

Abstract
The microbial metabolites known as the macrolides are some of the most successful natural products used to treat infectious and immune diseases. Describing the structures of these complex metabolites, however, is often extremely difficult due to the presence of multiple stereogenic centers inherent in this class of polyketide-derived metabolites. With the availability of genome sequence data and a better understanding of the molecular genetics of natural product biosynthesis, it is now possible to use bioinformatic approaches in tandem with spectroscopic tools to assign the full stereostructures of these complex metabolites. In our quest to discover and develop new agents for the treatment of cancer, we observed the production of a highly cytotoxic macrolide, neaumycin B, by a marine-derived actinomycete bacterium of the genus Micromonospora. Neaumycin B is a complex polycyclic macrolide possessing 19 asymmetric centers, usually requiring selective degradation, crystallization, derivatization, X-ray diffraction analysis, synthesis, or other time-consuming approaches to assign the complete stereostructure. As an alternative approach, we sequenced the genome of the producing strain and identified the neaumycin gene cluster (neu). By integrating the known stereospecificities of biosynthetic enzymes with comprehensive NMR analysis, the full stereostructure of neaumycin B was confidently assigned. This approach exemplifies how mining gene cluster information while integrating NMR-based structure data can achieve rapid, efficient, and accurate stereostructural assignments for complex macrolides.
Graphical Abstract
INTRODUCTION
Macrolides, a class of natural products produced by type I polyketide synthases, are one of the most important classes of microbially produced antibiotic, anticancer, and immunomodulatory drugs. Classic macrolides such as erythromycin and fidaxomicin and the semisynthetic macrolides azithromycin and clarithromycin have become a cornerstone in the treatment of Gram-positive bacterial infections,1,2 while the amphoterocin B and nystatin macrolides are widely used for persistent fungal infections.3 Other macrolides, such as tacrolimus (FK-506), have proven to be of immense utility in immunosuppression following organ transplant surgery.4–6 Although classically developed for infectious diseases, the macrolide ixabepilone, a semisynthetic analogue of epothilone B, has been developed for the treatment of breast cancer.7 Of course, the avermectins stand out as one of the most important macrolides ever discovered.8 They have found extensive commercial applications as pesticides and treatments for parasitic worms. Other recent studies of classic macrolides as well as newly discovered marine-derived macrolides indicate considerable unrecognized potential in the treatment of cancer.9,10 Assignment of the full absolute configuration for macrolides is often a complex and difficult task requiring a multitude of approaches including X-ray diffraction methods, extensive 2D NMR analysis, the preparation of chiral derivatives, partial degradation, and analogue and asymmetric synthesis. Consequently, the full structures of many macrolides have required extensive efforts often requiring many years of effort. As more is learned about macrolide biosynthesis, it has become clear that numerous enzymatic steps including those mediated by ketoreductases, dehydratases, and enoyl-reductases are highly specific in terms of stereochemical outcome. As biosynthetic predictions based on bioinformatic data become more confident, it is becoming increasingly feasible to use enzyme stereospecificity to help define the absolute configurations of polyketide metabolites. Here we demonstrate the application of this approach in the assignment of the full relative and absolute configuration at 19 chiral centers in neaumycin B (1), a member of the cytovaricin-ossamycin-oligomycin class macrolides.11–14 The history of neaumycin is complex. The first isolation of a neaumycin congener was reported by Shen and co-workers,15 but the structure was substantially revised (as neaumycin A) in 2015, albeit without stereochemistry and lacking crucial physical and optical data.16 A congener, neaumycin B (1), was reported at the same time, also as a planar macrolide and lacking substantial optical and spectral properties.16 Comparing our spectroscopic and physical data with this report shows considerable similarities; however given the massive number of possible stereoisomers associated with 19 chiral centers, we can only assume that our isolated macrolide is identical to neaumycin B (1).
EXPERIMENTAL PROCEDURES
General Experimental Information.
Optical rotations were measured using a JASCO P-2000 polarimeter with a 1 cm cell. UV spectra were measured with a Varian Cary UV–visible spectrophotometer with a path length of 1 cm, and IR spectra were acquired on a PerkinElmer 1600 FT-IR spectrometer. The 1D and 2D NMR spectroscopic data were obtained on Varian INOVA 500 and JEOL 500 NMR spectrometers. The chemical shift values are reported in ppm units, and coupling constants are reported in Hz. NMR chemical shifts were referenced to the residual solvent peaks (δH 7.25 and δC 77.2 for CDCl3). The high-resolution ESI-TOF mass spectral data were recorded on an Agilent 6530 HR-TOF LCMS. Preparative HPLC separations were performed using a Hewlett-Packard 1050 series with a reversed-phase C-18 column (Phenomenex Luna, 10.0 × 250 mm, 10 μm) at a flow rate of 2.0 mL/min. The HPLC/MS data were obtained using an Agilent 6530 Accurate-Mass Q-TOF spectrometer coupled to an Agilent 1260 LC system with a Phenomenex Luna C18 column (4.6 × 100 mm, 5 μm, flow rate 0.7 mL/min).
Isolation, Identification, and Cultivation of Micromonospora Strain CNY-010.
Micromonospora sp. (strain CNY-010) was isolated from the surface of the brown alga Stypopodium zonale, collected in the Bahamas Islands in 2010, by swabbing the algal surface. The strain was orange with black spores and did not require seawater for growth. BLAST analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) revealed that strain CNY-010 shared 99% 16S rRNA gene sequence identity with M. tulbaghiae strain TVU1 (GenBank accession no. KM485658), thus identifying it as a member of the genus Micromonospora. The strain was typically cultured in 36 × 1 L scale in 2.8 L Fernbach flasks each containing 1 L of a seawater-based medium (10 g of starch, 4 g of yeast, and 2 g of peptone) and shaken at 180 rpm at 27 °C. After 7 days of cultivation, sterilized XAD-7 resin (20 g/L) was added to adsorb the liberated organic materials, and the culture and resin were shaken at 180 rpm for 3 h. The resin and microbial cell mass were filtered through cheesecloth, washed with deionized water, and eluted with acetone. The acetone was next removed under reduced pressure, and the resulting complex aqueous layer extracted with ethyl acetate (2 × 300 mL). The ethyl acetate-soluble fraction was dried in vacuo to yield 4.5 g of organic extract from a 36 × 1 L culture.
Isolation and Purification of Neaumycin B (1).
The ethyl acetate extract (4.5 g) was subjected to silica vacuum flash chromatography, using a step gradient solvent system of n-hexane, EtOAc, and MeOH (1:0:0, 10:1:0, 5:1:0, 2:1:0, 1:1:0, 0:1:0, 0:10:1, 0:5:1, 0:2:1, and 0:0:1; each 200 mL) to yield 10 fractions. Fraction 8 was subjected to C-18 reversed phase Prep HPLC (Phenomenex Luna C-18 column, 250 × 10, 10 μm, 89% isocratic MeOH/H2O, 2 mL/min, UV detection at 280 nm), which resulted in the isolation of neaumycin B (20 mg). Neaumycin B (1): pale yellow, amorphous solid, 48.0 (c 0.1, CHCl3); IR (NaCl) υmax 3436 (br), 2940, 2866, 1739, 1470 cm−1; UV (MeOH) λmax (log ε) 274 (3.39) nm; 1H and 13C NMR data, see Tables S1 and S2; HR ESI-TOF m/z 897.5696 [M + Na]+ (calcd for C50H82O12Na, 897.5698).
