

Abstract
Background
Anti–tumour necrosis factor (TNF) agents are effective in treating people with rheumatoid arthritis (RA), but are associated with (dose‐dependent) adverse effects and high costs. To prevent overtreatment, several trials have assessed the effectiveness of down‐titration compared with continuation of the standard dose. This is an update of a Cochrane Review published in 2014.
Objectives
To evaluate the benefits and harms of down‐titration (dose reduction, discontinuation, or disease activity‐guided dose tapering) of anti‐TNF agents on disease activity, functioning, costs, safety, and radiographic damage compared with usual care in people with RA and low disease activity.
Search methods
We searched MEDLINE, Embase, Web of Science and CENTRAL (29 March 2018) and four trial registries (11 April 2018) together with reference checking, citation searching, and contact with study authors to identify additional studies. We screened conference proceedings (American College of Rheumatology and European League Against Rheumatism 2005‐2017).
Selection criteria
Randomised controlled trials (RCTs) and controlled clinical trials (CCTs) comparing down‐titration (dose reduction, discontinuation, disease activity–guided dose tapering) of anti‐TNF agents (adalimumab, certolizumab pegol, etanercept, golimumab, infliximab) to usual care/no down‐titration in people with RA and low disease activity.
Data collection and analysis
We used standard Cochrane methodology.
Main results
One previously included trial was excluded retrospectively in this update because it was not an RCT/CCT. We included eight additional trials, for a total of 14 studies (13 RCTs and one CCT, 3315 participants in total) reporting anti‐TNF down‐titration. Six studies (1148 participants) reported anti‐TNF dose reduction compared with anti‐TNF continuation. Eight studies (2111 participants) reported anti‐TNF discontinuation compared with anti‐TNF continuation (three studies assessed both anti‐TNF discontinuation and dose reduction), and three studies assessed disease activity–guided anti‐TNF dose tapering (365 participants). These studies included data on all anti‐TNF agents, but primarily adalimumab and etanercept. Thirteen studies were available in full text, one was available as abstract. We assessed the included studies generally at low to moderate risk of bias; our main concerns were bias due to open‐label treatment and unblinded outcome assessment. Clinical heterogeneity between the trials was high. The included studies were performed at clinical centres around the world and included people with early as well as established RA, the majority of whom were female with mean ages between 47 and 60. Study durations ranged from 6 months to 3.5 years.
We found that anti‐TNF dose reduction leads to little or no difference in mean disease activity score (DAS28) after 26 to 52 weeks (high‐certainty evidence, mean difference (MD) 0.06, 95% confidence interval (CI) −0.11 to 0.24, absolute risk difference (ARD) 1%) compared with continuation. Also, anti‐TNF dose reduction does not result in an important deterioration in function after 26 to 52 weeks (Health Assessment Questionnaire Disability Index (HAQ‐DI)) (high‐certainty evidence, MD 0.09, 95% CI 0.00 to 0.19, ARD 3%). Next to this, anti‐TNF dose reduction may slightly reduce the proportion of participants switched to another biologic (low‐certainty evidence), but probably slightly increases the proportion of participants with minimal radiographic progression after 52 weeks (moderate‐certainty evidence, risk ratio (RR) 1.22, 95% CI 0.76 to 1.95, ARD 2% higher). Anti‐TNF dose reduction may cause little or no difference in serious adverse events, withdrawals due to adverse events and proportion of participants with persistent remission (low‐certainty evidence).
Results show that anti‐TNF discontinuation probably slightly increases the mean disease activity score (DAS28) after 28 to 52 weeks (moderate‐certainty evidence, MD 0.96, 95% CI 0.67 to 1.25, ARD 14%), and that the RR of persistent remission lies between 0.16 and 0.77 (low‐certainty evidence). Anti‐TNF discontinuation increases the proportion participants with minimal radiographic progression after 52 weeks (high‐certainty evidence, RR 1.69, 95% CI 1.10 to 2.59, ARD 7%) and may lead to a slight deterioration in function (HAQ‐DI) (low‐certainty evidence). It is uncertain whether anti‐TNF discontinuation influences the number of serious adverse events (due to very low‐certainty evidence) and the number of withdrawals due to adverse events after 28 to 52 weeks probably increases slightly (moderate‐certainty evidence, RR 1.46, 95% CI 0.75 to 2.84, ARD 1% higher).
Anti‐TNF disease activity–guided dose tapering may result in little or no difference in mean disease activity score (DAS28) after 72 to 78 weeks (low‐certainty evidence). Furthermore, anti‐TNF disease activity–guided dose tapering results in little or no difference in the proportion of participants with persistent remission after 18 months (high‐certainty evidence, RR 0.89, 95% CI 0.75 to 1.06, ARD −9%) and may result in little or no difference in switching to another biologic (low‐certainty evidence). Anti‐TNF disease activity–guided dose tapering may slightly increase proportion of participants with minimal radiographic progression (low‐certainty evidence) and probably leads to a slight deterioration of function after 18 months (moderate‐certainty evidence, MD 0.2 higher, 0.02 lower to 0.42 higher, ARD 7% higher), It is uncertain whether anti‐TNF disease activity‐guided dose tapering influences the number of serious adverse events due to very low‐certainty evidence.
Authors' conclusions
We found that fixed‐dose reduction of anti‐TNF, after at least three to 12 months of low disease activity, is comparable to continuation of the standard dose regarding disease activity and function, and may be comparable with regards to the proportion of participants with persistent remission. Discontinuation (also without disease activity–guided adaptation) of anti‐TNF is probably inferior to continuation of treatment with respect to disease activity, the proportion of participants with persistent remission, function, and minimal radiographic damage. Disease activity–guided dose tapering of anti‐TNF is comparable to continuation of treatment with respect to the proportion of participants with persistent remission and may be comparable regarding disease activity.
Caveats of this review are that available data are mainly limited to etanercept and adalimumab, the heterogeneity between studies, and the use of superiority instead of non‐inferiority designs.
Future research should focus on the anti‐TNF agents infliximab and golimumab; assessment of disease activity, function, and radiographic outcomes after longer follow‐up; and assessment of long‐term safety, cost‐effectiveness, and predictors for successful down‐titration. Also, use of a validated flare criterion, non‐inferiority designs, and disease activity–guided tapering instead of fixed‐dose reduction or discontinuation would allow researchers to better interpret study findings and generalise to clinical practice.
Plain language summary
Lowering the dose of or stopping anti‐tumour necrosis factor drugs in people with rheumatoid arthritis who are doing well (low disease activity)
We conducted an updated review of studies in which treatment with anti‐tumour necrosis factor (anti‐TNF) drugs (adalimumab, certolizumab pegol, etanercept, golimumab, and infliximab) was lowered or stopped in people with rheumatoid arthritis (RA) who use anti‐TNF drugs and are doing well (low disease activity). Our systematic search up to March 2018 identified 14 studies (3315 participants). The included studies were performed at clinical centres around the world and included people with early as well as established RA, the majority of whom were female with mean ages varying between 47 and 60. Study durations ranged from 6 months to 3.5 years.
What is rheumatoid arthritis? What is stopping or lowering the dose of anti‐TNF drugs?
When you have RA, your immune system, which normally fights infection, attacks the lining of your joints. This makes your joints swollen, stiff, and painful. There is no cure for RA, so treatments aim to relieve pain and stiffness, improve ability to move, and prevent damage to the joints.
Anti‐TNF agents are biological drugs for RA. They lessen complaints by reducing inflammation in the joints, and they reduce radiographic joint damage. Reducing or stopping anti‐TNF treatment when disease activity is low might reduce dose‐dependent side effects (mainly infections) and costs.
Key results
Data were available for all anti‐TNF agents, but mostly for adalimumab and etanercept.
Disease activity
‐ People who lowered the dose of anti‐TNF showed little or no increase in disease activity compared with people who continued anti‐TNF (high‐certainty evidence).
‐ People who stopped anti‐TNF had a 0.96 unit increase in disease activity on a scale from 0.9 to 8 compared with people who continued anti‐TNF (absolute difference 14%, moderate‐certainty evidence).
‐ People who gradually lowered the dose of anti‐TNF showed little or no increase in disease activity compared with people who continued anti‐TNF (low‐certainty evidence).
Persistent remission
‐ There was little or no difference in the number of people who had persistent remission between those who lowered the dose of anti‐TNF compared with continuation of anti‐TNF (low‐certainty evidence).
‐ Data on how stopping anti‐TNF affects persistent remission were not pooled because results were not similar across studies (low‐certainty evidence). The absolute difference varied between 15% and 68% fewer people that remained in remission when stopping anti‐TNF compared to continuation of anti‐TNF.
‐ There was little or no difference in the number of people who had persistent remission between those who gradually lowered the dose of anti‐TNF compared with continuation of anti‐TNF (high‐certainty evidence).
X‐ray progression
‐ 24 more people per 1000 had a greater than 0.5 point progression of joint damage after a year when lowering the dose of anti‐TNF (scale 0 to 448) (absolute difference 2%, moderate‐certainty evidence).
‐ 73 more people per 1000 who stopped anti‐TNF had a greater than 0.5 point progression of joint damage after a year than people who continued anti‐TNF (absolute difference 7%, high‐certainty evidence).
‐ 110 more people per 1000 had greater than 0.5 or greater than 1.0 point progression of joint damage after 1.5 years when gradually lowering the dose of anti‐TNF (low‐certainty evidence).
Function
‐ People who lowered the dose of anti‐TNF had a 0.09 unit worsening of function (scale 0 to 3) compared with people who continued anti‐TNF (absolute difference 3%, high‐certainty evidence).
‐ People stopping anti‐TNF had a 0.18 unit worsening of function compared with people who continued anti‐TNF (absolute difference 6%, low‐certainty evidence).
‐ People gradually lowering the dose of anti‐TNF had a 0.2 unit worsening in function compared with people who continued anti‐TNF (absolute difference 7%, moderate‐certainty evidence).
Side effects
‐ There was little or no difference in number of serious adverse events in people lowering the dose of anti‐TNF compared to continuation anti‐TNF (low‐certainty evidence).
‐ It is uncertain whether gradually lowering the dose of or stopping anti‐TNF influences the number of serious adverse events (very low‐certainty evidence).
Summary of findings
Summary of findings for the main comparison. Anti‐TNF dose reduction versus anti‐TNF continuation.
