DOI] [PubMed] [Google Scholar]
- Guindon J., Hohmann A. G. Br. J. Pharmacol. 2008;153:319–334. doi: 10.1038/sj.bjp.0707531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettekoven M., Adam J. M., Bendels S., Bissantz C., Fingerle J., Grether U., Grìner S., Guba W., Kimbara A., Ottaviani G., Pìllmann B., Rogers-Evans M., Rçver S., Rothenhusler B., Schmitt S., Schuler F., Schulz-Gasch T., Ullmer C. ChemMedChem. 2016;11:179–189. doi: 10.1002/cmdc.201500218. [DOI] [PubMed] [Google Scholar]
- Ogawa S., Kunugi H. Curr. Neuropharmacol. 2015;13:760–775. doi: 10.2174/1570159X13666150612225212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn E. J., Hohmann A. G. Neurotherapeutics. 2009;6:713–737. doi: 10.1016/j.nurt.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleoni M., Sacerdote P. Biochim. Biophys. Acta, Mol. Basis Dis. 2010;1802:924–933. doi: 10.1016/j.bbadis.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Seltzer Z. E., Dubner R., Shir Y. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Decosterd I., Woolf C. J. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Malmberg A. B., Basbaum A. I. Pain. 1998;76:215–222. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Bennett G. J., Xie Y. K. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Kim S. H., Chung J. M. Pain. 1992;50:355–363. [Google Scholar]
- Liang Y. C., Huang C. C., Hsu K. S. Neuropharmacology. 2007;53:169–177. doi: 10.1016/j.neuropharm.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Turu G., Hunyady L. J. Mol. Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- Miller L. K., Devi L. A. Pharmacol. Rev. 2011;63:461–470. doi: 10.1124/pr.110.003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda L. A., Lolait S. J., Brownstein M. J., Young A. C., Bonner T. I. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Howlett A. C., Bidaut-Russell M., Devane W. A., Melvin L. S., Johnson M., Herkenham M. Trends Neurosci. 1990;13:420–423. doi: 10.1016/0166-2236(90)90124-s. [DOI] [PubMed] [Google Scholar]
- Daigle T. L., Kwok M. L., Mackie K. J. Neurochem. 2008;106:70–82. doi: 10.1111/j.1471-4159.2008.05336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H., Delgado-Peraza F., Mackie K., Kendall D. A., Yudowski G. A. Nat. Commun. 2014;5:4589. doi: 10.1038/ncomms5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson B. D., Hebert T. E., Kelly M. E. Mol. Pharmacol. 2010;77:1–9. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar A., Mackie K. J. Pharmacol. Exp. Ther. 2016;358:342–351. doi: 10.1124/jpet.116.232561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushlin I., Gupta A., Stockton S. D., Miller L. K., Devi L. A. PLoS One. 2012;7(12):e49789. doi: 10.1371/journal.pone.0049789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A., Zimmer A. M., Hohmann A. G., Herkenham M., Bonner T. I. Proc. Natl. Acad. Sci. U. S. A. 1999;96:5780–5785. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C., Valverde O., Cossu G., Petitet F., Aubert J. F., Beslot F., Bohme G. A., Imperato A., Pedrazzini T., Roques B. P., Vassart G., Fratta W., Parmentier M. Science. 1999;283:401–404. doi: 10.1126/science.283.5400.401. [DOI] [PubMed] [Google Scholar]
- Castañé A., Célérier E., Martín M., Ledent C., Parmentier M., Maldonado R., Valverde O. Neuropharmacology. 2006;50:111–122. doi: 10.1016/j.neuropharm.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Rácz I., Nent E., Erxlebe E., Zimmer A. Brain Res. Bull. 2015;114:42–48. doi: 10.1016/j.brainresbull.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Lim G., Sung B., Ji R. R., Mao J. Pain. 2003;105:275–283. doi: 10.1016/s0304-3959(03)00242-2. [DOI] [PubMed] [Google Scholar]
