

Abstract
Recent advances in metalloprotein engineering have led to the development of a myoglobin-based catalyst, Mb(H64V,V68A), capable of promoting the cyclopropanation of vinylarenes with high efficiency and high diastereo- and enantioselectivity. Whereas many enzymes evolved in nature often exhibit catalytic proficiency and exquisite stereoselectivity, how these features are achieved for a non-natural reaction has remained unclear. In this work, the structural determinants responsible for chiral induction and high stereocontrol in Mb(H64V,V68A)-catalyzed cyclopropanation were investigated via a combination of crystallographic, computational (DFT), and structure-activity analyses. Our results show the importance of steric complementarity and non-covalent interactions involving first-sphere active site residues, heme-carbene, and the olefin substrate, in dictating the stereochemical outcome of the cyclopropanation reaction. High stereocontrol is achieved through two major mechanisms. First, by enforcing a specific conformation of the heme-bound carbene within the active site. Second, by controlling the geometry of attack of the olefin on the carbene via steric occlusion, attractive van der Waals forces and protein-mediated π−π interactions with the olefin substrate. These insights could be leveraged to expand the substrate scope of the myoglobin-based cyclopropanation catalyst toward non-activated olefins and to increase its cyclopropanation activity in the presence of a bulky α-diazo-ester. This work sheds first light into the origin of enzyme-catalyzed enantioselective cyclopropanation, furnishing a mechanistic framework for both understanding the reactivity of current systems and guiding the future development of biological catalysts for this class of synthetically important, abiotic transformations.
Keywords: Olefin cyclopropanation, myoglobin, biocatalytic carbene transfer, heme carbenes, enantioselective cyclopropanation, Density Functional Theory
Graphical Abstract
Introduction
The development of engineered and artificial enzymes for ‘abiotic’ chemistry (i.e., reactions not occurring in nature) has recently created new opportunities toward expanding the range of bond-forming reactions accessible via biocatalysis.1–5 In particular, engineered heme-containing proteins have emerged as a promising class of biological catalysts for mediating selective carbene transfer reactions, including olefin cyclopropanation,6–15 Y—H insertion (where Y = N, S, or Si),16–20 C—H functionalization,21–23 aldehyde olefination,24, 25 and other relevant transformations.26, 27 Inspired by the exquisite chemo-, regio- and stereoselectivity of many naturally occurring enzyme-catalyzed reactions,28–30 efforts in this area have often involved re-engineering of the target protein and its active site to modulate the diastereoselectivity and/or stereoselectivity of the carbene transfer process. The asymmetric cyclopropanation of olefins is of particular interest as it provides access to conformationally constrained and stereochemically rich cyclopropane rings, which represent key building blocks for the discovery and development of drugs and other biologically active molecules.31 Using myoglobin (Mb) as hemoprotein scaffold, our group has recently reported the development of an engineered biocatalyst, Mb(H64V,V68A), which offers high catalytic activity (>10,000 turnovers) as well as excellent diastereo- and enantioselectivity (>95–99% detrans and ee1S,2S) for the cyclopropanation of vinylarenes in the presence of ethyl α-diazoacetate (EDA) as the carbene donor.8 This Mb variant, along with a stereocomplementary variant thereof, could be applied to the enantioselective synthesis of a panel of cyclopropane-containing drugs at the gram scale.9 More recently, the scope of the Mb(H64V,V68A) biocatalyst was extended to enable highly enantioselective and efficient olefin cyclopropanations in the presence of other acceptor-only diazo reagents, such as 2-diazotrifluoroethane32 and diazoacetonitrile.33 Because of its synthetic utility and ability to process a broad range of aryl-substituted olefins with a predictable trans-(1S,2S)-selectivity,8, 9, 32–34 Mb(H64V,V68A) represents an ideal system for investigating general stereoselectivity-determining factors in biocatalytic cyclopropanation beyond the confines of a single substrate-enzyme pair.
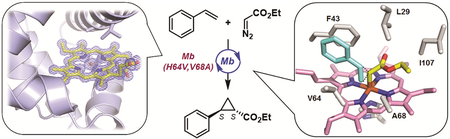
Based on previous studies, myoglobin-catalyzed cyclopropanation is predicated to proceed via the formation of a reactive heme-bound carbene intermediate, which arises from reaction of the catalytically active ferrous hemoprotein with the diazo reagent (Figure 1).8, 35–37 Recent experimental and computational analyses have further revealed that the reaction between this species and the olefin substrate involves a concerted, asynchronous olefin insertion process (Figure 1),36 which is distinct from the step-wise, radical cyclopropanation mechanism exhibited by other metalloporphyrin systems.38, 39 Furthermore, these studies elucidated the beneficial role of the axial imidazole ligand from the heme-ligating proximal histidine residue toward enhancing the cyclopropanation reactivity of this metalloprotein.36 In other studies, replacement of the axial histidine ligand with natural amino acids22 or the unnatural amino acid N-methyl-histidine40 was found to be beneficial toward increasing the carbene transfer activity of this myoglobin variant under non-reducing conditions.
Figure 1.

Mechanism of myoglobin-catalyzed olefin cyclopropanation with α-diazo esters.
Despite this progress, the structural determinants underlying protein-mediated chiral induction and stereocontrol in these reactions have remained largely unknown. Recently, a computational study was performed on two P450 variants with divergent cis:trans selectivity in styrene cyclopropanation with EDA, but these analyses provided only a qualitative assessment of a change in diastereoselectivity resulting from substitution of the proximal ligand (Cys vs. Ser).41 In this work, crystallographic, computational, and structure-activity analyses were carried out to clarify the basis of high diastereo- and enantiocontrol in Mb(H64V,V68A)-catalyzed olefin cyclopropanation. These studies shed first-time mechanistic insights into asymmetric cyclopropanation catalyzed by a metalloprotein, thus providing a basis for rationalizing the reactivity of current systems and guiding further reaction and catalyst development. Illustrating this point, the insights gained from these analyses could be leveraged to further improve the substrate scope of the myoglobin-based cyclopropanation catalyst across unactivated olefin substrates and a bulky diazo reagent.
Results and Discussion
Crystallographic analysis of Mb(H64V,V68A)
Compared to wild-type sperm whale myoglobin (Mb), Mb(H64V,V68A) features two amino acid substitutions at the level of the ‘distal’ histidine residue (His64) and a second residue (Val68) located on the distal side of the heme cofactor. Previous studies showed that the H64V mutation mainly enhances the catalytic activity of the metalloprotein in the cyclopropanation of styrene with ethyl α-diazo-acetate (EDA) with minimal effect on the diastereo- and enantioselectivity of the reaction (93% de, 10% ee with Mb(H64V) vs. 86% de, 6% ee with wild-type Mb for trans-(1S,2S)-configured product).9 The Val68Ala mutation, on the other hand, was found to play a key role in enhancing both the diastereo- and enantioselectivity of the metalloprotein in this reaction, as derived from direct comparison of the selectivity properties of Mb(H64V,V68A) (99.9% de and 99.9% ee) with those of Mb(H64V).9
To gain insights into the effects of these mutations on the protein structure, ferric Mb(H64V,V68A) (acquo-complex) was crystallized and its X-ray crystal structure was determined at 1.1 Å resolution. Superposition of the Mb(H64V,V68A) structure with that of wild type Mb42 (CO-complex) crystallized in the same space group (P6), showed an RMSD of 0.3 Å as calculated with Dali43, indicating that the protein three-dimensional fold is not greatly affected by the mutations. Furthermore, hydrogen bond distances between the protein and heme as well as a network of hydrophobic interactions appear to be maintained when comparing the Mb variant to the wild-type structure (PDB: 1JW8) (Figure 2A). At the same time, the two mutations in Mb(H64V,V68A) expand the volume of the heme distal pocket (Figure 2B), resulting in a solvent accessible volume of 243 Å3 for Mb(H64V,V68A) compared with 125 Å3 for wild-type Mb as calculated using DoGSiteScorer44 (Figure 3). In particular, the mutations create a larger void above the solvent-exposed half (= rings C/D) of the porphyrin (H64V) and above ring A and the meso position between rings A and D (V68A) (Figures 2C and 3A). This configuration allows greater substrate accessibility to the distal pocket in the hemoprotein (Figure 3A). Furthermore, the His64→Val mutation contributes to enhance the hydrophobicity near the heme-bound iron. Altogether, these structural features are likely beneficial for promoting binding to the diazo compound and olefin substrate, providing conditions conducive to the cyclopropanation reaction. Further inspection of the crystal structure showed that the side chains of four residues, i.e., Val64, Ala68, Phe43, and Ile107, define the space above the plane of the iron porphyrin (Figure 2C).