Genomic DNA isolation. Sequencing and Assembly.
High-purity genomic DNA was extracted by successive phenol/chloroform/isoamyl alcohol purification steps followed by precipitation with 2-propanol, treatment with RNase, and a final purification and precipitation step.17 Quantification was done in 1% agarose gel electrophoresis with a NanoDrop spectrometer (Saveen Werner, Sweden) and Qubit 2.0 analyzer (Invitrogen, UK). Genome sequencing was performed using Illumina HiSeq4000 and Pacific Biosciences RS single-molecule real-time (SMRT) sequencing platforms at the IGM Genomics Center, University of California, San Diego, La Jolla, CA, USA. Genome assembly was done with the hybridSPAdes algorithm.18 Final polishing steps (correction of ambiguous bases and circularization) were performed using PCR and Sanger sequencing. The genome was submitted to the National Center for Biotechnology Information (NCBI) database under the accession number CP024087. Biosynthetic gene clusters were identified in the assembled genome using AntiSMASH 4,19 and domains identified in the neaumycin gene cluster aligned using CLC Main Workbench version 7.8.1 (gap open cost: 10, gap extension cost: 1, end gap cost: as any other).
Cytotoxicity Bioassay Methods.
Human glioblastoma U87, ovarian cancer SKOV3, breast cancer MDA-MB-231, and colorectal carcinoma HCT116 were grown in RPMI medium (Corning) supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin/L-glutamine, nonessential amino acids, sodium pyruvate, and HEPES. For LD50 determination, cells were plated in low density and treated across a titration of specified compound (w/v) in biologic triplicate. After 5 days of treatment, cell viability was determined by following the alamarBlue (Thermo) cell viability protocol. Briefly, alamarBlue was loaded at 1:10 v/v dilution into each well and allowed to be properly metabolized by cells. After incubating for the appropriate length of time, 3–6 h, fluorescence intensity was measured using a Tecan plate reader (excitation: 560 nm, emission: 590 nm). Absorbance values were then normalized to the lowest concentration (1 × 10−5 μg/mL) to calculate percent survival. Normalized absorbance values were then used to calculate LD50 values using nonlinear regression analysis software (Prism).
RESULTS AND DISCUSSION
Discovery of Neaumycin B (1) and Preliminary Structure Analysis.
Pursuing our interest in exploring marine microorganisms for cancer drug discovery, we have continued to isolate and screen marine-derived actinomycete bacteria for their production of cell-cycle-inhibitory compounds. Our goal continues to be the discovery of new drug candidates that show significant levels of activity coupled with cancer-tissue-type selectivity in the NCI’s 60 cell line panel. In this regard, we focused our attention on an actinomycete strain, a Micromonospora sp. (strain CNY-010), which we isolated from the surface of the tropical brown alga Stypopodium zonale collected in the Bahamas Islands. The culture extract of this strain showed modest in vitro growth inhibition against HCT-116 colon carcinoma (IC50 = 1 μM) and was thus subjected to thorough bioassay-guided fractionation first by preparative methods and then by RP C-18 HPLC. Final purification yielded a complex macrolide, tentatively identified as neaumycin B (1), that analyzed for the molecular formula C50H82O12 by HRESIMS data ([M + Na]+, m/z = 897.5696, calcd 897.5698) in combination with 2D NMR analysis. In preliminary testing in the National Cancer Institute’s 60 cell line panel, significant activity and selectivity were observed against RPMI-8226, a major plasma cancer cell type involved in multiple myeloma (Figure S1), suggesting potential in cancer drug discovery.
Planar NMR Structure Analysis for Neaumycin B (1).
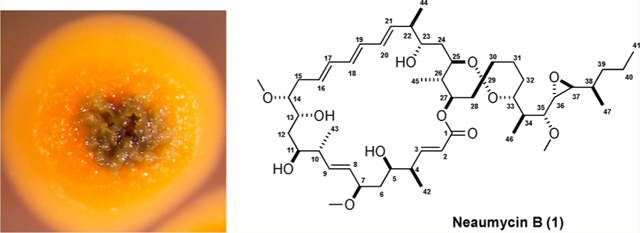
The UV spectrum of 1 in MeOH exhibited absorption bands at 262 (sh), 274, and 283 (sh) nm, suggestive of a triene moiety. An IR absorption band at 1739 cm−1 suggested the presence of an ester group, which was supported by the presence of one carbonyl signal at δC 165.9 in the 13C NMR spectrum. 13C NMR, DEPT, and HSQC spectral data (Tables S1 and S2 and Figures S10–S21) confirmed the presence of 50 carbons, including an ester carbonyl, an oxygenated quaternary carbon, 10 olefinic methines, 12 oxymethines, 10 methylenes, six aliphatic methines, seven methyl-bearing carbons, and three methoxy-bearing carbons. These carbon signals explained six of the 10 degrees of unsaturation inferred from the molecular formula, thus illustrating that 1 was composed of four rings. The 1H and HSQC NMR spectra (Figures S10 and S14) revealed 78 proton signals, including 10 downfield olefinic protons at δH 6.75–5.22, 12 oxymethine protons at δH 4.84–2.47, three methoxy groups at δH 3.54, 3.14, and 3.23, 10 methylene groups at δH 2.62–1.40, sixupfield methine protons at δH 2.31, 2.22, 2.16, 1.61, 1.47, and 1.36, and seven methyl groups at δH 1.11, 1.10, 1.02, 0.93, 0.91, 0.83, and 0.66. The planar structure of 1 was next established by interpretation of COSY, TOCSY, HSQC, and HMBC NMR correlations. First, interpretation of COSY and TOCSY spectral data for 1 showed four continuous spin systems (substructures A–D), as in Figure 2. These four substructures were assigned by detailed analysis of all 1D and 2D NMR spectral data including HMQC, HMBC, and other methods.
Figure 2.
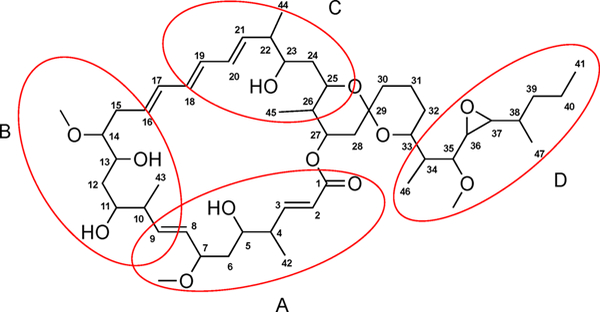
Substructures A–D and the full planar structure of neaumycin B (1) assigned by 2D NMR methods.
