

Abstract
In yeast, the Atg2-Atg18 complex regulates Atg9 recycling from phagophore assembly site during autophagy; their function in higher eukaryotes remains largely unknown. In a targeted screening in Drosophila melanogaster, we show that Mef2-GAL4-RNAi-mediated knockdown of Atg2, Atg9 or Atg18 in the heart and indirect flight muscles led to shortened healthspan (declined locomotive function) and lifespan. These flies displayed an accelerated age-dependent loss of cardiac function along with cardiac hypertrophy (increased heart tube wall thickness) and structural abnormality (distortion of the lumen surface). Using the Mef2-GAL4-MitoTimer mitochondrial reporter system and transmission electron microscopy, we observed significant elongation of mitochondria and reduced number of lysosome-targeted autophagosomes containing mitochondria in the heart tube but exaggerated mitochondrial fragmentation and reduced mitochondrial density in indirect flight muscles. These findings provide the first direct evidence of the importance of Atg2-Atg18/Atg9 autophagy complex in the maintenance of mitochondrial integrity and, regulation of heart and muscle functions in Drosophila, raising the possibility of augmenting Atg2-Atg18/Atg9 activity in promoting mitochondrial health and, muscle and heart function.
Keywords: mitophagy, autophagy related genes, indirect flight muscle, cardiac function, negative geotaxis, functional aging, healthspan, lifespan
Author Summary
A self-eating, autophagy process in cells is critical for normal cell function. We used a genetic approach to reduce autophagy genes in fruit fly muscle and heart to search for the ones with functional importance in maintaining healthy mitochondria, the power plants of cells. We found that reduced expression of Atg2, Atg9 or Atg18 led to impaired locomotor function, early death and signs of heart failure along with evidence of abnormal mitochondrial structure, indicating the importance of these autophagy genes in maintenance of mitochondrial function and healthy physiology.
1. Introduction
Macroautophagy, a bulk degradation process for cellular components and organelles, is a cell survival mechanism under nutrient-deficient conditions [1,2,3,4]. It is a conserved process that involves sequestering cytotoxic protein aggregates, senescent organelles and other cellular debris into double-membrane vesicles, which then fuse with lysosomes for a complete degradation [5,6,7]. Although early work had described autophagy as a non-specific homeostatic cellular process, recent studies have demonstrated selective autophagic targeting and degradation of organelles, such as mitochondria and endoplasmic reticulum (ER) [8]. Genetic screenings in yeast have identified many autophagy-related genes (Atgs), with the majority being conserved from yeast to mammals [9]. Generally, autophagy involves: 1) induction/nucleation; 2) expansion; and 3) maturation [10] with five core-complexes involved: the Atg1/Ulk1 protein-kinase complex, the Atg9.Atg2-Atg18 complex, the Vps34-Atg6/Beclin1 class III phosphoinositide 3-kinase (PI-kinase) complex, the Atg12 and Atg8/LC3 conjugation systems [11].
The functional importance of autophagy in mammals is best demonstrated in its role in maintaining cardiac homeostasis since disruption of this pathway leads to deleterious effects [12,13] and in the association of abnormal autophagy with various cardiovascular diseases, such as cardiac hypertrophy and heart failure [14,15]. For instance, Danon disease, a lethal cardiomyopathy, results from a deficiency of a principal lysosomal membrane protein lysosome-associated membrane protein-2 (LAMP2) [12]. Mice with cardiac-specific deficiency of Atg5, a key regulator of autophagy, have mitochondrial mis-alignment and aggregation in cardiac myocytes and develop heart failure with aging [13,16]. In a recent study, the ablation of myeloid cell leukemia-1 (Mcl-1, an anti-apoptotic Bcl-2 protein) in the adult heart in mice led to accumulation of dysfunctional mitochondria, impaired autophagy and rapid development of heart failure. Autophagy has also shown to be critical for normal skeletal muscle structure and function. Skeletal muscle-specific deletion of an essential autophagy gene, Atg7, resulted in accelerated age-dependent muscle atrophy and weakness along with accumulation of abnormal mitochondria measure redox status [17]. Interestingly, autophagy mediated by Atg7 has also been shown to protect mitochondria during physical activity [18]. These findings clearly demonstrate the importance of autophagy in maintaining normal muscle and heart functions, indicating a role in clearance of damaged/dysfunctional mitochondria. There has not been a study with systemic delineation of autophagy genes in an animal model.
Although the linear heart tube and indirect flight muscle (IFM) in Drosophila are less complex than the mammalian heart and skeletal muscle, the developmental process and control of function are highly conserved between Drosophila and human. Drosophila has been demonstrated to be a great model to study various human cardiac and skeletal muscle diseases with great feasibility of genetic interrogation [19,20,21]. In addition, we have recently developed a reporter gene, pMitoTimer, which encodes a mitochondrial targeted discosoma sp. red fluorescent protein (DsRed) mutant that fluoresce like the green fluorescent protein (GFP) when newly synthesized and irreversibly shifts to DsRed when oxidized [22]. We have generated inducible UAS-MitoTimer transgenic fly and demonstrated that we could measure Red:Green ratio of the MitoTimer signal as a parameter of mitochondrial oxidative stress and detect pure red puncta in the tissue of interest, which has been confirmed to be mitophagosome targeted by lysosome [22], hence allowing quantification of mitophagy. Therefore, we can quantify mitochondrial oxidative stress, structure and mitophagy in any tissue in vivo, including the heart and IFM [22].
To systemically investigate the role of autophagy in mitochondrial health and muscle and heart function, we took advantage of this reporter system and RNAi-mediated knockdown of Atgs in Drosophila heart and IFM and selectively screened for Atgs, of which knockdown leads to shortened lifespan with progressive decline of locomotive and cardiac function. Importantly, the UAS-MitoTimer reporter system allowed us to focus on mitochondria in these tissues when each of the genes was silenced. Our findings have for the first time demonstrated the important role of the Atg9•Atg2-Atg18 complex in maintaining mitochondrial structural integrity and muscle function in both heart and IFM, laying a solid foundation to pharmacologically target these genes to protect function in aging heart and skeletal muscle.