Figure 2. High-resolution crystal structure of sperm whale Mb(H64V,V68A) variant (top) and comparison to wild-type Mb (bottom; PDB:1JW8).

A) Arrangement of amino acids surrounding the heme cofactor. The axial water ligand in Mb(H64V,V68A) and axial CO ligand in Mb are displayed as sphere (red) and stick model, respectively. B) View of the molecular surface representation around the heme binding site. C) Top view of the bound heme and nearby amino acid residues, along with the labels for pyrrole N atoms and porphyrin rings. Carbon atoms are shown as yellow (heme), gray (double-mutant structure), cyan (wild-type structure) or salmon (mutated residues), nitrogens are blue and oxygens red. Inter-residue distances are measured in angstroms and shown as dashed lines. In the top panel of (C), riding model hydrogen atoms are shown as thin white sticks. The very high resolution of the data allowed for a limited number of hydrogens to be discerned in the electron density maps, and thus their positions were added throughout the model, except in the case of solvent molecules. The 2mFo-DFc electron density map surrounding the heme is shown at 1σ. See Table S1 for crystallographic parameters.
Figure 3.

Pocket cavity space (mesh) above the heme cofactor in (A) Mb(H64V,V68A) and (B) wild-type sperm whale myoglobin. The pocket volumes were calculated to correspond to 243 Å3 and 125 Å3, respectively, based on the crystallographic structure atom coordinates and a difference of Gaussian filter method (DoGSiteScorer).44
Model of Mb(H64V,V68A) active site and DFT method
To gain insights into the effect of the protein matrix in controlling the stereoselectivity of Mb-catalyzed cyclopropanation, a model of Mb(H64V,V68A) active site was built for performing computational analysis of the carbene intermediate and transition states via Density Functional Theory (DFT). Guided by crystallographic information, this model was chosen to include the full heme cofactor (with the propionic groups substituted for propyl groups to avoid artificial H-bonding interactions), the axial His93 ligand, and the ‘first-sphere active site residues’45 Leu29, Phe43, Phe46, Val64, Ala68, and Ile107 (Cβ(amino acid)···Fe(heme) distances from 5.1 Å (Ala68) to 13.2 Å (Phe46)); Figure 2A). Each residue was truncated at the Cα position and each Cα atom fixed at the position found in the reference X-ray crystal structure to mimic the protein environment, as done previously in the context of other metalloproteins.46, 47 The atoms in the models were allowed to be optimized using a range-separated hybrid DFT method with dispersion correction, ωB97XD,48 based on its excellent performance on heme carbenes and other catalytic systems from previous methodological studies.35, 36, 49–51 Considering the large size of this model, it was divided into high and low levels, with the former containing the core part of the reaction system (iron porphyrin, His93 ligand, [:CHCO2Et] carbene, and styrene) and the low level containing all other atoms. The basis set for the high level part of the system includes the effective core potential (ECP) basis LanL2DZ52 for iron and the triple-zeta basis 6–311G(d) for all other elements in this level, which was found to provide accurate predictions of various experimental reaction properties of heme carbenes.35, 36, 51 For all atoms in the low level, 6–31G(d) was used. More details about the DFT method can be found in the Supporting Information.
Analysis of carbene conformations
Using this system, an extensive conformational analysis was first conducted to identify the most favorable orientation(s) of the carbene moiety in the active site of Mb(H64V,V68A). Given the asymmetry of the protein active site and two possible alignments of carbene groups found in structurally characterized carbene-iron porphyrin complexes,53, 54 eight conformations were considered, in which the initial carbene plane is aligned either along each of the porphyrin N—Fe axes or in-between each of the two neighboring N—Fe axes. These conformations are defined by ∠N1FeC1C dihedral angle values range from −135° to +180° with an interval of 45°, where N1 is the nitrogen atom of pyrrole ring A of the heme, C1 is the iron-bound carbon atom, and C is the carbonyl carbon atom (see Table 1 for data entries and Figure 4A for atom labels). Since rotation of the carbene-borne carboxylate group yields two additional conformers, herein referred to as (+) and (−) based on ∠HC1CO1 dihedral angle (Figure 4), a total of sixteen conformations were calculated. After geometry optimization, the sixteen initial conformations could be divided into four groups (Table 1), with the carbene-borne ester group occupying four distinct inter-residue regions proximal to the porphyrin ring (Figure 2C), namely the regions between Ala68…Ile107, between Ile107…Phe43, between Phe43…Val64, or between Val64…Ala68 (Table 1). Analyses of the relative energies associated with these conformational isomers revealed that conformation 2(+) (Figure 5), in which the carbene ester group is located between Ala68 and Ile107, is most favorable, whereas other conformations have ~3–10 kcal/mol higher Gibbs free energies (ΔG) (Table 1). The only exception was conformation 1(+), which after optimization converges to a conformation geometrically and energetically identical to that derived from 2(+). These results indicate that steric effects exerted by the protein matrix play a major role in controlling the conformation of the heme-bound carbene within the protein active site, with the lowest energy conformer, 2(+), occupying the region with the second largest inter-residue distance (Figure 2C). Meanwhile, it became apparent that other factors also influence the orientation of the carbene moiety. Indeed, conformations 3(+) and 6(+) are energetically similar (ΔG = +3.4 vs. 3.2 kcal mol−1 relative to 2(+)), despite the fact that the carbene moiety in 3(+) occupies a sterically less hindered region (~2.6 Å wider inter-residue distance; Figure 2C). Conversely, the carbene groups in conformations 3(−) and 6(+) occupy regions of similar steric accessibility, yet the former has about 3 kcal/mol higher energy than the latter.
Table 1.
Relative energies and initial and optimized dihedral angles for the 16 carbene conformations within Mb(H64V,V68A) active site. The conformations are numbered 1 through 8 and (+)/(−) according to the initial ∠N1FeC1C and ∠HC1CO1 dihedral angles, respectively, and grouped according to the inter-residue region occupied by the carbene-borne ester group. The reported energy values are relative to the lowest-energy conformation, 2(+).