| Anti‐TNF dose reduction compared to anti‐TNF continuation for rheumatoid arthritis in patients with low disease activity | |||||||
| Patient or population: people with rheumatoid arthritis with low disease activity using a standard dose of anti‐TNF agents Setting: clinical research centres Intervention: anti‐TNF dose reduction Comparison: anti‐TNF continuation | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | What happens | |
| Risk with anti‐TNF continuation | Risk with anti‐TNF dose reduction | ||||||
| Disease activity score Assessed with: DAS28 Scale from 0.9 to 8; higher scores indicate worse disease activity Follow‐up: range 26 weeks to 52 weeks | The mean disease activity score was 2.34 | MD 0.06 higher (0.11 lower to 0.24 higher) | ‐ | 501 (2 RCTs) | ⊕⊕⊕⊕ HIGH | Absolute risk difference: 1% higher (95% CI 2% lower to 3% higher) Relative percentage change: 2% higher (95% CI 5% lower to 10% higher) |
Anti‐TNF dose reduction results in little or no difference in disease activity score (DAS28). |
| Proportion of participants with persistent remission Assessed with: DAS28 < 2.6 (remission) Follow‐up: 52 weeks | 653 per 1000 | 659 per 1000 (522 to 835) | RR 1.01 (0.80 to 1.28) | 612 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 | Absolute risk difference: 1% higher (95% CI 13% lower to 18% higher) Relative percentage change: 1% higher (95% CI 20% lower to 28% higher) NNTB: not applicable (not statistically significant) |
Anti‐TNF dose reduction may result in little or no difference in the proportion of participants with persistent remission (DAS28 < 2.6). |
|
Proportion of participants switched to another biologic Mean follow‐up of 3.5 ± 1.5 years |
110 per 1000 | 44 per 1000 (19 to 102) | RR 0.40 (0.17 to 0.93) | 323 (1 RCT) | ⊕⊕⊝⊝ LOW 2 | Absolute risk difference: 7% lower (95% CI 9% lower to 1% lower) Relative percentage change: 60% (95% CI 83% lower to 7% lower) NNTB: 15 (95% CI 12 to 100) |
Anti‐TNF dose reduction may slightly reduce the proportion of participants switched to another biologic. |
| Proportion of participants with minimal radiographic progression Assessed with: mSvdH score > 0.5 Follow‐up: 52 weeks | 105 per 1000 | 129 per 1000 (80 to 206) | RR 1.22 (0.76 to 1.95) | 553 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | Absolute risk difference: 2% higher (95% CI 3% lower to 10% higher) Relative percentage change: 22% higher (95% CI 34% lower to 95% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF dose reduction probably slightly increases the proportion of participants with minimal radiographic progression (mSvdH > 0.5) . |
| Function Assessed with: Health Assessment Questionnaire Scale from 0 to 3; higher scores indicate worse function Follow‐up: range 26 weeks to 52 weeks | The mean function was 0.52 | MD 0.09 higher (0 to 0.19 higher) | ‐ | 501 (2 RCTs) | ⊕⊕⊕⊕ HIGH | Absolute risk difference: 3% higher (95% CI 0% higher to 6% higher) Relative percentage change: 15% higher (95% CI 0% higher to 31% higher) |
Anti‐TNF dose reduction does not result in an important deterioration in function. |
|
Number of serious adverse events Follow‐up: range 26 weeks to 52 weeks (mean follow‐up for Raffeiner 2015 3.5 ± 1.5 years) |
52 per 1000 | 56 per 1000 (34 to 94) | RR 1.09 (0.65 to 1.82) | 1084 (5 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | Absolute risk difference: 0% (95% CI 2% lower to 4% higher) Relative percentage change: 9% higher (95% CI 35% lower to 82% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF dose reduction may cause little or no difference in the number of serious adverse events.. |
|
Withdrawals due to adverse events Follow‐up: 52 weeks (mean follow‐up for Raffeiner 2015 3.5 ± 1.5 years) |
31 per 1000 | 33 per 1000 (16 to 70) | RR 1.07 (0.51 to 2.24) | 937 (3 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | Absolute risk difference: 0% (95% CI 2% lower to 4% higher) Relative percentage change: 7% higher (95% CI 49% lower to 124% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF dose reduction may cause little or no difference in the number of withdrawals due to adverse events . |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DAS28: disease activity score in 28 joints; MD: mean difference; mSvdH: modified Sharp van der Heijde; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; RCT: randomised controlled trial: RR: risk ratio; TNF: tumour necrosis factor | |||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
1Downgraded two levels due to heterogeneity (I2=73%) 2Downgraded two levels due to concerns about study risk of bias (high risk of selection bias, performance bias, detection bias and other bias). 3Downgraded one level due to imprecision (insufficient sample size/low number of events). 4Downgraded one level due to concerns about study risk of bias (mainly due to high risk of attrition bias in Smolen 2013 (PRESERVE) and high risk of bias on several domains in Raffeiner 2015).
Summary of findings 2. Anti‐TNF discontinuation versus anti‐TNF continuation.
| Anti‐TNF discontinuation compared to anti‐TNF continuation for rheumatoid arthritis in patients with low disease activity | |||||||
| Patient or population: people with rheumatoid arthritis with low disease activity using a standard dose of anti‐TNF agents Setting: clinical research centres Intervention: anti‐TNF discontinuation Comparison: anti‐TNF continuation | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | What happens | |
| Risk with anti‐TNF continuation | Risk with anti‐TNF discontinuation | ||||||
| Disease activity score ‐ assessed with: DAS28 Scale from: 0.9 to 8; higher scores indicate worse disease activity follow up: range 28 weeks to 52 weeks |
Discontinuation without restarting, or with restarting and LOCF analysis Mean disease activity score was 2.82 |
MD 0.96 higher (0.67 higher to 1.25 higher) | ‐ | 733 (2 RCTs) | ⊕⊕⊕⊝ MODERATE 1 | Absolute risk difference: 14% higher (95% CI 9% higher to 18% higher) Relative percentage change: 25% (95% CI 18% higher to 33% higher) |
Anti‐TNF discontinuation probably increases the disease activity score slightly |
| Proportion of participants with persistent remission Assessed with: DAS28 < 2.6 (remission) Follow‐up: range 28 weeks to 52 weeks | RR values range from 0.16 to 0.77. Absolute risk differences range from 15% lower to 68% lower. | 1188 (6 RCTs) | ⊕⊕⊝⊝ LOW 2 | Data not pooled due to heterogeneity. | Anti‐TNF discontinuation may reduce the proportion of participants with persistent remission | ||
| Proportion participants that switched to another biologic due to loss of response ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | No studies were found that evaluated the proportion of participants that switched to another biologic due to persistent loss of response. | |
| Proportion participants with minimal radiographic progression Assessed with: mSvdH > 0.5 Follow‐up: mean 52 weeks | 105 per 1000 | 178 per 1000 (116 to 273) | RR 1.69 (1.10 to 2.59) | 549 (3 RCTs) | ⊕⊕⊕⊕ HIGH | Absolute risk difference: 7% higher (95% CI 1% higher to 17% higher) Relative percentage change: 69% higher (95% CI 10% higher to 159% higher) NNTH: 15 (95% CI 6 to 100) |
Anti‐TNF discontinuation increases the proportion participants with minimal radiographic progression > 0.5 mSvdH point. |
| Function Assessed with: Health Assessment Questionnaire Scale from 0 to 3; higher scores indicate worse functioning Follow‐up: range 28 weeks to 52 weeks | The mean function was 0.52 | MD 0.18 higher (0.05 higher to 0.31 higher) | ‐ | 1498 (4 RCTs) | ⊕⊕⊝⊝ LOW 2 | Absolute risk difference: 6% higher (95% CI 2% higher to 17% higher) Relative percentage change: 26% higher (95% CI 7% higher to 44% higher) |
Anti‐TNF discontinuation may lead to a slight deterioration in function. |
|
Number of serious adverse events Follow‐up: range 28 weeks to 52 weeks |
57 per 1000 | 74 per 1000 (47 to 116) | RR 1.29 (0.82 to 2.03) | 2095 (8 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 3 4 | Absolute risk difference: 2% higher (95% CI 1% lower to 6% higher) Relative percentage change: 29% higher (95% CI 18% lower to 103% higher) NNTH: not applicable (not statistically significant) |
It is uncertain whether anti‐TNF discontinuation influences the number of serious adverse events because the certainty of the evidence is very low and because of imprecision. |
|
Withdrawals due to adverse events Follow‐up: range 28 weeks to 52 weeks |
27 per 1000 | 39 per 1000 (20 to 76) | RR 1.46 (0.75 to 2.84) | 1116 (4 RCTs) | ⊕⊕⊕⊝ MODERATE 3 | Absolute risk difference: 1% higher (95% CI 1% lower to 5% higher) Relative percentage change: 46% (95% CI 25% lower to 184% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF discontinuation probably slightly increases the number of withdrawals due to adverse events. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DAS28: disease activity score in 28 joints; LOCF: last observation carried forward; MD: mean difference; mSvdH: modified Sharp van der Heijde; NNTH: number needed to treat for an additional harmful outcome; RCT: randomised controlled trial: RR: risk ratio; TNF: tumour necrosis factor | |||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
1Downgraded one level due to heterogeneity (I2=61% for disease activity score and I2=31% for number of serious adverse events). 2Downgraded two levels due to heterogeneity (I2=80% for proportion of participants with remission and I2=79% for function). 3Downgraded one level due to imprecision (low number of events). 4Downgraded one level due to concerns about study risk of bias (high risk of selection bias, detection bias, attrition bias and other bias).
Summary of findings 3. Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation.
| Anti‐TNF disease activity–guided dose tapering compared to anti‐TNF continuation for rheumatoid arthritis in patients with low disease activity | |||||||
| Patient or population: people with rheumatoid arthritis with low disease activity using a standard dose of anti‐TNF agents Setting: clinical research centres Intervention: anti‐TNF disease activity–guided dose tapering Comparison: anti‐TNF continuation | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | What happens | |
| Risk with anti‐TNF continuation | Risk with anti‐TNF disease activity–guided dose tapering | ||||||
| Disease activity score Assessed with: DAS28 Scale from 0.9 to 8; higher scores indicate worse disease activity Follow‐up: range 72 weeks to 78 weeks | The mean disease activity score was 2.34 | MD 0.25 higher (0.17 lower to 0.67 higher) | ‐ | 357 (3 RCTs) | ⊕⊕⊝⊝ LOW 1 | Absolute risk difference: 4% higher (95% CI 2% lower to 9% higher) Relative percentage change: 10% higher (95% CI 7% lower to 26% higher) |
Anti‐TNF disease activity–guided dose tapering may result in little or no difference in disease activity score. |
| Proportion of participants with persistent remission Assessed with: DAS28 < 2.6 (remission) Follow‐up: 18 months | 797 per 1000 | 709 per 1000 (597 to 844) | RR 0.89 (0.75 to 1.06) | 180 (1 RCT) | ⊕⊕⊕⊕ HIGH | Absolute risk difference: 9% lower (95% CI 20% lower to 5% higher) Relative percentage change: 10% higher (95% CI 7% lower to 26% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF disease activity–guided dose tapering results in little or no difference in the proportion of participants with persistent remission. |
| Proportion of participants switched to another biologic due to loss of response Follow‐up: 18 months | 76 per 1000 | 47 per 1000 (19 to 117) | RR 0.62 (0.25 to 1.54) | 317 (2 RCTs) | ⊕⊕⊝⊝ LOW 2 | Absolute risk difference: 3% lower (95% CI 6% lower to 4% higher) Relative percentage change: 38% higher (95% CI 75% lower to 54% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF disease activity–guided dose tapering may result in little or no difference in the proportion of participants that switch to another biologic. |
| Proportion of participants with minimal radiographic progression Assessed with: mSvdH score > 0.5 or > 1.0 Follow‐up: mean 18 months | 242 per 1000 | 352 per 1000 (187 to 662) | RR 1.45 (0.77 to 2.73) | 312 (2 RCTs) | ⊕⊕⊝⊝ LOW 3 4 | Absolute risk difference: 11% higher (95% CI 6% lower to 42% higher) Relative percentage change: 45% higher (95% CI 23% lower to 173% higher) NNTH: not applicable (not statistically significant) |
Anti‐TNF disease activity–guided dose tapering may slightly increase the proportion of participants with minimal radiographic progression (mSvdH > 0.5 or > 1.0). |
| Function Assessed with: Health Assessment Questionnaire Scale from 0 to 3; higher scores indicate worse function Follow‐up: mean 18 months | The mean function was 0.4 | MD 0.2 higher (0.02 lower to 0.42 higher) | ‐ | 123 (1 RCT) | ⊕⊕⊕⊝ MODERATE 4 | Absolute risk difference: 7% higher (95% CI 1% lower to 14% higher) Relative percentage change: 33% higher (95% CI 3% lower to 70% higher) |
Anti‐TNF disease activity–guided dose tapering probably leads to a slight deterioration of function. |
|
Number of serious adverse events Follow‐up: 18 months |
129 per 1000 | 160 per 1000 (54 to 477) | RR 1.24 (0.42 to 3.70) | 317 (2 RCTs) | ⊕⊝⊝⊝ VERY LOW 1 2 | Absolute risk difference: 3% higher (95% CI 8% lower to 35% higher) Relative percentage change: 24% higher (95% CI 58% lower to 270% higher) NNTH: not applicable (not statistically significant) |
It is uncertain whether anti‐TNF disease activity‐guided dose tapering influences the number of serious adverse events because the certainty of the evidence is very low and because of imprecision. |
| Withdrawals due to adverse events ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | No studies were found that evaluated the number of withdrawals due to adverse events. | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DAS28: disease activity score in 28 joints; MD: mean difference; mSvdH: modified Sharp van der Heijde; NNTH: number needed to treat for an additional harmful outcome; RCT: randomised controlled trial: RR: risk ratio; TNF: tumour necrosis factor | |||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | |||||||
1Downgraded two levels due to heterogeneity (I2=70% for disease activity score and I2=69% for number of serious adverse events). 2Downgraded two levels due to imprecision (insufficient sample size/low number of events). 3Downgraded one level due to heterogeneity (I2=59%). 4Downgraded one level due to imprecision (insufficient sample size).