Analysis of COSY and TOCSY data led to the assignment of substructure A (C-1 to C-9), Figure 2. The methoxy group (C-7-OCH3) and a methyl group (C-42) were placed at C-7 (δC 84.3) and C-4 (δC 44.1) by analysis of COSY and HMBC NMR data. HMBC NMR correlations of the H-2 olefinic proton to the C-1 quaternary carbon (δC 165.9) and C-4 and of H-3 to C-1, C-2 (δC 122.0), C-4, and C-42 (δC 16.1) illustrated that substructure A incorporated the lactone functional group. Substructure B (C-8 to C-15) was assigned on the basis of COSY, TOCSY, and HMBC data. C-11, C-13, and C-14 were oxygenated, based on their 13C chemical shifts, δC 70.1, 70.3, and 83.9, respectively. Substructure C (C-18 to C-25) was similarly assigned by interpretation of 2D NMR data. COSY and HMBC NMR data showed the two methyl groups, C-44 (δH 17.2) and C-45 (δH 13.7), were positioned at C-22 (δC 45.2) and C-26 (δC 41.8), respectively. Finally, substructure D (C-34 to C-38) was assembled by identical interpretation of 2D NMR data (Figure 2). The presence of a trans epoxide at C-36 (δ 2.94; δC 61.2) and C-37 (δH 2.47; δC 58.8) was inferred from the 1H and 13C chemical shifts and 1H–1H coupling constant (JH-36,H-37 = 2.3 Hz).20 Also, the trans epoxide was confirmed by ROESY correlations between H-37 and H-35 (δH 3.19) and between H-36 and H-39 (δH 1.40).
Subsequently, substructures A and B were linked through HMBC correlations between H-8 and C-10 (δc 41.7) and H-10 (δH 2.22) and C-8 (δC 129.8), as well as long-range 1H–1H correlations from H-8 to H-10 observed in the TOCSY spectrum. Substructures B and C were linked by a triene chain (C-16 to C-21). HMBC cross correlations from H-2, H-3, and H-27 (δH 4.84) to the C-1 ester carbonyl carbon established the ester linkage of spin systems A and C. Similarly, HMBC correlations from H2-28 (δH 2.62, 1.27) and H2-30 (δH 1.65, 1.39) to C-29 (δC 98.9) indicated connectivity between spin systems C and D and the presence of the bicyclic spiro ketal system. ROESY NMR analysis of the spiro ketal protons provided strong support for the relative configuration of the spiro ketal moiety (Figure 8). This assignment was also confirmed by comparison of the 1H and 13C NMR data of 1 with that of an analogous spiroketal constellation in maclafungin.21 Consequently, the planar structure 1 was defined as a 28-membered macrolide featuring a bicyclic spiro ketal.
Figure 8.
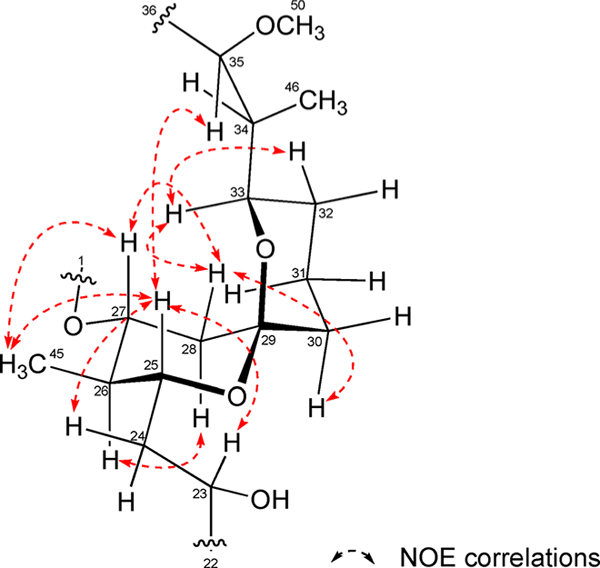
Absolute stereostructure of the spirobicyclic C-29 ketal in 1 as defined first by BGC analysis for the C-25 hydroxy group followed by assignment of the stereocenters at C-26, −27, −29, and −33 assigned in full by analysis of NOE NMR data.
The classic structural characteristics of neaumycin B obviously defined this compound as a polyketide derived from a modular type I polyketide synthase (PKS) biosynthetic pathway. Given the inherent difficulty in assigning the full absolute configuration of this macrolide, we sequenced the genome of strain CNY-010 with the aim of using bioinformatic approaches to facilitate full stereostructure elucidation.22,23 With this information in hand it was conceivable that considerable insight into the many stereogenic centers could be obtained. In many cases, as with neaumycin B, macrolides are not crystalline; hence their full stereostructures have been determined using laborious spectral methods, chemical degradation, and/or total synthesis, which is an extremely time-consuming approach.24–28 NMR methods such as J-based configuration analysis have been widely used to define the relative configurations of macrolides.29–31 However, this approach is lacking, as it fails to define the absolute configuration of the compound, and these assignments can have significant insecurity in flexible systems. In addition, remote, isolated stereocenters are almost impossible to assign since they cannot be linked to defined chiral centers.
Identification of the Type I PKS Biosynthetic Gene Cluster (BGC) Responsible for Neaumycin B Biosynthesis.
The full genome sequence of strain CNY-010 was obtained using Illumina HiSeq4000 and Pacific Biosciences RS SMRT sequencing platforms and evaluated for the presence of a BGC consistent with the 41-carbon PKS-derived backbone of neaumycin B. We employed a combination of long read PacBio and Illumina sequencing because of the modular nature of type I PKS genes, which include highly repetitive sequences that are notoriously difficult to assemble from short-read sequence data. The assembled genome, with a size of 7.2 Mb and 72.5% GC content, was analyzed for secondary metabolite BGCs using antiSMASH 4.19 Of the 16 BGCs identified (Figure 3), only one could be annotated as a modular type I PKS (BGC#16, Table S4), making it the only logical candidate for neaumycin B biosynthesis. This 126 kb neu BGC contains nine discrete PKS genes organized into 20 modules that contain 95 enzymatic domains, indicating it would be of sufficient complexity to encode the production of this 50-carbon macrolide (Figure 4).
Figure 3.
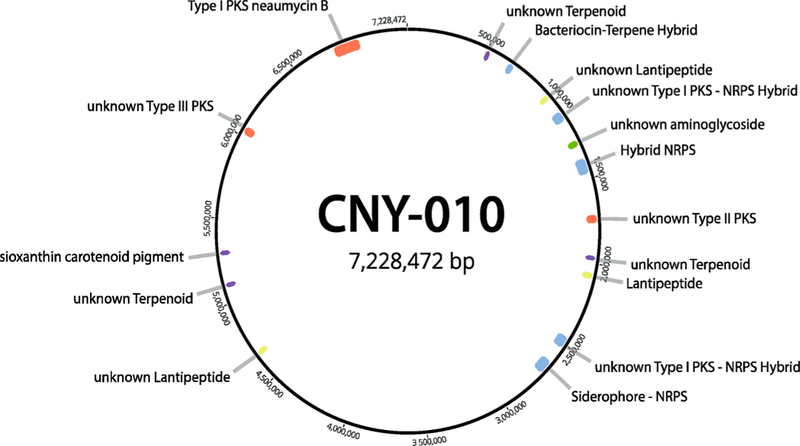
Graphic illustration of the Micromonospora CNY-010 genome showing the presence of 16 secondary metabolite biosynthetic gene clusters and one type 1 PKS cluster for neaumycin B biosynthesis. Additional details of the relationship of these clusters to known BGCs are described in Table S4.