| Region | Conformation | Initial ∠N1FeC1C | Optimized ∠N1FeC1C | ∠HC1CO1 | ΔE | ΔEZPE | ΔH | ΔG |
|---|---|---|---|---|---|---|---|---|
| (°) | (°) | (°) | (kcal/mol) | (kcal/mol) | (kcal/mol) | (kcal/mol) | ||
| I107…F43 | 1(−) | 0.0 | 15.4 | −113.2 | 3.5 | 3.2 | 1.4 | 10.4 |
| 2(−) | 45.0 | 44.8 | −97.7 | −1.1 | −1.8 | −3.4 | 4.3 | |
| 3(+) | 90.0 | 140.3 | 102.0 | 0.4 | 0.4 | −0.3 | 3.4 | |
| 4(+) | 135.0 | 133.1 | 97.3 | 2.5 | 2.6 | 0.9 | 8.8 | |
| F43…V64 | 3(−) | 90.0 | 125.0 | −98.0 | −2.2 | −2.2 | −4.2 | 6.1 |
| 4(−) | 135.0 | 124.3 | −100.6 | 0.6 | 0.8 | −0.2 | 6.2 | |
| 5(+) | 180.0 | −148.0 | 99.1 | 3.1 | 2.3 | 0.9 | 6.4 | |
| V64…A68 | 5(−) | 180.0 | 168.4 | −84.4 | 1.0 | 0.6 | −0.7 | 6.3 |
| 6(−) | −135.0 | −122.5 | −91.0 | 3.5 | 3.0 | 1.4 | 7.5 | |
| 6(+) | −135.0 | −96.2 | 92.2 | −0.9 | −1.4 | −2.5 | 3.2 | |
| 7(+) | −90.0 | −94.7 | 90.2 | 0.3 | −0.5 | −1.6 | 3.9 | |
| 8(+) | −45.0 | −101.9 | 84.8 | 0.6 | 0.6 | −1.3 | 6.3 | |
| A68…I107 | 7(−) | −90.0 | −70.0 | −104.6 | −2.3 | −2.6 | −3.8 | 2.7 |
| 8(−) | −45.0 | −63.3 | −102.0 | −2.3 | −2.8 | −5.2 | 7.0 | |
| 1(+) | 0.0 | 40.4 | 95.5 | −0.2 | −0.9 | −0.7 | 0.3 | |
| 2(+) | 45.0 | 43.1 | 92.9 | 0.0 | 0.0 | 0.0 | 0.0 |
Figure 4.

Conformations of the heme-bound carbene upon rotation of the ester groups. The two conformations are referred to as (+) (A) and (−) (B) in accordance to the sign of the ∠HC1CO1 dihedral angle.
Figure 5.

Side view (A) and top view (B) of the most favorable conformation 2(+) for the heme-bound carbene in Mb(H64V,V68A) active site.
A more detailed analysis of the lowest-energy conformers within each group indicated that van der Waals (vdW) interactions mediated by the carbene-borne ester group contribute to stabilization of the carbene conformations. Indeed, the most favorable conformation 2(+) encompasses a total of four short-distance contacts (2.4–2.6 Å) between the oxygen atoms of the ester group (O1 and O2) and C—H bonds of neighboring residues (Ala68, Ile107, Leu29, Phe43) (Figure 5A and Table S2). These distances are smaller than the sum of Bondi’s vdW radii for O and H (2.72 Å),55, 56 indicating the occurrence of favorable interatomic interactions. In comparison, 3(+) and 6(+), i.e., the lowest-energy conformations within the I107…F43 and V64…A68 regions, respectively, exhibit only two short-distance contacts of this type (Table S2). Conformer 3(−), i.e., the preferred conformer within the F43…V64 region, also displays two short O(carbene)····H(residue) contacts, but the corresponding distances (2.6–2.7 Å) are longer than those in 3(+) and 6(+), providing a plausible explanation for its lower stability compared to the latter. For all optimized conformations, no short contacts were observed between the carbene moiety and the side chains of Val64 and Phe46, indicating that Phe43, Leu29, Ala68, and Ile107 (Figure 2C) are primarily involved in affecting the orientation and stability of the heme-bound carbene.
These results are consistent with previous observations that the improved stereoselectivity of Mb(H64V,V68A), compared to wild-type Mb, is largely dictated by the V68A mutation rather than the H64V mutation.9 Altogether, the analyses above showed that the overall best conformation 2(+) arises from a good steric complementarity between the carbene group and the inter-residue space above the plane of the heme cofactor (Figure 5) combined with a larger number of favorable non-covalent interactions between the carbene and surrounding residues.
Stereodivergent pathways to cyclopropanation product
Having identified the most favorable orientation of the carbene group within the Mb(H64V,V68A) active site, we investigated the transition states leading to the four possible stereoisomers of the cyclopropanation product (i.e., TS1S,2S, TS1R,2R, TS1S,2R, TS1R,2S). For these analyses we used the singlet concerted reaction pathway, as the relevance of this mechanism for Mb-catalyzed cyclopropanation was established previously.36 The optimized structures for the four TS and corresponding ΔΔGǂ are shown in Figure 6 and Table 2, respectively. Based on these data, the Mb(H64V,V68A)-catalyzed reaction involving styrene and EDA is predicted to occur with a diastereomeric excess (de) of 99.9% and an enantiomeric excess (ee) of 99.8% for the trans-(1S,2S) configured cyclopropanation product. These values are in excellent agreement with the experimentally determined values of 99.9% de and 99.9% ee (Table 3, Entry 1).8 The trans isomers have significantly lower barriers than the cis isomers (ΔΔGǂ from −5.2 to −5.9 kcal mol−1; Table 2). This energetic difference was also observed in a protein-free model of heme-catalyzed cyclopropanation36 and originates from favorable π−π interactions and vdW contacts occurring in the TStrans vs. TScis. Whereas this result is consistent with the trans preference of iron-porphyrins in this reaction,57–59 this effect is greatly amplified within the Mb(H64V,V68A) active site due to steric clashes between the styrene ring and the side chain of Leu29, thus providing a basis for the significantly higher trans-diastereoselectivity exhibited by Mb(H64V,V68A) compared to hemin alone (99.9% vs. 87% de)8. To further probe this conclusion, a Mb(H64V,V68A)-derived variant was prepared in which the steric bulk at the level of Leu29 is reduced via substitution of this amino acid residue with alanine. The resulting Mb variant, Mb(L29A,H64V,V68A), was found to catalyze the cyclopropanation of styrene with EDA with 10-fold lower trans-diastereoselectivity compared to Mb(H64V,V68A) (50:1 vs. >500:1 trans:cis ratio; Table 3, Entry 10 vs. 1), thereby providing experimental support to the functional role of Leu29 side chain in controlling the diastereoselectivity (and enantioselectivity, vide infra) of the cyclopropanation reaction.
Figure 6.

Free energy diagram for the cyclopropanation steo and optimized structures for the transition states leading to the four possible stereoisomeric products in Mb(H64V,V68A)-catalyzed styrene cyclopropanation with EDA: (A) TS1S,2S, (B) TS1R,2R, (C) TS1S,2R, and (D) TS1R,2S.
Table 2.
Energy barriers (kcal/mol) for the four stereodivergent pathways in Mb(H64V,V68A)-catalyzed styrene cyclopropanation with EDA. ΔΔGǂ values are relative to TS1S,2S.
| ΔE≠ | ΔEZPE≠ | ΔH≠ | ΔG≠ | ΔΔG≠ | |
|---|---|---|---|---|---|
| TS1S,2S | −17.7 | −14.4 | −15.8 | 8.8 | 0.0 |
| TS1R,2R | −12.9 | −11.0 | −13.5 | 12.9 | 4.2 |
| TS1S,2R | −14.6 | −11.7 | −14.5 | 14.0 | 5.2 |
| TS1R,2S | −8.1 | −6.6 | −8.0 | 14.7 | 5.9 |
Table 3.
Activity, selectivity, and kinetic parameters for the cyclopropanation of styrene with α-diazoester derivatives catalyzed by Mb(H64V,V68A) and variants thereof.a
| Entry | Catalyst | Diazo | Yieldb | TON | % de | % ee |
kcatc (min−1) |
KMc (mM) |
|---|---|---|---|---|---|---|---|---|
| 1 | Mb(H64V,V68A) | 2a | 99% | >500 | 99.9 | 99.9 | 585 | 21.1 |
| 2 | Mb(H64V,V68A) | 2b | 84% | 420 | 93 | 83 | 103 | 7.3 |
| 3 | Mb(H64V,V68A) | 2c | 66% | 330 | 93 | 59 | 45 | 3.0 |
| 4 | Mb(H64V,V68A) | 2d | 99% | >500 | 83 | 92 | 295 | 3.2 |
| 5 | Mb(H64V,V68A) | 2e | 99% | >500 | 96 | 96 | 320 | 6.4 |
| 6 | Mb(H64V,V68A) | 2f | 41% | 205 | 99 | 84 | 62 | 8.1 |
| 7 | Mb(H64V,V68G) | 2a | 99% | >500 | 77 | 96 | n.d. | n.d. |
| 8 | Mb(H64V,V68G) | 2b | 77% | 385 | 69 | 76 | n.d. | n.d. |
| 9 | Mb(H64V,V68G) | 2c | 99% | >500 | 95 | 2 | n.d. | n.d. |
| 10 | Mb(L29A,H64V,V68A) | 2a | 99% | >500 | 96 | 84 | n.d. | n.d. |
Reaction conditions: 10 mM styrene (1), 20 mM diazo reagent (2a-2f), 10 mM Na2S2O4, 20 μM purified Mb variant in phosphate buffer (50 mM, pH 7.0), under anaerobic conditions, room temperature, 16 hours.
