

Abstract
Objectives:
Inflammation is a causal risk factor for cardiovascular disease (CVD). Group IIA secretory phospholipase A2 (sPLA2-IIA) plays an integral role in regulating vascular inflammation. While studies investigated sPLA2-IIA in secondary prevention, we prospectively evaluated sPLA2-IIA mass and genetic variants with CVD events in a primary prevention population with chronic inflammation.
Approach and Results:
The JUPITER trial (NCT00239681) randomized participants with LDLc <130 mg/dL and hsCRP ≥2 mg/L to high-intensity rosuvastatin vs placebo. Baseline and 1-year plasma sPLA2-IIA mass was measured (N=11,269 baseline; N=9,620 1-year). We also identified genetic variants influencing sPLA2-IIA using genome-wide association and examined them with CVD. 313 incident CVD events occurred during follow-up. Baseline sPLA2-IIA mass (median, 25th-75th percentile: 3.81, 2.49-6.03 ng/ml) was associated with increased risk of CVD: risk-factor adjusted HR (95% CI; p-value) per SD increment: 1.22 (1.08-1.38; p=0.002). This remained significant (1.18, 1.04-1.35; p=0.01) after incrementally adjusting for hsCRP. Similar estimates were observed in rosuvastatin and placebo groups (p-treatment interaction>0.05). The rs11573156C variant in PLA2G2A (encoding sPLA2-IIA) had the strongest effect on sPLA2-II: median (25th-75th percentile, ng/mL) for CC and GG genotypes: 2.79 (1.97-4.01) and 7.38 (5.38-10.19), respectively; and had non-significant trend for higher CVD risk (HR: 1.11, 95% CI 0.89-1.38, p=0.34).
Conclusions:
In the JUPITER population recruited on chronic inflammation, sPLA2-IIA mass was associated with CVD risk relating to vascular inflammation not fully reflected by hsCRP. Additional studies, including larger functional genetic and clinical studies, are needed to determine whether sPLA2-IIA may be a potential pharmacological target for primary prevention of CVD.
Keywords: Cardiovascular disease, statins, secretory phospholipase, prevention
Graphical Abstract
INTRODUCTION
A large body of evidence supports inflammation as a central causal risk factor in the progression of atherosclerotic cardiovascular disease (CVD).1 Most recently, a large randomized clinical trial of pharmacologic inhibition of the proinflammatory cytokine interleukin-1 beta by the monoclonal antibody canakinumab demonstrated that reducing systemic inflammation could reduce the risk of major adverse cardiovascular events including cardiovascular mortality.2 Importantly, the magnitude of trial participants’ reduction in the inflammatory acute phase protein high-sensitivity C-reactive protein (hsCRP) identified the group who experienced a reduction in CVD events on study drug.3 In light of these findings, there has been recent renewed interest in identifying pharmacologic targets for inflammation modification to prevent CVD events.
Secretory phospholipases A2 are members of the conserved super family of phospholipase A2 enzymes that catalyze the hydrolysis of the sn-2 fatty acyl ester bond of glycerophospholipids which are present in lipoproteins and membranes, liberating precursors of various pro-inflammatory cytokines such as bioactive free fatty acid and lysophospholipid. 4, 5 Several isoforms have been implicated in atherogenesis through pro-inflammatory roles, 4-6 including group IIA secretory phospholipase A2 (sPLA2-IIA), the most abundant secretory phospholipase A2 isoform. 5, 7 Experimental treatment with sPLA2-IIA leads to hydrolysis of lipoproteins, which renders low density lipoprotein (LDL) more susceptible to oxidative modification and increases its affinity for extracellular matrices of arterial intima, leading to the progressive formation of foam cells. 8-12 Genetically humanized mice that constitutively express sPLA2-IIA have also been shown to develop accelerated atherosclerosis, 13, 14 and human atherosclerotic lesions exhibit increased expression of sPLA2-IIA. 15, 16 Furthermore, epidemiologic studies have shown plasma mass of sPLA2-IIA to be increased in unstable angina patients, 17 and to be associated with recurrent cardiovascular events in patients with stable coronary heart disease 18, 19 and acute coronary syndromes (ACS). 17, 20, 21
While most studies to date have focused on examining associations between sPLA2-IIA and outcomes among patients with established CVD, the relation of sPLA2-IIA with incident CVD in a primary prevention setting has not been adequately evaluated. Despite the role of sPLA2-IIA in rendering LDL atherogenic, 11, 12 its association with CVD in statin-treated individuals has only been investigated in the setting of ACS and not in primary prevention. 22 To address the knowledge gap, we measured baseline and on-statin treatment levels of sPLA2-IIA in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial to examine: 1) the effect of high-intensity rosuvastatin treatment on sPLA2-IIA levels; 2) the association of sPLA2-IIA with incident CVD; and 3) the efficacy of rosuvastatin therapy in JUPITER by levels of sPLA2-IIA. Moreover, in order to evaluate whether an association of sPLA2-IIA with incident CVD events may be causal, we also performed a genome wide association study (GWAS) to identify genetic variants that determine sPLA2-IIA levels, and then in exploratory analyses assessed the effect of these variants on incident CVD events.
MATERIAL AND METHODS
Deidentified data that support the findings of this study are available from the corresponding author upon reasonable request from qualified researchers trained in human subject confidentiality protocols.
Study Population
JUPITER (NCT00239681) was a double-blind placebo controlled trial that evaluated rosuvastatin 20 mg daily versus placebo in the primary prevention of first major CVD events among 17 802 apparently healthy men ≥50 years and women ≥60 years with LDL-cholesterol <130mg/dl but who were at increased risk of cardiovascular events on the basis of elevated high sensitivity C-reactive protein (hsCRP ≥2 mg/L).23 Key exclusion criteria for JUPITER included previous or current use of lipid lowering therapy, current use of postmenopausal hormonal therapy, diabetes mellitus, and inflammatory conditions such as severe arthritis, lupus or inflammatory bowel disease or treatment with immunosuppressive medications. The trial protocol invited (but did not require) study participants to provide a baseline blood sample before randomization and after one year. In the present study, we analyzed a total of 11 269 participants who provided sufficient blood sample at baseline to enable measurements of sPLA2-IIA levels; of these, 9 620 participants had sufficient blood sample at both baseline and at one year.