Background
Description of the condition
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease characterised by symmetrical joint inflammation that often leads to joint damage. Tumour necrosis factor–blocking (anti‐TNF) agents have proved effective as therapies for RA (Blumenauer 2002; Blumenauer 2003; Navarro‐Sarabia 2005; Ruiz Garcia 2014; Singh 2009; Singh 2010). They improve clinical symptoms and functioning and inhibit joint destruction, and have become an important part of treatment prescribed for RA.
Description of the intervention
Treatment of individuals with RA has been evolving from traditional step‐up regimens to more aggressive step‐down strategies. Pivotal to these changes are the early start of treatment (hit early), the use of combination therapy including steroids with rapid escalation to biologics (hit‐hard), and, most important, frequent assessment of disease activity and treatment modification based on assessment (tight control). Strategies incorporating these concepts lead to the swift achievement of low disease activity or remission in most patients, which prevents joint damage and improves function and quality of life (Schipper 2010). An important disadvantage of the hit‐hard approach compared with the traditional step‐up approach, however, is that the former method does not allow for individual titration of the minimal effective treatment. Indeed, the traditional step‐up approach largely prevents overtreatment, but high(er) disease activity at the beginning of the disease has to be accepted. To prevent overtreatment when high‐dose or multidrug strategies are used, treatment must be tapered down when low disease activity is reached up to the point that disease activity increases again or medication can be stopped. In this way, the minimal effective dose is found and overtreatment is prevented. Optimal dosing of biologics is especially important because of the risk of dose‐dependent adverse effects and the risk of low cost‐effectiveness due to high cost (den Broeder 2010; Ramiro 2017; Singh 2011). The concept of dose reduction has been incorporated into current guidelines for the treatment of RA (Singh 2016; Smolen 2017).
The intervention that is the subject of this review is therefore dose reduction of anti‐TNF agents (by adaptation of dose or dosing interval) or discontinuation or both in people with RA and low disease activity status.
How the intervention might work
Successful dose reduction or discontinuation of anti‐TNF agents can be expected for several reasons. First, amongst patients who seem to respond to treatment with anti‐TNF agents are those who show spontaneous improvement (regression to the mean) (den Broeder 2010; van Vollenhoven 2004); this phenomenon applies to 10% to 30% of all patients, as was shown by proportions of placebo group response (Doherty 2009; St Clair 2004). Second, often concomitant medication is given that might induce a response. Both mechanisms are supported by the fact that a proportion of patients who seem to do well while taking the drug have (neutralising) antibodies (less than 5% to 43%) (Bartelds 2007; Klareskog 2011; Wolbink 2006). Finally, a substantial proportion of patients might need a lower than standard dose for a clinical response (Fautrel 2015; Verhoef 2017). Anti‐TNF agents are registered at the dose that shows the best response for the most patients (top of group level dose‐response curve). However, individual patients might respond to a lower dose as well, which is reflected in response percentages of lower doses in these initial trials (Genovese 2002; Maini 1998; Weinblatt 2003).
Uncontrolled research has shown that down‐titration of anti‐TNF agents can be successful in a relevant proportion of patients. Most data are available for infliximab, adalimumab, and etanercept, and most are derived from discontinuation studies (Brocq 2009; den Broeder 2002; Kavanaugh 2012; Nawata 2008; Saleem 2010; Tanaka 2010; Tanaka 2012; van den Bemt 2008; van der Bijl 2007; van der Maas 2012).
Why it is important to do this review
Although the adverse effects of anti‐TNF agents reported in clinical trials were generally mild in severity, these drugs are associated with unintended effects including increased risk of infection and perhaps a dose‐dependent increased risk of malignancy and rare severe adverse events (Bongartz 2006). The introduction of anti‐TNF agents ‐ and other biological drugs ‐ has also led to an increase in cost because they are much more expensive than traditional disease‐modifying antirheumatic drugs (DMARDs) (van Vollenhoven 2009).
It was appropriate at this time to conduct an update of this Cochrane Review of randomised controlled trials (RCTs) of anti‐TNF down‐titration as well as discontinuation studies, because several new RCTs on this topic are emerging, and additional information on the already included studies has been published.
Objectives
To evaluate the benefits and harms of down‐titration (dose reduction, discontinuation, or disease activity‐guided dose tapering) of anti‐TNF agents (adalimumab, certolizumab pegol, etanercept, golimumab, infliximab) on disease activity, functioning, costs, safety, and radiographic damage compared with usual care in people with RA and low disease activity.
Methods
Criteria for considering studies for this review
Types of studies
We considered all randomised controlled trials (RCTs) and controlled clinical trials (CCTs) (including cluster randomised and cross‐over trials) according to the Cochrane definition comparing down‐titration of tumour necrosis factor–blocking (anti‐TNF) agents versus usual care/no down‐titration for inclusion. The minimal required follow‐up was six months. Both superiority and non‐inferiority trials were included.
Types of participants
People with RA (1987, Arnett 1988, or 2010, Aletaha 2010 RA criteria, or both) American College of Rheumatology (ACR) criteria) using anti‐TNF agents in a standard (or lower) dosing regimen (adalimumab 40 mg every other week, etanercept 50 mg every week or 25 mg twice a week, infliximab 3 mg/kg every eight weeks, golimumab 50 mg every month, certolizumab pegol 200 mg every other week) for longer than six months and with a low disease activity state (clinical judgement of rheumatologist or disease activity score in 28 joints (DAS28) < 3.2; DAS < 2.4; Clinical Disease Activity Index (CDAI) < 10; Simplified Disease Activity Index (SDAI) < 11 or DAS28 < 2.6; DAS < 1.6; CDAI < 2.8; SDAI < 3.3, Aletaha 2005; Fransen 2005, or 2011 ACR/European League Against Rheumatism (EULAR) remission (Felson 2011)).
Types of interventions
Protocolised down‐titration or discontinuation of the anti‐TNF agent for optimal dose finding (not for other reasons, including reduction of side effects, availability, planned surgery, pregnancy). Non‐protocolised change in medication (DMARDs, non‐steroidal anti‐inflammatory drugs (NSAIDs), corticosteroids) was allowed. Comparison was usual care/no down‐titration/continuation of anti‐TNF.
Types of outcome measures
Major outcomes
Mean disease activity score; DAS28/DAS/CDAI/SDAI at six, 12, 18, and 24 months (Aletaha 2005; Prevoo 1995; Smolen 2003; van der Heijde 1990).
Proportion of participants with persistent remission (as specified above) after six, 12, 18, and 24 months.
Proportion of participants that switched to another biologic due to persistent loss of response, refractory to re‐instalment of the tapered anti‐TNF in the intervention group.
Proportion of participants with minimal radiographic progression, as measured by Larsen (Larsen 1973), Sharp (Sharp 1971), or modified Sharp‐van der Heijde score (mSvdH score) (van der Heijde 2000).
Function (as measured by Health Assessment Questionnaire (HAQ)/Arthritis Impact Measurement Scale (AIMS)).
Number of serious adverse events.
Withdrawals due to adverse events.
Minor outcomes
Proportion of participants with a flare (or loss of response) (defined as any composite disease activity index–based flare criteria) during follow‐up time.
Quality of life as measured by Short Form (SF) Health Survey‐12/36, Health Utilities Index (HUI), or EuroQoL Quality of Life Scale (EQ‐5D).
Costs (direct (e.g. medication, consultations, travel costs) and indirect (e.g. health‐related absenteeism)).
Decremental cost‐effectiveness ratio (difference in costs divided by difference in quality of life expressed as utility, thus the potential savings when accepting the loss of one quality‐adjusted life year (QALY)).
Time to flare.
Change in other medication (including DMARDs, NSAIDs, corticosteroids).
Search methods for identification of studies
Electronic searches
We searched the following electronic databases: MEDLINE (1946 to 29 March 2018), Embase (1974 to 29 March 2018), Web of Science (1945 to 2018) and the Cochrane Central Register of Controlled Trials (CENTRAL) 2018 issue 3. The specific search strategy for each of the databases is shown in the appendices (Appendix 1; Appendix 2; Appendix 3; Appendix 4). Our search was not limited by language, year of publication, or publication type. The search period for all databases extended from inception to September 2013 for the original review, and from 2013 to 29 March 2018 for the update.
Searching other resources
We searched proceedings of conferences from 2005 to 2017 of the ACR and from 2005 to 2017 of the European League Against Rheumatism (EULAR) for abstracts of RCTs and CCTs. We searched reference lists of identified clinical trials and performed citation tracking of the included trials in the ISI Web of Knowledge citation index. We searched trial registries for completed and ongoing trials (Appendix 5). We contacted experts (first authors of included studies) to ask about additional trials.
Data collection and analysis
Selection of studies
We selected studies based on the inclusion criteria outlined in the Criteria for considering studies for this review section. Two review authors (NvH and BJFvdB for the original review; LMV and BJFvdB for the update) independently screened titles and abstracts for inclusion, obtaining full articles if necessary. Any differences were resolved by discussion and consensus or by consultation with a third review author (AAdB) if needed. In case the same study population was described in more than one publication, all publications were used, but for the analysis, all were grouped, with the most informative publication as the primary reference and with other publications as secondary references. We recorded reasons for exclusion of studies.
Data extraction and management
Two review authors (NvH and BJFvdB for the original review; LMV and BJFvdB for the update) independently abstracted data from each study using a data extraction form. Any differences were resolved by discussion and consensus or by consultation with a third review author (AAdB) if needed. We pilot‐tested the data extraction form on a selection of trials. If necessary, we contacted the authors of a given study to ask for missing data.
We extracted the following data.
General study information: first author, author affiliation, publication source, publication year, and source of funding.
Study characteristics: design, setting, participant selection, method of randomisation, allocation procedure, blinding, inclusion/exclusion criteria, and study duration.
Population characteristics: age, sex, diagnostic criteria, disease duration, DMARD comedication, previous DMARD use, previous anti‐TNF use, rheumatoid factor status, anti–cyclic citrullinated peptide (CCP) status, disease activity state, total number of participants screened, total number of participants recruited, total number of participants randomly assigned, total number of participants followed, and numbers in each group.
Intervention characteristics: anti‐TNF agent, type of intervention (dose reduction/interval widening/discontinuation), treatment comparators.
Outcome measures as noted above.
Analysis: statistical technique used, intention‐to‐treat analyses and/or per‐protocol analyses used.
Results with number, mean and standard deviation.
Assessment of risk of bias in included studies
Two review authors (NvH and BJFvdB for the original review; LMV and BJFvdB for the update) assessed risk of bias in the included studies in accordance with the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Appendix 6) (Higgins 2011).
We assessed the following 'Risk of bias' domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment.
Incomplete outcome data.
Selective reporting.
Other sources of bias (baseline imbalance in possible prognostic variables: DMARD comedication, duration of anti‐TNF use, and disease duration).
We judged each of these domains as having low, high, or unclear risk of bias.
Measures of treatment effect
We analysed the results of the included studies using Review Manager 2014. Continuous data were expressed as mean differences (MDs) or standardised mean differences (SMDs). Dichotomous data were expressed as risk ratios (RRs). Rates were expressed as rate ratios (RaRs). We summarised data in meta‐analyses if the studies were sufficiently homogeneous, both clinically and statistically.
Unit of analysis issues
The participant was the unit of analysis. Post‐hoc, it was chosen to pool the data from the two dose reduction arms in the study by Ibrahim 2017 (OPTTIRA) for outcomes in which data from multiple studies could be pooled because this facilitated comparison with the 50% dose reduction applied in all other included dose reduction studies (mean dose reduction of 33% and 66% being 50%).
Dealing with missing data
We accepted missing clinical data in trials when they represented less than 20% of findings. We planned to perform a sensitivity analysis if more than 20% of the data from a given study were missing in order to explore the impact of including or excluding such studies. We attempted to obtain missing information on parameter variability by contacting the authors of the trial. In the event that study authors were not able or were unwilling to provide this information, it was estimated from ranges if provided or from comparable trials.
Assessment of heterogeneity
We evaluated heterogeneity first clinically by considering comparability across trials on the following variables: type of intervention (dose reduction/discontinuation/disease activity–guided dose tapering), type of anti‐TNF agent, duration of anti‐TNF use, baseline disease activity (low disease activity versus remission), disease duration, DMARD comedication, and presence of anti‐TNF rescue strategy. We examined forest plots and tested for heterogeneity using the Chi2 test with a P < 0.10 indicating significant heterogeneity. We used the I2 statistic to describe the percentage of variability in effect estimates that is due to heterogeneity rather than to chance (Higgins 2003). A value greater than 50% may indicate substantial heterogeneity (Higgins 2011). If we detected significant heterogeneity (I2 > 80%), we did not pool data but performed subgroup analyses in an attempt to explain the heterogeneity.
