

Abstract
Patients harboring homozygous c.498_499insC mutations in MFRP demonstrate hyperopia, microphthalmia, retinitis pigmentosa, retinal pigment epithelial atrophy, variable degrees of foveal edema, and optic disc drusen. The disease phenotype is variable, however, with some patients maintaining good central vision and cone function till late in the disease. A knock-in mouse model with the c.498_499insC mutation in Mfrp (Mfrp KI/KI) was developed to understand the effects of these mutations in the retina. The model shares many of the features of human clinical disease, including reduced axial length, hyperopia, retinal degeneration, retinal pigment epithelial atrophy, and decreased electrophysiological responses. In addition, the eyes of these mice had a significantly greater refractive error (p < 0.01) when compared to age-matched wild-type control animals. Administration of recombinant adeno-associated virus–mediated Mfrp gene therapy significantly prevented thinning from retinal neurodegeneration (p < 0.005) and preserved retinal electrophysiology (p < 0.001) when treated eyes were compared to contralateral sham-treated control eyes. The Mfrp KI/KI mice will serve as a useful tool to model human disease and point to a potential gene therapeutic approach for patients with preserved vision and electrophysiological responses in MFRP-related retinopathy.
Keywords: Gene therapy, MFRP mouse model, retinal degeneration, RPE, AAV
Introduction
Mutations in the membrane frizzled related protein (MFRP) gene have been reported to result in a wide variety of clinical manifestations, including hyperopia, microphthalmia, nanophthalmos, acute-angle closure glaucoma, retinitis pigmentosa, retinal pigment epithelial (RPE) atrophy, retinal folds, macular edema, retinal cysts, and retinal degeneration.1–3 A Mexican pedigree was previously reported with known clinical features of MFRP-associated eye disease but in whom two of the four family members maintained good central vision, even into their late 30s and early 40s. The affected cases were all found to carry a homozygous single base-pair insertion of a cytosine (c.498_499insC) in exon 5 of MFRP, leading to a frame shift and premature termination of transcription, resulting in lack of functional protein.4,5 Although a number of mutations have now been described, there is little knowledge about the long-term natural history of patients with MFRP-associated eye disease.
The MFRP gene encodes a 584 amino acid protein6,7 and is expressed as a bicistronic transcript with C1-tumor necrosis factor related protein 5 (C1QTNF5/CTRP5), a member of the adiponectin superfamily.8 MFRP is a type II transmembrane protein, expressed predominantly in the RPE and ciliary body within the eye.6,9 MFRP is structurally organized into three domains: a cysteine-rich domain (CRD) at the C-terminus, a type II transmembrane domain, and an extracellular region with tandem repeats of CUB-LDLR domains. The CUB-LDLR region of MFRP is known to interact with CTRP5.9,10 The CRD is homologous to the canonical Wnt receptor and binds Wnt,11,12 suggesting a role for MFRP in Wnt signaling. However, MFRP's functional role within the eye is yet to be fully established.
Mice with spontaneous mutations in the murine orthologous gene Mfrp have been described previously.13,14 The first Mfrp mouse model was the rd6 mouse, which results from a common, spontaneous 4 bp deletion in a splice donor site, leading to skipping of exon 4 of Mfrp.7 The mutation is predicted to result in the formation of a truncated protein with the loss of 58 amino acids located between the transmembrane and CUB-LDLR domains.7 A second mouse model resulting from a spontaneous c.174delG (rdx) mutation was also described.14 This mutation is predicted to form a truncated protein with the loss of both CRD and CUB-LDLR domains in MFRP. Both models were reported to have minimal levels of MFRP expression in the RPE and developed severe retinal degeneration.7,14 Initial reports did not note microphthalmia in either of the mice.7,13 However, more recent work has reported a microphthalmic phenotype in rd6 mice.3 Both mice developed a flecked retina with prominent white spots, and as a result, the rd6 mouse was initially proposed as a good model for other fleck retinal dystrophies, including Stargardt disease or fundus albipunctatus.15 Neither of the mutations has been reported in patients thus far, and currently there are no mouse models harboring MFRP gene mutations reported in patients.
The present study describes the long term follow-up of a pedigree with homozygous c.498_499insC mutation in MFRP. The pathogenicity of the mutation was validated by generating a mouse model harboring homozygous c.498_499insC in Mrfp and characterizing its ocular phenotype. Finally, the possibility of rescue of the retinal phenotype in this mouse model was evaluated using recombinant adeno-associated virus (rAAV)-Mfrp gene augmentation therapy.
Methods
Clinical evaluation of patients
Four siblings from a Mexican family underwent a full ophthalmic examination, including best-corrected visual acuity (BCVA) using a Snellen visual acuity chart, slit-lamp and dilated fundus examination, wide-field pseudocolor imaging of the fundus, wide-field scanning laser ophthalmoscopy, and spectral-domain optical coherence tomography (SD-OCT). Full-field electroretinography (ffERG; Metrovision ERG system, Perenchies, France) was performed in all the patients using the International Society for Clinical Electrophysiology of Vision (ISCEV) standard stimuli.16
Generation of Mfrp KI/KI mice
The targeting vector was constructed by subcloning an 11.15 kb region from a C57BL/6 BAC clone (RP23:270P20). This 11.15 kb region included exons 1–13 of the Mfrp gene. A single LoxP site was inserted upstream of exon 3 in introns 2–3 and a LoxP/FRT-flanked Neo cassette was inserted downstream of exon 9 in introns 9–10.The C-insertion mutation in exon 5 was generated by site-directed mutagenesis. The target region was 2.7 kb containing exons 3–9 of the Mfrp gene. The total size of the targeting construct (including the vector backbone) was15.25 kb.
The targeting vector (10 μg) was linearized by NotI and transfected into IC1 C57BL/6 embryonic stem (ES) cells. Recombinant ES clones were screened by polymerase chain reaction (PCR) following G418 antibiotic selection. The positive clone identified by PCR was further confirmed by Southern blotting analysis. The positive ES cell clones were injected into BALB/c blastocysts and implanted into pseudopregnant mice to generate chimeras. C57BL/6J mice were mated with these chimeras to transmit the targeting allele. Mfrp KI/KI mice were identified by PCR of tail-tip DNA. The Mfrp KI/KI were generated with assistance from inGenious Targeting Laboratory, Inc. (Stony Brook, NY).
In homozygous Mfrp KI mice, the kanamycin marker was flanked by an FRT sequence, and this was removed by breeding with FLP mice. Mice were tested for the rd8 mutation. Mice free of the rd8 mutation were utilized to generate the mouse colony with the homozygous c498_499insC mutation (Mfrp KI/KI).
The presence of a 1 bp insertion (cytosine) in Mfrp was confirmed by using a loxP specific forward and an endogenous Mfrp reverse primer (LOX1: CCAGACAGAGTTCTGTAATCCTGC; SDL2: AGCCCTCCCAGTTACTTACGAGCC; wild-type [WT] copy, 352 bp; mutant copy, 413 bp).
All animal maintenance and experimental procedures were performed in accordance with the protocols approved by the University of California San Diego Institutional Animal Care and Use committee.
Immunohistochemistry
The antibodies used for the studies were goat polyclonal anti-MFRP (1:250; catalog # AF3445; R&D Systems, Minneapolis, MN).9 Primary antibodies against S-opsin (sc-14363; Santa Cruz Biotechnology, Inc., Dallas, TX) and M-opsin (AB5405, MilliporeSigma, Burlington, MA) were used for the immunostaining (dilution 1:200). Immunohistochemistry was performed, as previously described, using standard procedures.9
Western blot analysis
Mouse RPE-choroid was used to make the total protein extract in 1 × RIPA buffer containing protease inhibitor cocktail (Sigma–Aldrich, St. Louis, MO), as described previously.17 Approximately 30 μg of protein was used in each lane for Western blot analyses. Rabbit polyclonal anti-β-actin (1:500; Cell signaling Technology, Danvers, MA) and anti-rabbit in donkey-HRP (1:1,000; Santa Cruz Biotechnology, Inc.) was used to detect MFRP expression. The expression level of MFRP was compared between mouse genotypes by normalizing against the intensity of β-actin protein.