Laboratory Methods
sPLA2-IIA was measured at Quest Diagnostics Nichols Institute (San Juan Capistrano, CA) with a commercially available enzyme immunoassay (Cayman assay; Cayman Chemical Co. Ann Arbor MI) based on a double-antibody sandwich technique that is specific for sPLA2-IIA and does not cross react with Group I, IV, V or X phospholipase A2 enzymes nor other inflammatory mediators such as tumor necrosis factor, interleukin-1, or platelet activating factor. The limit of detection for this assay is 0.46 ng/ml and the mean coefficient of variation for duplicate inter assay runs were 8.75, 5.16, and 7.66% at concentrations of 2.24, 8.52, and 15.80 ng/ml respectively. Lipids, hsCRP, and glucose measurements were performed on fasting samples in a central laboratory as part of the main trial protocol.
Outcomes
The primary outcome of the current study is the trial primary endpoint defined as non-fatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, arterial revascularization, or cardiovascular death. We additionally performed a sensitivity analysis using an expanded endpoint of the primary endpoint and all-cause death, consistent with prior analyses in JUPITER.24, 25
Statistical Analysis
sPLA2-IIA Mass, Rosuvastatin Therapy, and Incident CVD Risk
Baseline characteristics were expressed as means (25th to 75th percentiles) or percentages. χ2 was used to compare levels of sPLA2-IIA by clinical subgroups and spearman correlations were used to quantify associations of sPLA2-IIA with clinical risk factors. The Wilcoxon signed rank test was used to compare baseline and one year sPLA2-IIA by randomization arm. Changes in sPLA2-IIA levels after one year of study treatment were expressed as percentages within each arm. The effect of rosuvastatin versus placebo on sPLA2-IIA levels was assessed by Wilcoxon rank sum test. For comparison, similar analyses were performed for hsCRP. Person-time of follow-up was assessed from the time of randomization to the first occurrence of a primary outcome component, date of death, or last study visit. Absolute event rates were calculated per 100 person-years. Hazard ratios (HRs) and 95% confidence intervals (CIs) for the association of baseline and on-treatment sPLA2-IIA mass with CVD outcomes were quantified using Cox proportional hazards regression according to quartiles of sPLA2-IIA and per standard deviation (SD). Cox regressions were based on three sequentially adjusted models: model 1 adjusted for age, sex, and randomized treatment assignment; model 2 additionally adjusted for race, smoking, family history of premature coronary disease, body mass index, systolic blood pressure, glucose, high density lipoprotein cholesterol, LDL-cholesterol, and natural log-transformed triglycerides; and model 3 included model 2 variables plus natural log-transformed hsCRP.
We performed stratified analyses according to randomization arm and tested for treatment interaction by including a cross-product term between sPLA2-IIA and randomization treatment. We performed additional analyses to examine if the association of sPLA2-IIA with CVD was modified by sex, age (≤65 or >65 years), smoking (yes or no), family history of coronary heart disease (yes or no), metabolic syndrome (yes or no), time to event (≤24 or >24 months), or fasting glucose (<100 or ≥100 mg/dl). Finally, we examined whether rosuvastatin efficacy in reducing the primary outcome was modified by varying levels of sPLA2-IIA (quartiles). All probability values were 2-tailed, with values <0.05 considered statistically significant. Statistical analyses were performed with SAS version 9.3 (SAS Institute).
Genetic Analyses
Genetic analyses were performed in a subset of participants in the present study who consented to genetic analysis, had sPLA2-II measurements at baseline and were of verified self-reported European ancestry as previously described.26 GWAS of SNPs associated with sPLA2-IIA mass was performed using genotyped SNPs with minor allele frequency frequencies >1% and, Hardy-Weinberg equilibrium P<10−6. Association statistics were obtained from inverse-quantile normalized residualized values of sPLA2-II adjusted for age, sex, region and measures of subpopulation stratification. Genome wide significance was set at the conventional threshold of α=5×10−8. Conditional analysis was performed using the genome-wide complex trait analysis tool27 to determine the number of non-redundant loci contributing to the sPLA2-II association. For the 69 SNPs reaching genome-wide significance (p<5×10−8) in association with sPLA2-II, we assessed the association with incident CVD in a Cox regression model adjusting for age, sex, randomization treatment assignment, region and measures of subpopulation stratification. Additionally, we assessed interaction with rosuvastatin treatment on the associations between the SNPs with incident CVD by adding a SNP-by-allocation interaction term to the model. Statistical analyses were performed using PLINK, GCTA, and R.
Genotyping
Genotyping was performed on each study participant for a total of 1 006 348 single-nucleotide polymorphisms (SNPs) using the Omni 1M Quad platform and processed using GenomeStudio software v1.6.2 (both Illumina, San Diego, CA) by the manufacturer. SNPs with poor clustering metrics for parameters such as AbrMean (intensity), cluster separation, Hardy-Weinberg Equilibrium, and call frequency, were visually inspected and either annotated, removed, or manually edited. Markers were retained for the final data if the updated clusters met quality standards and the genotyping was successful >98.5% of the samples. Multidimensional scaling procedures implemented in PLINK were used for verification of self-reported European ancestry. Sub-European population stratification was estimated using EIGENSTRAT. First-degree relatives were identified by state clustering in PLINK and excluded.
RESULTS
Baseline Characteristics
Baseline characteristics of study participants with measured sPLA2-IIA were similar to those of the entire JUPITER cohort except that there were more white participants in the present study (Supplemental Table I). The median (25th to 75th percentile) sPLA2-IIA was 3.81 (2.49-6.03) ng/ml, and was higher in women, blacks, and participants with the metabolic syndrome (Table 1). sPLA2-IIA correlated positively with hsCRP (baseline r=0.31; on-treatment r=0.38, p<0.0001 for both), and inversely albeit more weakly with lipoprotein associated phospholipase A2 (Lp-PLA2) activity (baseline r=−0.17; on-treatment r=−0.02, p<0.05 for both); and with other patient characteristics and biomarkers (r=−0.16 to 0.21) (Supplemental Table II).
Table 1.