- Siegling A., Hofmann H. A., Denzer D., Mauler F., De Vry J. Eur. J. Pharmacol. 2001;415:R5–R7. doi: 10.1016/s0014-2999(01)00798-1. [DOI] [PubMed] [Google Scholar]
- Hama A., Sagen J. Brain Res. 2011;1412:44–54. doi: 10.1016/j.brainres.2011.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn E. J., Deng L., Thakur G. A., Vemuri K., Zvonok A. M., Lai Y. Y., Makriyannis A., Hohmann A. G. Mol. Pain. 2014;10:27. doi: 10.1186/1744-8069-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine P. G., Rosenfeld M. J. Curr. Pain Headache Rep. 2014;18:451. doi: 10.1007/s11916-014-0451-2. [DOI] [PubMed] [Google Scholar]
- Zuardi A. W. Braz J Psychiatry. 2006;28:153–157. doi: 10.1590/s1516-44462006000200015. [DOI] [PubMed] [Google Scholar]
- Elsohly M. A., Slade D. Life Sci. 2005;78:539–548. doi: 10.1016/j.lfs.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Brenneisen R., in Marijuana and the Cannabinoids Forensic Science and Medicine, ed. M. ElSohly, Humana Press, New York, NY, 2007, pp. 17–49, 10.1007/978-1-59259-947-9_2. [DOI] [Google Scholar]
- Gaoni Y., Mechoulam R. J. Am. Chem. Soc. 1964;86:1646–1647. [Google Scholar]
- Maione S., Piscitelli F., Gatta L., Vita D., De Petrocellis L., Palazzo E., de Novellis V., Di Marzo V. Br. J. Pharmacol. 2011;162:584–596. doi: 10.1111/j.1476-5381.2010.01063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K. P. JAMA, J. Am. Med. Assoc. 2015;313:2474–2483. doi: 10.1001/jama.2015.6199. [DOI] [PubMed] [Google Scholar]
- Whiting P. F., Wolff R. F., Deshpande S., Nisio M. D., Duffy S., Hernandez A. V., Keurentjes J. C., Lang S., Misso K., Ryder S., Schmidlkofer S., Westwood M., Kleijnen J. JAMA, J. Am. Med. Assoc. 2015;313:2456–2473. doi: 10.1001/jama.2015.6358. [DOI] [PubMed] [Google Scholar]
- Izzo A. A., Borrelli F., Capasso R., Di Marzo V., Mechoulam R. Trends Pharmacol. Sci. 2009;30:515–527. doi: 10.1016/j.tips.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Laprairie R. B., Bagher A. M., Kelly M. E., Denovan-Wright E. M. Br. J. Pharmacol. 2015;172:4790–4805. doi: 10.1111/bph.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Cheng K., Cui T., Godlewski G., Rice K. C., Xu Y., Zhang L. Nat. Chem. Biol. 2011;7:296–303. doi: 10.1038/nchembio.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W., Cui T., Cheng K., Yang F., Chen S. R., Willenbring D., Guan Y., Pan H. L., Ren K., Xu Y., Zhang L. J. Exp. Med. 2012;209:1121–1134. doi: 10.1084/jem.20120242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R., Feigenbaum J. J., Lander N., Segal M., Järbe T. U. C., Hiltunen A. J., Consroe P. Experientia. 1989;44:762–764. doi: 10.1007/BF01959156. [DOI] [PubMed] [Google Scholar]
- De Vry J., Kuhl E., Franken-Kunkel P., Eckel G. Eur. J. Pharmacol. 2004;491:137–148. doi: 10.1016/j.ejphar.2004.03.051. [DOI] [PubMed] [Google Scholar]
- Scott D. A., Wright C. E., Angus J. A. Pain. 2004;109:124–131. doi: 10.1016/j.pain.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Sain N. M. H., Liang A., Kane S. A., Urban M. O. Neuropharmacology. 2009;57:235–241. doi: 10.1016/j.neuropharm.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Fox A., Kesingland A., Gentry C., McNair K., Patel S., Urban L., James I. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- Herzberg U., Eliav E., Bennett G. J., Kopin I. J. Neurosci. Lett. 1997;221:157–160. doi: 10.1016/s0304-3940(96)13308-5. [DOI] [PubMed] [Google Scholar]
- Strangman N. M., Walker J. M. J. Neurophysiol. 1999;82:472–477. doi: 10.1152/jn.1999.82.1.472. [DOI] [PubMed] [Google Scholar]