Histology study
Mice aged from 21 days to 12 months were used to study the histology of Mfrp KI/KI mice and were compared with age-matched control. The eyes were fixed by immersion fixation in 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer. A graded series of ethanols was used to dehydrate the eye before the eyes were embedded in Embed812 (EMS, Hatfield, PA), as described previously.18 Micro-thick retinal sections were stained with hematoxylin and eosin and viewed under an Olympus IX81 motorized inverted microscope (Olympus, Tokyo, Japan). Thin sections were examined on a JEM 1200EX II electron (JOEL USA, Inc., Peabody, MA) microscope.
Autofluorescence and optical coherence tomography imaging
For in vivo imaging studies and for intraocular surgery, mice were anesthetized with an intraperitoneal loading dose of ketamine (93 mg/kg) and xylazine (8mg/kg). Next, 1% atropine and 0.5% tropicamide were used to dilate the pupil. A heating pad was used to maintain the body temperature at 37°C. SD-OCT imaging was performed using a Spectralis™ HRA + OCT (Heidelberg Engineering, Heidelberg, Germany). Mfrp KI/KI mice at 1, 2, and 3 months of age (n = 3 for each age). WT mice at 3 months of age (n = 3) were used as controls. The thickness of the retina was measured using Heidelberg eye explorer software. Three-month-old Mfrp KI/KI mice were used to study retinal autofluorescence and compared to age-matched WT controls. Autofluorescence imaging was performed using the same machine to study the autofluorescent spots in the eyes of Mfrp KI/KI mice using methods that have been previously described.19,20 A 55° angle lens was used to capture 10 frames per fundus.
Infrared imaging
Infrared imaging was used to measure the refractive power of the eye.21 The dioptric measurement obtained when the optic disc was in focus was used as a surrogate measure for refractive power provided by the Spectralis™ HRA + OCT (Heidelberg Engineering). Mfrp KI/KI mice aged 2 and 3 months were used to measure the diopteric power. WT controls were imaged for comparison at 3 months of age.
Quantitative reverse transcription PCR
Total RNA was extracted from RPE choroid and retina from 2-month-old mice (RNeasy kit; Bio-Rad Laboratories, Hercules, CA). cDNA was synthesized using a Bio-Rad cDNA synthesis kit (i-script). Quantitative PCR was performed using SYBR green PCR master mix (iQTM SYBR® green supermix; Bio-Rad). Rpe65, Zo-1, Mfrp, and Ctrp5 expression was normalized against the RpL19 housekeeping gene, as described previously.17
Electroretinography
ffERG were recorded to evaluate retinal function in Mfrp KI/KI mice and WT control mice at ages 3.5 and 6.5 months (n = 6 for each). ffERGs were obtained using similar methods to those described by the International Society for Clinical Electrophysiology of Vision.16 The mice were dark adapted overnight and anesthetized under dim red light. Pupils were dilated using a mixture of tropicamide (1%) and phenylephrine (2.5%). Proparacaine (0.5%) was used as a topical anesthetic in order to minimize blinking. ERGs were recorded using a looped thin stainless steel wire to make contact with the corneal surface through a thin layer of Genteal Tear Gel (Novartis Pharmaceuticals, Basel, Switzerland). Needle electrodes placed on the lower lip and tail served as reference and ground leads, respectively. Mice were then placed on a heating pad in a Ganzfeld bowl stimulator. Amplification with a Grass 15LT external amplifier (at 0.3–300 Hz, without notch filtering), stimuli presentation, and data acquisition were programmed and performed using the VERIS v6.0.9 Science software system (Electro-Diagnostic Imaging, Inc., Redwood City, CA). Different flash intensities for scotopic (−3.5, +1.09, and +2.00 cd.s/m2) and photopic (+1.09 and +2.00 cd.s/m2) conditions were used for stimulation. For photopic ERG responses, mice were light adapted for a minimum of 10 min prior to recording. The a-wave and b-wave amplitudes from each eye were determined and averaged for comparison of retinal function.
Assessment of cone survival
Frozen sections from 10-month-old rAAV-Mfrp-treated and sham-treated control eyes from the same animal were obtained for immunohistochemistry, as previously described.9 The whole-eye images were captured using the Keyence BZ-X710 fluorescence microscope (Keyence, Itasca, IL). ImageJ22 was used to divide the neuroretinal images into 200 micron sections, starting at the optic nerve (Fig. 1B). The number of cones immunostained with either S or M opsin were counted in the central, middle, and peripheral regions of the dorsal and ventral retina either side of the optic nerve, with the eye cut in coronal section (n = 3).
Figure 1.

Long-term effects of recombinant adeno-associated virus (rAAV) Mfrp treatment on the rescue of retinal function in Mfrp KI/KI mice. (A) Representative images of Mfrp immunostaining (green) in rAAV-Mfrp and sham-treated control eyes. The arrows point to the retinal pigment epithelial (RPE) layer. (B) Representative images of M-opsin staining (red) in a rAAV-Mfrp treated and sham-treated control eye from the same animal. The arrows highlight the opsin immunostaining. (C) Analysis of cone photoreceptor count across the retina 9 months after rAAV-Mfrp injection. Cone survival was analyzed by counting the number of cones using S and M opsin immunostaining. There was a significantly greater number of cones in dorsal central and ventral mid-peripheral regions of rAAV-Mfrp-treated eyes compared to sham-treated eyes (p < 0.032). (D) Analysis of cone photoreceptor lengths across the retina in eyes 9 months after rAAV-Mfrp treatment. rAAV-Mfrp-treated eyes (n = 3) were compared to sham-treated control eyes (n = 3). Cone photoreceptor opsin immunostaining length was significantly longer in treated eyes in the central region compared to control eyes (p < 0.01). Color images are available online.
Assessment of cone length
Frozen sections from 10-month-old rAAV-Mfrp-treated and sham-treated control eyes from the same animal were obtained for immunohistochemistry, as previously described.9 Primary antibodies against S opsin (sc-14363; Santa Cruz Biotechnology, Inc.) and M opsin (AB5405; MilliporeSigma) were used for immunostaining (dilution 1:200). The whole-eye images were captured using the Keyence BZ-X710 fluorescence microscope (Keyence). ImageJ22 was used to divide the neuroretinal images into 200 μm sections, starting at the optic nerve (Fig. 1B). ImageJ was employed to measure the length of cones from the apical terminus of the photoreceptor to the outer nuclear layer (ONL; 0, if cone within ONL or no cones). The cone length was measured in four different areas within each 200 μm section in the central, middle, and peripheral regions of the dorsal and ventral retina. The average of four measurements was utilized for the final graph (n = 3).
Subretinal delivery of rAAV in Mfrp KI/KI mice
A self-complimentary tyrosine-capsid mutant rAAV serotype 8 (rAAV8; Y733F) vector containing the ubiquitous small chicken β-actin promoter driving the WT mouse Mfrp gene was kindly provided by Astra Dinculescu (University of Florida, Gainesville, FL). Production and titration of recombinant virus was described previously.23 Subretinal injections were performed on a group of 15 mice at postnatal week 6 under anesthesia by first penetrating the sclera with a 30-gauge disposable needle nasal to the corneal limbus. A 33-gauge blunt-tip needle on a Hamilton syringe was introduced into the eye through the same incision. The needle was extended into the subretinal space under direct visualization. The treated eye received 1 μL of virus at a titer of 1 × 1012 genome copies/mL. The sham-treated eye was given 1 μL of buffer using an identical method.23 Imaging and ffERG analysis were performed on both eyes of treated mice 4 months post injection. A subset of mice (n = 3) were maintained till 10 months of age in order to see the long-term effects of rAAV-Mfrp treatment.
Statistical analysis
The data were compared using two-tailed independent Student's t-tests. The data are presented as the mean ± standard deviation. p-Values of <0.05, <0.01, and <0.001 were deemed significant. p-Values were calculated using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA).