Baseline sPLA2-IIA mass levels according to clinical subgroups
| N | Median (25th – 75th) |
P-value | ||
|---|---|---|---|---|
| Sex | Men | 7082 | 3.18 (2.15-4.77) | <0.0001 |
| Women | 4187 | 5.40 (3.57-8.29) | ||
| Treatment group | Placebo | 5645 | 3.79 (2.53-5.95) | 0.45 |
| Rosuvastatin | 5624 | 3.83 (2.46-6.10) | ||
| Race or ethnic group | <0.0001 | |||
| White | 9026 | 3.64 (2.41-5.59) | ||
| Black | 969 | 6.58 (3.69-11.9) | ||
| Asian | 171 | 3.04 (2.14-4.80) | ||
| Hispanic | 999 | 4.33 (2.70-6.73) | ||
| Other or unknown | 102 | 4.73 (2.85-7.64) | ||
| Current smoker | No | 9504 | 3.81 (2.48-6.03) | 0.92 |
| Yes | 1760 | 3.82 (2.53-5.98) | ||
| Family history of premature coronary disease | No | 9829 | 3.82 (2.50-6.03) | 0.73 |
| Yes | 1398 | 3.81 (2.47-6.02) | ||
| Metabolic syndrome | No | 6755 | 3.70 (2.39-5.94) | <0.0001 |
| Yes | 4351 | 4.00 (2.66-6.18) | ||
| Aspirin use | No | 9318 | 3.83 (2.50-6.07) | 0.13 |
| Yes | 1951 | 3.76 (2.47-5.82) |
P-values are obtained from X2 tests. sPLA2-IIA indicates group IIA secretory phospholipases A2
Effect of Rosuvastatin Therapy on sPLA2-IIA Mass
sPLA2-IIA decreased by a median (25th to 75th percentile) of −1.3 (−2.4 to −0.5) ng/ml and −1.6 (−2.9 to −0.7) ng/ml in the placebo and rosuvastatin groups, respectively, comparing one year with baseline values (P <0.0001 for one year versus baseline values in either treatment group, Supplemental Table 3), corresponding to a median percent change of −38% (−56 to −15%) and −46% (−60 to −26%) respectively (P comparing changes between treatment groups <0.0001; Figure 1 and Supplemental Table III). The corresponding percent changes in hsCRP were −20% (−50 to 21%) and −48% (−69 to −16%) respectively.
Figure 1.

Baseline to 1-year median percent change in sPLA2-IIA according to randomized treatment. Values obtained from individuals with both baseline and 1 year measurements (n=9 620). hsCRP indicates high-sensitivity C-reactive protein.*P values from the Wilcoxon signed rank test comparing baseline and year 1 values were statistically significant (P <0.0001) ¶P values from the Wilcoxon rank sum test comparing the change among the rosuvastatin group with the change among the placebo group were <0.0001.
sPLA2-IIA Mass and Incident CVD
Among the 11 269 participants with baseline measurement of sPLA2-IIA, 313 cases of the primary outcome were confirmed over a median follow-up of 1.9 (maximum, 5.0) years; 65% (201) and 35% (109) of these cases occurred in the placebo and rosuvastatin groups, respectively, similar to the proportions observed in the entire JUPITER trial. 23 In the multivariable adjusted model 2, HRs (95% CIs) for the primary endpoint for quartiles 2 to 4 of baseline sPLA2-IIA levels compared to quartile 1 as the reference were, 0.98 (0.70-1.37), 1.18 (0.85-1.64), 1.54 (1.10-2.15) (Plinear trend = 0.006); additional adjustment for hsCRP resulted in a slightly attenuated but statistically significant association: HR (95% CI) for quartile 4 versus 1 was 1.43 (1.01-2.03; Plinear trend = 0.03) (Table 2). The risk estimates per SD increase in baseline sPLA2-IIA were 1.22 (95% CI, 1.08-1.38; p=0.002) and 1.18 (95% CI, 1.04-1.35; p=0.01) in the corresponding multivariable adjusted models respectively (Table 2). There was no statistical interaction by randomized treatment assignment in either model 2 or 3 (P for interaction = 0.88 and 0.34) (Supplemental tables IV and V). Results were generally similar when levels of baseline sPLA2-IIA were examined in relation to the expanded primary endpoint that included all-cause death (Table 2, Supplemental tables IV and V).
Table 2.
Incidence rates and hazard ratios with 95% confidence intervals for baseline sPLA2-IIA mass levels and cardiovascular events overall (combined placebo and rosuvastatin)
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | P for trend | *HR per 1-SD | P-value | |
|---|---|---|---|---|---|---|---|
| sPLA2-IIA levels (ng/mL) | ≤ 2.49 | 2.50 – 3.81 | 3.82 – 6.03 | >6.03 | |||
| Primary Endpoint | |||||||
| # of events / N | 70/2823 | 69/2815 | 82/2827 | 92/2804 | 313/11269 | ||
| Incidence rate, per 100 person-years | 1.21 (0.96-1.53) |
1.18 (0.93-1.49) |
1.38 (1.11-1.71) |
1.61 (1.31-1.97) |
|||
| Model 1 | 1.00 | 0.98 (0.70-1.37) P=0.91 |
1.20 (0.87-1.66) P=0.27 |
1.52 (1.10-2.12) P=0.01 |
0.006 | 1.21 (1.07-1.37) |
0.002 |
| Model 2 | 1.00 | 0.98 (0.70-1.37) P=0.90 |
1.18 (0.85-1.64) P=0.33 |
1.54 (1.10-2.15) P=0.01 |
0.006 | 1.22 (1.08-1.38) |
0.002 |
| Model 3 | 1.00 | 0.97 (0.67-1.36) P=0.83 |
1.14 (0.82-1.59) P=0.44 |
1.43 (1.01-2.03) P=0.04 |
0.03 | 1.18 (1.04-1.35) |
0.01 |
| Primary Endpoint Plus Total Mortality | |||||||
| # of events / N | 114/2823 | 117/2815 | 140/2827 | 158/2804 | 529/11269 | ||
| Incidence rate, per 100 person-years | 1.98 (1.65-2.37) |
2.00 (1.67-2.39) |
2.35 (2.00-2.77) |
2.76 (2.37-3.22) |
|||
| Model 1 | 1.00 | 1.02 (0.79-1.32) P=0.89 |
1.24 (0.97-1.60) P=0.09 |
1.53 (1.18-1.97) P=0.001 |
0.0004 | 1.26 (1.15-1.38) |
<0.0001 |
| Model 2 | 1.00 | 1.02 (0.79-1.33) P=0.87 |
1.22 (0.95-1.58) P=0.12 |
1.50 (1.16-1.94) P=0.002 |
0.0008 | 1.25 (1.14-1.37) |
<0.0001 |
| Model 3 | 1.00 | 0.99 (0.76-1.29) P=0.95 |
1.14 (0.88-1.48) P=0.31 |
1.29 (0.99-1.69) P=0.06 |
0.04 | 1.17 (1.06-1.29) |
0.002 |
Model One: Adjusted for age, gender, race, and treatment group.
Model Two: Adjusted for age, gender, race, treatment group, smoking, family history of premature atherosclerosis, body mass index, systolic blood pressure, fasting glucose, high density lipoprotein-cholesterol, low density lipoprotein -cholesterol, and ln triglycerides
Model Three: Adjusted for age, gender, race, treatment group, smoking, family history of premature atherosclerosis, body mass index, systolic blood pressure, fasting glucose, high density lipoprotein-cholesterol, low density lipoprotein -cholesterol, ln triglycerides, ln high-sensitivity C-reactive protein. sPLA2-IIA indicates group IIA secretory phospholipases A2; HR, hazard ratio; SD, standard deviation; CVD, cardiovascular disease. P Interaction by randomization treatment assignment were >0.05.