GC yield.
Apparent kinetic parameters for the carbene donor at sub-saturating styrene concentration (20 mM) under aerobic conditions.
Side-by-side comparison of TS1S,2S vs. TS1R,2R structures provided further insights into the determinants responsible for the high (1S,2S)-enantioselectivity of Mb(H64V,V68A). Most notably, the TS1S,2S structure revealed a π−π stacking interaction between the phenyl ring of styrene and that of Phe43 (Figure 6, panel A, red line), which is absent in all of the other transition states (Figure 6, panels B and D). This interaction is made possible through a ~90° rotation of Phe43 phenyl ring around the Cβ-Cγ bond (χ2 bond), as evinced from comparison of the structure of TS1S,2S with the crystal structure of Mb(H64V,V68A) (Figure 6A vs. 2A). Intriguingly, this protein-olefin substrate interaction could explain the broad substrate scope of Mb(H64V,V68A) across aryl-substituted olefins,8 whereas no reactivity was observed with alkenes lacking an aromatic substituent (e.g., 1-octene),8, 9 despite the latter are viable substrates for hemoprotein-catalyzed cyclopropanation.13 In addition to the aforementioned π−π interaction, two vdW contacts between the carbene moiety and the styrene substrate are present in TS1S,2S (O1(carbene)…H(styrene): 2.5/2.5 Å), in contrast with only one in TS1R,2R (O2(carbene)…H(styrene): 2.6 Å). In TS1S,2S, other favorable vdW force involve the O2 atom and residue Phe43 (2.6 Å) and Leu29 (2.7 Å), compared to only one in TS1R,2R (O2…H(Ala68): 2.6 Å). Regarding the two transition states leading to the cis-configured cyclopropanation products, only TS1S,2R displays a favorable O1(carbene)…H(styrene) contact (2.6 Å) that stabilizes the substrate. While this interaction is similar to that present in TS1R,2R, TS1S,2R involves more important steric clashes between the carbene group and Ile107, as suggested by the shorter H(carbene)…H(Ile107) distance of 2.3 Å vs. 4.0 Å in TS1R,2R. Altogether, these analyses indicate that (i) stabilization of the carbene conformation as a result of residue-carbene interactions and (ii) close-range weak interactions between the active site residues and the styrene substrate, jointly contribute to direct and favor the attack of styrene to the si face of the heme-bound carbene, thereby dictating the high trans-diastereoselectivity and (1S,2S)-enantioselectivity of the Mb(H64V,V68A)-catalyzed cyclopropanation reaction. Furthermore, in TS1S,2S the phenyl ring of styrene is projected toward the opening connecting the heme pocket to the solvent, which is expanded by the H64V mutation (Figure 3A vs. 3B). This arrangement is expected to make the chirality induced by Mb(H64V,V68A) in the cyclopropanation reaction rather independent of structural variations at the level of the aromatic ring of the olefin, which can explain the preserved trans-(1S,2S) selectivity of Mb(H64V,V68A) across a broad range of aryl-substituted olefins as observed in previous studies.8
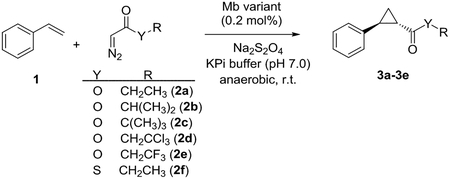
Structure-reactivity studies with α-diazoester derivatives
Whereas the mechanistic scenario presented above is able to explain the enantioselectivity of Mb(H64V,V68A) both qualitatively and quantitively, additional experiments were performed in order to probe its validity. To this end, the Mb(H64V,V68A)-catalyzed styrene cyclopropanation reaction was carried out with a series of probe diazo reagents (2b-2f, Table 3), which feature varying steric demands at the proximal (C1’) (2b, 2c) and distal carbon atom (C2’) of the alkyl group (2d), as well as isosteric substitutions within the ester group (2e, 2f). Compared to EDA (2a), the reactions with isopropyl (2b) and tert-butyl α-diazoacetate (2c) show a progressive decrease in yield, catalytic turnovers (>500 (3a) → 420 (3b) → 330 TON (3c); Table 3, Entries 2–3 vs. 1), turnover rate (kcat: 585 (3a) → 103 (3b) → 45 min−1 (3c)) and (1S,2S)-enantioselectivity (99% (3a) → 83% (3b) → 59% ee (3c) as the steric bulk at the C1’ atom is increased. In contrast, a more moderate reduction in all of these parameters was observed with 2’,2’,2’-trichloro-ethyl α-diazoacetate (2d), which bears increased steric bulk at the C2’ atom (Entry 4). These results are consistent with the preferred conformation of the heme-bound carbene, 2(+) (Figure 4), in which close carbene-residue (Ile107) contacts occur at the level of the C1’ atom, whereas a larger space is available around the C2’ atom. These structural differences are expected to impose a lower tolerance of the C1’ over the C2’ position toward substitution with larger group(s) (i.e., H→CH3/Cl) in the context of the cyclopropanation reaction, an effect clearly reflected by the experimentally observed trend in catalytic activity and selectivity across the diazoesters 2a-2d.
Based on the results above, it was surmised that increasing the active site space around residue 68 should better accommodate diazo reagents with a bulkier alkyl chain such as 2c. To probe these predictions, a variant carrying a Ala→Gly substitution at this position, Mb(H64V,V68G), was tested in the styrene cyclopropanation reaction in the presence of 2a, 2b, or 2c. Gratifyingly, the reaction with tert-butyl α-diazoacetate (2c) shows a significantly higher trans-diastereoselectivity compared to those with EDA (2a) or isopropyl α-diazoacetate (2b) (95% vs. 69–77% de; Table 3, Entries 7–9), indicating a better match between the bulkier tert-butyl group on the carbene and the further enlarged cavity between residue 68 and 107 (Figure S1), and consequently, a less optimal match between the latter and the smaller alkyl groups in the 2a- and 2b-derived carbenes. This effect is also evident from the enhanced styrene cyclopropanation activity of Mb(H64V,V68G) in the presence of tert-butyl α-diazoacetate compared to Mb(H64V,V68A) with respect to yield (99% vs. 66%) and TON (>500 vs. 330). At the same time, the enantioselectivity of this reaction is drastically reduced compared to that with the other α-diazo acetates, leading to formation of the two enantiomeric trans-cyclopropanes in nearly equal amounts (2% vs. 76–96% ee). These results can be rationalized in view of the calculated transition states leading to the (1S,2S)- and (1R,2R)-configured cyclopropane products (Figure 6, panels A and B). Indeed, the bulkier tert-butyl group significantly increases steric hindrance at the level of the si face of the heme-bound carbene compared to an ethyl or isopropyl group, as evidenced by a model of TS1S,2S involving heme-bound [:CHCO2(tBu)] carbene (Figure S1). This effect is expected to disfavor styrene attack to the si vs. the re face of the carbene, resulting in the drastically reduced enantioselectivity for this reaction as observed experimentally.