Results
Clinical findings in a pedigree with MFRP c.498_499insC mutation
A pedigree harboring homozygous c.498_499insC mutations in MFRP has previously been described4 (Supplementary Fig. S1). In this pedigree, four of six siblings of a consanguineous Mexican family were described with retinitis pigmentosa, foveoschisis, microphthalmos with hyperopia, and optic disc drusen. Rod function was lost early in all patients. However, it was uncertain how long cone function and central vision would remain intact amid reports of slow progression and phenotypic heterogeneity in MFRP-associated retinopathy patients.2 In order to understand the natural history of ophthalmic disease resulting from these mutations, the affected individuals were reexamined 12 years after this initial examination.
Examination revealed a progression of the clinical findings when compared to the initial evaluation (Table 1, Fig. 2, and Supplementary Figs. S2–S4).4 Best corrected visual acuity (BCVA) remained stable but extremely poor in patients 1 and 2 aged 61 and 57 years, respectively. Both cone and rod electrophysiological responses were completely extinguished in the latest evaluation in both patients 1 and 2 (Supplementary Fig. S7). Patients 3 and 4, aged 53 and 51 years, respectively, were noted to have moderate loss of BCVA when compared to the original examination. The visual acuity of patient 3 had reduced only minimally during the follow-up period, with the right eye remaining stable at 20/70 and the left eye falling to 20/90 (Table 1). In patient 4, the visual acuity had deteriorated to 20/90 OD and 20/100 OS. Ultra-wide-field pseudo-color images showed scattered pigmentation with mid-peripheral retinal atrophy and macular sparing (Fig. 2A and B). Scanning laser microscopy (SLO) showed marked mid-peripheral hypo-autofluorescence with some central macular sparing (Fig. 2C and D and Supplementary Fig. S3). SD-OCT showed foveal thickening bilaterally but a preserved central ellipsoid zone and external limiting membrane suggested an intact photoreceptor layer at the central macula in both patients (Fig. 2E and Supplementary Fig. S3). Electrophysiological scotopic responses were extinguished in both patients. However, there were residual photopic responses bilaterally in both patients 3 and 4, although these were attenuated (Supplementary Fig. S7).
Table 1.
Summary of the clinical findings of affected members of a pedigree with the homozygous mutation c498_499insC in MFRP
| Patient number | Age (years) | Sex | BCVA | Anterior segment findings | Posterior segment findings | Electrodiagnostics | Ocular coherence tomography |
|---|---|---|---|---|---|---|---|
| 1 | 61 | Female | 20/200 OD, 20/400 OS | Laser peripheral iridotomy for occludable angle in OU; posterior iris synechiae in OS secondary to acute unilateral non-granulomatous anterior uveitis; microcoria OU due to posterior synechiae; posterior subcapsular lens opacity (P3) OD, nuclear opalescence (NO4) lens opacity OS | Optic disc drusen, pigment clumping and bone-spicule pigmentation OU | Completely extinguished rod and cone responses OU | Diffuse macular thickening and foveoschisis OD; OCT assessment was unavailable in OS due to microcoria and lens opacity |
| 2 | 57 | Male | Hand motion OD, 20/200 OS | Laser peripheral iridotomy for occludable angle in OU, posterior iris synechiae OU secondary to acute bilateral non-granulomatous anterior uveitis; microcoria OU due to posterior synechiae, total lens opacity OD; nuclear opalescence (NO3) lens opacity OS | Optic disc drusen, pigment clumping and bone-spicule pigmentation OS; assessment of posterior segment OD was not possible due to lens opacity | Completely extinguished rod and cone responses OU | Diffuse macular thickening and foveoschisis OS; OCT assessment was unavailable in OD due to microcoria and lens opacity |
| 3 | 53 | Female | 20/70 OD, 20/90 OS | Laser peripheral iridotomy for occludable angle in bilaterally; nuclear opalescence (NO1) lens opacity bilaterally | Pigment clumping and bone-spicule pigmentation OU; absent optic disc drusen OU | Completely extinguished rod responses OU and markedly diminished cone responses OU | Diffuse macular thickening and foveoschisis bilaterally |
| 4 | 51 | Male | 20/90 OD, 20/100 OS | Laser peripheral iridotomy for occludable angle in bilaterally | Optic disc drusen, pigment clumping and bone-spicule pigmentation bilaterally | Completely extinguished rod responses OU and markedly diminished cone responses OU | Diffuse macular thickening and foveoschisis bilaterally |
BCVA, best-corrected visual acuity; OCT, optical coherence tomography.
Figure 2.

Retinal phenotype of patient with homozygous c.498_499 mutation in MFRP. (A and B) Pseudocolor wide-field fundus images of OD and OS demonstrating scattered mid-peripheral pigmentations and mid-peripheral and peripheral areas of hypopigmentation bilaterally suggesting retinal atrophy. (C and D) Scanning laser microscopy (SLO) wide-field fundoscopy images of OD and OS identify areas of reduced autofluorescence in the mid-peripheral retina with central macular sparing. (E) Spectral-domain optical coherence tomography (SD-OCT) cross-sectional image of OS across the fovea reveal macular thickening. However, a clear foveal external limiting membrane and ellipsoid zone line suggests that photoreceptors are still intact across the fovea. Color images are available online.
Development of a Mfrp KI/KI (Mfrp c498_499insC) mouse model
To understand the pathologic effects of the human mutation (c498_499insC) in MFRP identified in this pedigree better, a knock-in (KI) mouse model with the homozygous c498_499insC mutation (Mfrp KI/KI) on the C57BL/6 mouse genetic background was generated (Fig. 3A). The murine Mfrp gene shares >60% sequence homology with human MFRP (Supplementary Fig. S6). Genotyping of Mfrp KI/KI mice confirmed the presence of homozygous c498_499insC Mfrp mutations. Western blot analysis was performed to understand the effect of the mutations on MFRP expression. A Western blot analysis of posterior optic cup lysates identified a 120 kDa band, which corresponded to the predicted molecular weight of a MFRP dimer in the WT C57BL/623 (Fig. 3B). However, the corresponding band was absent in posterior optic cup lysates from Mfrp KI/KI mice at the age of 2 months, suggesting that Mfrp levels were undetectable in these mice.24 To confirm these findings further, immunostaining of the posterior eye cup was performed with antibody against MFRP. MFRP was identified in the apical RPE in WT mice (n = 3; Fig. 3C) In contrast, no immunostaining was noted in the RPE of Mfrp KI/KI mice at the age of 2 months (n = 3; Fig. 3C). However, it should be noted that the antibody used to detect MFRP recognizes the C terminal region, which is downstream of the premature stop caused by c498_499insC in Mfrp. Hence, it is unclear whether there is complete loss of MFRP protein expression or whether a truncated protein is still expressed as a result of this mutation.
Figure 3.

Schematic representation of the Mfrp KI/KI mouse development strategy and evaluation of MFRP protein expression. (A) Targeting vector with an insertion of 1 bp (c.498_499) in exon 5 of Mfrp indicated in the diagram (red arrow). The wild-type (WT) Mfrp allele with 1–14 exons are depicted as square boxes. The Neo gene cassette was introduced and flanked by RFT sequences. (B) Western blot analysis of MFRP and β-actin protein from the posterior eye cup lysate of Mfrp KI/KI and WT mice. (C) Immunohistochemical analysis of MFRP expression in WT and Mfrp KI/KI mice. The arrows identify the RPE layer. Color images are available online.
Accumulation of autofluorescent spots in Mfrp KI/KI mice
Patients with homozygous c.498_499insC mutations in MFRP were noted to have patches of RPE atrophy and optic disc drusen. To examine if similar features could be identified in the Mfrp KI/KI mice, performed autofluorescence imaging was using ophthalmic SLO, as described previously.17 SLO identified the presence of autofluorescent spots from 1 month of age in Mfrp KI/KI mice (Fig. 4A). The spots were distributed throughout the retina and were found to increase with age when compared to 3-month-old WT control retina (n = 3). In addition, B-scan imaging across the spots revealed a sub-retinal localization (Fig. 4F). The number of autofluorescent spots in the fundus of Mfrp KI/KI mice was significantly greater than in the fundus of age-matched WT mice at 3 months of age (p < 0.001; Fig. 4B).
Figure 4.