Hazard ratios are expressed per 1-SD increment in ln sPLA2-IIA
On-treatment sPLA2-IIA Mass and Residual CVD Risk
For the rosuvastatin-allocated group, the model 2 HRs (95% CIs) for the primary endpoint for quartiles 2 to 4 of on-treatment sPLA2-IIA mass compared to quartile 1 as reference were, 0.78 (0.38-1.59), 1.28 (0.67-2.44), 1.52 (0.78-2.93) (Plinear trend = 0.10); after additional adjustment for hsCRP the HR for quartile 4 versus 1 was 1.18 (0.58-2.39; Plinear trend = 0.43) (Table 3) The risk estimates per SD increment in on-treatment sPLA2-IIA were 1.19 (95% CI, 0.97-1.45; P=0.09) and 1.08 (95% CI, 0.87-1.34; P=0.50) in the corresponding multivariable adjusted models respectively. For the rosuvastatin-allocated group, on-treatment sPLA2-IIA was significantly associated with the expanded primary endpoint that included all-cause death [HR for quartile 4 versus 1 was 1.66 (95%CI: 0.95-2.89); Plinear trend = 0.02] or per SD increment [HR: 1.31 (95% CI: 1.11-1.55), P=0.001] (Table 3). This association was attenuated after adjustment for on-treatment hsCRP [HR for quartile 4 versus 1 was 1.25 (95%CI: 0.69-2.25); Plinear trend = 0.25] or per 1-SD increment (P=0.06) (Table 3).
Table 3.
Incidence rates and hazard ratios with 95% confidence intervals for on-treatment sPLA2-IIA mass and residual risk of cardiovascular events among participants randomly allocated to rosuvastatin
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | P for trend | HR per 1-SD* | P-value | |
|---|---|---|---|---|---|---|---|
| sPLA2-IIA levels (ng/mL) | ≤ 1.31 | 1.32 – 2.09 | 2.10 – 3.57 | >3.57 | |||
| Primary Endpoint | |||||||
| # of events / N | 19/1212 | 14/1194 | 23/1200 | 26/1200 | 82/4806 | ||
| Incidence rate, per 100 person-years | 0.76 (0.49-1.19) |
0.53 (0.31-0.89) |
0.87 (0.58-1.31) |
1.00 (0.68-1.47) |
|||
| Model 1 | 1.00 | 0.70 (0.35-1.40) P=0.31 |
1.24 (0.67-2.30) P=0.49 |
1.56 (0.83-2.91) P=0.16 |
0.07 | 1.21 (0.99-1.47) |
0.06 |
| Model 2 | 1.00 | 0.78 (0.38-1.59) P=0.49 |
1.28 (0.67-2.44) P=0.45 |
1.52 (0.78-2.93) P=0.22 |
0.10 | 1.19 (0.97-1.45) |
0.09 |
| Model 3 | 1.00 | 0.74 (0.36-1.52) P=0.42 |
1.10 (0.57-2.13) P=0.77 |
1.18 (0.58-2.39) P=0.64 |
0.43 | 1.08 (0.87-1.34) |
0.50 |
| Primary Endpoint Plus Total Mortality | |||||||
| # of events / N | 24/1212 | 21/1194 | 34/1200 | 41/1200 | 120/4806 | ||
| Incidence rate, per 100 person-years | 0.96 (0.65-1.43) |
0.79 (0.51-1.21) |
1.29 (0.92-1.80) |
1.58 (1.17-2.14) |
|||
| Model 1 | 1.00 | 0.80 (0.45-1.44) P=0.46 |
1.37 (0.80-2.32) P=0.25 |
1.73 (1.02-2.95) P=0.04 |
0.01 | 1.34 (1.14-1.58) |
0.0004 |
| Model 2 | 1.00 | 0.86 (0.47-1.57) P=0.62 |
1.38 (0.80-2.39) P=0.25 |
1.66 (0.95-2.89) P=0.07 |
0.02 | 1.31 (1.11-1.55) |
0.001 |
| Model 3 | 1.00 | 0.81 (0.44-1.48) P=0.50 |
1.18 (0.68-2.07) P=0.56 |
1.25 (0.69-2.25) P=0.46 |
0.25 | 1.19 (0.99-1.43) |
0.06 |
Model One: Adjusted for age, gender, and race.
Model Two: Adjusted for age, gender, race, smoking, family history of premature atherosclerosis, body mass index, systolic blood pressure, fasting glucose, high density lipoprotein-cholesterol, low density lipoprotein -cholesterol, and ln triglycerides
Model Three: Adjusted for age, gender, race, smoking, family history of premature atherosclerosis, body mass index, systolic blood pressure, fasting glucose, high density lipoprotein-cholesterol, low density lipoprotein -cholesterol, ln triglycerides, ln high-sensitivity C-reactive protein. sPLA2-IIA indicates group IIA secretory phospholipases A2; HR, hazard ratio; SD, standard deviation; CVD, cardiovascular disease. P Interaction by randomization treatment assignment were >0.05.
Hazard ratios are expressed per 1-SD increment in ln sPLA2-IIA
Efficacy of Rosuvastatin Therapy According to Baseline sPLA2-IIA Mass
In an analysis that examined participants based on categories that took both treatment assignment and baseline sPLA2-IIA mass into account, those who were on placebo and had sPLA2-IIA levels ≤2.49 ng/ml (1st quartile) were considered as the reference category. For both the placebo and rosuvastatin groups, there was a suggested trend of increasing risk with increasing baseline sPLA2-IIA levels; however, at each level the rosuvastatin group had lower risk (Figure 2). Consequently, rosuvastatin therapy had similar efficacy regardless of baseline sPLA2-IIA levels, as illustrated in Figure 3 (P for interaction=0.33), with estimates centered around the effect estimate reported in the JUPITER trial [HR: 0.56; 95% CI: 0.46-0.69)]. 23
Figure 2.

Fully adjusted Hazard ratios (95% CI) for the primary event according to sPLA2-IIA levels and treatment assignment, relative to subjects on placebo with the lowest quartile of baseline sPLA2-IIA levels.
Figure 3.

Efficacy of rosuvastatin for the primary event, stratified by baseline sPLA2-IIA mass.