Figure 4.
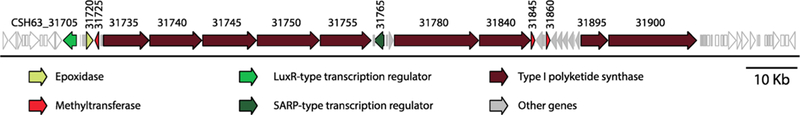
Neaumycin B (neu) BGC identified using antiSMASH analysis. Annotations are given for key biosynthetic genes; for complete gene annotation see Table S3.
To better understand the evolutionary history of the neaumycin B gene cluster (#16 Table S4), we used it as a multigene BLAST query against all 63 Micromonospora genome sequences available in the NCBI database (as of October 2017).32 This gene cluster was not detected in any of the Micromonospora genomes or in any reported bacterial genome based on NCBI BLAST analysis, indicating its rarity and the likelihood it was acquired by strain CNY-010 via horizontal gene transfer. The gene clusters for the other sources of this class of compound [Streptomyces NEAU-x21115 and Actinoplanes sp. ATCC 3307616] have apparently not been defined. Interestingly, the regions up- and downstream from the neu BGC are highly conserved among Micromonospora strains (Figure S2), which helped to define the BGC boundaries.
Assuming the precedent of linearity in the assembly of polyketides from type I modular PKSs,33,34 we next examined if the module and domain architecture of the PKS genes in BGC#16 (Table S4) conformed to the reported neaumycin B planar structure16 and that predicted by our NMR analysis (Figure 2). The loading/starter and termination modules could be inferred by the presence of ketosynthaseQ (KSQ) (locus_tag CSH63_31735) and thioesterase (TE) domains (CSH63_31900) at the N and C termini, respectively. The assembly of the linear polyketide backbone was then predicted using antiSMASH and manual analyses of sequence motifs associated with acyl transferase (AT) domain substrate specificity.35–37 For the AT domains associated with the 20 PKS modules, 11 are predicted to incorporate malonyl (modules 2, 4, 5, 6, 8, 10, 11, 12, 16, 17, and 19), eight methylmalonyl (modules LD, 1, 3, 7, 9, 14, 15, and 18), and one (module 13) methoxymalonyl CoA substrate (Figures 4 and 6A). These predictions matched perfectly with the NMR planar structure with the exception of module 14, which incorporates malonate instead of the predicted methylmalonate (methyl group at C-12, Figure 5). While this AT domain contained the amino acid signature for methylmalonate [YASH], two amino acid changes immediately upstream of this motif could explain the noncanonical substrate specificity (Figure S4).
Figure 6.
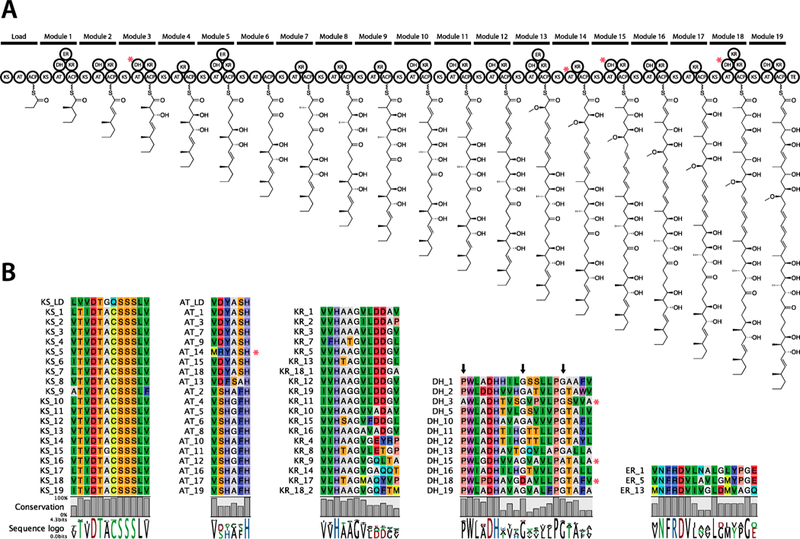
Neaumycin B BGC (neu). (A) Modular and domain architecture of the PKS genes and predicted structure of the growing polyketide. (B) Amino acid sequence alignments of the domain active sites. Red asterisks denote domains with mutations that may account for change of function. Black arrows denote specific amino acid positions where changes may account for domain inactivation. For full alignments see the Supporting Information, Figures S3–S7.
Figure 5.

Predicted planar structure of neaumycin B (1) based on bioinformatic analysis of the neu gene cluster. Note that the predicted C-12 methyl substituent based upon methylmalonate incorporation was not observed in the final macrolide.
Next, we further addressed the bioinformatic structure predictions based on the distribution of ketoreductase, dehydratase, and enoyl-reductase domains in the PKS modules.38 Module 6 lacks all three domains and thus accounts for the only carbonyl in the molecule (C-29), outside of that incorporated via the terminal module (C-1). The remaining 18 extender modules contain a KR domain, and all of these appear to be functional based on antiSMASH predictions, active site sequence alignments (Figure 6B), and the planar NMR structure. Twelve DH domains were identified, and likewise all appear to be functional based on antiSMASH and active site sequence alignments. However, the NMR structure reveals hydroxy groups present at C-5, C-11, and C-35 that were not dehydrated to the olefin as predicted (Figure 2). Further examination of the active site amino acid sequences for these three domains (modules 3, 15, and 18) reveals mutations that could account for inactivation (Figure 6B), thus explaining the deviations from the bioinformatic predictions. All three ER domains identified in the gene cluster appear functional based on sequence alignments (Figure S7), which is consistent with NMR data.
The remaining differences between the predicted linear structure defined by the PKS assembly line and the NMR- based structure can be accounted for by post-PKS tailoring enzymes observed in the BGC. More specifically, the three methyltransferases encoded within the BGC (CSH63_31725, CSH63_31845, CSH63_31860) could account for methylation of the hydroxy groups at C-7 and C-35. The discrepancy between the number of methyltransferases and positions methylated suggests that not all of these enzymes are functional. The epoxidase at locus CSH63_31720 could account for the olefin epoxidation between C-36 and C-37 (Figure 3). Overall, there is a remarkably strong correlation between the predicted bioinformatic planar structure and the NMR structure. Given this congruence and the observation that BGC#16 is the only type I PKS observed in the CNY-010 genome sequence, it can be established with a high degree of confidence as the biosynthetic origin of neaumycin B. The subtle disparities between bioinformatic prediction and NMR-based structure assignment demonstrate the value of combining both approaches and provide additional exceptions to bioinformatic paradigms.38
Bioinformatic Prediction of Absolute Configuration.
