

To the editor:
The field of genome editing is advancing rapidly1, most recently exemplified by the advent of base editors that enable changing single nucleotides in a predictable manner2,3,4. For the validation and characterization of genome editing experiments, targeted amplicon sequencing has become the gold standard. Here, we present a substantially updated version of our CRISPResso tool8 to facilitate the analysis of data that would be difficult to handle with existing tools5,6,7,8.
CRISPResso2 introduces five key innovations: (1) Comprehensive analysis of sequencing data from base editors; (2) A batch mode for analyzing and comparing multiple editing experiments; (3) Allele-specific quantification of heterozygous or polymorphic references; (4) A biologically-informed alignment algorithm; and (5) Ultra-fast processing time.
CRISPResso2 allows users to readily quantify and visualize amplicon sequencing data from base editing experiments. It takes as input raw FASTQ sequencing files and outputs reports describing frequencies and efficiencies of base editing activity, plots showing base substitutions across the entire amplicon region (Fig. 1a), and nucleotide substitution frequencies for a region specified by the user (Fig. 1b). Additionally, users can specify the nucleotide substitution (e.g., C->T or A->G) that is relevant for the base editor used, and publication-quality plots are produced for nucleotides of interest with heatmaps showing conversion efficiency.
Figure 1: Novel features of CRISPResso2.
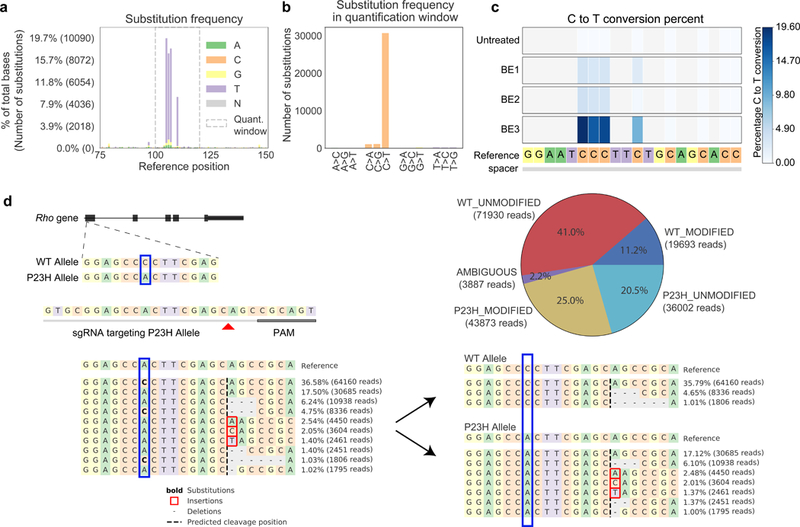
a-c) CRISPResso2 analysis of base editing data. a) Locations of substitutions across the FANCF reference sequence for the BE3 base editor2. At each position, the number of substitutions from the reference base to each non-reference base are shown. The quantification window is outlined by the dashed gray box. b) Barplot showing the frequency of substitution from a reference base to a non-reference base including only bases in the quantification window from part a. c) Batch output mode comparing the editing efficiencies of three base editors and an untreated control at the FANCF locus2. C>T conversion rates are shown at each cytosine overlapping the guide. d) Allele-specific editing outcomes of SaCas9-KKH editing of the Rho gene in P23H heterozygous mice 10. Reads (left) can be assigned to each allele using CRISPResso2 (right) to achieve accurate quantification of genome editing at genomic loci with multiple alleles. The pie chart shows the assignment of each read to the wild-type (red and dark blue) allele or to the P23H allele (yellow and light blue). Ambiguous alignments that could not be attributed uniquely to one of the alleles (e.g., due to a deletion at the SNP location) are shown in purple.
We also improved processing time and memory usage of CRISPResso2 to enable users to analyze, visualize and compare results from hundreds of genome editing experiments using batch functionality. This is particularly useful when many input FASTQ files must to be aligned to the same amplicon or have the same guides, and the genome editing efficiencies and outcomes can be visualized together. In addition, CRISPResso2 generates intuitive plots to show the nucleotide frequencies and indel rates at each position in each sample. This allows users to easily visualize the results and extent of editing in their experiments for different enzymes (Fig. 1c).
In cases where the genome editing target contains more than one allele (for example when heterozygous SNPs are present), genome editing on each allele must be quantified separately, although reads from both alleles are amplified and mixed in the same input FASTQ file. Current strategies are not capable of analyzing multiple reference alleles and may lead to incorrect quantification. CRISPResso2 enables allelic specific quantification by aligning individual reads to each allelic variant and assigning each read to the most closely-aligned allele. Downstream processing is performed separately for each allele so that insertions, deletions, or substitutions that distinguish each allele are not confounded with genome editing. To demonstrate the utility of our approach, we reanalyzed amplicon sequencing data from a mouse with a heterozygous SNP at the Rho gene where an engineered SaCas9-KKH nuclease was directed to the P23H mutant allele10. CRISPResso2 deconvoluted reads, quantified insertions and deletions from each allele, and produced intuitive visualizations of experimental outcomes (Fig. 1d).