- Costa B., Colleoni M., Conti S., Trovato A. E., Bianchi M., Sotgiu M. L., Giagnoni G. Br. J. Pharmacol. 2004;141:4–8. doi: 10.1038/sj.bjp.0705587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual D., Goicoechea C., Suardíaz M., Martín M. I. Pain. 2005;118:23–34. doi: 10.1016/j.pain.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Burgos E., Gomez-Nicola D., Pascual D., Martin M. I., Nieto-Sampedro M., Goicoechea C. Eur. J. Pharmacol. 2012;682:62–72. doi: 10.1016/j.ejphar.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Kunos G., Osei-Hyiaman D., Batkai S., Sharkey K. A., Makriyannis A. Trends Pharmacol. Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. Philos. Trans. R. Soc., B. 2012;367:3353–3363. doi: 10.1098/rstb.2011.0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Hitchcock S. A. Expert Opin. Invest. Drugs. 2007;16:951–965. doi: 10.1517/13543784.16.7.951. [DOI] [PubMed] [Google Scholar]
- Yu X. H., Cao C. Q., Martino G., Puma C., Morinville A., St-Onge S., Lessard E., Perkins M. N., Laird J. M. A. Pain. 2010;151:337–344. doi: 10.1016/j.pain.2010.07.019. [DOI] [PubMed] [Google Scholar]
- Redmond W. J., Cawston E. E., Grimsey N. L., Stuart J., Edington A. R., Glass M., Connor M. Br. J. Pharmacol. 2016;173:115–127. doi: 10.1111/bph.13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vry J., Denzer D., Reissmueller E., Eijckenboom M., Heil M., Meier H., Mauler F. J. Pharmacol. Exp. Ther. 2004;310:620–632. doi: 10.1124/jpet.103.062836. [DOI] [PubMed] [Google Scholar]
- Agarwal N., Pacher P., Tegeder I., Amaya F., Constantin C. E., Brenner G. J., Rubino T., Michalski C. W., Marsicano G., Monory K., Mackie K., Marian C., Batkai S., Parolaro D., Fischer M. J., Reeh P., Kunos G., Kress M., Lutz B., Woolf C. J., Kuner R. Nat. Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst M., Salim K., Burstein S., Conrad I., Hoy L., Schneider U. JAMA, J. Am. Med. Assoc. 2003;290:1757–1762. doi: 10.1001/jama.290.13.1757. [DOI] [PubMed] [Google Scholar]
- Burstein S. H., Karst M., Schneider U., Zurier R. B. Life Sci. 2004;75:1513–1522. doi: 10.1016/j.lfs.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Dyson A., Peacock M., Chen A., Courade J. P., Yaqoob M., Groarke A., Brain C., Loong Y., Fox A. Pain. 2005;116:129–137. doi: 10.1016/j.pain.2005.03.037. [DOI] [PubMed] [Google Scholar]
- Burstein S. AAPS J. 2005;7(1):E143–E148. doi: 10.1208/aapsj070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim K., Schneider U., Burstein S., Hoy L., Karst M. Neuropharmacology. 2005;48:1164–1171. doi: 10.1016/j.neuropharm.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Tepper M. A., Zurier R. B., Burstein S. H. Bioorg. Med. Chem. 2014;22:3245–3251. doi: 10.1016/j.bmc.2014.04.062. [DOI] [PubMed] [Google Scholar]
- Chorvat R. J. Bioorg. Med. Chem. Lett. 2013;23:4751–4760. doi: 10.1016/j.bmcl.2013.06.066. [DOI] [PubMed] [Google Scholar]
- Wu Y. K., Yeh C. F., Ly T. W., Hung M. S. Curr. Top. Med. Chem. 2011;11:1421–1429. doi: 10.2174/156802611795860997. [DOI] [PubMed] [Google Scholar]
- Dziadulewicz E. K., Bevan S. J., Brain C. T., Coote P. R., Culshaw A. J., Davis A. J., Edwards L. J., Fisher A. J., Fox A. J., Gentry C., Groarke A., Hart T. W., Huber W., James I. F., Kesingland A., Vecchia L. L., Loong Y., Lyothier I., McNair K., O'Farrell C., Peacock M., Portmann R., Schopfer U., Yaqoob M., Zadrobilek J. J. Med. Chem. 2007;50:3851–3856. doi: 10.1021/jm070317a. [DOI] [PubMed] [Google Scholar]
- Diouf O., Depreux P., Chavatte P., Poupaert J. H. Eur. J. Med. Chem. 2000;35:699–706. doi: 10.1016/s0223-5234(00)00163-x. [DOI] [PubMed] [Google Scholar]