SNPs Associated with sPLA2-IIA Mass
In 6 692 JUPITER participants who consented for genetic research (3 333 participants in the placebo group and 3 359 participants in the rosuvastatin group), a genome wide scan of 796 141 SNPs identified 69 SNPs that met genome wide significance threshold (P<5×10−8) for association with baseline sPLA2-IIA levels (Supplemental Table VI). Among the identified SNPs, all but one are located in a cluster of loci on chromosome 1 which are all members of the secretory phospholipase A2 family of genes (PLA2G2A, PLA2G2C, PLA2G2D,PLA2G2E, PLA2G2F, and PLA2G5 that encode for sPLA2-IIA, sPLA2-IIC, sPLA2-IID, sPLA2-IIE, sPLA2-IIF, and sPLA2-V, respectively), in addition to flanking genes OTUD3, TMCO4, RNF186, UBXN10; with the remaining identified SNP in a locus on chromosome 17, ABCA8. The 3 top SNPs (rs11573156 P=5.1 × 10-472; rs2307246 P=3.2 × 10−441; and rs4744 P=1.1 × 10−441) with the strongest magnitude of association with levels of sPLA2-IIA are located within PLA2G2A (encoding sPLA2-IIA). rs11573156, which was the lead SNP explained 28% of the variance in sPLA2-IIA levels. The median (25th to 75th percentile) sPLA2-IIA value for individuals homozygous for the common allele of rs11573156 (CC) was 2.79 ng/ml (1.97-4.01 ng/ml) while that for individuals homozygous for the rare allele (GG) was 7.38 ng/ml (5.38-10.19 ng/ml); similar values were obtained for the extreme genotypes of rs2307246 and rs4744. Conditional analysis from genome-wide complex trait analysis tool revealed 2 non-redundant signals on chromosome 1 rs2307246 and rs12023742 (PLA2G2A); 1 on chromosome 17 rs34931250 (ABCA8); and 1 on chromosome 12 rs7310409 (HNF1A) that did not meet initial genome wide significance (P=1.6 × 10−8).
Association of SNPs with Incident CVD
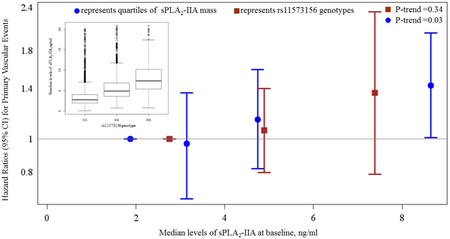
The lead SNP in the PLA2G2A locus, rs11573156, with a minor allele frequency of 0.23, had no significant association with traditional CVD risk factors including body mass index, systolic blood pressure, LDL-cholesterol, high density lipoprotein (HDL)-cholesterol, triglycerides, and hsCRP (Supplemental Table VII). Of the 6 692 participants included in the genetic analysis of CVD events, 218 developed incident CVD. The HR for rs11573156 with incident CVD was 1.11 (95%CI: 0.89-1.38; P=0.34), demonstrating a trend towards higher CVD risk in a manner that was consistent with the association of sPLA2-IIA mass with CVD risk, but which were not statistically significant (Graphic Abstract). Results were similar for other SNPs in PLA2G2A (Table 4) as well as the other genetic determinants of sPLA2-IIA (Supplemental Table VI). There was no evidence of treatment interaction for the association of any of the SNPs with incident CVD (Supplemental Table VI).
Table 4.
Hazard ratios with 95% confidence intervals of top three PLA2G2A variants influencing sPLA2-IIA mass levels and incident CVD
|
PLA2G2A variants (minor allele) |
Homozygous for non-effect allele |
Intermediate genotype | Homozygous for effect allele |
HR (95% CI) per minor allele |
P Value for additive model |
|---|---|---|---|---|---|
| rs11573156 (G) | 1 | 1.06 (0.80-1.40) | 1.36 (0.79-2.34) | 1.11 (0.89-1.38) | 0.34 |
| rs2307246 (T) | 1 | 1.12 (0.90-1.39) | 0.32 | ||
| rs4744 (T) | 1 | 1.11 (0.89-1.38) | 0.37 |
HRs (95% CI) are adjusted for age, sex, randomization treatment assignment, region and measures of subpopulation stratification HR denotes hazard ratio; and CI, confidence interval.
DISCUSSION
In the JUPITER trial – an at-risk primary prevention trial among individuals with evidence of systemic inflammation – elevated levels of baseline sPLA2-IIA mass were significantly associated with greater risk of the composite primary endpoint that included myocardial infarction, coronary revascularization, unstable angina, stroke, or fatal cardiovascular events, independent of traditional cardiometabolic risk factors. This association persisted after adjustment for hsCRP and lacked evidence of statistical heterogeneity by rosuvastatin therapy. The three SNPs in PLA2G2A (rs11573156, rs2307246, and rs4744) employed herein accounted for up to 28% of the variance in sPLA2-IIA levels, and demonstrated a non-significant trend towards higher CVD risk in a manner that mirrored the observational associations observed between sPLA2-IIA mass with CVD risk.
Multiple inflammatory pathways play key roles in the genesis and progression of atherosclerosis.1 Importantly, reducing inflammation with interleukin-1β inhibition among those with elevated hsCRP has recently been found to prevent CVD events and improve mortality. 2 As such, identifying clinically relevant vascular inflammatory pathways for further targeting has taken on critical importance. While several studies have demonstrated the prognostic usefulness of sPLA2-IIA in patients with existing CVD 15-18 there are limited data on the value of sPLA2-IIA for CVD risk assessment in primary prevention settings. We know of only one study that previously examined sPLA2-IIA in an apparently healthy population without baseline CVD – a nested case-control study in the European Prospective Investigation into Cancer (EPIC)-Norfolk cohort – where the study authors found sPLA2-IIA to be associated with risk of fatal and non-fatal coronary artery disease 28. While these two studies were largely performed in similar populations, key differences in study design were present (Supplemental Table VIII). Both studies nonetheless described associations between sPLA2-IIA and incident CVD, validating and extending each other’s findings. To our knowledge, the current study is the first to prospectively evaluate the association of sPLA2-IIA with the risk of incident CVD events in a primary prevention population recruited on the basis of chronic inflammation, finding that baseline measures of sPLA2-IIA are associated with a significant increase in the risk of incident CVD, even after adjustment for established clinical risk factors as well as other inflammatory markers, such as hsCRP. That such an association between sPLA2-IIA and CVD events remained significant after adjusting for hsCRP suggests that sPLA2-IIA could be an important parallel vascular inflammatory pathway in patients with inflammatory risk of CVD. The present study was conducted in a cohort selected on the basis of elevated hsCRP, and additional studies in other populations are needed.