2. Results
2.1. Knockdown of Atg2, Atg9 or Atg18 in muscle and heart leads to impaired negative geotaxis and a short lifespan
To ascertain which autophagy genes are critical for mitochondrial health in muscle and heart and determine their functional role in function and lifespan, we screened 12 autophagy RNAi lines for 9 Atgs (Table 1) by crossing with the combined line of Mef2-Gal4>>UAS-MitoTimer. Following an initial screening, only Atg2i (Mef2>>MitoTimer/UAS-Atg2 RNAi), Atg9i (Mef2>>MitoTimer/UAS-Atg9 RNAi) and Atg18i (Mef2>>MitoTimer/UAS-Atg18 RNAi) showed clear signs of altered mitochondrial structure measured by the morphology of MitoTimer fluorescence as well as reduced mitophagy as indicated by reduced number of MitoTimer pure red puncta in the heart tube at 40 days of age (Table 1). To ensure the efficacy of gene knockdown, we tested 3 RNAi lines by crossing them with GS-Tub5GS-Gal4 and Mef2-Gal4 line followed by analysis of mRNA expression in the whole body and in the heart, respectively, by quantitative RT-PCR. All 3 Mef2-Gal4 knockdown lines showed significant reduction in the target gene expression although the data from Atg9i line was quite variable (Fig. 1A). Similar findings were obtained in the whole-body knockdown flies (Supplemental Fig. S1). These data confirmed reliable RNAi-mediated knockdown for Atg2 with less consistent result for Atg9. Since previous yeast studies have shown that Atg2, Atg9 and Atg18 function in a complex that is critical for the formation of autophagosome [23,24], we chose to focus on these genes and conduct comprehensive analyses to ascertain their functional role in mitophagy, mitochondrial health, muscle and cardiac function as well as in functional aging and lifespan.
Table 1.
Summary results of the selective screening for RNAi-mediated knockdown of Atgs
| Line | Role in autophagy |
Phenotype | Mitochondrial structure |
Red/Green ratio |
Pure red puncta |
|---|---|---|---|---|---|
| Atg1i | Induction | Normal | Normal | ↑ | ↓ |
| Atg13i | Induction | Eclosion failed at the end of pupal stage | |||
| Atg6i | Nucleation | Normal | → | ↑ | |
| Atg14i | Nucleation | Eclosion failed at the end of pupal stage. | Normal | ||
| Atg2i | Autophagosome formation | Weak line and short lifespan | Abnormal | ↓ | ↓ |
| Atg9i | Autophagosome formation | Normal | Abnormal | → | ↓ |
| Atg18i | Autophagosome formation | Short lifespan | Abnormal | → | ↓ |
| Atg4i | Autophagosome formation | Impaired negative geotaxis at 25 days | Normal | ↑ | ↓ |
| Atg7i | Autophagosome formation | Normal | Normal | → | ↓ |
Fig.1. Knockdown of Atg2 or Atg18 in both IMF and heart leads to shortened lifespan and reduced locomotor function across the lifespan.
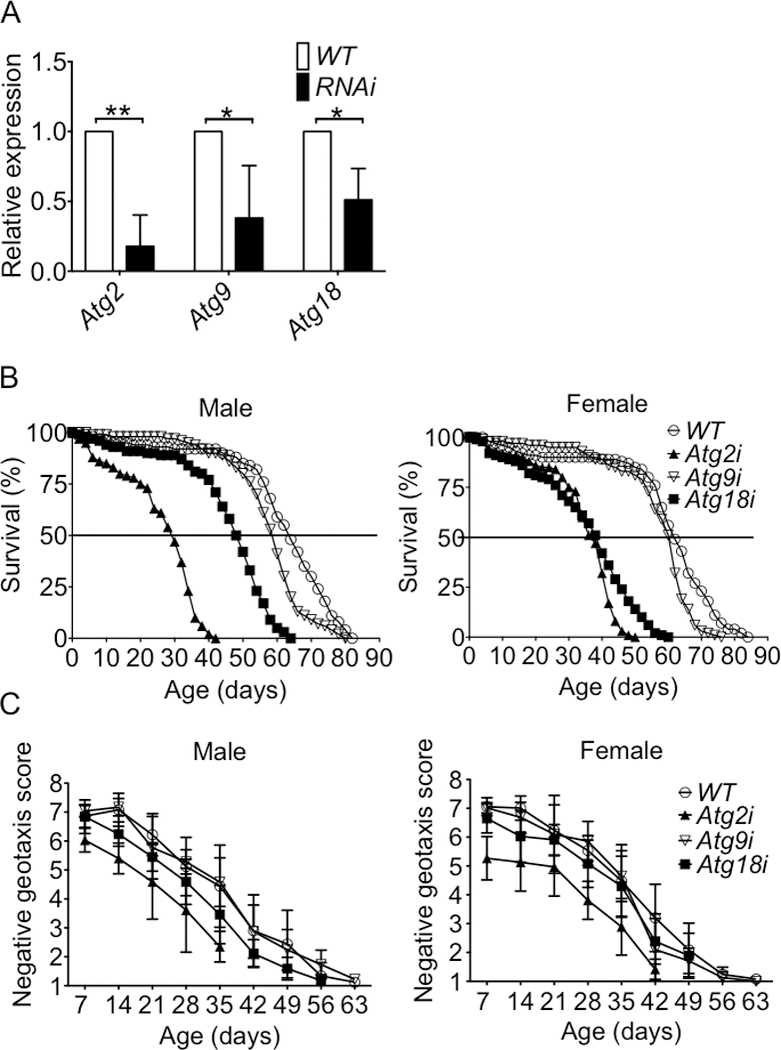
Atg2, Atg9, Atg18 mRNA expression in the heart tube and survival and negative geotaxis were measured in Atg2i, Atg9i and Atg18i flies compared with WT flies across the lifespan. A) One-step SYBR Green-based quantitative RT-PCR analysis of 60 isolated heart tubes. The relative gene expression from an average of three biological replications are normalized to WT (Mef2-Gal4>yw), and significance was determined via an unpaired t-test. B) Survival curve for male and female Atg2i, Atg9i, Atg18i and WT flies; and C) Negative geotaxis score for male and female Atg2i, Atg9i, Atg18i and WT flies. * and ** denote p < 0.05 and p < 0.01, respectively).