- Bell M. R., D'Ambra T. E., Kumar V., Eissenstat M. A., Herrmann J. L., Wetzel J. R., Rosi D., Philion R. E., Daum S. J. J. Med. Chem. 1991;34:1099–1110. doi: 10.1021/jm00107a034. [DOI] [PubMed] [Google Scholar]
- Huffman J. W., Dai D., Martin B. R., Compton D. R. Bioorg. Med. Chem. Lett. 1994;4:563–566. [Google Scholar]
- Brusberg M., Arvidsson S., Kang D., Larsson H., Lindstrom E., Martinez V. J. Neurosci. 2009;29:1554–1564. doi: 10.1523/JNEUROSCI.5166-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardin A., Kucher K., Kiese B., Appel-Dingemanse S. Drug Metab. Dispos. 2009;37:827–833. doi: 10.1124/dmd.108.024000. [DOI] [PubMed] [Google Scholar]
- Yu X. H., Cao C. Q., Martino G., Puma C., Morinville A., St-Onge S., Lessard E., Perkins M. N., Laird J. M. Pain. 2010;151:337–344. doi: 10.1016/j.pain.2010.07.019. [DOI] [PubMed] [Google Scholar]
- Groblewski T., Karlsten R., Segerdhal M., Kalliomäki J., Jonzon B., Bielenstein M., Cebers G., Swedberg M., Annas A., Christoph G., Tellefors P., Ståhle L., Bouw R., Fagerholm U., Berg A., Butler S., O'Malley M. and Anstrén G., ICRS, 2010.
- Schou M., Varnas K., Jucaite A., Gulyas B., Halldin C., Farde L. Nucl. Med. Biol. 2013;40:410–414. doi: 10.1016/j.nucmedbio.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Kalliomäki J., Segerdahl M., Webster L., Reimfelt A., Huizar K., Annas P., Karlsten R., Quiding H. Scand. J. Pain. 2013;4:17–22. doi: 10.1016/j.sjpain.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Kalliomaki J., Annas P., Huizar K., Clarke C., Zettergren A., Karlsten R., Segerdahl M. Clin. Exp. Pharmacol. Physiol. 2013;40:212–218. doi: 10.1111/1440-1681.12051. [DOI] [PubMed] [Google Scholar]
- Page D., Balaux E., Boisvert L., Liu Z., Milburn C., Tremblay M., Wei Z., Woo S., Luo X., Cheng Y. X., Yang H., Srivastava S., Zhou F., Brown W., Tomaszewski M., Walpole C., Hodzic L., St Onge S., Godbout C., Salois D., Payza K. Bioorg. Med. Chem. Lett. 2008;18:3695–3700. doi: 10.1016/j.bmcl.2008.05.073. [DOI] [PubMed] [Google Scholar]
- Cheng Y. X., Pourashraf M., Luo X., Srivastava S., Walpole C., Salois D., St-Onge S., Payza K., Lessard E., Yu X. H., Tomaszewski M. J. Bioorg. Med. Chem. Lett. 2012;22:1619–1624. doi: 10.1016/j.bmcl.2011.12.124. [DOI] [PubMed] [Google Scholar]
- Doan K. M. M., Humphreys J. E., Webster L. O., Wring S. A., Shampine L. J., Serabjit-Singh C. J., Adkison K. K., Polli J. W. J. Pharmacol. Exp. Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Adam J. M., Clark J. K., Davies K., Everett K., Fields R., Francis S., Jeremiah F., Kiyoi T., Maidment M., Morrison A., Ratcliffe P., Prosser A., Schulz J., Wishart G., Baker J., Boyce S., Campbell R., Cottney J. E., Deehan M., Martin I. Bioorg. Med. Chem. Lett. 2012;22:2932–2937. doi: 10.1016/j.bmcl.2012.02.048. [DOI] [PubMed] [Google Scholar]
- Seltzman H. H., Shiner C., Hirt E. E., Gilliam A. F., Thomas B. F., Maitra R., Snyder R., Black S. L., Patel P. R., Mulpuri Y., Spigelman I. J. Med. Chem. 2016;59:7525–7543. doi: 10.1021/acs.jmedchem.6b00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. Mol. Pharmacol. 2014;86:463–478. doi: 10.1124/mol.114.094342. [DOI] [PubMed] [Google Scholar]
- Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., Kobilka B. K. Nature. 2010;463:108–112. doi: 10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka B. K. Trends Pharmacol. Sci. 2011;32:213–218. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. R., Baillie G. L., Thomas A., Stevenson L. A., Easson M., Goodwin R., McLean A., McIntosh L., Goodwin G., Walker G., Westwood P., Marrs J., Thomson F., Cowley P., Christopoulos A., Pertwee R. G., Ross R. A. Mol. Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Ross R. A., Baillie G. L., Pertwee R. G. FASEB J. 2012;26:836. [Google Scholar]