Assessment of reporting biases
Publication bias implies that studies that report favourable results are more likely to be published than those describing negative or inconclusive (non‐significant) results, leading to a bias in the overall published literature. To minimise the effect of selective reporting of results, we searched trial registries for completed but unpublished studies. We planned to use a funnel plot to assess potential publication bias. However, due to the small number of studies, the funnel plot was not informative. We also searched the trial registries for ongoing studies that are potentially interesting for a future update of this review (see Characteristics of ongoing studies for details), and for additional data on included studies.
We assessed reporting bias at the outcome level by using published protocols of the studies along with published results of the study to compare outcomes intended to be analysed with those actually analysed.
Data synthesis
When possible, we analysed data using an intention‐to‐treat model and, for non‐inferiority studies, by also using a per‐protocol model. Our reason for this was that intention‐to‐treat analyses can lead to false conclusions of non‐inferiority in non‐inferiority trials. We analysed outcomes of included studies using a random‐effects model.
Subgroup analysis and investigation of heterogeneity
We planned that if sufficient data were available we would perform subgroup analyses for the following candidate effect modifiers: type of intervention (dose reduction/discontinuation/disease activity–guided dose tapering), type of anti‐TNF agent, duration of anti‐TNF use, baseline disease activity (low disease activity versus remission), disease duration, DMARD comedication, and presence of anti‐TNF rescue strategy.
Sensitivity analysis
We planned to perform the following sensitivity analyses when possible.
Effect of risk of bias of included studies.
Effect of imputation of missing data or statistical transformations.
'Summary of findings' tables
We completed three separate 'Summary of findings' tables included in Review Manager 2014 to improve the readability of the review. We examined seven outcomes in a table for each of the three subgroups of down‐titration: (1) dose reduction, (2) discontinuation, and (3) disease activity–guided dose tapering. The study population consisted of people with RA with low disease activity using a standard dose of anti‐TNF. The intervention provided was down‐titration (dose reduction, discontinuation, or disease activity–guided dose tapering). The intervention was compared with usual care (continuation or no formalised dose reduction of anti‐TNF). In addition to the absolute and relative magnitude of effect, the number needed to treat for an additional beneficial outcome (NNTB) and number needed to treat for an additional harmful outcome (NNTH) were calculated by comparing the intervention group with the control group. We used GRADEpro 2015 to conduct an overall grading of the quality of evidence.
The GRADE approach specifies four levels of certainty (high, moderate, low, and very low). The highest certainty rating is given for randomised trial evidence. Randomised trial evidence can be downgraded to moderate, low, or very low depending on the presence of five factors.
Limitations in the design and implementation of available studies suggesting high likelihood of bias.
Indirectness of evidence.
Unexplained heterogeneity or inconsistency of results.
Imprecision of results.
High probability of publication bias.
Results
Description of studies
The results of the search are presented in Figure 1 and are described in detail in the following sections of the review.
1.

Flow chart of study selection.
Results of the search
The previous version of this review included seven studies. Database searches for this update (2013 to March 2018) resulted in 2352 records, and after de‐duplication 1565 search results. Reference checking, contact with experts, and performing additional searches in congress abstract databases and trial registers resulted in 42 additional records. After title and abstract screening of these 1607 records, 21 studies remained. After assessing these 21 studies for eligibility, we identified eight new studies for inclusion in the review. One of the previously included studies, Harigai 2012 (BRIGHT), was retrospectively excluded for this updated version of the review because we considered their method of allocation (at the discretion of the physician) as not random or quasi‐random, which is a prerequisite for the classification as RCT or CCT. Newly found studies that used allocation based on physician or patient preference were also not included in this updated version (Tanaka 2013 (HONOR); Tanaka 2014 (HOPEFUL‐2)). Finally, a total of 14 studies were included in this update of the systematic review, consisting of six old studies and eight new studies. All of the old studies were now available as full text. Of the eight new studies, one was published as abstract and seven as full text.
We contacted the authors of 11 studies to obtain missing data or to clarify methods/results. We received a response from authors of 10 studies.
The total number of participants in the studies included in this review was 3315. Most participants (2111) were included in the eight studies comparing anti‐TNF discontinuation versus anti‐TNF continuation. Six studies (1148 participants) compared anti‐TNF dose reduction versus continuation. Three studies (365 participants) compared disease activity–guided anti‐TNF dose tapering versus continuation. Eleven studies used a superiority design; two studies used a non‐inferiority design; and one study reported an equivalence design.
Included studies
Anti‐TNF dose reduction versus anti‐TNF continuation studies
Design
Six studies compared anti‐TNF fixed‐dose reduction versus anti‐TNF continuation (El Miedany 2016; Ibrahim 2017 (OPTTIRA); Raffeiner 2015; Smolen 2013 (PRESERVE); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). Weinblatt 2017 (C‐EARLY), Smolen 2013 (PRESERVE), and van Vollenhoven 2016 (DOSERA) were randomised, blinded, placebo‐controlled, superiority studies that reported three arms (discontinuation, dose reduction, and continuation). The randomisation ratio was 1:1:1 for Smolen 2013 (PRESERVE) and van Vollenhoven 2016 (DOSERA); for Weinblatt 2017 (C‐EARLY) this was 2:3:2 (stop; dose reduction; continuation). El Miedany 2016, Raffeiner 2015, and Ibrahim 2017 (OPTTIRA) were open‐label superiority studies. The study by Raffeiner 2015 was reported as a prospective long‐term follow‐up study; randomisation was done in a consecutive manner (alternation) in a ratio 1:1, which we defined as quasi‐random, making the study a CCT. The randomisation ratio for Ibrahim 2017 (OPTTIRA) was 1:1:2, and for El Miedany 2016 it was 1:1:1:1:1 (only group 1 and group 5 were relevant for this review).
The duration of the included studies was 6 months in Ibrahim 2017 (OPTTIRA); 40 weeks in van Vollenhoven 2016 (DOSERA); 52 weeks in Smolen 2013 (PRESERVE), El Miedany 2016, and Weinblatt 2017 (C‐EARLY); and a mean follow‐up of 3.5 ± 1.5 years in Raffeiner 2015. The study by Smolen 2013 (PRESERVE) had a total follow‐up of 88 weeks, however 52 weeks of follow‐up were provided after randomisation for dose reduction or continuation of etanercept. The total follow‐up for van Vollenhoven 2016 (DOSERA) was 48 weeks, and 40 weeks of follow‐up were provided after randomisation for dose reduction or continuation of etanercept. The study by Weinblatt 2017 (C‐EARLY) describes period 2 of the C‐EARLY study with a duration of 52 weeks, which was a re‐randomisation of participants from the first period, which also lasted 52 weeks.
Sample size
The sample size for this comparison varied from 50 participants in the study by van Vollenhoven 2016 (DOSERA) (73 participants in total study due to multiple intervention arms) to 404 participants in Smolen 2013 (PRESERVE) (604 participants in total study due to multiple intervention arms).
Setting
Ibrahim 2017 (OPTTIRA) reported that participants were screened at 20 centres in the United Kingdom. The study by Raffeiner 2015 was reported as a single‐centre study in Italy. Smolen 2013 (PRESERVE) was reported to have been conducted in 80 centres in Europe, Latin America, Asia, and Australia. The study by van Vollenhoven 2016 (DOSERA) was performed in 16 rheumatology units in Sweden (5), Denmark (2), Finland (2), Norway (3), Hungary (3), and Iceland (1). Weinblatt 2017 (C‐EARLY) reported that it recruited participants at 103 centres in in Europe, Australia, North America, and Latin America. El Miedany 2016 did not report a specific setting.
Participants
El Miedany 2016 did not provide information on participant characteristics. Most participants were female in the studies by van Vollenhoven 2016 (DOSERA), Smolen 2013 (PRESERVE), Weinblatt 2017 (C‐EARLY), and Raffeiner 2015. Mean age was approximately 47 years in Smolen 2013 (PRESERVE); 49 years in Weinblatt 2017 (C‐EARLY); 56 years in Raffeiner 2015; and 57 years in van Vollenhoven 2016 (DOSERA) and Ibrahim 2017 (OPTTIRA). Disease duration ranged from around 2.6 months in Weinblatt 2017 (C‐EARLY) (median disease duration at baseline of C‐EARLY period 1) to 14 years in Raffeiner 2015 and van Vollenhoven 2016 (DOSERA). Duration of anti‐TNF agents had to be > 3 months in Ibrahim 2017 (OPTTIRA); ≥ 6 months in El Miedany 2016; ≥ 12 months in Raffeiner 2015; and ≥ 14 months in van Vollenhoven 2016 (DOSERA). Smolen 2013 (PRESERVE) started the anti‐TNF agent at study start 36 weeks before randomisation for dose reduction or discontinuation. In the study by Weinblatt 2017 (C‐EARLY), all participants had started certolizumab pegol treatment one year earlier (period 1 of C‐EARLY). El Miedany 2016 and Ibrahim 2017 (OPTTIRA) did not report previous use of DMARDs. Participants in Raffeiner 2015 and Smolen 2013 (PRESERVE) were biologic disease‐modifying antirheumatic drug (bDMARD) naive before the study. Raffeiner 2015 reported a mean (standard deviation (SD)) of 2.4 (1.1) previously used DMARDs in the dose reduction group and 2.4 (1.3) in the continuation group. Participants in Weinblatt 2017 (C‐EARLY) were bDMARD and conventional synthetic disease‐modifying antirheumatic drug (csDMARD) naive. van Vollenhoven 2016 (DOSERA) described that 66% of the participants had used a DMARD other than methotrexate (MTX) before the study.
In all included studies, participants had to have low disease activity, Ibrahim 2017 (OPTTIRA); Smolen 2013 (PRESERVE); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY), or remission, El Miedany 2016; Raffeiner 2015. Duration of low disease activity/remission had to be > 3 months in Ibrahim 2017 (OPTTIRA); ≥ 6 months in El Miedany 2016; ≥ 12 months in Raffeiner 2015; or ≥ 11 months in van Vollenhoven 2016 (DOSERA). Participants in the study by Smolen 2013 (PRESERVE) had to have a mean DAS28 ≤ 3.2 in the 24‐week period before randomisation and a DAS28 ≤ 3.2 at the moment of randomisation. In the study by Weinblatt 2017 (C‐EARLY), participants needed to have a DAS28 ≤ 3.2 12 weeks before randomisation and at the moment of randomisation. All included studies used a DAS28‐based criterion to define low disease activity or remission.
Intervention and comedication
Raffeiner 2015 reported etanercept dose reduction by comparing etanercept 25 mg twice a week versus etanercept 25 mg once a week. Smolen 2013 (PRESERVE) and van Vollenhoven 2016 (DOSERA) reported etanercept dose reduction (25 mg/week) compared with etanercept continuation (50 mg/week). Ibrahim 2017 (OPTTIRA) reported 33% and 66% dose reduction of adalimumab and etanercept versus 100%. El Miedany 2016 reported 50% dose reduction of bDMARDs versus continuation. Weinblatt 2017 (C‐EARLY) reported 50% dose reduction of certrolizumab pegol (200 mg/4 weeks) versus continuation (200 mg/2 weeks). Participants were required to use MTX comedication (dose ranged from 7.5 to 25 mg/week) in Smolen 2013 (PRESERVE) and van Vollenhoven 2016 (DOSERA). In Raffeiner 2015, steroids, NSAIDs, and DMARDs were continued at the same dosages. No intra‐articular steroids were permitted during the study period. Smolen 2013 (PRESERVE) allowed up to three intra‐articular corticosteroid injections during the study. In the study by van Vollenhoven 2016 (DOSERA), participants continued MTX and other medications at the same dose. Participants in Weinblatt 2017 (C‐EARLY) used MTX in the maximum tolerated ("optimised") dose throughout the study. Use of intra‐articular, intramuscular, or intravenous corticosteroids at any dose was prohibited. The maximum allowed dose of oral corticosteroids during the study was ≥ 10 mg/day prednisone or equivalent, and no changes in dose were allowed during the study period. In the study of El Miedany 2016, participants in the relevant study arms used a stable dose of a csDMARD during the trial. No intramuscular or local steroid joint injections were allowed. In five studies (El Miedany 2016; Ibrahim 2017 (OPTTIRA); Raffeiner 2015; van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)), participants could return to their initial dose of anti‐TNF after disease flare. In Smolen 2013 (PRESERVE), no attempt was made to recapture low disease activity by reintroducing etanercept in participants whose condition had deteriorated after etanercept withdrawal.