Analysis of fundus autofluorescence, determination of retinal thickness, and photoreceptor survival of Mfrp KI/KI mice. (A) Fundus SLO autofluorescence images from Mfrp KI/KI (1, 2, and 3 months; n = 3 for each age point) compared to WT mice (3 months; n = 3). (B) Quantification of autofluorescent white spots in 3-month-old Mfrp KI/KI mice with 3-month-old age-matched WT mice. (C) SD-OCT B-scan images from Mfrp KI/KI mice (1, 2, and 3 months; n = 3 for each age point) compared to WT mice (3 months; n = 3). (D) Total retinal thickness measurements in Mfrp KI/KI mice with age and compared to the control WT mice. (E) Measurement of outer nuclear layer (ONL) thickness in Mfrp KI/KI mice of 1, 2 and 3 months of age when compared to 3-month-old WT mice. (F) Analysis of pseudodrusen in a Mfrp KI/KI mouse using SD-OCT. The en-face autofluorescence reference image demonstrates marked pseudodrusen across the fundus. The pseudodrusen (blue arrowheads) are situated either side of a retinal vessel (red arrowhead) on the scan line in the left panel. The B-scan SD-OCT image demonstrates sub-retinal localization of pseudodrusen (blue arrowheads) with the blood vessel (red arrowhead) as a reference to demonstrate correspondence with the en-face image indicated in the left panel. Color images are available online.
Decrease in retinal thickness in Mfrp KI/KI mice
Progressive retinal degeneration is one of the predominant clinical features observed in patients with homozygous c.498_499insC mutations in MFRP. Evaluation of the retinal phenotype of Mfrp KI/KI mice using SD-OCT also revealed a significant reduction in neuroretinal thickness as early as 1 month of age when compared with 3-month-old WT control mice (Fig. 4C). Total retinal thickness was significantly decreased with age at 1, 2, and 3 months in Mfrp KI/KI mice (p < 0.001; Fig. 4D). In addition, measurement of ONL thickness from SD-OCT also revealed a significant reduction starting from 1 month of age when compared to 3-month-old WT control mice ONL (Fig. 4E; n = 3; p < 0.001).
Histological examination of retina of Mfrp KI/KI mice
Retinal morphology was studied to gain a more detailed insight into retinal pathology in Mfrp KI/KI mice. The retina of Mfrp KI/KI mice of different ages (21 days and 1, 2, 3, 6, and 12 months; n = 3 at each age point) were assessed by histology. Twenty-one-day-old and 12-month-old WT mice (n = 3) were used for comparison. Analysis of hematoxylin and eosin–stained sections revealed a progressive decrease in ONL thickness in Mfrp KI/KI mice when compared to controls (Fig. 5A). A significant decrease in ONL thickness was observed as early as 1 month of age (p < 0.05) when compared to 12-month-old WT control retina. Normally, retinae of WT mice possess a mean of 12 ± 2 rows of nuclei in their ONL. By contrast, in 1-month-old Mfrp KI/KI mice, only 6.3 ± 0.6 rows of nuclei were seen in ONL. With age, the number of rows of nuclei in ONL decreased in Mfrp KI/KI mice as follows: 3.3 ± 0.6 rows of nuclei at 2 months, 2.3 ± 0.6 rows at 3 months, 1.7 ± 0.6 rows at 6 months, and 1.3 ± 0.57 rows of nuclei at 12 months (Fig. 5C). Although the first signs of a decrease in ONL thickness were apparent by 1 month of age, pathology was noted as early as postnatal day 21.
Figure 5.

Study of retinal histology of Mfrp KI/KI mice. Retinal histology was studied in Mfrp KI/KI mice from age 21 days and 1, 2, 3, 6, and 12 months (n = 3 at each age point) and compared to age-matched control mice (21 days and 12 months; n = 3) histology. (A) There was a gradual decrease in ONL nuclei of Mfrp KI/KI mice from 1 month to 12 months. The number of rows of nuclei in the ONL at 1 month was 6–7, at 2 months was 3–4, at 3 months was 2–3, at 6 months was 1–2, and at 12 months was 1.The number of rows of nuclei in the ONL in WT mice at 12 months was 10–14. There was a decrease in outer segment thickness by 21 days in Mfrp KI/KI mice when compared to 21-day WT mice. Magnification: 20 × . (B) Ultrastructural analysis of RPE and sub-RPE areas from Mfrp KI/KI and WT mice. Areas enclosed in boxes in (A–C) are magnified in panels A′–C′. RPE from control mice (C, C′) showed no abnormalities at 12 months of age. The basal infoldings of the RPE (black brackets in A′, B′, and C′) are organized and filled with a homogenous substance. Bruch's membrane (asterisk) is mildly disrupted in Mfrp KI/KI mice at both 2 and 10 months of age. All markings are identical in all panels. Magnification 5,000 × with bar = 2 μm in the top row and 12,000 × with bar = 500 nm in the bottom row. (C) Quantification of rows of nuclei in the ONL in Mfrp KI/KI at 1, 2, 3, 6, and 12 months compared to 12-month-old WT mice. (D) Analysis of cone survival in Mfrp KI/KI mice. Cone survival was analyzed by immunostaining 1-, 4-, and 10-month-old Mfrp KI/KI eyes and 10-month-old WT frozen sections for S (green) and M opsin (red). Color images are available online.
Ultrastructural analyses of RPE and sub-RPE regions in Mfrp KI/KI and WT mice were also performed using electron microscopy. The RPE of Mfrp KI/KI mice (marked as B, B′; C, C′ in Fig. 5B) had structural aberrations beginning as early as 2 months of age. By 10 months of age, the RPE basal infoldings were disorganized, and numerous pockets of undigested membranous debris were seen in the basal RPE. The thickness of the photoreceptor layer was also decreased when compared to the control group, with disorganized rod and cone outer segments (Fig. 5B). In order to study photoreceptor survival in Mfrp KI/KI mice, immunohistochemistry was performed on frozen retinal sections. Photoreceptors expressing M and S opsins were analyzed (n = 3). A regional distribution of M and S opsins was noted, with S opsin expressing photoreceptors localized more ventrally, while M opsin expressing photoreceptors were found more dorsally. This was similar to previous reports in the murine retina (Supplementary Figure S5).17 Subjectively, decreased numbers of photoreceptors were observed in Mfrp KI/KI mice visually from 1 month of age when compared to 10-month-old WT controls (Fig. 5D), indicating a loss of photoreceptors as well as a shortening of photoreceptor outer segments with time in the model when compared to WT mice photoreceptor outer segments.
Increased hyperopia in Mfrp KI/KI mice eyes
To investigate whether the hyperopia identified in patients was noted in mouse eyes, infrared imaging was performed on Mfrp KI/KI mice at 2 and 3 months of age (n = 3). Three-month-old WT controls were used for comparison. The optical location of the retinal surface in diopters was used as a surrogate for measuring refractive error. A significantly higher diopteric measurement was required to focus the optic nerve in the Mfrp KI/KI mice when compared to WT mice (p < 0.01; Fig. 6A). Similarly, retinal cross sections stained with S and M opsin also showed a decrease in the overall diameter of the eye (Fig. 6B), corroborating earlier observations.
Figure 6.

Determination of the refractive power of the eye in Mfrp KI/KI mice. Diopteric power of Mfrp KI/KI mice at age 2 and 3 months of age were compared to those from WT mice (3 months; n = 3). There was a significantly greater diopteric measure in Mfrp KI/KI mice at 2 months of age (p < 0.05) and 3 months of age (p < 0.01), suggesting an increase in refractive power of the eye. (B) Representative images from eye cross-sections of 1- and 4-month-old Mfrp KI/KI mice and 10-month-old WT mice immunostained for S (green) and M opsin (red) shows a decrease in overall size of Mfrp KI/KI mice eyes when compared to WT type controls. DAPI (blue) was used to stain the nuclei.