Atg2i, Atg9i and Atg18i flies were short lived. Specifically, median survival of Atg2i or Atg18i flies were significantly reduced compared to wild type flies for both genders, while flies showed a similar trend (Fig. 1B and Table 2). Pairwise log-rank analysis showed significant differences for all 3 knockdown lines compared to wild-type flies. Since Mef2 is expressed in both cardiac and IFM, we used TinC-GAL4 and Act5C88F-GAL4 to knockdown these Atgs specifically in the heart and IFM, respectively. Interestingly, both cardiac-specific Atg2i and Atg18i flies had significantly reduced median survival compared to WT flies, but only IFM-specific Atg2i flies showed significantly reduced lifespan by log-rank (Supplemental Fig. S2 and Supplemental Table 1), suggesting that the heart is more vulnerable to the impairment of autophagy. Consistent with Mef2-mediated knockdown, Atg9i flies showed a trend. Again, pairwise log-rank analysis showed significant differences for both cardiac-specific and IFM-specific knockdown flies except for IFM-specific Atg9i line. To investigate the impact of Atg2i, Atg9i or Atg18i on muscle function, we performed negative geotaxis test across the lifespan. Both male and female Atg2i flies showed significantly reduced negative geotaxis across the whole lifespan (Fig. 1C). Male, but not female, Atg18i flies had significantly impaired negative geotaxis compared to WT flies, while Atg9i flies (both genders) showed no significant impairment. Thus, Atg2i flies have greater impairment of locomotive function than Atg18i and Atg9i flies. These findings demonstrate for the first time the importance of Atg2, Atg9 and Atg18 in maintaining normal muscle and cardiac function and lifespan in Drosophila.
Table 2.
Lifespan in RNAi-mediated knockdown of Atg2, Atg9 and Atg18 flies compared with WT flies
| Line | Genotype | Median survival (days) |
% Change |
Maximal lifespan (days) |
% Change |
n of tubes |
n of flies |
p for log- rank curve |
|---|---|---|---|---|---|---|---|---|
| Male | ||||||||
| WT | Mef2>>MitoTimer/+ | 66±6 | 75±8 | 10 | 237 | |||
| Atg2i | Mef2>>MitoTimer/UAS-Atg2 RNAi | 31±3* | −52% | 40±4* | −46% | 8 | 246 | < 0.0001 |
| Atg9i | Mef2>>MitoTimer/UAS-Atg9 RNAi | 60±4 | −9% | 70±8 | −7% | 8 | 192 | < 0.0001 |
| Atg18i | Mef2>>MitoTimer/UAS-Atg18 RNAi | 47±6* | −29% | 56±7* | −26% | 12 | 275 | < 0.0001 |
| Female | ||||||||
| WT | Mef2>>MitoTimer/+ | 63±5 | 75±7 | 8 | 184 | |||
| Atg2i | Mef2>>MitoTimer/UAS-Atg2 RNAi | 36±4* | −43% | 44±4* | −41% | 6 | 146 | < 0.0001 |
| Atg9i | Mef2>>MitoTimer/UAS-Atg9 RNAi | 59±4 | −7% | 70±4 | −7% | 6 | 121 | < 0.0001 |
| Atg18i | Mef2>>MitoTimer/UAS-Atg18 RNAi | 38±6* | −40% | 55±5* | −27% | 8 | 192 | < 0.0001 |
Note: Male and female Atg knockdown and WT flies were aged at 25°C with 20–25 flies per tube containing normal fly food. Median survival was calculated as the time when 50% of the flies were alive.
denotes p < 0.001.
2.2. Knockdown of Atg2, Atg18 or Atg9 causes cardiac hypertrophy
Autophagy in the heart is critical for protein quality control, cellular function and survival [13], and inhibition of autophagy, such as deletion of the Atg5 gene, has been shown to lead to progressive cardiac hypertrophy and heart failure [13,16]. To dissect the role of Atg2, Atg9 and Atg18 in the heart, we assessed cardiac function in Atg2i, Atg18i or Atg9i flies using Mef2>MitoTimer/+ as wild type control (WT) by video-based cardiac function analysis (systolic dimension, diastolic dimension and fractional shortening) [25,26]. In young and middle-aged flies (5-day and 20-day, respectively), knockdown of these Atg genes did not show any significant impact on cardiac function compared with WT flies. However, old (40-days) Atg2i and Atg18i flies showed significantly reduced fractional shortening (29.5±1.3% and 29.8±1.1% for Atg2i and Atg18i flies, respectively, with p < 0.05 for both vs. 37.2±1.9% for WT) (~20% reduction) with a trend of reduction for Atg9i flies (33.0±3.5%) (Fig. 2A and 2B, Supplemental Fig. S3A and 3B). Video imaging analysis showed thicker heart wall in all 3 Atg knockdown flies even at 20 days of age (Fig.2C, Supplemental Fig. S3C). To examine the cardiac structure in more details, HE staining and histological analysis were performed for the heart longitudinal and transverse sections. All 3 Atg knockdown flies (40-day old) showed significant increases of the heart wall thickness compared with WT flies (17.8 ±2.1 μm, 15.1±0.3, 20.0±1.0, in Atg2i, Atg9i and Atg18i flies with p < 0.001, 0.01 and 0.001, respectively, vs. 9±0.5 in WT flies) (Fig. 2D and 2E). The heart tube of WT flies showed relatively smooth lumen surface, while Atg knockdown flies showed various degree of distortion, making the lumen surface rough, with Atg2i and Atg18i being the most severe (Fig. 2D). These results demonstrate that knocking down Atg2i, Atg18i or Atg9i promotes cardiac hypertrophy and structural abnormalities, leading to impaired cardiac function in an age-dependent manner.
Fig. 2. Knockdown of Atg2, Atg9 or Atg18 leads to age-dependent cardiac dysfunction and hypertrophy.