Outcomes
All studies reported a primary outcome measure. Three studies reported proportion of participants with low disease activity or remission as the primary outcome. Raffeiner 2015 and El Miedany 2016 used DAS28 ≤ 2.6, and Smolen 2013 (PRESERVE) used DAS28 ≤ 3.2. The primary outcome in the study by van Vollenhoven 2016 (DOSERA) was proportion of non‐failures for etanercept 50 mg/week versus placebo. The primary outcome for Ibrahim 2017 (OPTTIRA) was reported to be time to flare. Weinblatt 2017 (C‐EARLY) reported maintenance of low disease activity (disease activity score in 28 joints using erythrocyte sedimentation rate (DAS28‐ESR) of ≤ 3.2) for all 5 consecutive study visits to week 52 without flares as the primary outcome measure. Secondary outcomes reported in the included studies were very different. None of the included studies provided data on costs or change in comedication. All studies were analysed with a (modified) intention‐to‐treat approach.
Anti‐TNF discontinuation versus anti‐TNF continuation studies
Design
Eight of the included studies reported anti‐TNF discontinuation compared with anti‐TNF continuation (Chatzidionysiou 2016 (ADMIRE); Ghiti Moghadam 2016 (POEET); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY); Yamanaka 2016 (ENCOURAGE)). All included studies were randomised controlled superiority studies comparing anti‐TNF discontinuation versus continuation. Smolen 2014 (OPTIMA), Smolen 2013 (PRESERVE), van Vollenhoven 2016 (DOSERA), Pavelka 2017, and Weinblatt 2017 (C‐EARLY) were blinded placebo‐controlled studies. The other studies were open‐label studies (Chatzidionysiou 2016 (ADMIRE); Ghiti Moghadam 2016 (POEET); Yamanaka 2016 (ENCOURAGE)). Chatzidionysiou 2016 (ADMIRE) was reported to be a pilot study. Smolen 2013 (PRESERVE), van Vollenhoven 2016 (DOSERA), and Weinblatt 2017 (C‐EARLY) reported three arms (both discontinuation and dose reduction compared with continuation).
Smolen 2013 (PRESERVE) and van Vollenhoven 2016 (DOSERA) reported a 1:1:1 randomisation ratio, and Weinblatt 2017 (C‐EARLY) a randomisation ratio of 2:3:2. Chatzidionysiou 2016 (ADMIRE), Pavelka 2017, Smolen 2014 (OPTIMA), and Yamanaka 2016 (ENCOURAGE) reported a 1:1 randomisation ratio. Ghiti Moghadam 2016 (POEET) randomised in a ratio of 2:1 (discontinuation versus continuation). Smolen 2013 (PRESERVE), Smolen 2014 (OPTIMA), Pavelka 2017, and van Vollenhoven 2016 (DOSERA) reported a "run‐in" period in which anti‐TNF treatment was given open‐label, before randomisation was provided for anti‐TNF continuation, discontinuation, or dose reduction in a double‐blind phase.
The duration of the included studies was 48 weeks for van Vollenhoven 2016 (DOSERA) (40 weeks double‐blind period); 52 weeks for Chatzidionysiou 2016 (ADMIRE), Ghiti Moghadam 2016 (POEET), and Pavelka 2017 (28 weeks double‐blind period). Weinblatt 2017 (C‐EARLY) reported a total follow‐up of 104 weeks, in which the second 52‐week double blind period was of interest for this review. Smolen 2014 (OPTIMA) and Smolen 2013 (PRESERVE) reported a total follow‐up of 78 weeks and 88 weeks, respectively; however, both described 52‐week follow‐up after randomisation for discontinuation or continuation of the anti‐TNF agent. Yamanaka 2016 (ENCOURAGE) described a period of one year in which participants were treated with open‐label etanercept and MTX before they were randomised to open‐label continuation or discontinuation.
Sample size
The sample size varied from 31 participants in Chatzidionysiou 2016 (ADMIRE) to 817 in Ghiti Moghadam 2016 (POEET).
Setting
All eight studies were reported as multicentre studies. Chatzidionysiou 2016 (ADMIRE) was performed in several hospitals in Sweden, and Ghiti Moghadam 2016 (POEET) in 47 rheumatology centres throughout the Netherlands. Smolen 2013 (PRESERVE) reported that the study was conducted in 80 centres in Europe, Latin America, Asia, and Australia. van Vollenhoven 2016 (DOSERA) recruited participants at 16 rheumatology units in Sweden (5), Denmark (2), Finland (2), Norway (3), Hungary (3), and Iceland (1). Pavelka 2017 was conducted at 61 centres in 19 countries in Africa, Asia, Central and Eastern Europe, Latin America, and the Middle East. Smolen 2014 (OPTIMA) reported 161 sites around the world. Weinblatt 2017 (C‐EARLY) was conducted at 103 participating sites in Europe, Australia, North America, and Latin America. Yamanaka 2016 (ENCOURAGE) was a co‐operation of rheumatology institutes/departments in Japan and Korea.
Participants
Six studies reported a minimum age of 18 years for inclusion (Chatzidionysiou 2016 (ADMIRE); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). Ghiti Moghadam 2016 (POEET) was reported to include people 18 years of age or older. Smolen 2013 (PRESERVE) reported an upper age limit (70 years) for inclusion. Yamanaka 2016 (ENCOURAGE) did not report any age criteria. The mean age of participants varied from around 47 in Pavelka 2017 and Smolen 2013 (PRESERVE) to early 60s in Chatzidionysiou 2016 (ADMIRE) and Ghiti Moghadam 2016 (POEET). Most participants in the included studies were female. Mean disease duration ranged from seven to 14 years, except in Smolen 2014 (OPTIMA), in which the mean disease duration was only 3.9 months; Weinblatt 2017 (C‐EARLY), in which median disease duration was around 2.7 months (measured one year before randomisation); and Yamanaka 2016 (ENCOURAGE), in which mean disease duration was two years. Duration of the anti‐TNF agent had to be ≥ 6 months in Chatzidionysiou 2016 (ADMIRE); ≥ 1 year in Ghiti Moghadam 2016 (POEET); and ≥ 14 months in van Vollenhoven 2016 (DOSERA). Pavelka 2017, Smolen 2014 (OPTIMA), Smolen 2013 (PRESERVE), and Yamanaka 2016 (ENCOURAGE) started the anti‐TNF agent at study start, 24 weeks, 26 weeks, 36 weeks, and 1 year, respectively before randomisation for dose reduction or discontinuation. In the study by Weinblatt 2017 (C‐EARLY), participants were treated with certolizumab pegol (blinded) one year before randomisation for dose reduction or discontinuation. Participants in Smolen 2013 (PRESERVE), Smolen 2014 (OPTIMA), and Weinblatt 2017 (C‐EARLY) were bDMARD naive before study start. Smolen 2014 (OPTIMA) reported that 8.8% of participants in the discontinuation group and 9.5% in the continuation group had used ≥ 1 DMARD. Chatzidionysiou 2016 (ADMIRE) reported a median of 1 (interquartile range (IQR) 0 to 1) number of previous bDMARDs and 2 (IQR 1 to 3) previous csDMARDs. In the study by Ghiti Moghadam 2016 (POEET), 13.4% of participants in the discontinuation group and 15% in the continuation group had previously used a bDMARD. In Pavelka 2017, 34% of participants in the discontinuation group had previously used a csDMARD versus 38% in the continuation group. van Vollenhoven 2016 (DOSERA) reported that 66% of all participants had used a DMARD other than MTX before study start. Yamanaka 2016 (ENCOURAGE) did not report on prior DMARD use.
Participants in all included studies had to have low disease activity, Ghiti Moghadam 2016 (POEET); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY), or remission, Chatzidionysiou 2016 (ADMIRE); Yamanaka 2016 (ENCOURAGE). The duration of low disease activity had to be 4 weeks in Smolen 2014 (OPTIMA); ≥ 3 months in Chatzidionysiou 2016 (ADMIRE); ≥ 6 months in Ghiti Moghadam 2016 (POEET); or ≥ 11 months in van Vollenhoven 2016 (DOSERA). Participants in the study by Smolen 2013 (PRESERVE) had to have a mean DAS28 ≤ 3.2 in the 24‐week period before randomisation and a DAS28 ≤ 3.2 at the moment of randomisation. In the study by Weinblatt 2017 (C‐EARLY), participants needed to have a DAS28 ≤ 3.2 12 weeks before randomisation and at the moment of randomisation. In Yamanaka 2016 (ENCOURAGE), participants had to have a DAS < 2.6 at 6 and 12 months after study start. Pavelka 2017 reported that participants had to have low disease activity after period 1 (24 weeks after study start). All included studies used a DAS28‐based criterion to define low disease activity or remission.
Intervention and comedication
Smolen 2013 (PRESERVE), van Vollenhoven 2016 (DOSERA), Yamanaka 2016 (ENCOURAGE), and Pavelka 2017 reported etanercept discontinuation compared with etanercept continuation. The studies by Chatzidionysiou 2016 (ADMIRE) and Smolen 2014 (OPTIMA) reported adalimumab discontinuation compared with adalimumab continuation. Ghiti Moghadam 2016 (POEET) reported discontinuation of all anti‐TNF agents versus anti‐TNF continuation. Weinblatt 2017 (C‐EARLY) reported discontinuation of certolizumab pegol compared to continuation of the standard dose.
Participants in most included studies were required to use MTX comedication (dose ranged from 6 to 25 mg/week). Ghiti Moghadam 2016 (POEET) included participants using any csDMARD comedication. Participants included in Smolen 2014 (OPTIMA) were MTX naive at the start of the study (26 weeks before randomisation for discontinuation or continuation of adalimumab). Seven studies stated that participants could restart the anti‐TNF after disease flare (Chatzidionysiou 2016 (ADMIRE); Ghiti Moghadam 2016 (POEET); Pavelka 2017; Smolen 2014 (OPTIMA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY); Yamanaka 2016 (ENCOURAGE)). The study by Smolen 2013 (PRESERVE) allowed up to three intra‐articular corticosteroid injections during the study; however, no attempt was made to recapture low disease activity by reintroducing etanercept in participants whose condition had deteriorated after etanercept withdrawal.
Outcomes
All studies reported a primary outcome measure; for most studies this was proportion of participants with low disease activity or remission. All studies used DAS28‐based criteria, but different definitions were employed. Chatzidionysiou 2016 (ADMIRE) and Yamanaka 2016 (ENCOURAGE) used DAS28 remission (< 2.6). Smolen 2013 (PRESERVE) and Pavelka 2017 used DAS28 low disease activity (≤ 3.2 for Smolen 2013 (PRESERVE) and < 3.2 for Pavelka 2017). Weinblatt 2017 (C‐EARLY) reported maintenance of low disease activity (DAS28‐ESR of ≤ 3.2) for all 5 consecutive study visits to week 52 without flares as the primary outcome measure. Ghiti Moghadam 2016 (POEET) reported proportion of participants with a flare (DAS28 ≥ 3.2 plus an increase > 0.6) as the primary outcome. The primary outcome in the study by van Vollenhoven 2016 (DOSERA) was proportion of non‐failure. The primary outcome in Smolen 2014 (OPTIMA) was the proportion of participants with both low disease activity and radiographic non‐progression; however, this concerned a comparison of study groups that was not of interest for this review (adalimumab continuation versus methotrexate monotherapy). Secondary outcomes reported in the included studies concerned many different domains, including participant‐reported outcomes (function, quality of life), radiographic outcomes, number of flares, relapse‐free survival, and safety outcomes. None of the included studies provided data on costs or change in comedication. All studies were analysed with a (modified) intention‐to‐treat approach.