- Abood M. E. J. Med. Chem. 2016;59:42–43. doi: 10.1021/acs.jmedchem.5b01824. [DOI] [PubMed] [Google Scholar]
- Morales P., Goya P., Jagerovic N., Hernandez-Folgado L. Cannabis Cannabinoid Res. 2016;1:22–30. doi: 10.1089/can.2015.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. Proc. Natl. Acad. Sci. U. S. A. 2012;109:20781–20782. doi: 10.1073/pnas.1218529110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H., Mahmoud M. M., Shim J. Y., Kendall D. A. J. Biol. Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Qiu Y., Jing L., Thorn D. A., Zhang Y., Li J. X. Pharmacol. Res. Perspect. 2014;2(6):e00069. doi: 10.1002/prp2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing L., Qiu Y., Zhang Y., Li J. X. Drug Alcohol Depend. 2014;143:251–256. doi: 10.1016/j.drugalcdep.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage T. F., Ignatowska-Jankowska B. M., Wiley J. L., Abdelrahman M., Trembleau L., Greig I. R., Thakur G. A., Tichkule R., Poklis J., Ross R. A., Pertwee R. G., Lichtman A. H. Behav. Pharmacol. 2014;25:182–185. doi: 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T., German N., Decker A. M., Li J. X., Wiley J. L., Thomas B. F., Kenakin T. P., Zhang Y. Bioorg. Med. Chem. 2015;23:2195–2203. doi: 10.1016/j.bmc.2015.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M. M., Ali H. I., Ahn K. H., Damaraju A., Samala S., Pulipati V. K., Kolluru S., Kendall D. A., Lu D. J. Med. Chem. 2013;56:7965–7975. doi: 10.1021/jm4009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli F., Ligresti A., La Regina G., Coluccia A., Morera L., Allara M., Novellino E., Di Marzo V., Silvestri R. J. Med. Chem. 2012;55:5627–5631. doi: 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- Khurana L., Ali H. I., Olszewska T., Ahn K. H., Damaraju A., Kendall D. A., Lu D. J. Med. Chem. 2014;57:3040–3052. doi: 10.1021/jm5000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C. J., Ali H. I., Ahn K. H., Kolluru S., Kendall D. A., Lu D. Eur. J. Med. Chem. 2016;121:517–529. doi: 10.1016/j.ejmech.2016.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig I. R., Baillie G. L., Abdelrahman M., Trembleau L., Ross R. A. Bioorg. Med. Chem. Lett. 2016;26:4403–4407. doi: 10.1016/j.bmcl.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Laprairie R. B., Kulkarni A. R., Kulkarni P. M., Hurst D. P., Lynch D., Reggio P. H., Janero D. R., Pertwee R. G., Stevenson L. A., Kelly M. E., Denovan-Wright E. M., Thakur G. A. ACS Chem. Neurosci. 2016;7:776–798. doi: 10.1021/acschemneuro.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horswill J. G., Bali U., Shaaban S., Keily J. F., Jeevaratnam P., Babbs A. J., Reynet C., In P. W. K. Br. J. Pharmacol. 2009;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German N., Decker A. M., Gilmour B. P., Gay E. A., Wiley J. L., Thomas B. F., Zhang Y. J. Med. Chem. 2014;57:7758–7769. doi: 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska-Jankowska B. M., Baillie G. L., Kinsey S., Crowe M., Ghosh S., Owens R. A., Damaj I. M., Poklis J., Wiley J. L., Zanda M., Zanato C., Greig I. R., Lichtman A. H., Ross R. A. Neuropsychopharmacology. 2015;40:2948–2959. doi: 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poklis J. L., Clay D. J., Ignatowska-Jankowska B. M., Zanato C., Ross R. A., Greig I. R., Abdullah R. A., Mustafa M. A., Lichtman A. H., Poklis A. J. Anal. Toxicol. 2015;39:353–358. doi: 10.1093/jat/bkv015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie R., Kulkarni P., Cascio M., Pertwee R., Kelly M., Denovan-Wright E., Thakur G. FASEB J. 2015;29(1):770.13. [Google Scholar]