Considering the arduous and time-consuming approaches involved in relying solely on chemical methods to assign absolute configuration, we returned to the neu BGC to help guide NMR-based structure analysis. In regard to the methyl groups associated with the incorporation of methylmalonyl-CoA, AT domains are specific for the 2S-isomer with the subsequent KS-catalyzed condensation reaction determining whether this configuration is retained or inverted.39 In cases where a ketoreductase (KR) domain is present, the beta-ketone is reduced to the corresponding hydroxy group in a stereospecific manner during chain elongation, with the KR referred to as type A or B depending upon the resulting orientation. KR domains can also act as epimerases, thereby impacting C2 methyl configuration.40,41 Based on amino acid sequence alignments and antiSMASH analyses, six of the KR domains in this BGC could be assigned to type A with no epimerase activity (subtype A1, modules 4, 8, 9, 14, 17, and 18), eight to type B with no epimerase activity (subtype B1, modules 1, 2, 3, 5, 11, 12, 13, and 19), and one each to subtypes B2 (type B with epimerase activity) and C2 (epimerase but no ketoreductase activity, modules 7 and 18, respectively). Given that module 18 contains two KR domains (type A1 and C2), stereochemical predictions were not be made for the C-4 methyl and C-5 hydroxy groups. To confirm the presence of two KR domains in this module, which to the best of our knowledge is unprecedented, we PCR amplified across both domains and showed that the genome assembly was correct (Figure S8). In the case of module 15, the KR domain presents an [FDD] motif. While similar to the type-B [LDD] motif, it is not clear if this would confer a type B absolute configuration; thus the absolute configurations for the C-10 methyl and C-11 hydroxy groups could not be predicted. A prediction was also not made for the C-22 methyl group incorporated by module 9 given evidence that some type A1 KR domains can in fact have epimerase activity.39,40
Given these bioinformatic assessments, the hydroxy groups at carbons C-7, C-13, C-25, C-27, C-33, and C-35 could be assigned R configurations based on the KR domains in modules 17, 14, 8, 7, 4, and 3, respectively, while C-23 was assigned an S configuration based on module 9. Furthermore, the methoxy functionality at C-14 (module 13) was assigned an R configuration, while the methyl groups associated with the incorporation of methylmalonyl CoA were assigned R configurations at C-26 (module 7) and C-38 (module 1) and an S configuration at C-34 (module 3). In all 11 of the cases where predictions could be made, they were confirmed by NMR-based analysis (Figure 7).
Figure 7.
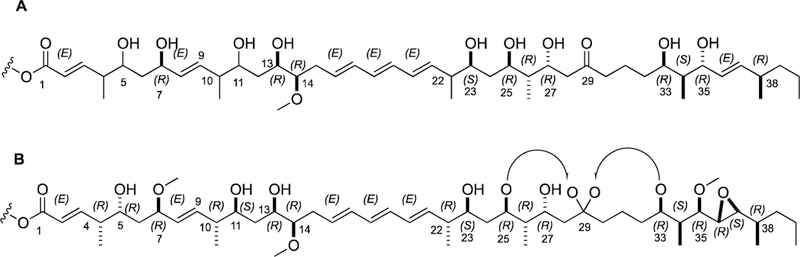
Comparison of the absolute configuration for neaumycin B (1) based on bioinformatic prediction (A) and NMR-based assignments (B).
Some of the hydroxy groups in the neaumycin B polyketide were further converted to olefins by dehydratase (DH) activity. While cis (Z) double bonds have been occasionally observed in polyketides, it has been suggested that all PKS DHs produce trans (E) double bonds and that changes in configuration arise from mechanisms other than direct DH-catalyzed dehydration.39 For this reason, the olefinic bonds at C-2, C-8, C-16, C- 18, C-20, and C-36 were predicted to be in E configurations, an assignment that was confirmed by the NMR coupling constant data derived by 2D J-resolved methods (Figure S18).
Enoyl reductase (ER) domains are responsible for the reduction of double bonds to saturated methylene groups during polyketide biosynthesis. When methylmalonyl CoA is incorporated, enoyl reduction has stereochemical consequences, producing both the 2R and 2S configurations.42 For the ER domains present in modules 1 and 13 (which incorporate methylmalonate), module 1 possesses the conserved tyrosine (Y44, ER alignment numbering in Figure S7) signature for the 2S-methyl configuration, while module 13 has a valine (V44) in this position, indicating a 2R-methyl configuration. The predicted stereochemistry was observed for module 13 (C-14) but not for module 1 (C-38), which we ultimately assigned an R configuration based on 2D NMR data. Although recent advances have been made in the relationships between ER domain structure and stereochemical control, it remains apparent that other factors are important in determining absolute configuration.39 We could find no gene-sequence-based explanation as to why the predicted orientation was not observed.
Combining Gene Cluster-Derived and NMR-Based Structural Information.
It was immediately clear from the BGC that neaumycin B was off-loaded from module 19 in typical fashion as a 28-membered macrolactone. NMR analysis illustrated that the lactonization occurred with the C-27 = R hydroxy group. With the extensive genomic stereochemical predictions as a guide, we approached assigning the full relative and absolute configuration of 1 using a variety of NMR experiments. The linear carbon backbone was readily confirmed by analysis of COSY and HMBC NMR correlations. What became immediately obvious was the lack of a ketone functionality in 1 that was predicted at C-29 by BGC analysis. From our NMR data it became clear that the ketone had cyclized to form a spiro-bicyclic ketal involving the C-25 and C-33 hydroxy groups. The rigid spiroketal functionality was readily defined by a combination of 2D NMR data including ROeSy NMR data, using the predicted C-25 = R assignment from the BGC analysis as a guide. The rigidity of the bicyclic spiroketal allowed NOE-based analysis that confirmed the C-33 carbon to also have an R configuration. Further COSY NMR and NOE-based analysis of the bicyclic spiroketal component allowed the full absolute configuration of this bicyclic ketal to be assigned (Figure 8, Table S2, Figure S20).
To correlate all stereogenic centers in neaumycin B, we began by using the gene cluster assignment of the absolute configuration at C-7 as R and integrating the other predicted stereocenters to construct molecular models. Consequently, interpretation of NMR data involving J-based HETLOC configurational analysis, along with interpretation of ROESY NMR correlations, led to confident assignments.
Relative Configuration by NMR for Neaumycin B (1).