Consistent with the effect of steric bulk at the level of the carbene ester group on the catalyst stereoselectivity, the cyclopropanation reaction with 2’,2’,2’-trifluoro-ethyl α-diazoacetate (2e) was found to proceed with comparably high efficiency and trans-(1S,2S)-selectivity as with 2a (Table 3, Entry 5 vs. 1), as expected given the comparable size of the trifluoroethyl group vs. ethyl group. At the same time, kinetic analyses showed that the apparent Michealis-Menten constant (KM) of Mb(H64V,V68A) for 2e is nearly 4-fold lower than that for EDA (6.4 vs. 21.1 mM; Table 3). This effect is consistent with the trifluoroethyl group projecting toward the hydrophobic core of the protein as in 2(+). Indeed, while being isosteric to EDA, the increased lipophilicity of 2e should lead to a tighter interaction with the hemoprotein than EDA, a phenomenon that would be reflected by a decrease in the corresponding KM value. Corroborating this trend and thus further substantiating the mechanistic model presented above, a progressive decrease in KM was observed for the Mb(H64V,V68A)-catalyzed cyclopropanation reaction with ethyl (2a) vs. isopropyl (2b) vs. tert-butyl α-diazoacetate (21.1 → 7.3 → 3.0 mM; Table 3). Furthermore, a reasonably good correlation (R2: 0.63) could be found between the KM values of Mb(H64V,V68A) for 2a-2f and the hydrophobicity of these molecules, as estimated based on the octanol:water partition coefficient of the corresponding acetate (thio)esters (Figure S3).
Finally, the functional role of the ester group in protein-induced asymmetric induction was probed using 2f, i.e., an EDA analog in which the ester oxygen atom (O1) is substituted for sulfur. Despite this subtle (isosteric) substitution, the Mb(H64V,V68A)-catalyzed styrene cyclopropanation reaction with 2f shows a significant reduction in yield (41% vs. 99%) and kcat (62 vs. 585 min−1), along with a 20-fold lower selectivity for formation of the (1S,2S) enantiomer compared to that with EDA (e.r. 11:1 (84% ee) vs. 200:1 (99.9% ee); Table 3, Entry 6 vs. 1). To illuminate the basis of this effect, a model of the Mb(H64V,V68A) active site with heme-bound [:CHCOSEt] carbene was built with the COSEt group located within the A68…I107 region (same as the ethyl ester group in 2(+)), followed by optimization (Figure S2A). Based on the computed transition state energies for the four stereoisomers in Table S3, the predicted de and ee values for this reaction are 99.9% and 85%, respectively, which are in excellent agreement with the experimental data (99% de and 84% ee). While the optimized TS1S,2S structure (Figure S2B) was found to display some basic features of TS1S,2S obtained for the reaction with EDA, such as the π-π interaction between styrene and Phe43 and attractive O…H interactions between carbene and active site residues, only two attractive O(carbene)…H or S(carbene)…H interactions are established with Phe43 (2.7 Å) and styrene (2.9 Å) respectively, as opposed to four in the TS1S,2S structure for the reaction with EDA (Table S2). Furthermore, the contact distances with the [:CHCOSEt] group are longer than those with the EDA-derived carbene. These relatively less favorable interactions may contribute to the increased computed ΔGǂ for TS1S,2S in the cyclopropanation reaction with [:CHCOSEt] compared to that with [:CHCO2Et]. This difference is consistent with the experimentally observed lower catalytic activity and rate of the Mb(H64A,V68A)-catalyzed cyclopropanation reaction with 2f compared to those with EDA (Table S4). In contrast with this reduced reactivity for TS1S,2S due to the O→S substitution, the formation of TS1R,2R (Figure S2C) actually becomes more favorable (Tables 2 and S3) as a result of three attractive O(carbene)…H interactions with styrene (2.6/2.6 Å) and Ile107 (2.6 Å) compared to only two of such interactions found for TS1R,2R in the reaction with EDA. Altogether, these effects reduce the difference between the TS1S,2S and TS1R,2R barrier leading to reduced enantioselectivity for the (1S,2S) product, as observed experimentally. Overall, these results corroborate the involvement of the ester group in influencing the stereochemical outcome of the reaction as revealed by the computational analyses for EDA and highlight the importance of weak interactions between the carbene and the surrounding protein environment in controlling reactivity and selectivity in the cyclopropanation reaction.
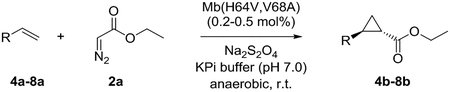
Expanding the substrate scope of Mb(H64V,V68A)
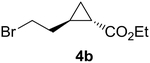
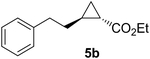
Previous studies showed Mb(H64V,V68A) catalyzes the cyclopropanation of a broad range of vinyl arenes, but lacks activity on olefin substrates such as 1-octene and cyclohexene.8 These findings previously suggested that the substrate scope of this biocatalyst may be limited to electronrich aryl-substituted olefins, owing to their higher reactivity toward the electrophilic heme-carbene intermediate mediating these reactions.8, 35–37 However, in view of the now revealed role of protein-mediated π−π interactions in favoring the cyclopropanation reaction with styrene, we hypothesized that unactivated olefins bearing an aromatic substituent could also constitute viable substrates for this biocatalyst. In agreement with our hypothesis, a Mb(H64V,V68A)-catalyzed reaction in the presence of 4-phenyl-but-1-ene (5a) and EDA showed that this substrate can be accepted by the Mb variant, yielding the corresponding cyclopropanation product 5a with high trans-selectivity (95% de; 75% ee; Table 4, Entry 2). In contrast, no activity was detected with 4-bromo-but-1-ene (4a). Since the C=C double bonds in 4a and 5a share similar electronic properties, the activity observed with 5a can be directly ascribed to the beneficial effect of the aromatic substituent in favoring the Mb(H64V,V68A)-mediated cyclopropanation of the unactivated olefin. Building upon these findings, the substrate scope of the carbene transferase could be further extended to the transformation of other non-styrenyl olefin substrates such as 6a-8a, enabling the synthesis of the corresponding cyclopropanation products 6b-8b in modest to good yields (18–65%), and good to excellent diastereo- and enantioselectivity (80–98% de; 74–99% ee) (Table 4, Entries 3–5). Altogether, these results demonstrate how the mechanistic insight gained from the present studies have enabled expansion of the substrate profile of Mb(H64V,V68A) as a cyclopropanation biocatalyst.
Table 4.
Activity and selectivity for Mb(H64V,V68A)-catalyzed cyclopropanation of non-styrenyl olefins with EDA.a
| Entry | Olefin | Product | Yieldb | TON | % de | % ee |
|---|---|---|---|---|---|---|
| 1 |  |
 |
0% | 0 | - | - |
| 2 | 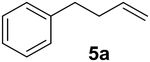 |
 |
13% | 25 | 95 | 75 |
| 3 | 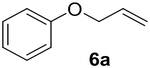 |
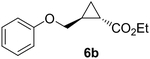 |
18% | 32 | 95 | 86 |
| 4c |  |
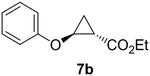 |
65% | 325 | 98 | 99 |
| 5c |  |
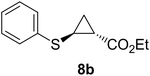 |
40% | 200 | 80 | 74 |
Reaction conditions: 20 mM olefin, 10 mM EDA, 10 mM Na2S2O4, 50 μM purified Mb variant in phosphate buffer (50 mM, pH 7.0), under anaerobic conditions, room temperature, 16 hours.
GC yield.
Using 20 μM catalyst.