Decreased ERG response in Mfrp KI/KI mice
As the patients in the cohort were noted to have a marked early reduction in rod photoreceptor dominated scotopic responses followed by a later reduction in cone photoreceptor dominated photopic responses during electrophysiology, the study investigated whether diminished photoreceptor function was also seen in the Mfrp KI/KI mouse model. ffERG was performed to analyze retinal function in Mfrp KI/KI mice at 3.5 and 6.5 months of age (Fig. 7A and B). There was a significant decrease in both scotopic a- and b-wave responses when compared with 3.5-month-old age-matched WT mice, indicating a loss of rod photoreceptor function (p < 0.001; Fig. 7B). Photopic ERG responses also showed a significant reduction in b-wave amplitude for 3.5- and 6.5-month-old Mfrp KI/KI mice (p < 0.001). However, differences in photopic a-wave did not significantly decrease with age (Fig. 7B). These results indicate that the c498_499insC Mfrp mutation is associated with progressive functional neurodegeneration in the Mfrp KI/KI model.
Figure 7.

Full-field electroretinography (ffERG) responses in Mfrp KI/KI mice. (A) Representative averaged waveforms following stimulation under both scotopic and photopic conditions from 3.5-month-old WT mice compared to 3.5- and 6.5-month-old Mfrp KI/KI mice. (B) Graph of mean ffERG waveform amplitude under scotopic and photopic conditions depicting a- and b-wave responses to −3.5, −1.09, −2.00, and +1.09, +2.00 cd.s/m2 stimulation intensities, respectively, in Mfrp KI/KI compared to WT mice. Each point represents the mean, with error bars representing the standard deviation (n = 6).
Decreased expression of RPE cell markers in Mfrp KI/KI mice
MFRP-protein is predominantly expressed by the RPE and ciliary body within the eye.9 Early structural aberrations were observed in RPE cells in the Mfrp KI/KI mice in ultrastructural studies. To investigate the effect of the c498_499insC Mfrp mutation on the RPE further, the expression of RPE markers Rpe65 and Zo-1 were measured and compared to expression from WT control mice posterior eye cup lysates. The level of expression of Rpe65 transcripts were reduced twofold (−2.15 ± 0.22) in 2-month-old Mfrp KI/KI mice when compared to age-matched WT control mice (Fig. 8; p < 0.01). Similarly, a 3.7-fold (−3.69 ± 0.19) reduction was observed with Zo-1 expression in Mfrp KI/KI mice when compared to WT mice (Fig. 8; p < 0.001). Levels of expression of Mfrp and its dicistronic partner Ctrp5 were 5.5-fold (−5.54 ± 2.82) and twofold (−2.18 ± 0.58) lower, respectively (p < 0.01 and p < 0.05, respectively) in 2-month-old Mfrp KI/KI mice when compared to WT mice (Fig. 8). The conserved CSD domain of MFRP is predicted to interact with the Wnt ligand in signaling processes. Therefore, the expression of wnt-1 and wnt-10b was evaluated in the retinae of WT and Mfrp KI/KI mice. A 5–7 fold (−5.75 ± 0.36) decrease in the levels of wnt-1 expression (p < 0.001) was found in Mfrp KI/KI mice when compared to WT mice (Fig. 8). However, wnt-10b expression was not significantly altered in mutant mice when compared to the expression in WT mice.
Figure 8.

Assessment of mRNA expression levels from the posterior eye cups of Mfrp KI/KI mice. Relative fold change of Rpe65, Zo-1, Mfrp, Ctrp5, Wnt1,and Wnt10b mRNA expression analyzed by quantitative reverse transcription polymerase chain reaction in the posterior eye cup lysates derived from the eyes of Mfrp KI/KI mice (2 months old; n = 3; n = 2 for Ctrp5) when compared to expression in posterior eye cup lysates from age-matched WT control mice (n = 3). Color images are available online.
MFRP expression after rAAV-mediated gene therapy in Mfrp KI/KI mice
The expression of MFRP was undetectable in the eyes of the Mfrp KI/KI mouse model. This suggested a loss of functional MFRP as the cause of the ocular phenotype in Mfrp KI/KI mice. Having established the Mfrp KI/KI mouse as a good model of human clinical disease, next the study investigated whether the ocular phenotype of Mfrp KI/KI mouse could be rescued using Mfrp gene replacement. To confirm this in this model and to see if the ocular phenotype could be rescued, one eye of Mfrp KI/KI mice was treated with whole Mfrp gene therapy delivered by sub-retinal injection at postnatal week 6. The contralateral eye was treated with a sham injection consisting of the virus vector vehicle.
The sham-treated control eyes of Mfrp KI/KI mice showed no MFRP expression (Fig. 9A and B). Following treatment with sub-retinal rAAV-Mfrp, expression of MFRP was confirmed using a Western blot analysis of lysate from posterior eye cups of 10-month-old Mfrp KI/KI mice (Fig. 9A). A further confirmation of MFRP expression was demonstrated by marked immunostaining of MFRP in the RPE of eyes in 10-month-old mice 9 months after treatment using sub-retinal rAAV-Mfrp (Fig. 9B). However, significant staining was also observed around the corneal region, which probably resulted from nonspecific binding of anti-MFRP antibody. This is determined by absence of such staining when immunohistochemistry was performed with secondary antibodies alone (negative control).
Figure 9.
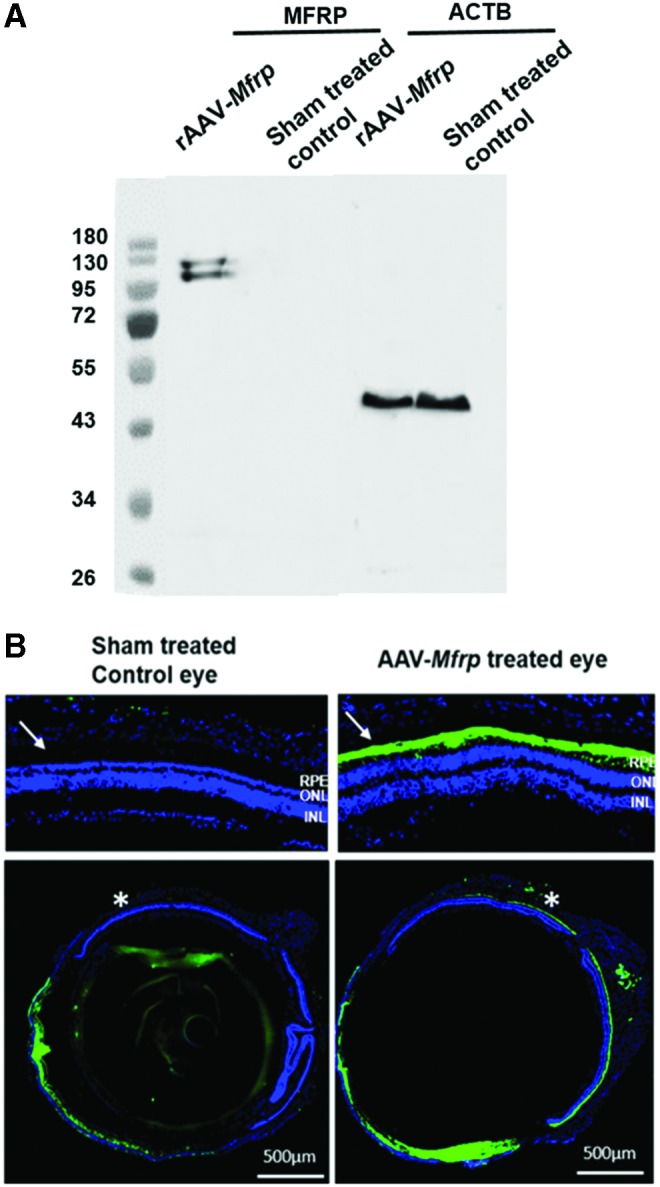
Expression of rAAV-Mfrp in Mfrp KI/KI mice. (A) Western blot of posterior eye cup tissue using antibody to detect MFRP expression in rAAV-Mfrp injected and control eyes in Mfrp KI/KI mice. (B) Immunohistochemistry using anti-MFRP in both rAAV-Mfrp injected and sham-treated control eyes. MFRP immunostaining (green) was predominant in the RPE in WT mice but was undetectable in Mfrp KI/KI mice. The upper images are magnified images of the retina from the regions marked with an asterisk.