The relative configurations of the four substructures in 1 (substructures A–D, Figure 2) were determined by HETLOC and ROESY NMR analyses (Figure 9). The relative configuration from C-4 to C-5 was determined by assignment of the vicinal methine carbons. A small homonuclear coupling constant between H-4 and H-5 (2.0 Hz) suggested that H-4 and H-5 were gauche. A large heteronuclear two-bond C–H coupling constant measured for C-5/H-4 (8.6 Hz) indicated that H-4 was gauche to the electronegative oxygen substituent at C-5. These data were consistent with two rotamer systems, threo or erythro; however, the observed ROESY correlations between H-4 and H-5 and between H-5 and H-42 were consistent with a threo rotamer system. On the basis of the relative configuration of a (4,5)-syn relationship, we determined it to be 4,5,7-syn/anti of C-4 to C-7 configurations, which was located between two trans double bonds through the ROESY correlations of H-2/H-42, H-42/H-5, H-5/H-7, and H-7/H-9 (Figure 9A). The relative stereochemistry of C-10 to C-14, containing a methyl group, two hydroxyl groups, and a methoxy group, was assigned an all-syn relationship except for the methyl group. First, the C-11/C-12 configuration was based on a small 3JH,H of H-11 with H-12h (2.3 Hz), a large 3JH,H of H-11 with H-121 (10.5 Hz), and a small 2JH,C of C-11 with H-12 (<2.0 Hz). These J values supported only one rotameric system. Second, the configuration at C-13/C-14 was established on the basis of a small 3JH,H of H-13 with H-14 (2.7 Hz) and small 2JH,C couplings of C-13 with H-14 (<2.0 Hz) and C-14 with H-13 (<2.0 Hz). Only one of four threo H/H-gauche orientations matched all these data. Third, the C-10 methyl group was assigned based on relevant ROESY correlations between two vicinal methine protons with two protons on the E double bond (H-8 with H-10 and H-10 with H-11). All of these data demonstrated a 10,11,13,14-anti/syn/syn relative configuration from C-10 to C-14. This was further supported by ROESY correlations of H-8/H-10, H-9/H-11, H-10/H-11, H-12l/H-14, and H-13/H-14 (Figure 9B). For the C-22–C-23 bond, a small vicinal coupling constant between H-22 and H-23 (2.3 Hz), a small 2JH,c of C-23 with H-22 (<2.0 Hz), and a large 2JH,c of C-22 with H-23 (<8.2 Hz) were observed. These J values indicated two possible rotameric orientations. However, key ROESY correlations of H-22 with H-23 and of H-23 and H3-44 allowed the relative configuration of C-22–C-23 to be assigned as the erythro gauche rotamer. Additionally, ROESY correlations between H-20/H-22, H-21/H-241, and H-23/H-25 established the relative configuration of C-20 to C-25 to be 22,23,25-anti/syn, although the key 2,3JH,C values were not detected in the HETLOC experiment (Figure 9C).
Figure 9.
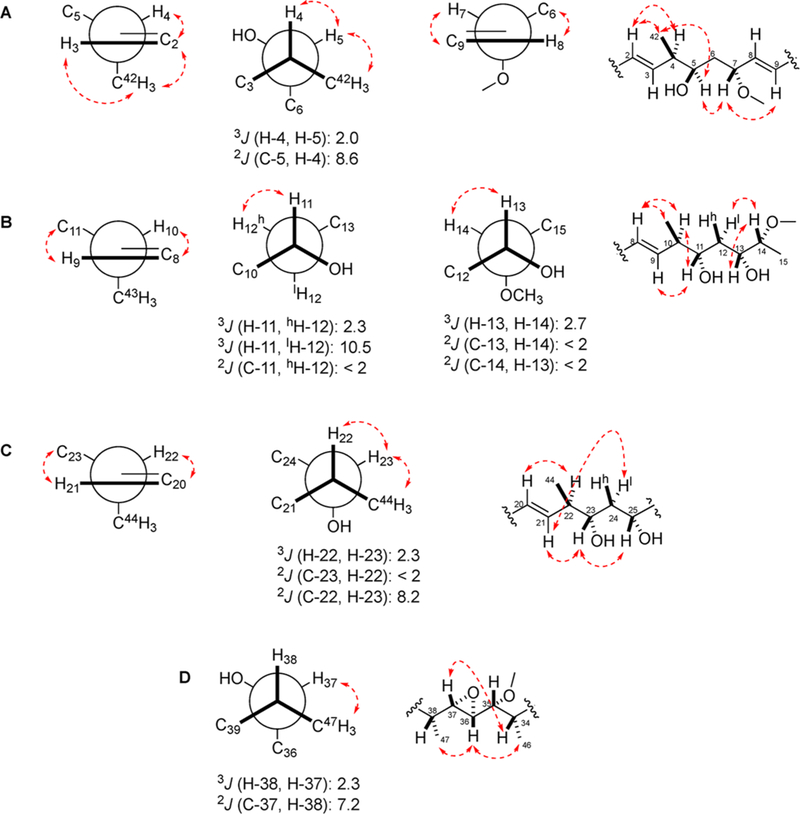
Connectivities and relative configurations for four individual spin systems (substructures A–D) by homo- and heteronuclear coupling constant analysis and NOE correlations. Substructures: (A) C-1 to C-9; (B) C-8 to C-15; (C) C-20 to C-25; and (D) C-34 to C-38. 2J and 3J values are in hertz; dashed red lines are NOE correlations.
The relative stereochemistry of C-34 to C-38, which included the trans epoxide unit, was deduced by a threo gauche rotamer (C-37/C-38) and relevant ROESY correlations. A small 3JH,H of H-38 with H-37 (2.3 Hz) and a large 3JH,C of H-38 with C-37 (7.2 Hz) suggested a threo system, and a continuous trans epoxide together with ROESY correlations between H-34/H-37, H-36/H3-46, and H-36/H3-47 established the relative stereochemistry depicted in Figure 9D. Thus, the relative configuration of the 28-membered macrolide, neaumycin B (1), containing 19 chiral centers was determined by NMR methods including J-based configuration analysis and interpretation of NOE correlations.
Correlation of Bioinformatic Structure Information with NMR Analysis to Derive the Full Absolute Stereostructure of Neaumycin B (1).
The confident interpretation of KR-induced hydroxy assignments by BGC analysis was next combined with the relative configuration defined by NMR analysis to assign the entire absolute configuration of neaumycin B (1). The 28-membered ring system of 1, which was based on the result of relative configuration, was opened in silico to display a linear structure at the lactonization position (C-27). The spiro-bicyclic ketal moiety was positioned at C-29, which confirmed the ketone functionality defined by BGC analysis. The relative configuration of the linear structure of 1 matched perfectly with the absolute configurations at carbons assigned by BGC analysis. To confirm the absolute configuration of the remaining eight stereogenic centers that were not confidently assigned, we carefully interpreted the NMR data along with absolute configurations assigned at numerous centers by BGC analysis.
The absolute configuration at C-7 = R, defined by BGC analysis, and J-based NMR and NOE NMR data for a 4,5,7-syn/anti configuration of C-4 to C-7, indicated the C-4 and C-5 could be assigned in 1 as R and R, respectively. Similarly, the BGC analysis assignments for C-13 and C-14 as R and NMR data analysis for 10,11,13,14-anti/syn/syn relative configurations from C-10 and C-11 in 1 allowed assignment of R and S configurations, respectively. The absolute configuration of the C-22 methyl carbon as R, as defined by BGC analysis for C-23, was also confirmed by diagnostic 2D ROESY NMR data.
The epoxide ring carbons (C-36 and C-37), produced by an epoxidase enzymatic domain, were confirmed as R and S, respectively, using the BGC assignments for C-35 and C-38 as R and by analysis of NOE correlations. Finally, the chiral spiroketal carbon at C-29 was defined as S on the basis of NOE correlations. By combining the confident assignment of numerous stereocenters based on bioinformatic specificity data, the full absolute configuration of 1 was confidently assigned.