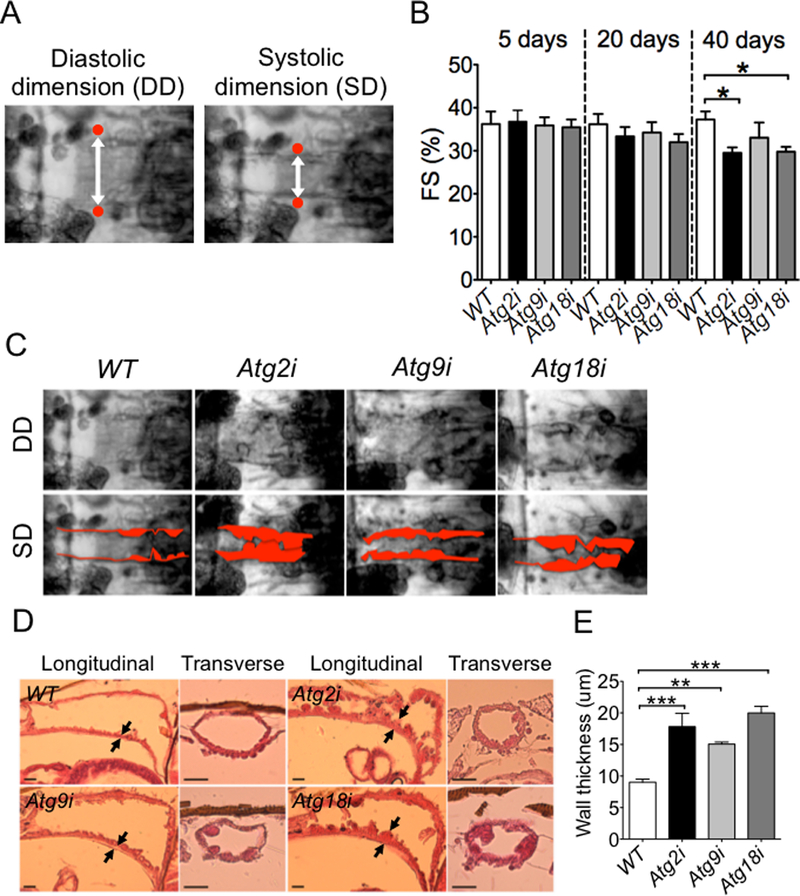
Cardiac function was measured by video-based imaging analysis at 5, 20 and 40 days of age, and heart wall thickness and structure were measure by H&E staining of the heart tube at 40 days of age. A) Captured frames of video images of a fly heart tube at the second abdominal segment. White double head arrows indicate the diastolic and systolic dimensions of the heart tube. Red dots indicate the heart tube edges; B) Quantification of fractional shortening in Atg2i, Atg9i, Atg18i and WT flies (n = 11–15). * denotes p < 0.05; C) Representative light microscope images of 40-day old hearts at the second abdominal segment during a cardiac contraction cycle. Pseudo-red color highlights the heart tube wall; D) H&E stained longitudinal and transverse sections show heart wall thickness. Arrows indicate where the wall thickness was measured. Scale bar = 20 μm; and E) Quantification of the heart wall thickness (n = 4–8). ** and *** denote p < 0.01 and p < 0.001, respectively.
2.3. Knockdown of Atg2, Atg18 or Atg9 in the heart leads to exaggerated mitochondrial elongation
Autophagy is important for structural integrity and function of mitochondria in the heart as genetic deletion of the Atg5 gene results in significant mitochondrial fragmentation and heart failure in mice [13,16]. Here, we investigated mitochondrial structure, oxidative stress and mitophagy in Atg2i, Atg18i and Atg9i flies in the background of newly established UAS-MitoTimer transgenic flies [22]. The Red/Green ratio of MitoTimer fluorescent signals indicates mitochondrial oxidative stress, while the pure red puncta represents mitochondria in autophagosome targeted by lysosome (mitophagolysosme). Forty days after eclosion, all Atg knockdown flies showed fused/enlarged mitochondrial network with significantly increased occupancy of mitochondria per unit cardiac myocyte area (mitochondrial volume density) (Fig. 3A, 3B and Supplemental Fig. S4B). However, the number of pure red puncta was significantly decreased compared with WT flies with no significant increases in Red/Green ratio, suggesting reduced mitophagy and no change in mitochondrial oxidative stress, respectively (Fig. 3C). At young age (20-day old), the mitochondrial network in all knockdown flies was more like WT flies (Supplemental Fig. S4A), indicating age-dependent loss of function. Transmission electron microscopy revealed abnormally elongated but narrowed mitochondria in cardiomyocytes compared with WT flies (Fig. 3D and Supplemental Fig. S4C). Moreover, real-time PCR of Drp1 and Mfn2 in 3 knockdown flies demonstrated that both genes were significantly decreased in Atg2i and Atg18i knockdown flies, but not in Atg9i flies (Fig. 3E and 3F), consistent with the findings of impaired mitochondrial dynamics. Finally, real-time PCR analysis of the Drosophila PGC-1 family homologue, spargel, in the hearts of Atg2i, Atg9i or Atg18i flies did not show significant changes from the WT flies (Fig. 3G), suggesting that the increased mitochondrial volume density is not due to increased mitochondrial biogenesis. However, the precise underlying mechanism remains to be ascertained in the future. Nevertheless, these data may suggest that Atg2, Atg18 and Atg9 are all important for maintaining mitochondrial structural integrity, and a lack of one of these autophagy genes may impair mitochondrial dynamics and reduce mitophagy in cardiomyocytes. Although we have not measured mitochondrial respiratory function directly, the phenotypic changes are consistent with impaired mitochondrial function in the heart.
Fig. 3. Knockdown of Atg2, Atg18 or Atg9 leads to reduced mitophagy and mitochondrial abnormalities in cardiomyocytes.