Conclusions
To conclude, using crystallographic, computational, and reactivity/mutagenesis analyses, we have gained first-time insights into the origin and structural determinants responsible for chiral induction and high stereocontrol in olefin cyclopropanation catalyzed by an engineered metalloprotein. These studies show that the excellent diastereo- and enantioselectivity of Mb(H64V,V68A)-catalyzed cyclopropanation is granted through protein-mediated control on both (i) the conformation of the heme-bound carbene; and (ii) the geometry and spatial orientation of the transition state, via a combination of steric effects (i.e., shape complementarity) as well as non-covalent vdW and π−π interactions between first-sphere active site residues, the heme-bound carbene, and the olefin substrate. Importantly, the mechanistic scenario developed here is able to reproduce experimentally observed enantioselectivities and can explain structure-reactivity trends with different protein variants and diazo reagents. In addition, the mechanistic insights gained through these studies could be leveraged to (i) expand the substrate scope of the Mb(H64V,V68A) carbene transferase to include unactivated olefins (5a, 6a) and (ii) design an alternative Mb-based biocatalyst, Mb(H64V,V68G), with higher reactivity toward a bulkier diazo reagent than EDA (tert-butyl α-diazoacetate). The present findings enhance our understanding of enzyme-catalyzed asymmetric cyclopropanation and are expected to have important implications toward guiding the future development of biological catalysts for this important class of C—C bond forming reactions.
EXPERIMENTAL DETAILS
General Information.
All chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, ACS Scientific, Alfa Aesar, J.T. Baker) and used without any further purification, unless otherwise stated. All reactions were carried out under argon pressure in oven-dried glassware with magnetic stirring using standard gas-tight syringes, cannulae and septa. 1H, 13C and 19F NMR spectra were measured on a Bruker DPX-400 instrument (operating at 400 MHz for 1H, 100 MHz for 13C, and 375 MHz for 19F) or a Bruker DPX-500 instrument (operating at 500 MHz for 1H and 125 MHz for 13C). Tetramethylsilane (TMS) was used as the internal standard (0 ppm) for 1H NMR spectra, CDCl3 was used as the internal standard (77.0 ppm) for 13C NMR spectra, and trifluorotoluene served as the internal standard (0 ppm) for 19F NMR spectra. Flash column chromatography purification was carried out using AMD Silica Gel 60 Å 230–400 mesh. Thin Layer Chromatography (TLC) was carried out using Merck Millipore TLC silica gel 60 F254 glass plates.
Molecular Cloning.
pET22b(+) (Novagen) was used as the recipient plasmid vector for expression of all of the myoglobin variants. In this construct, the gene encoding for sperm whale myoglobin carrying a neutral D122N mutation60 is C-terminally fused to a polyhistidine tag and it is under the control of an IPTG-inducible T7 promoter. The preparation of plasmids encoding for Mb(H64V,V68A) and Mb(H64V,V68G) was described previously and that encoding for Mb(L29A,H64V,V68A) was prepared using a similar procedure8, 9 and the mutagenizing primers in Table S5. A pET22-based plasmid containing the gene encoding for Mb(H64V,V68A) without the poly-histidine tag was prepared in a similar manner using Myo_XbaI_for and XhoI_NoHisTag_rev primers.
Protein Expression and Purification.
Engineered Mb variants were expressed in E. coli C41(DE3) cells as described previously.9 Briefly, cells were grown in TB medium (ampicillin, 100 mg L−1) at 37 °C (200 rpm) until OD600 reached 1.0–1.2. Cells were then induced with 0.25 mM β-d-1-thiogalactopyranoside (IPTG) and 0.3 mM δ-aminolevulinic acid (ALA). After induction, cultures were shaken at 180 rpm and 27 °C and harvested after 18–20 hours by centrifugation at 4000 rpm at 4 °C. After cell lysis by sonication, the cell lysate was loaded on a Ni-NTA column equilibrated with Ni-NTA Lysis Buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH 8.0). After washing with 50 mL Ni-NTA Lysis Buffer and 50 mL of Ni-NTA WashBuffer (50 mM KPi, 250 mM, NaCl, 20 mM imidazole, pH 8.0), the protein was eluted with Ni-NTA Elution Buffer (50 mM KPi, 250 mM, NaCl, 250 mM histidine, pH 7.0). The protein solution was buffer exchanged against potassium phosphate buffer (50 mM, pH 7.0) and the protein concentration was determined using ε410 = 156 mM−1 cm−1 as the extinction coefficient.
Protein Expression and Purification for Crystallization.
Freshly transformed BL21(DE3) cells expressing HisTag-free Mb(H64V,V68A) were used to inoculate 5 mL LB medium containing ampicillin (100 mg/L), followed by growth overnight at 37°C with shaking. The overnight culture was used to inoculate 1L of Terrific Broth medium containing ampicillin and supplemented with 0.3 mM delta-aminolevulinic acid, followed by incubation at 37°C with shaking (180 rpm). At OD600 = 0.6–0.8, cells were induced with 0.5 mM IPTG and incubated at 27°C with shaking (180 rpm) for 20–24 hours. Cells were harvested by centrifugation (4,000 rpm, 4°C, 20 minutes) and then resuspended in 20 mL Tris HCl buffer (20 mM, pH 8.0) containing 1 mM EDTA. After sonication and clarification of the lysate via centrifugation (14000 rpm, 4°C, 20 minutes), the protein was purified using a three-step purification process according to a modified version of a reported procedure.61 An initial salting out purification was performed by slowly adding solid ammonium sulfate to the clarified lysate in an Erlenmeyer flask with stirring on ice. At 65% m/v saturation, the solution was incubated for one hour in ice, followed by centrifugation (14000 rpm, 4°C, 45 minutes). The supernatant was transferred to a clean Erlenmeyer flask and solid ammonium sulfate was slowly added to the supernatant to reach 95% m/v saturation with constant stirring on ice, followed by incubation for one hour in ice and centrifugation (14000 rpm, 4°C, 45 minutes). The red-colored pellet containing precipitated Mb(H64V,V68A) was resuspended in a minimal volume of Tris HCl buffer (pH 8.0) containing 1 mM EDTA. The resuspended pellet was then dialyzed against 20 mM Tris HCl buffer (pH 8.0) and 1 mM EDTA using a 5 kDa cut-off membrane overnight. The protein was then purified by gel filtration chromatography using a Superdex 75 10/300 GL column (GE Healthcare) and isocratic elution with 20 mM Tris HCl buffer (pH 8.4) containing 1 mM EDTA, following absorbance at 409 nm. The protein was then further purified via ion-exchange chromatography using a DEAE Sepharose column (GE Healthcare) under isocratic elution with Tris HCl (pH 8.4) containing 1 mM EDTA. Purified Mb(H64V,V68A) was concentrated using a 10 kDa cut-off centrifugal filter to a final concentration of 4 mM and its purity was confirmed by SDS-PAGE.
Crystallization, Data collection, Processing, and Refinement.
A crystal of the ferric Mb(H64V,V68A) complexed with water was grown at room temperature using the hanging-drop vapor-diffusion method by mixing 1 μL of reservoir buffer (2 M ammonium sulfate, 0.2 M Tris pH 8.6) with 1 μL of protein in buffer (20 mM Tris pH 8.0, 1 mM EDTA). The crystal was cryoprotected by soaking it in a drop containing reservoir buffer supplemented with 25% sucrose for 5 minutes prior to being flash-cooled in liquid nitrogen. Data were collected at Cornell High Energy Synchrotron Light Source (CHESS) F1 station using a Pilatus3 6M detector. Data was integrated using iMosflm62 and scaled and merged using Scala63. Rigid body fitting of the crystallographic structure 1JW842 was used for initial molecular replacement phase calculations (PHENIX). This was followed by further rounds of refinement using PHENIX64 and COOT65. Data processing and refinement statistics are presented in Table S1. Structure coordinates were deposited with the Protein Data Bank under the accession number 6M8F. For comparative purposes (Fig. 1), a structure of wild type sperm whale Mb (ferrous CO-complex) crystallized in the same space group (P6) was chosen in consideration of subtle conformational differences of Mb observed under different crystalline environments.42 This structure presents two alternate orientations of the distal histidine residue (His64), both of which are shown in Figure 1.
Synthetic Procedures.
Detailed procedures for the synthesis of 2b-2f, 3b-3e, and 5b-8b are provided in the Supporting Information.
Cyclopropanation Reactions.