CONCLUSIONS
The study described herein illustrates the utility of applying bioinformatics approaches to assign the full stereostructures of complex macrolides. Using the known stereospecificities of type I PKS enzymes and carefully avoiding the use of weak assignments, we were able to make assignments in 11 of the 19 stereocenters present in neaumycin B. We then used these confident compositional markers using models and integrating comprehensive 2D NMR data. The remaining eight centers, including those associated with a bicyclic spiroketal formed via the cyclization of a BGC-predicted ketone functionality at C-29, were confidently assigned by 2D NMR NOE methods. Similar investigations using bioinformatics data to assist in the assignment of absolute configuration in macrolides have been reported. The total structure assignment of the myxobacterial macrolide etnangien was partially assigned by this method but also required derivatization and chemical transformations to achieve success.43,44 Similarly, structural components of the macrolide strombomycins and the macrolide salinilactam were partially assigned based upon BGC bioinformatic enzyme specificities.45,46
Since these earlier reports the utilization of BGC enzyme specificity data has increased in overall reliability; however caution must still be exercised to carefully analyze biosynthetic enzyme sequence data. In this study, the instances where these two approaches failed to agree demonstrate the continued need to improve our understanding of biosynthetic enzyme function. In this case, the unprecedented detection of two KR domains in module 18, the unusual amino acid sequence of the KR domain in module 15, and the unpredictability of epimerization by the type A1 KR domain in module 9 were some of the challenges faced. Despite these uncertainties, detailed analyses of the amino acid sequences associated with each enzymatic domain provided opportunities to develop hypotheses for why some did not function as predicted.
Neaumycin B is a 28-membered macrolide broadly related to the cytovaricin-ossamycin-oligomycin class macrolides.11–14 Neaumycin B is produced in low yield by an undescribed member of the genus Micromonospora (Figure S9) and was shown in preliminary testing to be active against the blood borne cancer cell lines in the NCI 60 cell line panel (Figure S1) with moderate selectivity toward RPMI-8226, a myeloma cell line involved in multiple myeloma. Unfortunately, neaumycin B has limited stability, and this has hampered acquiring larger quantities of the purified macrolide for in vivo testing. However, we recently completed in-house assays against several cancer cell lines not represented within the NCI panel. These new tests uncovered significant potency and selectivity toward U87 human glioblastoma, a highly difficult cancer to treat (Figure 10). Glioblastoma is the most malignant type of glioma and are are considered lethal neoplasms with poor survival even when extensive treatments involving surgical removal combined with various combinations of radiotherapy, chemotherapy, and possible immuno-therapy.47 In replicate analyses, the LD50 of neaumycin B was found to be 5.6 × 10−5 μg/mL (6.4 × 10−8 μM). This level of potency and selectively indicates that neaumycin B may form a lead structure for the design of drugs useful in glioblastoma treatment.
Figure 10.
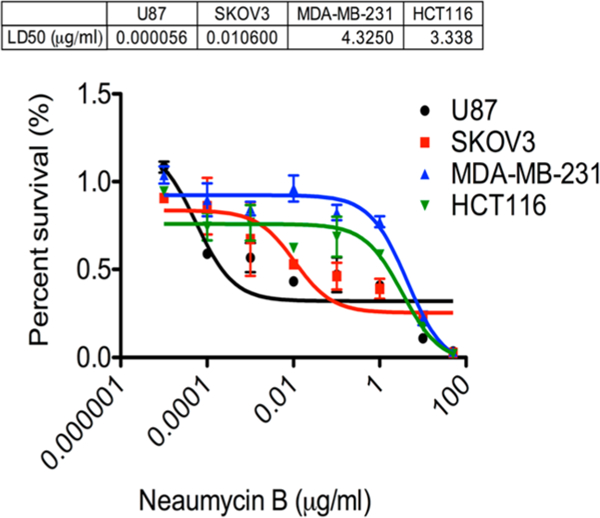
Dose response curves for neaumycin B (1) against four cancer cell lines illustrating the significant selectivity toward U87 human glioblastoma.
Supplementary Material
Figure 1.
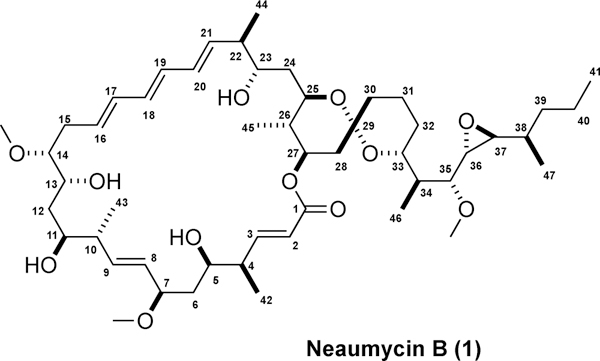
Final gene cluster bioinformatics- and NMR-based structure and absolute configuration assignment for neaumycin B (1).
ACKNOWLEDGMENTS
This study was supported by the NIH under grants R37 CA044848 to W.F. and R01 GM085770 to P.R.J. We thank Natalie Millán-Aguiñaga and Julia Busch, SIO, for their assistance with phylogenetic analysis of strain CNY-010 and for performing the HCT-116 cytotoxicity bioassays. We greatly appreciate the assistance of Dr. Jeff Rodvold, UCSD Cancer Center, in providing U87 glioblastoma cytotoxicity data.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b04848.
Tables of spectral and bioinformatics data, along with all NMR and mass spectra derived from neaumycin B (PDF)
Author Contributions
The authors declare no competing financial interest.