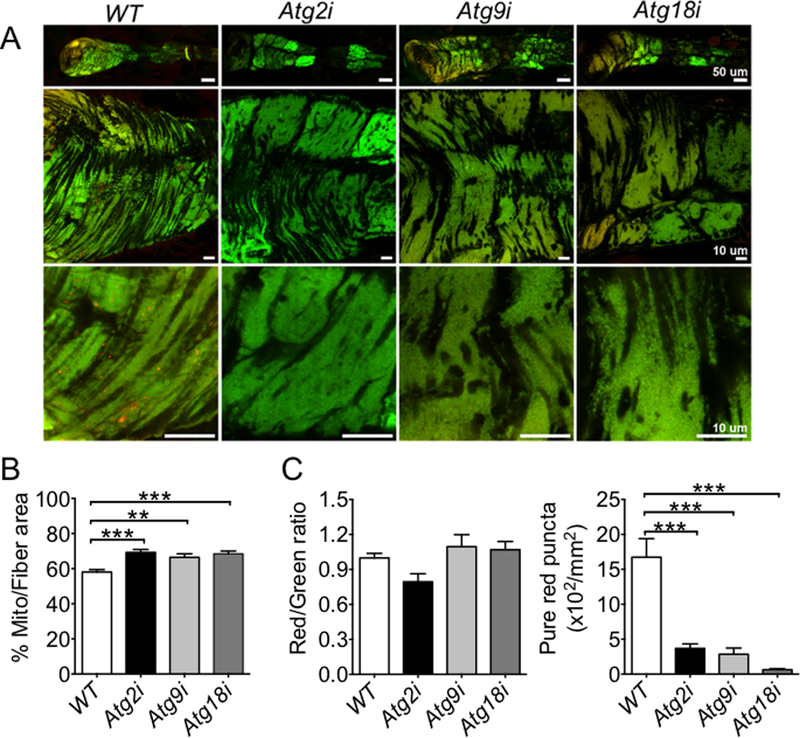
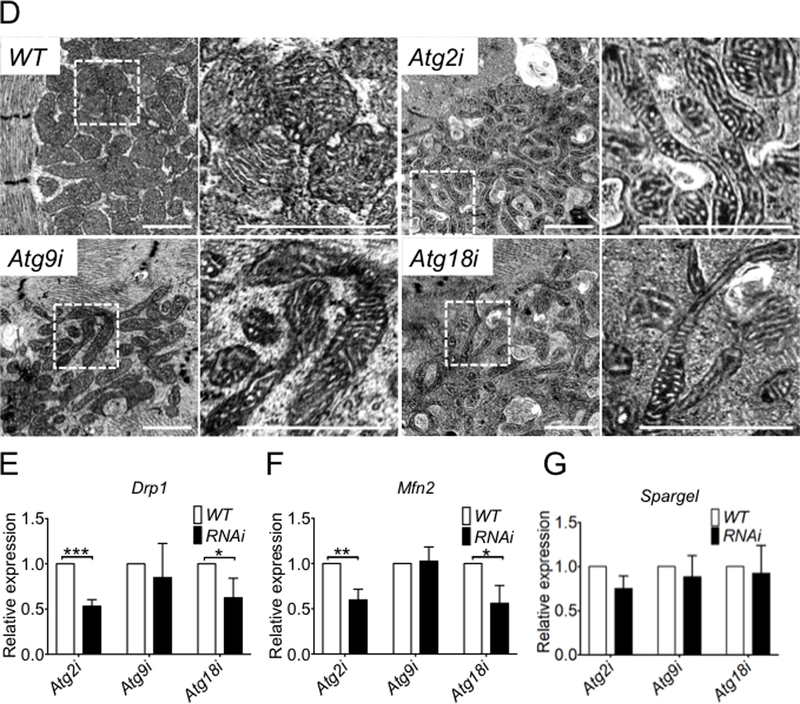
Confocal microscopy and transmission electron microscopy were performed for the heart tubes in Atg knockdown and WT control flies at 40 days of age. A) Representative images of MitoTimer signals (merged image of both GFP and DsRed channels) in cardiomyocyte for assessment of the structure of mitochondrial network and mitochondrial oxidative stress. B) Quantification of mitochondrial volume density (% occupancy of MitoTimer signal) in the A1 heart tube segment (n = 13–20). *** denotes p < 0.001; C) Quantification of Red:Green ratio and number of pure red puncta (n = 13–20). *** denotes p < 0.001; and D) Representative images of transmission electron micrographs. Scale bars = 1 μm; and E, F and G) RT-PCR analysis of Drp1, Mfn2 and Spargel mRNA in Atg2, Atg9, and Atg18 in the heart tubes of 60 knockdown flies (n = 3). *, ** and *** denote p < 0.05, p < 0.01 and p < 0.001, respectively.
2.4. Knockdown of Atg2, Atg18 or Atg9 in IFM leads to mitochondrial abnormalities and reduced mitochondrial density
Since Mef2-GAL4-RNAi-mediated knockdown also reduces the expression of the target gene in IFM, we dissected IFM and visualized MitoTimer signal by fluorescence microscopy. Interestingly, all 3 Atg knockdown flies showed increased fragmentation and reduced mitochondrial density in IFM (Fig. 4A and 4B). Among the Atg knockdown flies, Atg2i flies showed the most severely damaged mitochondrial network (enlarged or hollow mitochondria) with significantly variable mitochondrial sizes, which is very similar to Parkin knockdown flies, and Atg9i showed the least changes (Fig.4A). These findings are consistent with the finding that Atg2i flies had the most impaired negative geotaxis among Atg2, Atg9 and Atg18 knockdown flies (Fig. 1C) and only IMF-specific Atg2i showed significantly reduced lifespan parameters (Supplemental Fig. S2). Again, the phenotypic changes we observed are consistent with impaired mitochondrial function despite the fact that we have not measured mitochondrial respiratory function directly.
Fig. 4. Knockdown of Atg2, Atg9 or Atg18 results in reduced mitochondrial density and abnormal mitochondrial structure in IFM.
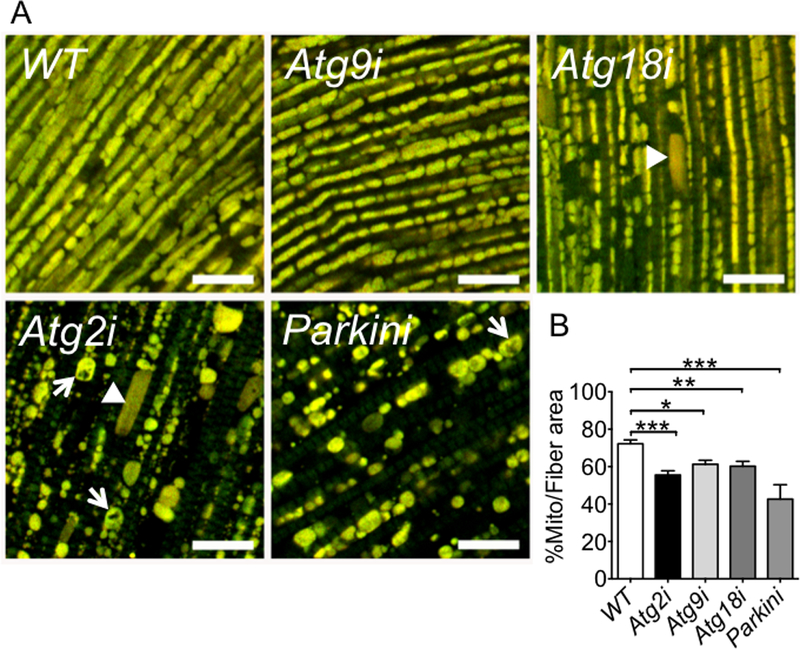
Confocal microscopy was performed for IFMs in Atg knockdown and WT control flies at 40 days of age along with Parkin knockdown flies as positive control. A) Representative images of MitoTimer signals (merged image of both GFP and DsRed channels) in IFM. Knockdown of Atg2 show increased numbers of fragmented (arrow head) and hollow mitochondria (arrows), similar to Parkin knockdown flies. Scale bar =10 μm; and B) Quantification of mitochondrial density in IFM (n = 14. *, ** and *** denote p < 0.05, p < 0.01 and p < 0.001, respectively.