Biocatalytic reactions were carried out at a 400 μL scale using 20 μM purified Mb variant, 10 mM styrene, 20 mM EDA, and 10 mM sodium dithionite. In a typical procedure, a solution containing sodium dithionite (100 mM stock solution) in potassium phosphate buffer (KPi 50 mM, pH 7.0) was degassed by bubbling argon into the mixture for 3 min. in a sealed glass crimp vial. A buffered solution containing myoglobin was degassed in a similar manner in a separate glass crimp vial. The two solutions were then mixed together via cannula. Reactions were initiated by addition of 5 μL of styrene (from a 0.8 M stock solution in ethanol), followed by the addition of 10 μL of EDA (from a 0.8 M stock solution in ethanol) with a syringe, and the reaction mixture was stirred for 16 hours at room temperature, under positive argon pressure.
Product Analysis.
The reactions were analyzed by adding 20 μL of internal standard (benzodioxole, 100 mM in methanol) to the reaction mixture, followed by extraction with 400 μL of dichloromethane (DCM) and analyzed by GC-FID (see Analytical Methods section in the Supporting Information). Calibration curves of the different cyclopropane products were constructed using synthetically produced authentic standards (see Synthetic Procedures in the Supporting Information). All measurements were performed at least in duplicate. For each experiment, negative control samples containing either no enzyme or no reductant were included. For stereoselectivity determination, the samples were analyzed by chiral GC-FID or chiral SFC as described in the Supporting Information.
Michaelis-Menten Kinetic Analyses.
Kinetic experiments were carried out using 1–10 μM Mb(H64V,V68A), 10 mM sodium dithionite, 20 mM styrene, and varying concentrations of diazo compound (0.5–80 mM) in KPi buffer (50 mM, pH 7.0) at room temperature and in open vessels (aerobic conditions). Initial rates were measured based on the amount of cyclopropanation product formed after 30 seconds as determined by GC-FID analysis. The reactions were quenched with 100 μL 2 N HCl and immediately extracted with 400 μL of dichloromethane, followed by the addition of 20 μL of benzodioxole as internal standard. The kinetic parameters Vmax, kcat, and KM were obtained by fitting the resulting plot of initial velocity (V) vs. diazo compound concentration to the Michaelis-Menten equation using SigmaPlot software. Experiments were performed in triplicate and representative curves are shown in Figure S5.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the U.S. National Institute of Health grant GM098628 (R.F) and GM124847 (N.A.) and the U.S. National Science Foundation grant CHE-1300912 (Y.Z). Diffraction data were collected at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the NSF & NIH/NIGMS via NSF award DMR-1332208, and the MacCHESS resource is supported by NIH/NIGMS award GM-103485. A. T. and E. J. M. acknowledge support from the Ford Foundation Graduate Fellowship Program and NIH Graduate Training Grant T32GM118283, respectively. The authors are grateful to Dr. Phil Jeffrey, Susannah Shoemaker, and Syed Muhammad Saad Imran for assistance with crystallization and data collection.
Footnotes
Supporting Information
Supporting information includes crystallographic data, synthetic procedures, analytical methods, compound characterization data, cartesian coordinates for DFT Models, and Supplementary Tables and Figures.
REFERENCES
- (1).Schwizer F, Okamoto Y, Heinisch T, Gu YF, Pellizzoni MM, Lebrun V, Reuter R, Kohler V, Lewis JC, and Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies, Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (2).Brandenberg OF, Fasan R, and Arnold FH Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions, Curr. Opin. Biotech 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hyster TK, and Ward TR Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions, Angew. Chem. Int. Ed 2016, 55, 7344–7357. [DOI] [PubMed] [Google Scholar]
- (4).Gober JG, and Brustad EM Non-natural carbenoid and nitrenoid insertion reactions catalyzed by heme proteins, Current Opinion in Chemical Biology 2016, 35, 124–132. [DOI] [PubMed] [Google Scholar]
- (5).Renata H, Wang ZJ, and Arnold FH Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution, Angew. Chem. Int. Ed 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Coelho PS, Brustad EM, Kannan A, and Arnold FH Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes, Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- (7).Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, and Brustad EM A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo, Nat. Chem. Biol 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bordeaux M, Tyagi V, and Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts, Angew. Chem. Int. Ed 2015, 54, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bajaj P, Sreenilayam G, Tyagi V, and Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity, Angew. Chem. Int. Ed 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, and Brustad EM Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation, Chembiochem 2016, 17, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gober JG, Ghodge SV, Bogart JW, Wever WJ, Watkins RR, Brustad EM, and Bowers AA P450-Mediated Non-natural Cyclopropanation of Dehydroalanine-Containing Thiopeptides, ACS Chem. Biol 2017, 2, 1726–1731. [DOI] [PubMed] [Google Scholar]
- (12).Ohora K, Meichin H, Zhao LM, Wolf MW, Nakayama A, Hasegawa J, Lehnert N, and Hayashi T Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis due to Rapid Formation of the Carbene Species, J. Am. Chem. Soc 2017, 139, 17265–17268. [DOI] [PubMed] [Google Scholar]
- (13).Knight AM, Kan SBJ, Lewis RD, Brandenberg OF, Chen K, and Arnold FH Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes, ACS Central Sci. 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Moore EJ, Steck V, Bajaj P, and Fasan R Chemoselective Cyclopropanation over Carbene Y-H Insertion Catalyzed by an Engineered Carbene Transferase, J. Org. Chem 2018, 83, 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Brandenberg OF, Prier CK, Chen K, Knight AM, Wu Z, and Arnold FH Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes, ACS Catal. 2018, 8, 2629–2634. [Google Scholar]
- (16).Wang ZJ, Peck NE, Renata H, and Arnold FH Cytochrome P450-catalyzed insertion of carbenoids into N-H bonds, Chem. Sci 2014, 5, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sreenilayam G, and Fasan R Myoglobin-catalyzed intermolecular carbene N-H insertion with arylamine substrates, Chem. Commun 2015, 51, 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tyagi V, Bonn RB, and Fasan R Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalysts, Chem. Sci 2015, 6, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kan SBJ, Lewis RD, Chen K, and Arnold FH Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life, Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wolf MW, Vargas DA, and Lehnert N Engineering of RuMb: Toward a Green Catalyst for Carbene Insertion Reactions, Inorg. Chem 2017, 56, 5623–5635. [DOI] [PubMed] [Google Scholar]
- (21).Dydio P, Key HM, Nazarenko A, Rha JY, Seyedkazemi V, Clark DS, and Hartwig JF An artificial metalloenzyme with the kinetics of native enzymes, Science 2016, 354, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sreenilayam G, Moore EJ, Steck V, and Fasan R Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts, Adv. Synth. Cat 2017, 359, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vargas DA, Tinoco A, Tyagi V, and Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles, Angew. Chem. Int. Ed 2018, 57, 9911–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tyagi V, and Fasan R Myoglobin-Catalyzed Olefination of Aldehydes, Angew. Chem. Int. Ed 2016, 55, 2512–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Weissenborn MJ, Low SA, Borlinghaus N, Kuhn M, Kummer S, Rami F, Plietker B, and Hauer B Enzyme-Catalyzed Carbonyl Olefination by the E. coli Protein YfeX in the Absence of Phosphines, Chemcatchem 2016, 8, 1636–1640. [Google Scholar]