Disease activity–guided dose tapering until stop versus anti‐TNF continuation studies
Design
Three studies compared disease activity–guided anti‐TNF dose tapering with anti‐TNF continuation (Bejerano 2016 (OPTIBIO) (abstract only); Fautrel 2016 (STRASS); van Herwaarden 2015 (DRESS)). All studies were open‐label RCTs. van Herwaarden 2015 (DRESS) and Bejerano 2016 (OPTIBIO) were reported to be non‐inferiority studies. Fautrel 2016 (STRASS) reported an equivalence design. Randomisation ratio was 2:1 (dose tapering versus continuation) in van Herwaarden 2015 (DRESS) and 1:1 in Fautrel 2016 (STRASS) and Bejerano 2016 (OPTIBIO). Study duration was 1 year for Bejerano 2016 (OPTIBIO) and 18 months for van Herwaarden 2015 (DRESS) and Fautrel 2016 (STRASS).
Sample size
The sample size varied from 48 in Bejerano 2016 (OPTIBIO) (66 in the total study, which also included other biologics besides anti‐TNF) to 180 in van Herwaarden 2015 (DRESS). The projected sample size for the study by Fautrel 2016 (STRASS) was 250 participants; however, only 137 participants were included. The abstract on Bejerano 2016 (OPTIBIO) reported preliminary data.
Setting
van Herwaarden 2015 (DRESS) and Fautrel 2016 (STRASS) were reported to be multicentre studies. van Herwaarden 2015 (DRESS) included patients from two hospitals in the Netherlands, and Fautrel 2016 (STRASS) recruited participants at 22 rheumatology departments in France and one department in Monaco. Bejerano 2016 (OPTIBIO) was a monocentre study conducted in a hospital in Spain.
Participants
The abstract by Bejerano 2016 (OPTIBIO) provided no information on participant characteristics of anti‐TNF users only. The mean age of participants was 56 years in the study by Fautrel 2016 (STRASS) and 59 years in the study by van Herwaarden 2015 (DRESS). Most participants were female in Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS). Mean disease duration at baseline was about 10 years for both Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS). Participants in van Herwaarden 2015 (DRESS) had a median of 2 (IQR 1 to 3) previous DMARDs and 0 (IQR 0 to 1) previous anti‐TNF agents. Fautrel 2016 (STRASS) reported a mean (SD) of 2.7 (1.7) previous DMARDs, and 24% of participants had previously used a bDMARD.
The duration of anti‐TNF agents had to be ≥ 6 months in van Herwaarden 2015 (DRESS) and > 1 year in Fautrel 2016 (STRASS). Bejerano 2016 (OPTIBIO) reported no minimal duration of anti‐TNF use. Participants in Bejerano 2016 (OPTIBIO) had to have clinical remission (DAS < 2.6, SDAI < 5, or ACR/EULAR 2011 criteria) for ≥ 6 months. Participants in Fautrel 2016 (STRASS) needed to have a DAS28 ≤ 2.6 for ≥ 6 months with no structural damage progression. Participants in van Herwaarden 2015 (DRESS) had to have stable low disease activity (DAS28 < 3.2) at two subsequent visits.
Intervention and comedication
All three studies reported disease activity–guided dose tapering. Bejerano 2016 (OPTIBIO) included all anti‐TNF agents, while Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS) included adalimumab and etanercept. Dose tapering in Fautrel 2016 (STRASS) was done by increasing the interval between two subcutaneous injections by 50% every three months up to a complete stop in the fourth step; if DAS28 remission (DAS28 ≤ 2.6) was not maintained, dose tapering was suspended or was reversed to the previous interval based on DAS28 level. The dose reduction strategy in van Herwaarden 2015 (DRESS) consisted of stepwise increases of the time interval between injections every three months until complete stop in the third step. In the instance of a flare (∆DAS28‐CRP score > 1.2, or ∆DAS28‐CRP > 0.6, and a current score of ≥ 3.2), the last effective interval was reinstated. The dose reduction strategy in Bejerano 2016 (OPTIBIO) consisted of a stepwise increase in interval every year with withdrawal as the third step. In case of flare (DAS28 > 2.6 or SDAI > 5 or ACR/EULAR criteria not fulfilled), participants returned to the standard dose. In all studies, the dose‐tapering intervention was compared with unchanged continuation of the anti‐TNF agents.
Outcomes
All studies reported a primary outcome measure that was based on the DAS28 score. Fautrel 2016 (STRASS) reported standardised difference of DAS28 slopes based on a linear mixed‐effects model as the primary outcome compared to an equivalence margin of ±30%. For van Herwaarden 2015 (DRESS), this was difference in proportions of participants with major flare (DAS28‐CRP‐based flare longer than three months) compared with a non‐inferiority margin of 20%. The primary outcome measure in Bejerano 2016 (OPTIBIO) was the proportion of participants that maintained clinical remission after one year. The abstract for this study did not report on secondary outcome measures. Several secondary measures were reported in Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), including function, radiographic progression, and adverse events. Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS) primarily performed a per‐protocol analysis and additionally performed an intention‐to‐treat analysis. Bejerano 2016 (OPTIBIO) did not specify their analysis approach, which was therefore labelled as intention‐to‐treat.
Excluded studies
We excluded 29 articles from this review (15 for the original publication and 14 from the updated version). Fourteen articles (concerning 13 studies) reported anti‐TNF down‐titration without an anti‐TNF continuation control arm (Awan 2011; Bejarano 2010; Detert 2013 (HIT‐HARD); Emery 2013 (PRIZE); Heimans 2016 (IMPROVED); Klarenbeek 2011; Oba 2017 (RRRR study); Quinn 2005; Seddighzadeh 2014 (NORD‐STAR); Smolen 2012 (CERTAIN); van den Broek 2011; van der Kooij 2009; Villeneuve 2012; Wiland 2016 (PRIZE)). In four studies, allocation to anti‐TNF continuation or discontinuation was based on patient or physician preference (Harigai 2012 (BRIGHT); Rakieh 2013; Tanaka 2013 (HONOR); Tanaka 2014 (HOPEFUL‐2)), therefore these study were not classified as RCT or CCT. Tada 2012 (PRECEPT) reported low‐dose versus standard‐dose etanercept from study start. In the study by Haschka 2016 (RETRO), participants were randomised to dose reduction or discontinuation of all DMARDs, therefore the intervention was too broad for this review. The studies by Kobelt 2011 and Kobelt 2014 provided data from a Markov model. Aletaha 2010, Ichikawa 2007, and Keystone 2003 were overview articles. Ramírez‐Herráiz 2013 was a retrospective study; CADTH Report 2014 described a literature study; and Greenberg 2014 was a cohort study. In the study by Haraoui 2014, no doses below standard dose were investigated.
See Characteristics of excluded studies for more information.
Risk of bias in included studies
See Characteristics of included studies for 'Risk of bias' tables with information on all aspects of risk of bias. Graphic summaries of the risk of bias in included studies are shown in Figure 2 and Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Seven included studies described an adequate random sequence generation and allocation concealment procedure, resulting in an assessment of low risk of selection bias (Fautrel 2016 (STRASS); Ibrahim 2017 (OPTTIRA); Pavelka 2017; Smolen 2013 (PRESERVE); van Herwaarden 2015 (DRESS); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). The precise method of random sequence generation was not described in three studies (Chatzidionysiou 2016 (ADMIRE); Smolen 2014 (OPTIMA); Yamanaka 2016 (ENCOURAGE)). Ghiti Moghadam 2016 (POEET) did not describe allocation concealment. The methods of randomisation and allocation concealment were not described in the abstract by Bejerano 2016 (OPTIBIO) and the study by El Miedany 2016. The study by Raffeiner 2015 described alternation as the method of randomisation, which resulted in a judgement of high risk of selection bias.
Blinding
Five studies were reported to be placebo controlled (Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). The remaining nine studies were open‐label (Bejerano 2016 (OPTIBIO); Chatzidionysiou 2016 (ADMIRE); El Miedany 2016; Fautrel 2016 (STRASS); Ghiti Moghadam 2016 (POEET); Ibrahim 2017 (OPTTIRA); Raffeiner 2015; van Herwaarden 2015 (DRESS); Yamanaka 2016 (ENCOURAGE)); five of these described blinding of X‐ray reading (Fautrel 2016 (STRASS); Ibrahim 2017 (OPTTIRA); Raffeiner 2015; van Herwaarden 2015 (DRESS); Yamanaka 2016 (ENCOURAGE), and the study by Fautrel 2016 (STRASS) also reported blinded DAS28 measurements, which resulted in an assessment of low risk of detection bias.
Incomplete outcome data
We used three criteria for judging this item: intention‐to‐treat analyses, imputation of missing data, and attrition rate.
Most studies performed an intention‐to‐treat analysis (Chatzidionysiou 2016 (ADMIRE); Fautrel 2016 (STRASS); Ghiti Moghadam 2016 (POEET); Ibrahim 2017 (OPTTIRA); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Herwaarden 2015 (DRESS); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY); Yamanaka 2016 (ENCOURAGE)). The abstract by Bejerano 2016 (OPTIBIO) and the studies by Raffeiner 2015 and El Miedany 2016 did not report on the type of analysis. Five studies did not report any imputation of missing data (El Miedany 2016; Fautrel 2016 (STRASS); Ibrahim 2017 (OPTTIRA); Raffeiner 2015; van Herwaarden 2015 (DRESS)). Smolen 2013 (PRESERVE) reported a modified non‐responder imputation analysis in which participants who discontinued early due to poor efficacy were imputed as non‐responders for all time points; all other participants were analysed by the last observation carried forward (LOCF) method. All other postbaseline analyses were based on the LOCF method (except radiographic endpoints). Chatzidionysiou 2016 (ADMIRE) used non‐responder imputation for participants with no available DAS28 at the time of the primary outcome (this included most participants who had a flare in the adalimumab discontinuation group). Ghiti Moghadam 2016 (POEET) reported imputation of DAS28 components based on the expectation‐maximisation algorithm using the participant's values of the remaining components of the DAS28. Pavelka 2017 reported that efficacy analyses were conducted in the full analysis set population in each period using the last observation before rescue carried forward approach. The study by Smolen 2014 (OPTIMA) used non‐responder imputation for the primary endpoint, and non‐responder imputation and LOCF, or both, for additional clinical outcomes; LOCF was used for functional outcomes. Markov chain Monte Carlo method was used to impute missing radiographic data 10 times (multiple imputation). Weinblatt 2017 (C‐EARLY) reported that missing data from participants who entered period 2 but withdrew before the end of the study were imputed using non‐responder imputation for the primary and key secondary endpoints. Radiographic analyses used linear extrapolation. In post hoc analyses, LOCF imputation was used for the proportions of participants achieving low disease activity, remission, and normative physical function. The study by Yamanaka 2016 (ENCOURAGE) described LOCF to impute missing data. van Vollenhoven 2016 (DOSERA) reported that a non‐responder imputation was applied for dichotomous clinical outcomes. The abstract by Bejerano 2016 (OPTIBIO) did not describe the procedure for handling missing data. Most studies reported some participants that were lost to follow‐up (Chatzidionysiou 2016 (ADMIRE); El Miedany 2016; Fautrel 2016 (STRASS); Ghiti Moghadam 2016 (POEET); Ibrahim 2017 (OPTTIRA); Pavelka 2017; Raffeiner 2015; Smolen 2014 (OPTIMA); van Herwaarden 2015 (DRESS); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). The study by Smolen 2013 (PRESERVE) reported that fewer participants completed the study in the placebo group than in the etanercept 50 mg and 25 mg groups (141 versus 181 and 175 participants). Yamanaka 2016 (ENCOURAGE) reported high dropout rates in both groups (16/49 in the continuation group and 16/50 in the discontinuation group). The abstract by Bejerano 2016 (OPTIBIO) did not describe completion rate.
Selective reporting
Most studies, with the exception of Raffeiner 2015 and El Miedany 2016, had a study protocol that was available. Bejerano 2016 (OPTIBIO) was published as abstract only, and therefore did not report all prespecified outcomes. All other studies reported the prespecified outcomes (Chatzidionysiou 2016 (ADMIRE); Fautrel 2016 (STRASS); Ghiti Moghadam 2016 (POEET); Ibrahim 2017 (OPTTIRA); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Herwaarden 2015 (DRESS); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY); Yamanaka 2016 (ENCOURAGE)).