3. Discussion
In this study, we took advantage of RNAi-mediated knockdown and mitochondrial reporter gene technology in Drosophila heart and IMF in a targeted screening for Atg genes with functional role in mitochondrial homeostasis and muscle and cardiac function. Our initial screening showed the functional importance of Atg2, Atg18 and Atg9 in mitochondrial integrity in the heart and lifespan. We then focused on the autophagy complex that is composed of Atg2, Atg18 and Atg9 with essential function in autophagosome formation [23,24] and obtained evidence that this complex is critical for mitochondrial structural homeostasis in both heart and indirect flight muscle and plays a key role in normal muscle and cardiac function and lifespan in Drosophila. These findings are consistent with previous findings of reduced Atg2 and Atg18 gene expression in neuronal tissues with aging [27], altogether suggesting that the Atg9•Atg2-Atg18 complex in muscle and heart plays a pivotal role in health and aging.
The Atg9•Atg2-Atg18 complex is comprised of Atg9/mAtg9, Atg2, and Atg18/WIPI-1 (please see review [11]). Atg9 is the sole transmembrane protein integrated into Golgi derived vesicles [24,28,29]. The roles of Atg9 have been investigated in yeast [30], Drosophila [31,32] and mammals [33,34,35](25–27). In mammalian cells, Atg9 appears to be recruited to damaged mitochondria during selective autophagic degradations [36]. In Saccharomyces cerevisiae, pre-autophagosome structure (PAS, phagophore assembly site) provides the structural framework for autophagosome formation. Atg2 and Atg18 form a complex and are interdependently localized to the PAS afterward, which is essential for autophagy [37,38]. In fact, the Atg2-Atg18 complex may regulate Atg9 recycling from PAS during autophagy in yeast [37,39]. However, the function of this complex in higher eukaryotes remains unclear.
The Atg9•Atg2-Atg18 complex is required for PAS assembly and is a downstream element of Atg1/ULK1 protein-kinase complex [11,37][40]. In our screening, we focused on Atg genes with functional roles in mitochondrial homeostasis. Deficiency of Atg9•Atg2-Atg18 complex showed the most obvious and interesting phenotype, while knockdown of other Atg genes, like Atg1i and Atg6i, did not show significant mitochondrial changes compared with wild type control flies. These findings suggest that the Atg9•Atg2-Atg18 complex plays a particularly important role in mitochondrial integrity. It is of note that among Atg2i, Atg18i and Atg9i knockdown flies, Atg9i demonstrated the weakest phenotypic changes in all the parameters that we measured, including lifespan, negative geotaxis, and Drp1 and Mfn2 mRNA levels. These findings are consistent with the results of more variable knockdown of the Atg9 gene in both tissue-specific and whole-body knockdown lines. We interpret the findings as a reflection of the fact that Atg9i line did not have efficient knockdown of the gene; however, we cannot completely exclude the possibility that Atg9 is relatively less important comparing to Atg2 and Atg18. Previous studies have shown that effective targeting of Atg2 to the PAS can compensate for loss of Atg18 function in autophagy [41], suggesting more potent role of Atg2 in autophagy/mitophagy.
Reduced autophagy is associated with aging [42,43]. In Drosophila, Atg2 and Atg18 gene expression decreases significantly by 3 weeks of age and remained suppressed thereafter, whereas over-expression of Atg8 in the nervous system extends lifespan [27]. In mice, conditional deletion of the Atg5 gene early in embryogenesis results in impaired cardiac function at 10 weeks of age [13]. These studies show the age-dependent nature of autophagy function. Interestingly, we showed here that Atg2i, Atg9i or Atg18i flies didn’t show significant impairment of cardiac function and mitochondrial structure until 40 days of age. It is conceivable that accumulation of dysfunctional organelles, such as mitochondria, and toxic proteins due to impaired autophagy in the cardiomyocytes leads to accelerated loss of cardiac function with aging.
We employed MitoTimer reporter flies in this study as we have previously shown that this model is particularly suitable for detecting mitochondrial oxidative stress and mitophagy in fly heart during aging or in response to mitochondrial respiratory chain inhibitor treatment [22]. Interestingly, in all three Atg knockdown hearts, the number of pure red puncta is significantly reduced concurrent with clear evidence of elongation of mitochondrial network, but no change in mitochondrial oxidative stress, compared with wild type control flies. The reduction of pure red puncta along with increased mitochondrial volume density in cardiomyocytes is consistent with reduced mitophagy as we did not detect changes in mRNA expression of spargel, the homolog of PGC1-alpha, the mammalian master regulator of mitochondrial biogenesis (Fig. 3G). These morphological changes could potentially be due to knockdown of Atg genes-mediated reduction of mitophagy and compensatory mitochondrial fusion (Fig 5A). We did not measure mitophagy flux directly in fly heart as it is still technically challenging, which is certainly a potential limitation of the present study. Elongation of mitochondria in the knockdown flies may also help explain why the knockdown flies have the same MitoTimer Red/Green ratio (mitochondrial oxidative stress) (Fig. 3C) as mitochondrial fusion helps to dilute the oxidative stress caused by the dysfunctional part of the mitochondrial network. The morphological changes of the mitochondrial network observed by fluorescence microscopy and under the TEM support that inhibition of the Atg9•Atg2-Atg18 complex promotes mitochondrial fusion as a possible compensatory protection.
Surprisingly, all three Atg knockdown flies showed various degree of abnormal mitochondrial morphology concurrent with reduced mitochondrial volume density in IFM, which was opposite of what were observed in the heart. In particular, Atg2i flies showed fragmented mitochondria with great variability in size, very similar to Parkin knockdown flies (Fig. 4A). Similar, but less severe mitochondrial morphological changes were found for Atg18i and Atg9i flies. These findings suggest that under the condition of autophagy inhibition IFM has normal/enhanced mitochondrial fission due to mitochondrial stress and damage. Therefore, the finding of reduced mitochondrial density could be due to a combination of mitochondrial fission and Atg2/Atg9/Atg18-indepdent mitophagy activity in IFM. Interestingly, previous studies show that by suppressing mitochondrial fusion in either IFM or heart, unhealthy mitochondria may be prevented from fusing with the normal ones and be rescued from mitochondrial dysfunction induced by Parkin deficiency [19,21]. It is, however, questionable if inhibition of mitochondrial fusion would be beneficial as the Parkin deficiency flies may have normal mitophagy while our Atg knockdown flies have impaired mitophagy. Nevertheless, the findings in the heart and IFM strongly support the importance of the Atg9•Atg2-Atg18 complex in maintaining mitochondrial structural integrity and function in muscle and heart.