- (26).Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, and Fasan R Biocatalytic Synthesis of Allylic and Allenyl Sulfides through a Myoglobin-Catalyzed Doyle-Kirmse Reaction, Angew. Chem. Int. Ed 2016, 55, 13562–13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chen K, Huang XY, Kan SBJ, Zhang RK, and Arnold FH Enzymatic construction of highly strained carbocycles, Science 2018, 360, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lin CI, McCarty RM, and Liu HW The Enzymology of Organic Transformations: A Survey of Name Reactions in Biological Systems, Angew. Chem. Int. Ed 2017, 56, 3446–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Reetz MT Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future, J. Am. Chem. Soc 2013, 135, 12480–12496. [DOI] [PubMed] [Google Scholar]
- (30).Honig M, Sondermann P, Turner NJ, and Carreira EM Enantioselective Chemo- and Biocatalysis: Partners in Retrosynthesis, Angew. Chem. Int. Ed 2017, 56, 8942–8973. [DOI] [PubMed] [Google Scholar]
- (31).Talele TT The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules, J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- (32).Tinoco A, Steck V, Tyagi V, and Fasan R Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene, J. Am. Chem. Soc 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Chandgude AL, and Fasan R Highly Diastereo- and Enantioselective Synthesis of Nitrile-Substituted Cyclopropanes by Myoglobin-Mediated Carbene Transfer Catalysis, Angew. Chem. Int. Ed 2018, 57, 15852–15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Moore EJ, Zorine D, Hansen WA, Khare SD, and Fasan R Enzyme stabilization via computationally guided protein stapling, Proc. Natl. Acad. Sci. USA 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Khade RL, and Zhang Y Catalytic and Biocatalytic Iron Porphyrin Carbene Formation: Effects of Binding Mode, Carbene Substituent, Porphyrin Substituent, and Protein Axial Ligand, J. Am. Chem. Soc 2015, 137, 7560–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wei Y, Tinoco A, Steck V, Fasan R, and Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution, J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Lewis RD, Garcia-Borras M, Chalkley MJ, Buller AR, Houk KN, Kan SBJ, and Arnold FH Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase, Proc. Natl. Acad. Sci. USA 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Dzik WI, Xu X, Zhang XP, Reek JNH, and de Bruin B ‘Carbene Radicals’ in CoII(por)-Catalyzed Olefin Cyclopropanation, J. Am. Chem. Soc 2010, 132, 10891–10902. [DOI] [PubMed] [Google Scholar]
- (39).Lu H, Dzik WI, Xu X, Wojtas L, de Bruin B, and Zhang XP Experimental Evidence for Cobalt(III)-Carbene Radicals: Key Intermediates in Cobalt(II)-Based Metalloradical Cyclopropanation, J. Am. Chem. Soc 2011, 133, 8518–8521. [DOI] [PubMed] [Google Scholar]
- (40).Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, and Hilvert D Capture and characterization of a reactive haem-carbenoid complex in an artificial metalloenzyme, Nature Catal. 2018, 1, 578–584. [Google Scholar]
- (41).Su H, Ma G, and Liu Y Theoretical Insights into the Mechanism and Stereoselectivity of Olefin Cyclopropanation Catalyzed by Two Engineered Cytochrome P450 Enzymes, Inorg. Chem 2018, 57, 11738–11745. [DOI] [PubMed] [Google Scholar]
- (42).Kondrashov DA, Zhang W, Aranda R. t., Stec B, and Phillips GN Jr. Sampling of the native conformational ensemble of myoglobin via structures in different crystalline environments, Proteins 2008, 70, 353–362. [DOI] [PubMed] [Google Scholar]
- (43).Holm L, and Sander C Protein-Structure Comparison by Alignment of Distance Matrices, J. Mol. Biol 1993, 233, 123–138. [DOI] [PubMed] [Google Scholar]
- (44).Volkamer A, Kuhn D, Rippmann F, and Rarey M DoGSiteScorer: a web server for automatic binding site prediction, analysis and druggability assessment, Bioinformatics 2012, 28, 2074–2075. [DOI] [PubMed] [Google Scholar]
- (45).Zhang K, Shafer BM, Demars MD 2nd, Stern HA, and Fasan R Controlled oxidation of remote sp3 C-H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity, J. Am. Chem. Soc 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yang L, Ling Y, and Zhang Y HNO binding in a heme protein: structures, spectroscopic properties, and stabilities, J. Am. Chem. Soc 2011, 133, 13814–13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Span I, Wang K, Eisenreich W, Bacher A, Zhang Y, Oldfield E, and Groll M Insights into the binding of pyridines to the iron-sulfur enzyme IspH, J. Am. Chem. Soc 2014, 136, 7926–7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Chai JD, and Head-Gordon M Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections, Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (49).Yang K, Zheng JJ, Zhao Y, and Truhlar DG Tests of the RPBE, revPBE, tau-HCTHhyb, omega B97X-D, and MOHLYP density functional approximations and 29 others against representative databases for diverse bond energies and barrier heights in catalysis, J. Chem. Phys 2010, 132, 164117. [DOI] [PubMed] [Google Scholar]
- (50).Khade RL, Fan WC, Ling Y, Yang L, Oldfield E, and Zhang Y Iron Porphyrin Carbenes as Catalytic Intermediates: Structures, Mossbauer and NMR Spectroscopic Properties, and Bonding, Angew. Chem. Int. Ed 2014, 53, 7574–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Khade RL, and Zhang Y C-H Insertions by Iron Porphyrin Carbene: Basic Mechanism and Origin of Substrate Selectivity, Chem. Eur. J 2017, 23, 17654–17658. [DOI] [PubMed] [Google Scholar]
- (52).Hay PJ, and Wadt WR Abinitio Effective Core Potentials for Molecular Calculations - Potentials for the Transition-Metal Atoms Sc to Hg, J. Chem. Phys 1985, 82, 270–283. [Google Scholar]
- (53).Li Y, Huang JS, Zhou ZY, Che CM, and You XZ Remarkably stable iron porphyrins bearing nonheteroatom-stabilized carbene or (Alkoxycarbonyl) carbenes: Isolation, X-ray crystal structures, and carbon atom transfer reactions with hydrocarbons, J. Am. Chem. Soc 2002, 124, 13185–13193. [DOI] [PubMed] [Google Scholar]
- (54).Liu YL, Xu W, Zhang J, Fuller W, Schulz CE, and Li JF Electronic Configuration and Ligand Nature of Five-Coordinate Iron Porphyrin Carbene Complexes: An Experimental Study, J. Am. Chem. Soc 2017, 139, 5023–5026. [DOI] [PubMed] [Google Scholar]
- (55).Bondi A Van Der Waals Volumes + Radii, J. Phys. Chem 1964, 68, 441–442. [Google Scholar]
- (56).Rowland RS, and Taylor R Intermolecular nonbonded contact distances in organic crystal structures: Comparison with distances expected from van der Waals radii, J. Phys. Chem 1996, 100, 7384–7391. [Google Scholar]
- (57).Wolf JR, Hamaker CG, Djukic JP, Kodadek T, and Woo LK Shape and Stereoselective Cyclopropanation of Alkenes Catalyzed by Iron Porphyrins, J. Am. Chem. Soc 1995, 117, 9194–9199. [Google Scholar]
- (58).Lai TS, Chan FY, So PK, Ma DL, Wong KY, and Che CM Alkene cyclopropanation catalyzed by Halterman iron porphyrin: participation of organic bases as axial ligands, Dalton T. 2006, 4845–4851. [DOI] [PubMed] [Google Scholar]
- (59).Carminati DM, Intrieri D, Caselli A, Le Gac S, Boitrel B, Toma L, Legnani L, and Gallo E Designing ‘Totem’ C2-Symmetrical Iron Porphyrin Catalysts for Stereoselective Cyclopropanations, Chemistry 2016, 22, 13599–135612. [DOI] [PubMed] [Google Scholar]
- (60).Liong EC, Dou Y, Scott EE, Olson JS, and Phillips GN Waterproofing the heme pocket - Role of proximal amino acid side chains in preventing hemin loss from myoglobin, J. Biol. Chem 2001, 276, 9093–9100. [DOI] [PubMed] [Google Scholar]
- (61).Springer BA, and Sligar SG High-Level Expression of Sperm Whale Myoglobin in Escherichia-Coli, Proc. Natl. Acad. Sci. USA 1987, 84, 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Powell HR, Battye TGG, Kontogiannis L, Johnson O, and Leslie AGW Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm, Nat. Protoc 2017, 12, 1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Evans P Scaling and assessment of data quality, Acta Crystallogr. D Biol. Crystallogr 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (64).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. D Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Emsley P, and Cowtan K Coot: model-building tools for molecular graphics, Acta Crystallogr. D Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.