While the relative risk reduction with rosuvastatin vs placebo in JUPITER was similar among those with and without elevated sPLA2-IIA, the absolute risk was higher among those with high sPLA2-IIA, hence the benefit is greater for these individuals on an absolute risk scale. This higher inflammatory risk may also potentially be reduced further with other anti-inflammatory therapies beyond statins. The value of sPLA2-IIA for assessing “residual risk” in statin-treated individuals therefore warrants further investigation in other populations and with newer anti-inflammatory therapies.
The decline in hsCRP and sPLA2-IIA in the placebo group at one year may be at least in part due to regression to the mean which is expected when screened subjects with low inflammation are excluded from a study as is the case in JUPITER 23. Periodic medical surveillance of all JUPITER participants as mandated in the trial protocol may have also resulted in improved overall risk factor control even among participants allocated to placebo. Furthermore, the Hawthorne Effect – whereby knowing that they are being monitored can lead participants to modify their behavior – may have led to improved compliance with medication and lifestyle recommendations. These factors may also contribute to the decline in sPLA2-IIA levels after one year particularly, given the degree of correlation we observed between both biomarkers at study entry.
We observed a genome-wide significant gene-dose effect between PLA2G2A variants and sPLA2-IIA levels, and in turn found that these variants also demonstrated a non-significant trend towards association with increased risk of CVD, although this analysis was exploratory and underpowered. We, however, note that a meta-analyses of data from European subjects in the general population which did not include JUPITER study participants also found rs11573156 polymorphisms located in PLA2G2A to be the most genome-wide significant determinant of sPLA2-IIA levels but found no evidence of an association between this SNP and incident CVD.29 In our study, we similarly did not observe a significant association between genetic variants in sPLA2-IIA and CVD events, although this should be examined in larger primary prevention cohorts to confirm an absence of this association.
The VISTA-16 trial testing the initiation of varespladip methyl, a very potent pan sPLA2 inhibitor, within 96 hours of ACS, was terminated after an interim analyses owing to futility and a signal towards harm accounted for by a significant increase in the risk of myocardial infarction.30 Prior studies suggested that some isoforms of sPLA2, such as sPLA2-X, may in fact be athero-protective, and hence the potential beneficial effects of varespladip on sPLA2-IIA might have been offset by deleterious effects on other sPLA2 isoforms.31 In addition, the balance of pro- and anti-inflammatory cytokines after an ACS may significantly impact near term prognosis. 32 On the other hand, in individuals with no history of CVD but with an elevated degree of inflammation, we did not find genetic polymorphisms influencing the sPLA2-IIA isoform to be associated with an increased risk of CVD. Rather our results suggest that pharmacological blockade specific to the sPLA2-IIA isoform may be a potential strategy for preventing the onset of CVD, in particular if enzyme activity is specifically inhibited at the arterial wall, the site of atherosclerotic lesion formation.33
Furthermore, pharmacological blockade of Lp-PLA2 – an inflammatory mediator functionally similar to sPLA2-IIA involved in modification of lipoproteins and liberation of pro-inflammatory free lipid – did not show benefit when tested in ACS patients.34 However, there was a significant reduction in vascular risk for the individual endpoints of total and major coronary events when tested in patients with stable coronary heart disease.35 Nonetheless, the value of pharmacologically blocking sPLA2-IIA, and not other sPLA2 isoforms, for the prevention of a first cardiovascular event is yet to be tested. The challenge with sPLA2 enzymes in drug discovery is their similar protein structure, since almost all the sPLA2 isoforms share the same catalytic domain. This has made it difficult to generate specific small chemical inhibitors. Development of novel therapeutic agents with the potential for sPLA2-IIA selectivity amongst other sPLA2 isoforms may represent a novel pharmacological strategy for the prevention of CVD, particularly among those with a high burden of systemic vascular inflammation.
The inflammatory cascade is a complex network that involves sequential activation of effector proteins with a tendency for downstream proteins to overlap functionally. In this regard, transgenic mice expressing sPLA2-IIA demonstrated greater susceptibility to atherosclerosis when compared with wild type.13 Interestingly, reduced levels of HDLs and increased levels of LDLs were also detected, suggesting changes in lipoproteins as a potential intermediate mechanism. This potential link was strengthened by the observation that murine sPLA2 also expression influenced HDL particle size and subclass composition, and that sPLA2 may be involved in mediating the interaction between changes in HDL and inflammatory stimuli.36 Our results suggest that humans who already have an elevated level of inflammation but enriched in polymorphisms that increase the expression of sPLA2-IIA exhibited an increased risk of CVD. However, the polygenic inheritance of inflammation and the functionally redundant nature of several mediators of the inflammatory cascade, further argues for a well-powered study to confirm that the increased risk of first CVD that we observed for variants of PLA2G2A is statistically significant. This will be important to further investigate the usefulness of sPLA2-IIA for the management of CVD beyond that of a biomarker role.
A major strength of the current study is the large number of men and women who were randomly allocated to a potent statin or placebo, had both baseline and on-treatment (statin and placebo) measurement of sPLA2-IIA mass and genotyping; and were followed for CVD events. Our study is limited by the JUPITER entry criteria which excluded participants with low hsCRP, high LDL-cholesterol, high triglycerides, and known CVD or diabetes; therefore our findings cannot be generalized to dissimilar populations. The limited years of follow-up and the reduced number of CVD events in the rosuvastatin group may have limited our ability to detect a meaningful association for on-treatment sPLA2-IIA or for the genotype with CVD events. We also measured only sPLA2-IIA mass in this study, and hence cannot assess comparisons with sPLA2-IIA activity, although experimental studies have found that inhibition of sPLA2-II activity leads also to a decrease in sPLA2-IIA mass.37With respect to Lp-PLA2, inhibition of the enzyme activity does not change the mass/protein levels.
In sum, sPLA2-IIA may be a measurable biomarker to assess the prognostic importance of inflammation in primary prevention patients at increased inflammatory risk for CVD. Whether sPLA2-IIA may be a potential pharmacological target for reducing CVD risk in primary prevention settings warrants further study.
Supplementary Material
Highlights.
Group IIA secretory phospholipase A2 (sPLA2-IIA) plays an integral pro-inflammatory role in regulating vascular inflammation.
In the JUPITER primary prevention population recruited on the basis of chronic inflammation, elevated levels of baseline sPLA2-IIA mass were significantly associated with greater risk of incident cardiovascular disease (CVD) events.
This association persisted after adjustment for clinical factors and hsCRP, suggesting that sPLA2-IIA mass identifies vascular inflammation not fully reflected by hsCRP.
Similar association was observed in the randomized rosuvastatin and placebo groups.