In summary, we have found that 1) Atg2i or Atg18i leads to a short lifespan along with impaired negative geotaxis; 2) Atg2i, Atg18i or Atg9i in the heart leads to age-dependent hypertrophy and impaired cardiac function along with an accumulation of abnormally elongated mitochondria; and 3) Atg2i, Atg18i or Atg9i in indirect flight muscles results in significant mitochondrial fragmentation with reduced mitochondrial density. Our findings suggest that deficiency of Atg2, Atg9 or Atg18 inhibits mitophagy, subsequently leading to exaggerated accumulation of abnormal mitochondria and impaired function of the targeted tissues.
4. Material and Methods
4.1. Fly strains and culture
The UAS-MitoTimer transgenic fly line was established using a commercial embryo injection facility (Genetic Services, Inc). Wild type (w1118), park RNAi (37509), GAL4-mef2, tincΔ4-GAL4, Atg1 Trip (26731) and Atg6 Trip (28060) fly lines were obtained from the Bloomington Drosophila Stock Center (Indiana University, IN). GS-Tub5GS-Gal4 is a generous gift from Dr. Scott Pletcher at University of Michigan. The p{tinC-GFP};p{tinC-GAL4} and act5C88F-Gal4 are gifts from Dr. Matthew Wolf. Atg1i (v16133), Atg2i (v108447), Atg4i (v107317), Atg6i (v22122, v22123), Atg7i (v45558), Atg9i (v10045), Atg13i (v103381), Atg14i (108559) and Atg18i (v105366) RNAi lines were obtained from the Vienna Drosophila RNAi Center (VDRC). UAS-MitoTimer flies were crossed with Mef2-GAL4 to obtain homozygous Mef2-GAL4>UAS-MitoTimer flies (Mef2>>MitoTimer), and then female of these flies were crossed with male of different Atg RNAi flies to generate Atg RNAi line (Mef2>>MitoTimer/UAS-Atg RNAi). Mef2>>MitoTimer/+ flies were used as wild type control. Two days after eclosion once-mated Males and females were collected under light CO2 anesthesia, and housed separately in groups of 20–25 per vial. Flies were transferred to fresh medium and dead flies were scored every 2–3 days. Flies were maintained on a standard corn flour, yeast, and agar medium in a humidified, temperature-controlled incubator at 25°C on a 12:12-h light-dark cycle.
4.2. Negative geotaxis
A test vial is divided into eight octant of equal height of 1 cm. After flies were transfer to the test vials without anesthesia, the test tubes were sharply tapped down 5 times, and the climbing performance was recorded by a video camera. The picture frame at 10 seconds from the last tapping-down was analyzed. Flies that climb to the topmost segment receive a score of 8, flies in the next segment receive a 7, and so on. Negative geotaxis score was calculated as described previously [44]. To assess the impact of aging, we performed this test of each genotype every week throughout a period of 9 weeks. The same experiments were repeated two times at different periods of the year.
4.3. Video-based cardiac function analysis
Semi-intact Drosophila hearts were prepared as described by Ocorr et al. [25,26]. Briefly, flies were anesthetized by Fly Nap (Carolina Biological Supply Co.) for 2–5 minutes, and then dissected in artificial hemolymph buffer at room temperature (~25°C). The surrounding fat and tissue were removed using a pulled glass capillary pipette. The heart beating was recorded for 10 seconds with a high-speed EM-CCD camera. Systolic (SD) and diastolic dimensions (DD) of the heart tube were measured from the movies using our custom-designed MATLAB-based algorithm. A timecourse of overall image brightness was generated for each movie, as described previously [45]. Then, the ‘findpeaks’ algorithm was used to identify minima and maxima in image brightness that corresponds to systole and diastole, respectively. The algorithm randomly selected three systolic and three diastolic images, from which SD and DD heart tube dimensions were quantified in a condition-blinded manner. Fractional shortening (FS) was calculated as (DD-SD)*100/DD.
4.4. Histological analysis
Fly heart wall thicknesses were measured as described by Yu et al. [46]. Forty-day old female flies (adults) were collected and fixed in Telly’s fixation buffer (60% ethanol, 3.33% formalin, 4% glacial acetic acid) for 1 week at 4°C. The samples were embedded in paraffin after ethanol dehydration through sequential gradients and xylenes washes. Paraffin blocks were sectioned serially at 8-μm thickness in longitudinal or transverse orientation. Sections were rehydrated, stained with hematoxylin and eosin, and imaged using a microscope equipped with a digital camera. The heart wall thickness was measured by ImageJ software.
4.5. Confocal imaging of MitoTimer.
The dissected adult fly hearts were fixed in 4% paraformaldehyde for 20 minutes at room temperature. Immediately following fixation, the hearts were whole mounted with 50% glycerol in PBS. Confocal images were obtained within abdominal segment A1 with a set of fixed acquisition parameters for both GFP (Ex/Em 448/518 nm) and DsRed (Ex/Em 558/583 nm) channels (Olympus Fluoview FV1000). The images per heart for analysis were 4–6 nm from the top section. The analysis of MitoTimer signals was previous described [22]. Briefly, positive pixels were thresholded at a value of 1.5 times the mean gray value of above background pixels for each channel. This made the threshold robust against differences in fiber size between images. Saturated pixels (gray level=255) were removed from analysis. In addition, the pixels with red to green ratio of >2.5 are removed for the ratiometric analysis. The ratio of red to green was calculated as a mean value of red to green ratio for all the remaining positive pixels in the image. The number of red dots in the images was calculated by counting areas with clusters of ≥5 pixels with high fluorescence signal (>175) and a red to green ratio of >2.5. The red dot counting was done on images including saturated pixels.
4.6. Transmission electron microscopy
We collected and placed fly abdomens in fixative buffer (3% paraformaldehyde, 2% glutaraldehyde, 100 mM sucrose, 100 mM sodium phosphate buffer, pH 7.2, 2 mM EGTA) at 4°C for overnight. After 45-min wash in 100 mM sodium phosphate buffer (pH 7.2), the hearts were dissected and treated with a post-fixative buffer (1.0% osmium, 100 mM sodium phosphate, pH 7.2) for 2 h at 4°C. Then samples were washed in distilled water for 45 min at 4°C followed by dehydration in increasing concentrations of ethanol. Finally, the samples were embedded in EPON after a sequential incubation in Acetone/EPON. The processing of the samples and image acquisition were performed according to the method describe previously [47].