- Fernandez-Ruiz J., Lastres-Becker I., Cabranes A., Gonzalez S., Ramos J. A. Prostaglandins, Leukotrienes Essent. Fatty Acids. 2002;66:257–267. doi: 10.1054/plef.2001.0350. [DOI] [PubMed] [Google Scholar]
- Navarro H. A., Howard J. L., Pollard G. T., Carroll F. I. Br. J. Pharmacol. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie R. B., Dupre D. J., Kelly M. E. M., Denovan-Wright E. M. FASEB J. 2013;27(1_supplement):lb553. [Google Scholar]
- Baillie G. L., Horswill J. G., Anavi-Goffer S., Reggio P. H., Bolognini D., Abood M. E., McAllister S., Strange P. G., Stephens G. J., Pertwee R. G., Ross R. A. Mol. Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Peraza F., Ahn K. H., Nogueras-Ortiz C., Mungrue I. N., Mackie K., Kendall D. A., Yudowski G. A. Mol. Pharmacol. 2016;89:618–629. doi: 10.1124/mol.115.103176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal K. M., Bohn L. M. Handb. Exp. Pharmacol. 2014;219:427–443. doi: 10.1007/978-3-642-41199-1_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprairie R. B., Bagher A. M., Kelly M. E., Denovan-Wright E. M. Mol. Pharmacol. 2016;89:364–375. doi: 10.1124/mol.115.101980. [DOI] [PubMed] [Google Scholar]
- Denovan-Wright E. M., Robertson H. A. Neuroscience. 2000;98:705–713. doi: 10.1016/s0306-4522(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Glass M., Dragunow M., Faull R. L. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Van Laere K., Casteels C., Dhollander I., Goffin K., Grachev I., Bormans G., Vandenberghe W. J. Nucl. Med. 2010;51:1413–1417. doi: 10.2967/jnumed.110.077156. [DOI] [PubMed] [Google Scholar]
- Sim-Selley L. J., Martin B. R. J. Pharmacol. Exp. Ther. 2002;303:36–44. doi: 10.1124/jpet.102.035618. [DOI] [PubMed] [Google Scholar]
- Blair R. E., Deshpande L. S., Sombati S., Elphick M. R., Martin B. R., DeLorenzo R. J. Neuropharmacology. 2009;57:208–218. doi: 10.1016/j.neuropharm.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W. A., Hanus L., Breuer A., Pertwee R. G., Stevenson L. A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Kondo S., Kondo H., Nakane S., Kodaka T., Tokumura A., Waku K., Sugiura T. FEBS Lett. 1998;429:152–156. doi: 10.1016/s0014-5793(98)00581-x. [DOI] [PubMed] [Google Scholar]
- Dowie M. J., Scotter E. L., Molinari E., Glass M. Pharmacol. Ther. 2010;128:305–323. doi: 10.1016/j.pharmthera.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Dowie M. J., Howard M. L., Nicholson L. F., Faull R. L., Hannan A. J., Glass M. Neuroscience. 2010;170:324–336. doi: 10.1016/j.neuroscience.2010.06.056. [DOI] [PubMed] [Google Scholar]
- Khajehali E., Malone D. T., Glass M., Sexton P. M., Christopoulos A., Leach K. Mol. Pharmacol. 2015;88:368–379. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- Cawston E. E., Redmond W. J., Breen C. M., Grimsey N. L., Connor M., Glass M. Br. J. Pharmacol. 2013;170:893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond W. J., Cawston E. E., Grimsey N. L., Stuart J., Edington A. R., Glass M., Connor M. Br. J. Pharmacol. 2015;173:115–127. doi: 10.1111/bph.13341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay J. F., Farrens D. L. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8469–8474. doi: 10.1073/pnas.1500895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T., Christopoulos A. Nat. Rev. Drug Discovery. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]