Three SNPs in PLA2G2A accounted for up to 28% of the variance in sPLA2-IIA mass, and demonstrated a non-significant trend towards higher CVD risk in a manner that mirrored the observational associations observed between sPLA2-IIA mass with CVD risk.
Acknowledgments
Sources of Funding: JUPITER was financially supported by AstraZeneca, which collected trial data and monitored sites but had no role in the decision to submit the manuscript for publication. Quest Diagnostics Nichols Institute in San Juan Capistrano, CA, absorbed the cost of performing the sPLA2-IIA measurements and performed them in a blinded manner. Research reported in this publication was supported in part by the National Heart, Lung, and Blood Institute of the National Institutes of Health under award numbers R01HL117861, R01134811, and K24 HL136852, and by support from the Molino Family Trust to Dr Mora. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Drs. Akinkuolie and Lawler were supported by the National Heart, Lung, and Blood Institute (T32 HL007575).
Abbreviation list:
- ACS
acute coronary syndrome
- CVD
cardiovascular disease
- hsCRP
high-sensitivity C-reactive protein
- JUPITER
Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin
- LDL
low density lipoprotein
- sPLA2-IIA
group IIA secretory phospholipase A2
Footnotes
JUPITER Trial (NCT00239681): URL http://clinicaltrials.gov/ct/show/NCT00239681
Disclosures: Dr. Caulfield is an employee of Quest Diagnostics. Dr. Chu is employed by Merck Research Laboratories. Drs. Hurt-Camejo, Ding, and Nyberg are employees of AstraZeneca. Dr. Nyberg holds shares in AstraZeneca. Dr Ridker has received research grant support from AstraZeneca, Novartis, Amgen, Kowa, and the National Heart, Lung, and Blood Institute and has served as a consultant to Genzyme, Janssen, Aegerion, ISIS, Vascular Biogenics, Boehringer, Pfizer, and Merck. Dr Ridker is listed as a coinventor on patents held by the Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in CVD that have been licensed to AstraZeneca and Siemens. Dr Mora has received institutional research support from AstraZeneca, Atherotech Diagnostics, and NIH; served as a consultant to Genzyme, Quest Diagnostics, Pfizer, and Cerenis Therapeutics; and received speaker honoraria from AstraZeneca, and the National Lipid Association for educational (nonpromotional) activities. The other authors report no conflicts.
Contributor Information
Akintunde O. Akinkuolie, Center for Lipid Metabolomics, Division of Preventive Medicine, Brigham and Women’s Hospital, Department of Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA.
Patrick R. Lawler, Center for Lipid Metabolomics, Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, MA; Peter Munk Cardiac Centre, Toronto General Hospital, and the Heart and Stroke/Richard Lewar Centre for Excellence in Cardiovascular Research, University of Toronto, Toronto, ON, Canada.
Audrey Y. Chu, Merck Research Laboratories, Boston, MA
Michael Caulfield, Department of Endocrinology & CVD, Quest Diagnostics Nichols Institute, San Juan Capistrano, CA.
Jianying Mu, Department of Endocrinology & CVD, Quest Diagnostics Nichols Institute, San Juan Capistrano, CA.
Bo Ding, Medical Evidence & Observational Research, Global Medical Affairs, AstraZeneca R&D, Mölndal, Sweden.
Fredrik Nyberg, Medical Evidence & Observational Research, Global Medical Affairs, AstraZeneca R&D, Mölndal, Sweden; Occupational and Environmental Medicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden.
Robert J. Glynn, Center for Lipid Metabolomics, Division of Preventive Medicine, Brigham and Women’s Hospital, Department of Biostatistics, Harvard School of Public Health, Boston, MA
Paul M Ridker, Center for Lipid Metabolomics, Division of Preventive Medicine and Cardiovascular Medicine, Brigham and Women’s Hospital, Boston, MA.
Eva Hurt-Camejo, Cardiovascular & Metabolic Diseases, Innovative Medicines, AstraZeneca R&D, Mölndal, Sweden; Division of Clinical Chemistry, Department of Laboratory Medicine, Karolinska Institute, Stockholm, Sweden.
Daniel I. Chasman, Center for Lipid Metabolomics, Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, MA
Samia Mora, Center for Lipid Metabolomics, Division of Preventive Medicine and Cardiovascular Medicine, Brigham and Women’s Hospital, Boston, MA.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. New Engl J Med. 2005;352:1685–1695. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Group CT. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018;391:319–328. [DOI] [PubMed] [Google Scholar]
- 4.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mallat Z, Lambeau G, Tedgui A. Lipoprotein-associated and secreted phospholipases A2 in cardiovascular disease: Roles as biological effectors and biomarkers. Circulation. 2010;122:2183–2200. [DOI] [PubMed] [Google Scholar]
- 6.Quach ND, Arnold RD, Cummings BS. Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol. 2014;90:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibeas E, Fuentes L, Martin R, Hernandez M, Nieto ML. Secreted phospholipase A2 type iia as a mediator connecting innate and adaptive immunity: New role in atherosclerosis. Cardiovasc Res. 2009;81:54–63. [DOI] [PubMed] [Google Scholar]
- 8.Rosenson RS, Hurt-Camejo E. Phospholipase A2 enzymes and the risk of atherosclerosis. Eur Heart J. 2012;33:2899–2909. [DOI] [PubMed] [Google Scholar]
- 9.Rosenson RS, Gelb MH. Secretory phospholipase A2: A multifaceted family of proatherogenic enzymes. Current Cardiology Reports. 2009;11:445–451. [DOI] [PubMed] [Google Scholar]
- 10.Jaross W, Eckey R, Menschikowski M. Biological effects of secretory phospholipase A2 group iia on lipoproteins and in atherogenesis. Eur J Clin Invest. 2002;32:383–393. [DOI] [PubMed] [Google Scholar]
- 11.Oorni K, Kovanen PT. Lipoprotein modification by secretory phospholipase A2 enzymes contributes to the initiation and progression of atherosclerosis. Curr Opin Lipidol. 2009;20:421–427. [DOI] [PubMed] [Google Scholar]
- 12.Cavigiolio G, Jayaraman S. Proteolysis of apolipoprotein A-I by secretory phospholipase A2: A new link between inflammation and atherosclerosis. J Biol Chem. 2014;289:10011–10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivandic B, Castellani LW, Wang XP, Qiao JH, Mehrabian M, Navab M, Fogelman AM, Grass DS, Swanson ME, de Beer MC, de Beer F, Lusis AJ. Role of group II secretory phospholipase A2 in atherosclerosis: 1. Increased atherogenesis and altered lipoproteins in transgenic mice expressing group IIa phospholipase A2. Arterioscler Thromb Vasc Biol. 1999;19:1284–1290. [DOI] [PubMed] [Google Scholar]