Phenotypic rescue of Mfrp KI/KI mice using rAAV-Mfrp mediated gene therapy
Analysis of fundus autofluorescence in sham-control-treated and rAAV-Mfrp eyes indicated a significantly decreased number of autofluorescent spots in rAAV-Mfrp-treated eyes when compared to sham-treated eyes at the age of 5.5 months (n = 3; p < 0.01; Fig. 10A and B). Interestingly, fewer dots were observed in sham-treated eyes of 5.5-month-old mice when compared to 3-month-old untreated control mice. However, the difference was not statistically significant (p < 0.07). This may be a result of the sham sub-retinal treatment in the control eyes suppressing the rate of autofluorescent spot generation, as the spots are located between the photoreceptor and RPE layer, which is the layer detached in order to apply the viral vector. Considering this fact, we employed sham treatment to compare the effect of rAAV-mediated gene therapy in the experiments, which has not been used in previous studies on gene therapy using the rd6 mouse model. Next, retinal thickness was evaluated by SD-OCT (n = 3). A significantly thicker neuroretina was noted in rAAV-Mfrp-treated eyes when compared to sham-treated control eyes (p < 0.01; Fig. 10C and D). To investigate whether there was a functional rescue, ERG responses were evaluated (Fig. 10E). ERGs were performed 4 months after rAAV-Mfrp treatment (n = 6) at postnatal age 5.5 months in both control and treated eyes. A significantly greater a- and b-wave response was found under scotopic conditions in treated eyes when compared to sham-treated eyes (p < 0.001; Fig. 10F). Additionally, photopic b-wave response amplitudes were significantly greater (p < 0.001) than in sham-treated control eyes. However, photopic a-wave responses were not significantly different.
Figure 10.

Rescue of retinal structure function in Mfrp KI/KI mice following rAAV-Mfrp treatment. (A) SLO autofluorescence images from the rAAV-Mfrp-treated and sham-treated control treated eyes of Mfrp KI/KI mice. (B) Comparison of the number of autofluorescent white spots in SLO images from the fundus of rAAV-Mfrp-treated (n = 3) and sham-treated control (n = 3) eyes. (C) SD-OCT images comparing retinal thickness from a rAAV-Mfrp-treated and sham-treated control eye. The representative image shows an increase in neuroretinal thickness and the preservation of layers in the rAAV-Mfrp injected eye. (D) Bar chart depicting mean retinal thickness in rAAV-Mfrp-treated eyes (n = 3) and sham-treated control eyes (n = 3). Each point represents the mean ± SD. (E) Measurement of the amplitude of ffERG response functions for rAAV-Mfrp-injected and sham-treated control eyes under scotopic and photopic conditions (n = 6). (F) Line graph of mean ffERG a- and b-wave responses to −3.5, −1.09, −2.00, and +1.09, +2.00 cd.s/m2 stimulation intensities, respectively, under scotopic and photopic conditions. Each point represents the mean ± SD. Color images are available online.
A subset of treated Mfrp KI/KI mice (n = 3) underwent imaging at 10 months of age in order to investigate the long-term effect of gene therapy and to check for signs of toxicity. Examination revealed no major areas of RPE loss or signs of retinal toxicity, even 8.5 months after initial treatment (Fig. 1A). Furthermore, significant rescue was observed in both number and length of cones 9 months after rAAV-Mfrp treatment in Mfrp KI/KI mice, indicating sustained preservation of photoreceptors predominantly in the central retina, which corresponded to the location of maximal MFRP expression post treatment (Fig. 1B; p < 0.01). Taken together, the results presented here indicate that there was a significant rescue in retinal structure and function following sub-retinal injection with rAAV-Mfrp in Mfrp KI/KI mice.
Discussion
In this study, a clinical characterization was performed of the ocular phenotype of patients homozygous for c.498_499insC mutations in MFRP in order to study the long-term natural history of the disease in this pedigree. In addition, a new mouse model was generated and characterized to study the underlying disease mechanism associated with pathology resulting from c.498_499insC mutations in Mfrp. Finally, the effect of gene therapy in this new mouse model was tested.
All affected members of the pedigree described in this study harbored homozygous c.498_499insC mutations in MFRP.4 The c.498_499insC change, located in exon 5 of MFRP, leads to a frame-shift mutation (P165fsX198), which is predicted to result in the loss of both the CUB-LDL and CRD domains. Alternatively, this mutation may also result in the loss of MFRP. All affected patients had a progression of their ocular phenotype over the 12 years of follow-up, with increased retinal pigment clumps and bone-spicule pigmentation seen in the retina. In addition, BCVA in at least one eye of each of the patients and electrophysiological responses in both eyes of all patients were noted to deteriorate when compared to the initial clinical examination.4 Imaging with higher resolution SD-OCT confirmed macular thickening and foveoschisis or foveal thickening in all patients, which was previously suggested using lower resolution time-domain OCT (Fig. 2E and Supplementary Figs. S2 and S4). It is unclear how much the chronic macular thickening and foveoschisis contributed to clinical progression and deterioration in BCVA. The patients were also not treated with topical carbonic anhydrase inhibitors, which have traditionally been used to treat macular edema associated with cone-rod dystrophy and have previously been shown to be successful in reducing macular edema in MFRP-associated retinopathy.25 In the original examination, patients 1 and 2, aged 49 and 45 years, respectively, were already noted to have severe sight impairment, with a BCVA ≤20/200. Patient 1 initially reported symptoms of progressive vision loss and nyctalopia at the age of 24 years that suggested a rapid loss of vision. However, patients 3 and 4 were noted to have good central vision and photopic electrophysiological responses at the initial examination at the ages of 41 and 39 years, respectively. In the most recent examination, they have maintained good central vision into their sixth decade, with preserved photopic electrophysiological responses at the ages of 53 and 51 years, respectively (Supplementary Fig. S7). This suggests that a subset of patients have a much slower rate of progression of disease and that there is heterogeneity in the rate of progression, even among patients harboring the same mutation.
A detailed genotype–phenotype correlation study has yet to be performed for mutations in MFRP. To the authors' knowledge, 18 unrelated pedigrees have been reported with MFRP mutations.2,3,26–28 The mutations observed in MFRP were nonsense or frame-shift mutations, except for five missense mutations. However, due to the lack of clinical information and compound heterozygosity, it is difficult to assess phenotypic differences resulting from nonsense and missense mutations. One of the largest reviews looked at four unrelated pedigrees.2 In 4/7 patients, BCVA was maintained at 20/200 or better, and in two of these cases, this was maintained until the sixth decade of life. However, phenotypic heterogeneity was found between families with a 34-year-old patient showing foveoschisis and hypermetropia but not describing nyctalopia. Another 50-year- old patient maintained 20/30 vision in one eye and 20/40 in the other and also denied nyctalopia, while her sibling aged 47 years, harboring the same mutations, was unable to perceive light in one eye or see hand movements in the other eye. It is unclear what factors cause the heterogeneity in phenotype in these pedigrees and in the one described in this study. As yet unidentified genetic modifiers may play a part. Nishina et al. identified modifiers on chromosome 1, 6, and 11 using quantitative trait analysis in RD6 mice.29 They predicted that the modifiers contributed 28% of phenotypic variation in photoreceptor cell loss in these mice. Similar modifying loci may also contribute to variation in human MFRP gene associated pathology. Taken together, the evidence presented suggests a wide phenotypic variability among patients affected by MFRP-associated retinopathy. Identification of factors responsible for the phenotype variation could be helpful in developing therapies to reduce progression of MFRP-associated retinopathy.
There is a relative dearth of information regarding progression in MFRP-associated ocular disease. Previous studies have reported short follow-up data from affected individuals. Mukhopadhyay et al. reported on the 3-year follow-up of a 41-year-old patient whose electrophysiological findings did not significantly change.2 Neri et al. performed a 30-month follow-up of a single 33-year-old case whose BCVA deteriorated from 20/100 to 20/125 bilaterally and whose electrophysiological responses deteriorated under both scotopic and photopic conditions.28 Among the patients reported with mutations in MFRP, the majority of older patients were reported to have nanophthalmos and RP, while most of the younger individuals presented with nanophthalmos without RP.28 In one pedigree, a 4-year-old patient was reported to have nanophthalmos without RP, while older relatives >30 years of age were diagnosed with both nanophthalmos and RP.28 Nanophthalmos has been reported as a risk factor for developing retinal folds and degeneration.30 It is likely that younger patients with MFRP mutations may develop retinal abnormalities at an older age as a consequence of nanophthalmos. Reevaluation of the retinal phenotype of younger patients with nanophthalmos without RP may provide a better understanding of the progression of the clinical phenotype associated with MFRP mutations.