REFERENCES
- (1).Ōmura S Macrolide Antibiotics, 2nd ed; Academic Press: San Diego, CA, 2002. [Google Scholar]
- (2).Zhanel GC; Dueck M; Hoban DJ; Vercaigne LM; Embil JM; Gin AS; Karlowsky JA Drugs 2001, 61, 443. [DOI] [PubMed] [Google Scholar]
- (3).Odds FC; Brown AJP; Gow NAR Trends Microbiol. 2003, 11, 272. [DOI] [PubMed] [Google Scholar]
- (4).Kino T; Hatanaka H; Hashimoto M; Nishiyama M; Goto T; Okuhara M; Kohsaka M; Aoki H; Imanaka HJ Antibiot. 1987, 40, 1249. [DOI] [PubMed] [Google Scholar]
- (5).Staatz CE; Tett SE Clin. Pharmacokinet. 2004, 43, 623. [DOI] [PubMed] [Google Scholar]
- (6).Abou-Jaoude MM; Naim R; Shaheen J; Naufal N; Abboud S; AlHabash M; Darwish M; Mulhem A; Ojjeh A; Almawi WY Transplant. Proc. 2005, 37, 3025. [DOI] [PubMed] [Google Scholar]
- (7).Denduluri N; Low JA; Lee JJ; Berman AW; Walshe JM; Vatas U; Chow CK; Steinberg SM; Yang SX; Swain SM J. Clin. Oncol. 2007, 25, 3421. [DOI] [PubMed] [Google Scholar]
- (8).Burg RW; Miller BM; Baker EE; Birnbaum J; Currie SA; Hartman R; Kong Y-L; Monaghan RL; Olson G; Putter I; Tunac JB; Wallick H; Stapley EO; Oiwa R; Omura S Antimicrob. Agents Chemother. 1979, 15, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Napolitano JG; Daranas AH; Norte M; Femández JJ Anti-Cancer Agents Med. Chem. 2009, 9, 122. [DOI] [PubMed] [Google Scholar]
- (10).Qi Y; Ma S ChemMedChem 2011, 6, 399. [DOI] [PubMed] [Google Scholar]
- (11).Kihara TH; Kusakabe N; Nakamura G; Sakurai T; Isono KJ Antibiot. 1981, 34, 1073. [DOI] [PubMed] [Google Scholar]
- (12).Kirst HA; Mynderse JS; Martin JW; Baker PJ; Paschal JW; Rios Steiner JL; Lobkovsky E; Clardy JJ Antibiot. 1996, 49, 162. [DOI] [PubMed] [Google Scholar]
- (13).Smith RM; Peterson WH; McCoy E Antibiot. Chemother. 1954, 4, 962. [PubMed] [Google Scholar]
- (14).Carter GT J. Org. Chem. 1986, 51, 4264. [Google Scholar]
- (15).Huang S-X; Wang X-J; Yan Y; Wang J-D; Zhang J; Liu C-X; Xiang W-S; Shen B Org. Lett. 2012, 14, 1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Simone M; Maffioli SI; Tocchetti A; Tretter S; Cattaneo M; Biunno I; Gaspari E; Donadio SJ Antibiot. 2015, 68, 406. [DOI] [PubMed] [Google Scholar]
- (17).Sambrook J; Russel DW Molecular Cloning: A Laboratory Manual; Cold Spring Harbour Laboratory Press: New York; 2001. [Google Scholar]
- (18).Antipov D; Korobeynikov A; McLean JS; Pevzner PA Bioinformatics 2016, 32, 1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blin K; Wolf T; Chevrette MG; Lu X; Schwalen CJ; Kautsar SA; Suarez Duran HG; de Los Santos ELC; Kim HU; Nave M; Dickschat JS; Mitchell DA; Shelest E; Breitling R; Takano E; Lee SY; Weber T; Medema MH Nucleic Acids Res. 2017, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nayler P; Whiting MC J. Chem. Soc. 1955, 0, 3037. [Google Scholar]
- (21).Mukhopadhyay T; Nadkarni SR; Patel MV; Bhat RG; Desikan KR; Ganguli BN; Rupp RH Tetrahedron 1998, 54, 13621. [Google Scholar]
- (22).Hoffmann TM; Müller S; Nadmid S; Garcia R; Müller R J. Am. Chem. Soc. 2013, 135, 16904. [DOI] [PubMed] [Google Scholar]
- (23).Laureti L; Song L; Huang S; Corre C; Leblond P; Challis GL; Aigle B Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Peng J; Place AR; Yoshida W; Anklin C; Hamann MT J. Am. Chem. Soc. 2010, 132, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Warabi K; Hamada T; Nakao Y; Matsunaga S; Hirota H; van Soest RWM; Fusetani NJ Am. Chem. Soc. 2005, 127, 13262. [DOI] [PubMed] [Google Scholar]
- (26).Luesch H; Yoshida WY; Moore RE; Paul VJ; Corbett TH J. Am. Chem. Soc. 2001, 123, 5418. [DOI] [PubMed] [Google Scholar]
- (27).Rychnovsky SD; Skalitzky DJ; Pathirana C; Jensen PR; Fenical WJ Am. Chem. Soc. 1992, 11, 671. [Google Scholar]
- (28).Smith AB; Ott GR J. Am. Chem. Soc. 1996, 118, 13095. [Google Scholar]
- (29).Matsumori N; Kaneno D; Murata M; Nakamura H; Tachibana KJ Org. Chem. 1999, 64, 866. [DOI] [PubMed] [Google Scholar]
- (30).Kawahara T; Izumikawa M; Takagi M; Shin-ya K Org. Lett. 2012, 14, 4434. [DOI] [PubMed] [Google Scholar]
- (31).Uhrín D; Batta G; Hruby VJ; Barlow PN; Kövér KJ Magn. Reson. 1998, 130, 155. [DOI] [PubMed] [Google Scholar]
- (32).Medema MH; Takano E; Breitling R Mol. Biol. Evol. 2013, 30, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Donadio S; Staver M; McAlpine J; Swanson S; Katz L Science 1991, 252, 675. [DOI] [PubMed] [Google Scholar]
- (34).Walsh CT Science 2004, 303, 1805. [DOI] [PubMed] [Google Scholar]
- (35).Haydock SF; Aparicio JF; König A; Molnár I; Schwecke T; Marsden AFA; Galloway IS; Staunton J; Leadley PF FEBS Lett. 1995, 374, 246. [DOI] [PubMed] [Google Scholar]
- (36).Minowa Y; Araki M; Kanehisa MJ Mol. Biol. 2007, 368, 1500. [DOI] [PubMed] [Google Scholar]
- (37).Yadav G; Gokhale RS; Mohanty D. J. Mol. Biol. 2003, 328, 335. [DOI] [PubMed] [Google Scholar]
- (38).Fischbach MA; Walsh CT Chem. Rev. 2006, 106, 3468. [DOI] [PubMed] [Google Scholar]
- (39).Weissman KJ Beilstein J. Org. Chem. 2017, 13, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Siskos AP; Baerga-Ortiz A; Bali S; Stein V; Mamdani H; Spiteller D; Popovic B; Spenser JB; Staunton J; Weissman KJ; Leadley PF Chem. Biol 2005, 12, 1145. [DOI] [PubMed] [Google Scholar]
- (41).Valenzano CR; Lawson RJ; Chen AY; Khosla C; Cane DE J. Am. Chem. Soc. 2009, 13, 18501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kwan DH; Sun Y; Schulz F; Hong H; Popovic B; Sim-Stark JCC; Haydock SF; Leadlay PF Chem. Biol. 2008, 15, 1231. [DOI] [PubMed] [Google Scholar]
- (43).Menche D; Arikan F; Perlova O; Horstmann N; Ahlbrecht W; Wenzel SC; Jansen R; Irchik H; Müller RJ Am. Chem. Soc. 2008, 130, 14234. [DOI] [PubMed] [Google Scholar]
- (44).Menche D Nat. Prod. Rep. 2008, 25, 905. [DOI] [PubMed] [Google Scholar]
- (45).Udwary DW; Zeigler L; Asolkar RN; Singan V; Lapidus A; Fenical W; Jensen PR; Moore BS Proc. Natl Acad. Sci. U. S. A. 2007, 104, 10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Laureti L; Song L; Huang S; Corre C; Leblonde P; Challis GL; Aigle B Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ostrom QT; Bauchet L; Davis FG; Deltour I; Fisher JL; Langer CE; Pekmezci M; Schwartzbaum JA; Turner MC; Walsh KM; Wrensch MR; Barnholtz-Sloan JS Neuro Oncol 2014, 16, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.