- 14.Ghesquiere SA, Gijbels MJ, Anthonsen M, van Gorp PJ, van der Made I, Johansen B, Hofker MH, de Winther MP. Macrophage-specific overexpression of group IIa spla2 increases atherosclerosis and enhances collagen deposition. J Lipid Res. 2005;46:201–210. [DOI] [PubMed] [Google Scholar]
- 15.Menschikowski M, Kasper M, Lattke P, Schiering A, Schiefer S, Stockinger H, Jaross W. Secretory group II phospholipase A2 in human atherosclerotic plaques. Atherosclerosis. 1995;118:173–181. [DOI] [PubMed] [Google Scholar]
- 16.Sartipy P, Johansen B, Gasvik K, Hurt-Camejo E. Molecular basis for the association of group IIa phospholipase A2 and decorin in human atherosclerotic lesions. Circ Res. 2000;86:707–714. [DOI] [PubMed] [Google Scholar]
- 17.Kugiyama K, Ota Y, Sugiyama S, Kawano H, Doi H, Soejima H, Miyamoto S, Ogawa H, Takazoe K, Yasue H. Prognostic value of plasma levels of secretory type II phospholipase A2 in patients with unstable angina pectoris. Am J Cardiol. 2000;86:718–722. [DOI] [PubMed] [Google Scholar]
- 18.Liu PY, Li YH, Tsai WC, Chao TH, Tsai LM, Wu HL, Chen JH. Prognostic value and the changes of plasma levels of secretory type II phospholipase A2 in patients with coronary artery disease undergoing percutaneous coronary intervention. Eur Heart J. 2003;24:1824–1832. [DOI] [PubMed] [Google Scholar]
- 19.Kugiyama K, Ota Y, Takazoe K, Moriyama Y, Kawano H, Miyao Y, Sakamoto T, Soejima H, Ogawa H, Doi H, Sugiyama S, Yasue H. Circulating levels of secretory type II phospholipase A2 predict coronary events in patients with coronary artery disease. Circulation. 1999;100:1280–1284. [DOI] [PubMed] [Google Scholar]
- 20.Koenig W, Vossen CY, Mallat Z, Brenner H, Benessiano J, Rothenbacher D. Association between type II secretory phospholipase A2 plasma concentrations and activity and cardiovascular events in patients with coronary heart disease. Eur Heart J. 2009;30:2742–2748. [DOI] [PubMed] [Google Scholar]
- 21.Mallat Z, Steg PG, Benessiano J, et al. Circulating secretory phospholipase A2 activity predicts recurrent events in patients with severe acute coronary syndromes. J Am Coll Cardiol. 2005;46:1249–1257. [DOI] [PubMed] [Google Scholar]
- 22.Ryu SK, Mallat Z, Benessiano J, Tedgui A, Olsson AG, Bao W, Schwartz GG, Tsimikas S, Myocardial Ischemia Reduction With Aggressive Cholesterol Lowering Trial I. Phospholipase A2 enzymes, high-dose atorvastatin, and prediction of ischemic events after acute coronary syndromes. Circulation. 2012;125:757–766. [DOI] [PubMed] [Google Scholar]
- 23.Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ, Group JS. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New Engl J Med.2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 24.Mora S, Glynn RJ, Boekholdt SM, Nordestgaard BG, Kastelein JJ, Ridker PM. On-treatment non-high-density lipoprotein cholesterol, apolipoprotein B, triglycerides, and lipid ratios in relation to residual vascular risk after treatment with potent statin therapy: JUPITER. J Am Coll Cardiol. 2012;59:1521–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mora S, Glynn RJ, Ridker PM. High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation. 2013;128:1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chasman DI, Giulianini F, Macfadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: The Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin trial. Circ Cardiovasc Genet. 2012;5:257–264. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Lee SH, Goddard ME, Visscher PM. Gcta: A tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boekholdt SM, Keller TT, Wareham NJ, Luben R, Bingham SA, Day NE, Sandhu MS, Jukema JW, Kastelein JJ, Hack CE, Khaw KT. Serum levels of type II secretory phospholipase A2 and the risk of future coronary artery disease in apparently healthy men and women: The EPIC-Norfolk prospective population study. Arterioscler Thromb Vasc Biol. 2005;25:839–846. [DOI] [PubMed] [Google Scholar]
- 29.Holmes MV, Simon T, Exeter HJ,et al. Secretory phospholipase A2-IIa and cardiovascular disease: A Mendelian randomization study. J Am Coll Cardiol. 2013;62:1966–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicholls SJ, Kastelein JJ, Schwartz GG, Bash D, Rosenson RS, Cavender MA, Brennan DM, Koenig W, Jukema JW, Nambi V, Wright RS, Menon V, Lincoff AM, Nissen SE. Varespladib and cardiovascular events in patients with an acute coronary syndrome: The VISTA-16 randomized clinical trial. JAMA. 2014;311:252–262 [DOI] [PubMed] [Google Scholar]
- 31.Ait-Oufella H, Herbin O, Lahoute C, et al. Group X secreted phospholipase A2 limits the development of atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2013;33:466–473. [DOI] [PubMed] [Google Scholar]
- 32.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. [DOI] [PubMed] [Google Scholar]
- 33.Giordanetto F, Pettersen D, Starke I, Nordberg P, Dahlstrom M, Knerr L, Selmi N, Rosengren B, Larsson LO, Sandmark J, Castaldo M, Dekker N, Karlsson U, Hurt-Camejo E. Discovery of AZD2716: A novel secreted phospholipase A2 inhibitor for the treatment of coronary artery disease. ACS Med Chem Lett. 2016;7:884–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Donoghue ML, Braunwald E, White HD,et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. JAMA. 2014;312:1006–1015. [DOI] [PubMed] [Google Scholar]
- 35.White HD, Held C, Stewart R, et al. Darapladib for preventing ischemic events in stable coronary heart disease. New Engl J Med. 2014;370:1702–1711. [DOI] [PubMed] [Google Scholar]
- 36.Tietge UJ, Maugeais C, Lund-Katz S, Grass D, deBeer FC, Rader DJ. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA-I in response to inflammation in human apoa-I transgenic mice. Arterioscler Thromb Vasc Biol. 2002;22:1213–1218. [DOI] [PubMed] [Google Scholar]
- 37.Hurt-Camejo E, Gautier T, Rosengren B, Dikkers A, Behrendt M, Grass DS, Rader DJ, Tietge UJ. Expression of type IIa secretory phospholipase A2 inhibits cholesteryl ester transfer protein activity in transgenic mice. Arterioscler Thromb Vasc Biol. 2013;33:2707–2714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.