Other potential sources of bias
Eight studies appeared to be free of other potential sources of bias (Chatzidionysiou 2016 (ADMIRE); El Miedany 2016; Ibrahim 2017 (OPTTIRA); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); van Herwaarden 2015 (DRESS); van Vollenhoven 2016 (DOSERA)). There was insufficient information in the abstract by Bejerano 2016 (OPTIBIO) to assess this domain. The study by Ghiti Moghadam 2016 (POEET) reported a different flare criterion in their final publication compared to the information in the trial register. No study protocol was present for Raffeiner 2015, but information from an earlier abstract indicated that the inclusion criteria, outcome measures, and duration of follow‐up had changed over time. A lower than anticipated number of participants was included in Yamanaka 2016 (ENCOURAGE), Weinblatt 2017 (C‐EARLY), and Fautrel 2016 (STRASS).
Effects of interventions
See: Table 1; Table 2; Table 3
We have presented study results by type of intervention: (1) dose reduction, (2) discontinuation, and (3) disease activity–guided dose tapering.
Anti‐TNF dose reduction versus anti‐TNF continuation
Major outcomes
See Table 1.
Mean disease activity: Of the six studies included for this comparison (1148 participants), two studies, Smolen 2013 (PRESERVE) and Ibrahim 2017 (OPTTIRA), with 501 participants provided data on mean disease activity (DAS28). Anti‐TNF dose reduction resulted in little or no difference in mean disease activity score after 26 to 52 weeks' follow‐up (mean difference (MD) 0.06, 95% confidence interval (CI) −0.11 to 0.24). We pooled data from the two dose reduction arms in the study by Ibrahim 2017 (OPTTIRA) for this outcome. Analysis 1.1
Proportion persistent remission: Of the six studies included for this comparison (1148 participants), two studies, Smolen 2013 (PRESERVE) and Weinblatt 2017 (C‐EARLY), with 612 participants provided data on persistent remission. Anti‐TNF dose reduction may result in little or no difference in the proportion of participants with persistent remission (DAS28 < 2.6) after 52 weeks (risk ratio (RR) 1.01, 95% CI 0.80 to 1.28). Analysis 1.2
Proportion of participants that switched to another biologic: Of the six studies included for this comparison (1148 participants), only one study with 323 participants provided data on this outcome (Raffeiner 2015). The data showed that anti‐TNF dose reduction may slightly reduce the proportion of participants who are switched to another biologic (RR 0.40, 95% CI 0.17 to 0.93; mean follow‐up period 3.5 ± 1.5 years). This result might be explained by a difference in treatment strategy after flare in the two treatment groups. In the continuation group, a flare resulted in a switch of biologic treatment, while in the dose reduction group the standard dose of etanercept was reinstated first (Raffeiner 2015). Analysis 1.3
Proportion of participants with minimal radiographic progression: Of the six studies included for this comparison (1148 participants), two studies provided data on radiographic progression, Smolen 2013 (PRESERVE) and Weinblatt 2017 (C‐EARLY), with 553 participants. Anti‐TNF dose reduction probably slightly increases the proportion of participants with minimal radiographic progression (mSvdH > 0.5) after 52 weeks (RR 1.22, 95% CI 0.76 to 1.95). Analysis 1.4
Function: Of the six studies included for this comparison (1148 participants), two studies, Smolen 2013 (PRESERVE) and Ibrahim 2017 (OPTTIRA), with 501 participants provided data on this outcome. Anti‐TNF dose reduction does not result in an important deterioration in function (HAQ Disability Index (HAQ‐DI)) after 26 to 52 weeks' follow‐up (MD 0.09, 95% CI 0.00 to 0.19). We pooled data from the two dose reduction arms in the study by Ibrahim 2017 (OPTTIRA) for this outcome. Analysis 1.5
Number of serious adverse events: Of the six studies included for this comparison (1148 participants), five studies with 1084 participants provided data on this outcome (Ibrahim 2017 (OPTTIRA); Raffeiner 2015; Smolen 2013 (PRESERVE); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). Anti‐TNF dose reduction may cause little or no difference inthe number of serious adverse events after 26 to 52 weeks' follow‐up (RR 1.09, 95% CI 0.65 to 1.82). We pooled data from the two dose reduction arms in the study by Ibrahim 2017 (OPTTIRA) for this outcome. Analysis 1.6
Withdrawals due to adverse events: Of the six studies included for this comparison (1148 participants), three studies with 937 participants provided data on this outcome (Raffeiner 2015; Smolen 2013 (PRESERVE); Weinblatt 2017 (C‐EARLY)). Anti‐TNF dose reduction may cause little or no difference in the number of withdrawals due to adverse events after 52 weeks (RR 1.07, 95% CI 0.51 to 2.24). Analysis 1.7
1.1. Analysis.
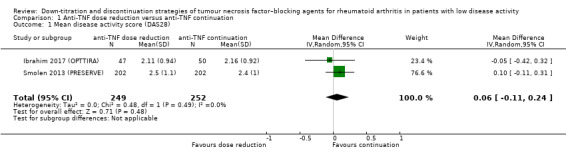
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 1 Mean disease activity score (DAS28).
1.2. Analysis.
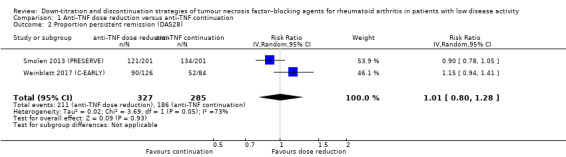
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 2 Proportion persistent remission (DAS28).
1.3. Analysis.
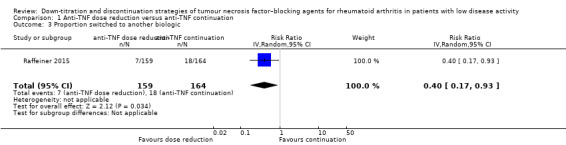
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 3 Proportion switched to another biologic.
1.4. Analysis.
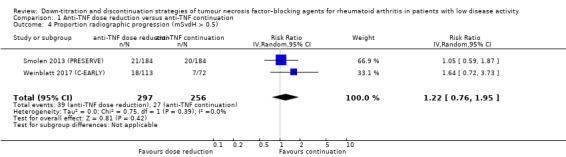
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 4 Proportion radiographic progression (mSvdH > 0.5).
1.5. Analysis.
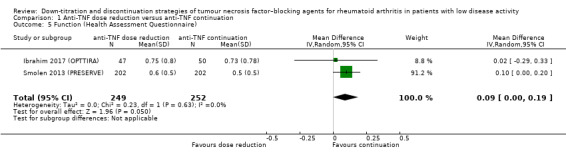
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 5 Function (Health Assessment Questionnaire).
1.6. Analysis.
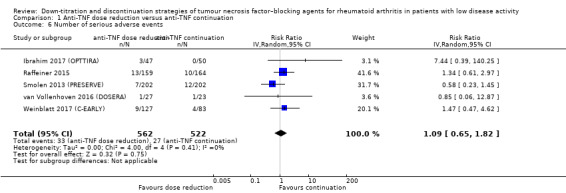
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 6 Number of serious adverse events.
1.7. Analysis.
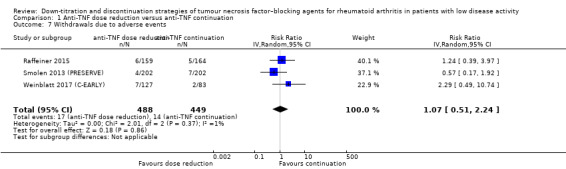
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 7 Withdrawals due to adverse events.
Minor outcomes
Proportion of participants with a flare: Of the six included studies for this comparison (1148 participants), three studies with 357 participants provided data on this outcome (Ibrahim 2017 (OPTTIRA); van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)). The three studies used different criteria for flare. Ibrahim 2017 (OPTTIRA) defined a flare as an increase in DAS28 scores ≥ 0.6 resulting in a DAS28 > 3.2 together with an increase in the swollen joint count; both had to be present on two occasions at least one week apart. An increase in DAS28 score ≥ 1.2 resulting in DAS28 > 3.2 was defined as flare irrespective of changes in swollen joints. van Vollenhoven 2016 (DOSERA) defined a flare as (a) a DAS28‐ESR > 5.1; (b) a DAS28‐ESR > 3.2 and an increase ≥ 1.2 from baseline; (c) DAS28‐ESR > 3.2 and an increase in DAS28 ≥ 0.6 from baseline on two consecutive visits at least one to three weeks apart; or (d) disease progression as determined by either the investigator or disease flare as experienced by the participant. Weinblatt 2017 (C‐EARLY) stated that participants reporting a flare also had to meet the following three criteria at two consecutive visits two weeks apart: 1) an increase in the DAS28‐ESR of ≥ 0.6 above the DAS28‐ESR at week 52; 2) a DAS28‐ESR of > 3.2; and 3) in the investigator’s judgement, an increase in the participant’s RA activity. Furthermore, Ibrahim 2017 (OPTTIRA) included two intervention groups: 33% and 66% dose reduction. Due to this heterogeneity data were not pooled. The studies did not show a difference between the anti‐TNF dose reduction group(s) and the continuation group; risk ratios were found between 0.29 and 1.79.
Quality of life: Of the six studies included for this comparison (1148 participants), two studies, Smolen 2013 (PRESERVE) and Ibrahim 2017 (OPTTIRA), with 501 participants provided data on this outcome. Anti‐TNF dose reduction resulted in little or no difference in mean EQ‐5D after 26 to 52 weeks' follow‐up (MD 0.00, 95% CI −0.04 to 0.03). We pooled data from the two dose reduction arms in the study by Ibrahim 2017 (OPTTIRA) for this outcome.
Costs: None of the six included studies provided data on this outcome.
Decremental cost‐effectiveness ratio: None of the six included studies provided data on this outcome.
Time to flare: Of the six studies included for this comparison (1148 participants), one study with 50 participants provided data on this outcome (van Vollenhoven 2016 (DOSERA)). Median time to failure was 48 weeks in the etanercept 50 mg/week continuation group and 36 weeks in the etanercept 25 mg/week dose reduction group, but no SDs were available.
Change in other medication: None of the six included studies reported data on this outcome.
Anti‐TNF discontinuation versus anti‐TNF continuation
See Table 2.
Major outcomes
Mean disease activity: Of the eight studies (2111 participants) included for this comparison, three studies, Smolen 2013 (PRESERVE), Ghiti Moghadam 2016 (POEET), and Pavelka 2017, with 402, 692, and 331 participants, respectively, provided data on mean disease activity. We pooled data from Pavelka 2017 and Smolen 2013 (PRESERVE). Anti‐TNF discontinuation probably increases the mean disease activity score (DAS28) slightly after 28 to 52 weeks' follow‐up (MD 0.96, 95% CI 0.67 to 1.25). We considered the study by Ghiti Moghadam 2016 (POEET) to be different since participants could return to standard dose in case of flare, and no LOCF was described, therefore the results will reflect the effect of a discontinuation and restarting strategy. This strategy resulted in a small, possibly unimportant increase in mean disease activity score after 52 weeks (MD 0.29, 95% CI 0.14 to 0.44). Analysis 2.1
Proportion persistent remission: Of the eight studies reporting on this comparison (2111 participants), six studies with 1188 participants provided data on the proportion of participants with persistent remission (Chatzidionysiou 2016 (ADMIRE); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); Weinblatt 2017 (C‐EARLY); Yamanaka 2016 (ENCOURAGE)). We were unable to pool data due to heterogeneity. The RR after 28 to 52 weeks varied between 0.16 in Chatzidionysiou 2016 (ADMIRE) and 0.77 in Smolen 2014 (OPTIMA) and Weinblatt 2017 (C‐EARLY). The absolute risk difference varied between 15% fewer in Weinblatt 2017 (C‐EARLY) and 68% fewer in Chatzidionysiou 2016 (ADMIRE). Analysis 2.2
Proportion of participants that switched to another biologic due to persistent loss of response (refractory to re‐instalment of the tapered anti‐TNF in the intervention group): None of the eight included studies provided data on this outcome. Smolen 2013 (PRESERVE) reported that no attempt was made to recapture low disease activity by reintroducing etanercept in participants whose condition had deteriorated after etanercept withdrawal, raising some ethical issues in our view.