The Mfrp KI/KI mouse model shares key features of the clinical phenotype. The frame-shift mutation in Mfrp is predicted to result in a premature stop 32 amino acids downstream of the mutation. A Western blot and quantitative reverse transcription PCR analyses revealed a significant reduction in Mfrp expression in the posterior eye cup tissue of Mfrp KI/KI mice, and similarly immunohistochemistry did not identify MFRP following immunostaining. However, it is unclear whether a truncated protein was still expressed as the antibody used targeted Gln91-Pro584 of MFRP, which also includes the region downstream of the frame-shift mutation. The Mfrp KI/KI mice developed a progressive retinal degeneration, with a loss of photoreceptors, reduced retinal function, and eyes with decreased axial length. This phenotype was rescued following sub-retinal delivery of rAAV-Mfrp. Taken together, the evidence points toward the homozygous c.498_499insC mutations in Mfrp as the cause of the ocular phenotype in Mfrp KI/KI mice and that Mfrp KI/KI mice are a good model of human disease sharing many of the key features of the human clinical phenotype.
Animal models of MFRP-associated eye disease have been described previously. The rd6 mouse model results from a natural mutation originally seen in the C3HfB/GaCas1b mouse strain.13 The rd6 mouse model appears to have a slower progression of disease when compared to the Mfrp KI/KI mice, with ERG responses extinguished at 70 weeks of age.13 The Mfrp KI/KI mice were already noted to have an extinguished response by 10 months (40 weeks) of age. The rdx mice were identified when studying mice of a 129/Sv background.14 These mice were crossed with C57Bl/6J mice to form hybrids with homozygous Mfrp 174delG mutations. The rdx mice also developed flecked retina, but these occurred slightly later at 3 months of age. Disease progression was more similar to the Mfrp KI/KI mice, as the ONL was reduced to two nuclei thickness by 7 months in the rdx mice. Optic disc drusen were not noted in either mouse, but RPE atrophy was a prominent feature in older rdx mice. Initial reports noted no ocular axial length abnormalities in rd6 and rdx mouse models.13,14 However, in a more recent paper, the rd6 mice were demonstrated to have a reduced axial length, which could be corrected with gene therapy.3 Microphthalmia was also noted to be a feature of a zebrafish model deficient in Mfrp.31 The variability in progression and phenotype in mice may result from the differing genetic backgrounds of the models.29 However, all three mouse models, including the Mfrp KI/KI model, develop early-onset progressive retinal degeneration, suggesting functional conservation of MFRP between mice and humans, therefore opening up an avenue for further exploration of MFRP-associated retinal pathology utilizing these mouse models.
There is increasing evidence that MFRP deficiency results in RPE dysfunction. MFRP is primarily expressed in the RPE and ciliary body within the eye.7 First, animal models and human studies demonstrate fundus RPE atrophy.14,32 A reduction in expression of RPE markers was also noted in the mouse model presented in this study, and ultrastructural studies have also noted early changes to RPE integrity. Evidence from other mouse models and human iPSC-RPE deficient in MFRP suggest that there are significant changes to RPE microvilli and organization resulting from deficiency.14,33 Additionally, photoreceptor outer segments accumulate in the RPE in vivo.7 In the Mfrp KI/KI model, as well as other models of rd6 and rdx, an increase in autofluorescent deposits/dots was noted in the retina. These autofluorescent spots in rd6 and rdx mice showed significant immunoreactivity with macrophage markers, indicating an accumulation of sub-retinal macrophages resulting in the formation of autofluorescent flecks.13,14 Although similar studies were not performed on Mfrp KI/KI mice, it is likely that the autofluorescent spots observed in Mfrp KI/KI mice may also have a similar origin. In this study, these dots were identified as being localized in the sub-retina by SD-OCT, which resembles pseudodrusen seen in other diseases in which RPE dysfunction is thought to be prominent34–36 (Fig. 4F). Reports from younger patients with MFRP mutations describe similar white flecks.28,32 Unfortunately, high-resolution SD-OCT was not performed in the area where these flecks were seen in these patients to confirm retinal sub-localization. The evidence suggests that MFRP deficiency also leads to RPE dysfunction, which is likely to play a part in disease pathogenesis.
rAAV-mediated gene therapy has recently been approved by the Food and Drug Administration for the treatment of another recessive retinal dystrophy: Leber's congenital amaurosis.37 A similar approach to treat patients with MFRP-associated retinopathy is also tempting. The findings in this study are similar to those seen in previous treatment studies using the rd6 mouse but differ in a number of ways. First, sham-treated eyes were used as controls in the same animals. Second, subretinal rAAV-Mfrp injections were performed later at 6 weeks of age in this study. Previous studies treated very young mice at post natal day 5 (P5)3,33 and P14.23,38 In the present study, rAAV-Mfrp treatment in Mfrp KI/KI exhibited approximately 95% and 48% of rescue in scotopic and photopic ERG responses, respectively, when treated eyes were compared to sham-control treated eyes. In addition to these observations, analysis of ERG responses in treated eyes of Mfrp KI/KI mice demonstrated approximately 35% and 51% response under scotopic and photopic conditions, respectively, when compared to the eyes of 6.5-month-old WT mice, indicating robust rescue of retinal function. The current study shows significant rescue, even when the rAAV-Mfrp was administered at a later time when there is already clear evidence of degeneration, suggesting that a similar rescue may be performed in later human disease. In this article investigating long-term progression of patients with homozygous c.498_499insC mutations in MFRP, two out of four of the affected individuals in this pedigree were shown to have good vision and continued to manifest photopic ERG responses. Although MFRP-associated retinopathy is variable, some patients appear to retain cone function up to the ages of 20 and 50 years.28 These patients are potentially amenable to gene therapy in order to preserve their central vision. At present, however, photopic ERG response may be the best indicator of potential therapeutic rescue without any clear functional studies specifically designed for rod-cone dystrophies at present. Longitudinal treatment studies in mice may be helpful in identifying the extent of the therapeutic rescue window. In Mfrp KI/KI mice, a gradual shortening of photoreceptor outer segment length was found with time. It would be interesting to see whether the outer segments would regenerate with rAAV-Mfrp treatment if administered at later stages of disease and whether this would also result in a similar improvement in ERG responses. Interestingly, earlier treatment in patients may have further benefits beyond retinal rescue. Velez et al. showed that sub-retinal treatment at an early age in mice could also rescue axial length abnormalities resulting from MFRP deficiency.3 A similar early treatment in younger patients may also have the same effect, as most of the deficit in shortening appears to occur at a young age.3 A further difference in this study compared to previous investigations is that follow-up was extended to 9 months post treatment, whereas in previous Mfrp gene therapy studies, the follow-up was much shorter, the longest being 2 months. A strong long-term expression of MFRP 9-months was observed after the initial rAAV-Mfrp injection, with expression distributed across the retinal sections in the RPE in the treated eye, resulting in robust phenotypic rescue with no notable toxic effects. However, significant staining was also observed around the cornea, which is likely to have resulted from nonspecific binding of MFRP antibody.
The Mfrp KI/KI mouse model harbors homozygous c.498_499InsC mutations in Mfrp. The model shares many features of clinical pathology seen in human subjects with homozygous c.498_499InsC, thus validating the mutation in MFRP as the cause of their ocular disease. This model can serve as a valuable model for understanding the etiology of human MFRP-related disease and for evaluating potential therapeutic strategies. Patients in the pedigree described in this study showed marked heterogeneity in disease progression, which suggests that some patients may have a wide therapeutic window. The rescue of the retinal phenotype in response to rAAV-Mfrp gene therapy in this new Mfrp KI/KI model provides further support to the utility of this strategy as a therapeutic option for treating the ocular phenotype in appropriate patients with MFRP-associated eye disease for the long-term rescue of vision.