4.7. qRT-PCR
Total RNA was prepared from 60 age-matched fly hearts of indicated genotype using Trizol (Invitrogen, Carlsbad, USA). One-step SYBR Green-based quantitative RT-PCR was run by an ABI 7000 SDS (Applied Biosystems). Expression of Act5C was used for normalization. Primer sequences are Atg2 (5’-CCCGCTAATCGAAAAATGTG and 3’- GGAAACGGAAGGGAATTGA), Atg9 (5’-AACTTTACGTGGCACGAGGT and 3’- GGAACCTTACAGGCAGCAGT), Atg18 (5’ –AGGTGACCGACGTGTTTAGC and 3’ –ACGGTGGGAATGGAATACAC), Atg1 (5’-ACGGCGGACAAGATTCTCTA and 3’-GCTGCTGCAATATGCTCAAA), Drp1 (5’ –CTCAGATTTCGTCGCACAGT and 3’ –ACGTGGTTCTGCTCATGC), Mfn2 (5’ –ACGAATTGCTTCTGCCAAGT and 3’ –TGATGTTCACCACATTTAGCTTC), and Spargel (5’- GGATTCACGAATGCTAAATGTGTTCC and 3’- GATGGGTAGGATGCCGCTCAG).
4.8. Statistics
Comparisons among different Atg knockdown flies and wild type flies were determined by one-way analysis of variances (ANOVA) with Dunnett’s Multiple Comparison post Test. Students’s t-test was used for 2-group comparisons. Pairwise log rank was performed for comparison of survival between each of the knockdown line with the appropriate wild type control. GraphPad Prism (GraphPad Software, Inc.) and Microsoft Excel statistical software were used for all analyses.
Supplementary Material
Acknowledegements
We thank Dr. Scott Pletcher at University of Michigan for providing us with GS-Tub5GS-Gal4 line for checking global RNAi-mediated knockdown of genes. Research reported in this publication was partially supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR050429 to ZY and the National Institute of Aging of the National Institute of Health under the Award Number of R21AG055712 to RJW. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest
None
References
- [1].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290: 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kim J, Klionsky DJ. Autophagy, cytoplasm-to-vacuole targeting pathway, and pexophagy in yeast and mammalian cells. Annu Rev Biochem 2000; 69: 303–342. [DOI] [PubMed] [Google Scholar]
- [3].Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20: 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284: 12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Noda T, Suzuki K, Ohsumi Y. Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Biol 2002; 12: 231–235. [DOI] [PubMed] [Google Scholar]
- [6].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9: 1102–1109. [DOI] [PubMed] [Google Scholar]
- [7].Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14: 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. Int J Cell Biol 2012; 2018:219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol 2016; 428: 1681–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamamoto A, Simonsen A. The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 2011; 43: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tanida I Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 2011; 14: 2201–2214. [DOI] [PubMed] [Google Scholar]
- [12].Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406: 906–910. [DOI] [PubMed] [Google Scholar]
- [13].Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13: 619–624. [DOI] [PubMed] [Google Scholar]
- [14].Kirshenbaum LA. Regulation of autophagy in the heart in health and disease. J Cardiovasc Pharmacol 2011; 60: 109. [DOI] [PubMed] [Google Scholar]
- [15].Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368: 651–662. [DOI] [PubMed] [Google Scholar]
- [16].Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010; 6: 600–606. [DOI] [PubMed] [Google Scholar]
- [17].Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging (Albany NY) 2009; 1: 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lo Verso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 2014; 10: 1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW 2nd. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 2014; 114: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Taghli-Lamallem O, Plantié E, Jagla K. Drosophila in the Heart of Understanding Cardiac Diseases: Modeling Channelopathies and Cardiomyopathies in the Fruitfly. J Cardiovasc Dev 2016; 3: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 2008; 105: 14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem 2014; 289: 12005–12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang CW, Kim J, Huang WP, Abeliovich H, Stromhaug PE, Dunn WA, et al. Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J Biol Chem 2001; 276: 30442–30451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 2012; 198: 219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A 2007; 104: 3943–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vogler G, Ocorr K. Visualizing the beating heart in Drosophila. J Vis Exp 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008; 4: 176–184. [DOI] [PubMed] [Google Scholar]
- [28].Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012; 23: 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell 2013; 154: 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 2014; 53: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nagy P, Varga A, Pircs K, Hegedus K, Juhasz G. Myc-driven overgrowth requires unfolded protein response-mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLoS Genet 2013; 9: e1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pircs K, Nagy P, Varga A, Venkei Z, Erdi B, Hegedus K, et al. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PLoS One 2012; 7: e44214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–1036. [DOI] [PubMed] [Google Scholar]
- [34].Webber JL, Young AR, Tooze SA. Atg9 trafficking in Mammalian cells. Autophagy 2007; 3: 54–56. [DOI] [PubMed] [Google Scholar]
- [35].Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 2009; 106: 20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125: 1488–1499. [DOI] [PubMed] [Google Scholar]
- [37].Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 2007; 12: 209–218. [DOI] [PubMed] [Google Scholar]
- [38].Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem 2008; 283: 23972–23980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Reggiori F, Tucker KA, Stromhaug PE, Klionsky DJ. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell 2004; 6: 79–90. [DOI] [PubMed] [Google Scholar]
- [40].Suzuki K, Akioka M, Kondo-Kakuta C, Yamamoto H, Ohsumi Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J Cell Sci 2013; 126: 2534–2544. [DOI] [PubMed] [Google Scholar]
- [41].Kobayashi T, Suzuki K, Ohsumi Y. Autophagosome formation can be achieved in the absence of Atg18 by expressing engineered PAS-targeted Atg2. FEBS Lett 2012; 586: 2473–2478. [DOI] [PubMed] [Google Scholar]
- [42].Vellai T, Takacs-Vellai K, Sass M, Klionsky DJ. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol 2009; 19: 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Toth ML, Sigmond T, Borsos E, Barna J, Erdelyi P, Takács-Vellai K, et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008; 4: 330–338. [DOI] [PubMed] [Google Scholar]
- [44].Bazzell B, Ginzberg S, Healy L, Wessells RJ. Dietary composition regulates Drosophila mobility and cardiac physiology. J Exp Biol 2013; 216: 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P, Izpisua Belmonte JC, Giles W, et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques 2009; 46: 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu L, Lee T, Lin N, Wolf MJ. Affecting Rhomboid-3 function causes a dilated heart in adult Drosophila. PLoS Genet 2010; 6: e1000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Miller S, Howell D. Viral infections in the acquired immunodeficiency syndrome. J Electron Microsc Tech 1988; 8: 41–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.