Existing amplicon sequencing analysis toolkits ignore the biological understanding of genome editing and instead optimize the alignment based only on sequence identity. However, this can lead to incorrect quantification of indel events, especially in sequences with short repetitive subsequences where the location of indels may be ambiguous due to multiple alignments with the same best score. In such cases, it is reasonable to assume that indels should overlap with the predicted nuclease cleavage site. Our improved alignment algorithm extends the Needleman-Wunsch algorithm with a mechanism to incentivize the assignment of insertions or deletions to specific indices in the reference amplicon sequence. These indices are chosen based on guide sequence, predicted cleavage site and nuclease properties (Supplementary note 1). This approach increases the accuracy of indel calling and produces alignments that reflect our current understanding of the editing mechanism. We compared our improved alignment algorithm to those used in other amplicon-based genome editing analysis software and found that our algorithm avoids the incorrect alignment to regions distal from the predicted cut site observed for other software tools (Supplementary note 2).
To study putative off-targets, it is often necessary to analyze large-scale pooled sequencing datasets that profile hundreds of sites to assess the potential safety of genome editing interventions9. These and other large datasets have created a need for faster, more accurate and efficient analysis tools. To accelerate performance and decrease processing time, we designed an efficient implementation of our biologically-informed alignment algorithm. Further optimization of other components of the processing pipeline has reduced processing time ten-fold for large datasets, so that an experiment analyzed using modern high-throughput sequencing technologies can be processed in under a minute (Supplementary Fig. 1). We tested the accuracy of our improved alignment algorithm and other optimizations using an extensive set of simulations with various mutational profiles and in the presence of sequencing errors and found that CRISPResso2 accurately recovered editing events with a negligible false-positive rate (<0.01) limited only by current sequencing technologies (Supplementary note 3).
In summary, CRISPResso2 is a software tool for the comprehensive analysis, visualization and comparison of sequencing data from genome editing experiments. In addition to accurate indel analysis from nucleases such as Cas9, CRISPResso2 offers analysis tools for recent base editors, support for multiple alleles, increased computational speed, an improved alignment algorithm, and a batch functionality for analyzing and comparing genome editing experiments (Supplementary note 4).
Supplementary Material
Acknowledgements
D.R.L. is supported by DARPA HR0011–17-2–0049; U.S. NIH RM1 HG009490, R01 EB022376, and R35 GM118062; and HHMI. J.K.J. is supported by DARPA HR0011–17-2–0042, NIH R35 GM118158 and NIH RM1 HG009490. D.E.B. is supported by NIDDK (R03DK109232), NHLBI (DP2OD022716, P01HL32262), Burroughs Wellcome Fund, Doris Duke Charitable Foundation, and St. Jude Children’s Research Hospital Collaborative Research Consortium. L.P. is supported by NHGRI (R00HG008399), DARPA HR0011–17-2–0042, and the Centers for Excellence in Genomic Science of the National Institutes of Health under award number RM1HG009490 through a new collaborator grant subaward.
Footnotes
Competing Interests
At the time of manuscript preparation, J.M.G. was a consultant for Beam Therapeutics, and now is employed by Beam Therapeutics. J.K.J. has financial interests in Beam Therapeutics, Editas Medicine, Endcadia, EpiLogic Therapeutics, Pairwise Plants, Poseida Therapeutics and Transposagen Biopharmaceuticals. J.K.J.'s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. J.K.J. is a member of the Board of Directors of the American Society of Gene and Cell Therapy. J.M.G. and J.K.J. are co-inventors on patents and patent applications that describe gene editing technologies. D.R.L. is a consultant and cofounder of Editas Medicine, Pairwise Plants and Beam Therapeutics, companies that use genome editing.
Code availability.
CRISPResso2 is available online at http://crispresso2.pinellolab.org, where users can run up to 4 samples simultaneously. The command line version without any limitations and with additional tools is available as a docker image at: https://hub.docker.com/r/pinellolab/crispresso2/ (Supplementary Note 4). The source code is available as Supplementary Software.
Data Availability
Figures 1a-c show data available as SRR3305546 (untreated), SRR3305543 (BE1), SRR3305544 (BE2), and SRR3305545 (BE3)2. Figure 1d shows data obtained from the authors10.
References
- 1.Tsai SQ & Joung JK Nature Reviews Genetics 17, 300–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YB et al. Nature Biotechnology 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komor AC et al. Science Advances 3, (2017). [Google Scholar]
- 5.Wang X et al. Bioinformatics 33, 3811–3812 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Park J, Lim K, Kim J-S & Bae S Bioinformatics 33, 286–288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindsay H et al. Nature Biotechnology 34, 701–702 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Pinello L et al. Nature Biotechnology 34, 695–697 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akcakaya P et al. Preprint at https://www.biorxiv.org/content/early/2018/02/27/272724 (2018) [Google Scholar]
- 10.Li P et al. The CRISPR Journal 1, 55–64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.