Supplementary Material
Acknowledgments
We thank the Department of Ophthalmology, UCSD Histology and Imaging core facilities supported by P30 EY022589 core grant. We would also like to thank Rachel Poleman, Stephanie Landeros, and John Quach for their excellent support with histology, mice imaging, and maintenance. R.A. was supported by The Foundation Fighting Blindness, Research to Prevent Blindness (NIH-EY21237, P30-EY22589). A.D. was supported by NIH grant EY026559, M2017035 Bright Focus Foundation, and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology of University of Florida (RPB). S.B. was supported by the Fulbright Fight for Sight program and by grants from the Global ophthalmology awards program and is supported by a Foundation Fighting Blindness career development award. MMJ was supported by a grant from Research to Prevent Blindness to Hamilton Eye Institute.
Author Disclosure
No competing financial interests exist.
References
- 1. Sundin OH, Leppert GS, Silva ED, et al. Extreme hyperopia is the result of null mutations in MFRP, which encodes a frizzled-related protein. Proc Natl Acad Sci U S A 2005;102:9553–9558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mukhopadhyay R, Sergouniotis PI, Mackay DS, et al. A detailed phenotypic assessment of individuals affected by MFRP-related oculopathy. Mol Vis 2010;16:540–548 [PMC free article] [PubMed] [Google Scholar]
- 3. Velez G, Tsang SH, Tsai YT, et al. Gene therapy restores mfrp and corrects axial eye length. Sci Rep 2017;7:16151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ayala-Ramirez R, Graue-Wiechers F, Robredo V, et al. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis 2006;12:1483–1489 [PubMed] [Google Scholar]
- 5. Crespi J, Buil JA, Bassaganyas F, et al. A novel mutation confirms MFRP as the gene causing the syndrome of nanophthalmos-renititis pigmentosa-foveoschisis-optic disk drusen. Am J Ophthalmol 2008;146:323–328 [DOI] [PubMed] [Google Scholar]
- 6. Katoh M. Molecular cloning and characterization of MFRP, a novel gene encoding a membrane-type frizzled-related protein. Biochem Biophys Res Commun 2001;282:116–123 [DOI] [PubMed] [Google Scholar]
- 7. Kameya S, Hawes NL, Chang B, et al. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet 2002;11:1879–1886 [DOI] [PubMed] [Google Scholar]
- 8. Schaffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab 2012;23:194–204 [DOI] [PubMed] [Google Scholar]
- 9. Mandal MN, Vasireddy V, Jablonski MM, et al. Spatial and temporal expression of MFRP and its interaction with CTRP5. Invest Ophthalmol Vis Sci 2006;47:5514–5521 [DOI] [PubMed] [Google Scholar]
- 10. Shu X, Tulloch B, Lennon A, et al. Disease mechanisms in late-onset retinal macular degeneration associated with mutation in C1QTNF5. Hum Mol Genet 2006;15:1680–1689 [DOI] [PubMed] [Google Scholar]
- 11. Rattner A, Hsieh JC, Smallwood PM, et al. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc Natl Acad Sci U S A 1997;94:2859–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 2006;127:469–480 [DOI] [PubMed] [Google Scholar]
- 13. Hawes NL, Chang B, Hageman GS, et al. Retinal degeneration 6 (rd6): a new mouse model for human retinitis punctata albescens. Invest Ophthalmol Vis Sci 2000;41:3149–3157 [PubMed] [Google Scholar]
- 14. Fogerty J, Besharse JC. 174delG mutation in mouse MFRP causes photoreceptor degeneration and RPE atrophy. Invest Ophthalmol Vis Sci 2011;52:7256–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sundin OH. The mouse's eye and Mfrp: not quite human. Ophthalmic Genet 2005;26:153–155 [DOI] [PubMed] [Google Scholar]
- 16. McCulloch DL, Marmor MF, Brigell MG, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol 2015;130:1–12 [DOI] [PubMed] [Google Scholar]
- 17. Chavali VR, Khan NW, Cukras CA, et al. A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum Mol Genet 2011;20:2000–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandal MN, Vasireddy V, Reddy GB, et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci 2006;47:5505–5513 [DOI] [PubMed] [Google Scholar]
- 19. Seeliger MW, Beck SC, Pereyra-Munoz N, et al. In vivo confocal imaging of the retina in animal models using scanning laser ophthalmoscopy. Vis Res 2005;45:3512–3519 [DOI] [PubMed] [Google Scholar]
- 20. von Ruckmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 1995;79:407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Llorente L, Barbero S, Cano D, et al. Myopic versus hyperopic eyes: axial length, corneal shape and optical aberrations. J Vis 2004;4:288–298 [DOI] [PubMed] [Google Scholar]
- 22. Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dinculescu A, Estreicher J, Zenteno JC, et al. Gene therapy for retinitis pigmentosa caused by MFRP mutations: human phenotype and preliminary proof of concept. Hum Gene Ther 2012;23:367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Won J, Smith RS, Peachey NS, et al. Membrane frizzled-related protein is necessary for the normal development and maintenance of photoreceptor outer segments. Vis Neurosci 2008;25:563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zacharias LC, Susanna R, Jr, Sundin O, et al. Efficacy of topical dorzolamide therapy for cystoid macular edema in a patient with MFRP-related nanophthalmos-retinitis pigmentosa-foveoschisis-optic disk drusen syndrome. Retin Cases Brief Rep 2015;9:61–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mameesh M, Ganesh A, Harikrishna B, et al. Co-inheritance of the membrane frizzled-related protein ocular phenotype and glycogen storage disease type Ib. Ophthalmic Genet 2017;38:544–548 [DOI] [PubMed] [Google Scholar]
- 27. Xu Y, Guan L, Xiao X, et al. Identification of MFRP mutations in Chinese families with high hyperopia. Optom Vis Sci 2016;93:19–26 [DOI] [PubMed] [Google Scholar]
- 28. Neri A, Leaci R, Zenteno JC, et al. Membrane frizzled-related protein gene-related ophthalmological syndrome: 30-month follow-up of a sporadic case and review of genotype-phenotype correlation in the literature. Mol Vis 2012;18:2623–2632 [PMC free article] [PubMed] [Google Scholar]
- 29. Won J, Charette JR, Philip VM, et al. Genetic modifier loci of mouse Mfrp(rd6) identified by quantitative trait locus analysis. Exp Eye Res 2014;118:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carricondo PC, Andrade T, Prasov L, et al. Nanophthalmos: a review of the clinical spectrum and genetics. J Ophthalmol 2018;2018:2735465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collery RF, Volberding PJ, Bostrom JR, et al. Loss of zebrafish Mfrp causes nanophthalmia, hyperopia, and accumulation of subretinal macrophages. Invest Ophthalmol Vis Sci 2016;57:6805–6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zenteno JC, Buentello-Volante B, Quiroz-Gonzalez MA, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis 2009;15:1794–1798 [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y, Wu WH, Hsu CW, et al. Gene therapy in patient-specific stem cell lines and a preclinical model of retinitis pigmentosa with membrane frizzled-related protein defects. Mol Ther 2014;22:1688–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gliem M, Muller PL, Mangold E, et al. Reticular pseudodrusen in Sorsby fundus dystrophy. Ophthalmology 2015;122:1555–1562 [DOI] [PubMed] [Google Scholar]
- 35. Cukras C, Flamendorf J, Wong WT, et al. Longitudinal structural changes in late-onset retinal degeneration. Retina 2016;36:2348–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Bats F, Wolff B, Mauget-Faysse M, et al. Association of reticular pseudodrusen and early onset drusen. ISRN Ophthalmol 2013;2013:273085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Voretigene neparvovec-rzyl (Luxturna) for inherited retinal dystrophy. Med Lett Drugs Ther 2018;60:53–55 [PubMed] [Google Scholar]
- 38. Dinculescu A, Min SH, Deng WT, et al. Gene therapy in the rd6 mouse model of retinal degeneration. Adv Exp Med Biol 2014;801:711–718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.