Proportion of participants with minimal radiographic progression: Of the eight studies (2111 participants) included for this comparison, three studies, Smolen 2013 (PRESERVE), Weinblatt 2017 (C‐EARLY), and Yamanaka 2016 (ENCOURAGE)), with 549 participants provided data on this outcome. The meta‐analysis showed that anti‐TNF discontinuation increases the proportion of participants with minimal radiographic progression > 0.5 mSvdH point after 52 weeks (RR 1.69, 95% 1.10 to 2.59). Analysis 2.3
Function: Of the eight studies (2111 participants) included for this comparison, four studies with 1498 participants provided data on this outcome (Ghiti Moghadam 2016 (POEET); Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA)). The results showed that anti‐TNF discontinuation may lead to a slight deterioration in function after 28 to 52 weeks' follow‐up (MD 0.18, 95% CI 0.05 to 0.31). Analysis 2.4
Number of serious adverse events: All eight studies included for this comparison provided data on this outcome, with 2095 participants. Due to the very low certainty of the evidence and imprecision of the results, it is uncertain whether anti‐TNF discontinuation influences the number of serious adverse events after 28 to 52 weeks (RR 1.29, 95% CI 0.82 to 2.03). Analysis 2.5
Withdrawals due to adverse events: Of the eight studies (2111 participants) included for this comparison, four studies with 1116 participants provided data on this outcome (Pavelka 2017; Smolen 2013 (PRESERVE); Smolen 2014 (OPTIMA); Weinblatt 2017 (C‐EARLY)). Anti‐TNF discontinuation probably slightly increases the number of withdrawals due to adverse events after 28 to 52 weeks (RR 1.46, 95% CI 0.75 to 2.84). Analysis 2.6
2.1. Analysis.
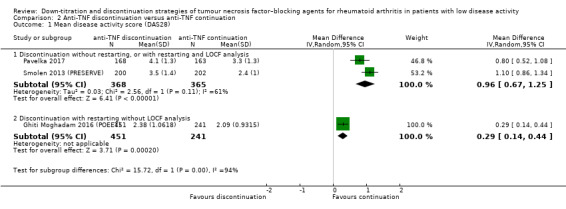
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 1 Mean disease activity score (DAS28).
2.2. Analysis.
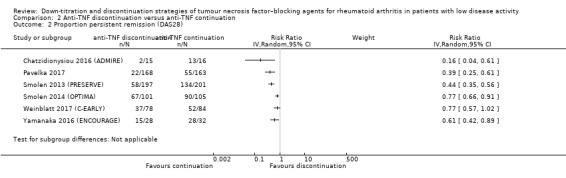
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 2 Proportion persistent remission (DAS28).
2.3. Analysis.
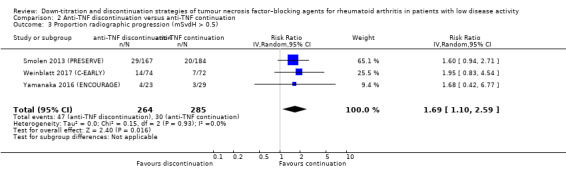
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 3 Proportion radiographic progression (mSvdH > 0.5).
2.4. Analysis.
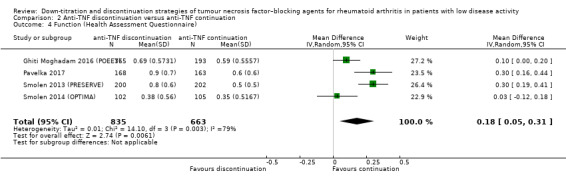
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 4 Function (Health Assessment Questionnaire).
2.5. Analysis.
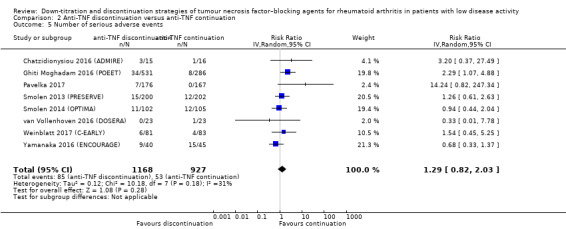
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 5 Number of serious adverse events.
2.6. Analysis.
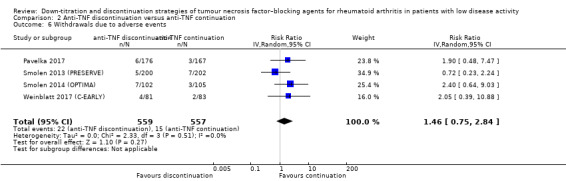
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 6 Withdrawals due to adverse events.
Minor outcomes
Proportion of participants with a flare: Of the eight studies (2111 participants) included for this comparison, five studies provided data on this outcome (Chatzidionysiou 2016 (ADMIRE); Ghiti Moghadam 2016 (POEET); Pavelka 2017; van Vollenhoven 2016 (DOSERA); Weinblatt 2017 (C‐EARLY)), with 31, 46, 331, 817, and 163 participants, respectively. We did not pool data because of clinical and statistical heterogeneity. The study by Chatzidionysiou 2016 (ADMIRE) defined flare as DAS28 ≥ 2.6 or an increase of more than 1.2 from baseline. After 28 weeks, proportion of flare in the adalimumab discontinuation group was not statistically significantly different from that in the adalimumab continuation group (RR 1.6, 95% CI 0.92 to 2.78). van Vollenhoven 2016 (DOSERA) defined flare as (a) a DAS28‐ESR > 5.1; (b) a DAS28‐ESR > 3.2 and an increase ≥ 1.2 from baseline; (c) DAS28‐ESR > 3.2 and an increase in DAS28 ≥ 0.6 from baseline on two consecutive visits at least one to three weeks apart; or (d) disease progression as determined by either the investigator or disease flare as experienced by the participant. After 48 weeks' follow‐up, the proportion of participants with flare was higher in the discontinuation group compared to the continuation group (RR 1.82, 95% CI 1.15 to 2.87). Ghiti Moghadam 2016 (POEET), Pavelka 2017, and Weinblatt 2017 (C‐EARLY) used the same criterion for flare: DAS28 ≥ 3.2 and an increase of 0.6 or more. Ghiti Moghadam 2016 (POEET) found that the proportion of participants with flare was higher in the anti‐TNF discontinuation group than in the continuation group (after 24 weeks RR 3.37, 95% CI 2.42 to 4.70; after 52 weeks RR 2.82, 95% CI 2.17 to 3.65). Pavelka 2017 found a higher proportion of flare in the participants that stopped anti‐TNF after 28 weeks (RR 1.53, 95% CI 1.30 to 1.81). Weinblatt 2017 (C‐EARLY) found no difference in the proportion of participants with flare between the anti‐TNF discontinuation group and the continuation group (RR 1.52, 95% CI 0.61 to 3.80).
Quality of life: Of the eight studies (2111 participants) included for this comparison, two studies, Smolen 2013 (PRESERVE) and Pavelka 2017, with 733 participants provided data on this outcome. Anti‐TNF discontinuation led to a deterioration in quality of life after 28 to 52 weeks (MD −0.10, 95% CI −0.13 to −0.07).
Costs: None of the eight included studies provided data on direct or indirect costs.
Decremental cost‐effectiveness ratio: None of the eight included studies provided data on this outcome.
Time to flare: Of the eight studies included for this comparison (2111 participants), two studies, Chatzidionysiou 2016 (ADMIRE) and van Vollenhoven 2016 (DOSERA), provided data on this outcome, with 31 and 46 participants, respectively. These two studies used different flare/failure criteria. Chatzidionysiou 2016 (ADMIRE) reported that survival curves suggested higher flare‐free survival over time in participants randomised to continue treatment with adalimumab, but that the difference did not reach statistical significance (P = 0.07). The study by van Vollenhoven 2016 (DOSERA) reported a median time to failure of 48 weeks in the etanercept 50 mg/week continuation group and six weeks in the etanercept discontinuation (placebo) group, but no SDs were available.
Change in other medication: None of the eight included studies provided data on change in other medication.
Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation
See Table 3.
Primary outcomes
Mean disease activity: All three studies included in this comparison reported on this outcome, with 357 participants (Bejerano 2016 (OPTIBIO) (abstract only); Fautrel 2016 (STRASS); van Herwaarden 2015 (DRESS)). Anti‐TNF disease activity–guided dose tapering may result in little or no difference in mean disease activity score (MD 0.25, 95% CI −0.17 to 0.67). Analysis 3.1
Proportion persistent remission: Of the three studies included in this comparison (365 participants), one study with 180 participants reported on this outcome (van Herwaarden 2015 (DRESS)). Anti‐TNF disease activity–guided dose tapering resulted in little or no difference in the proportion of participants with persistent remission (DAS28 < 2.6) after 18 months (RR 0.89, 95% CI 0.75 to 1.06). Analysis 3.2
Proportion of participants that switched to another biologic due to persistent loss of response (refractory to re‐instalment of the tapered anti‐TNF in the intervention group): Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), with 317 participants reported on this outcome. Anti‐TNF disease activity–guided dose tapering may result in little or no difference in the proportion of participants that switch to another biologic after 18 months (RR 0.62, 95% CI 0.25 to 1.54). Analysis 3.3
Proportion of participants with minimal radiographic progression: Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), with 312 participants reported on this outcome. Although Fautrel 2016 (STRASS) used a cut‐off value of 1 point and van Herwaarden 2015 (DRESS) a cut‐off value of 0.5 point mSvdH score, data could be pooled. Anti‐TNF disease activity–guided dose tapering may slightly increase the proportion of participants with minimal radiographic progression (mSvdH > 0.5 or > 1.0) after 18 months (RR 1.45, 95% CI 0.77 to 2.73). Analysis 3.4
Function: Of the three studies included in this comparison (365 participants), one study with 123 participants reported on this outcome (Fautrel 2016 (STRASS)). IAnti‐TNF disease activity–guided dose tapering probably leads to a slight deterioration in function after 18 months (MD 0.20, 95% CI −0.02 to 0.42). Analysis 3.5
Number of serious adverse events: Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), reported on this outcome, with 137 and 180 participants, respectively. Due to the very low certainty of the evidence and imprecision, it is uncertain whether anti‐TNF disease activity‐guided dose tapering influences the number of serious adverse events after 18 months. Analysis 3.6
Withdrawals due to adverse events: None of the three included studies provided data on this outcome.
3.1. Analysis.
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 1 Mean disease activity score (DAS28).
3.2. Analysis.
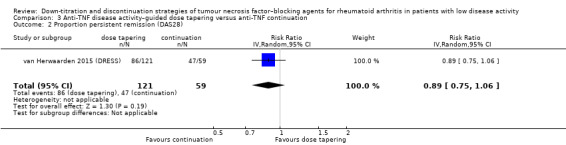
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 2 Proportion persistent remission (DAS28).
3.3. Analysis.
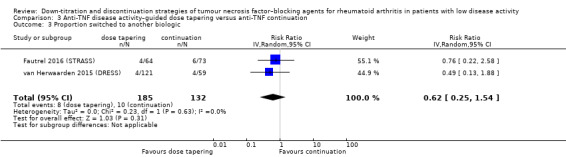
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 3 Proportion switched to another biologic.
3.4. Analysis.
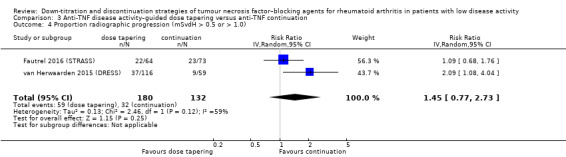
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 4 Proportion radiographic progression (mSvdH > 0.5 or > 1.0).
3.5. Analysis.

Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 5 Function (Health Assessment Questionnaire).
3.6. Analysis.
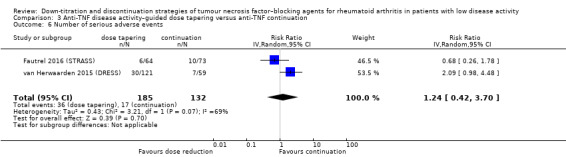
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 6 Number of serious adverse events.
Minor outcomes
Proportion of participants with a flare: All three studies (365 participants) included in this comparison reported on this outcome. The studies used different criteria for flare. In Bejerano 2016 (OPTIBIO) (abstract only), participants had a flare if DAS28 > 2.6; SDAI > 5; or when ACR/EULAR criteria were not fulfilled. Bejerano 2016 (OPTIBIO) (abstract only) found no difference in the proportion of participants with a flare between the anti‐TNF disease activity‐guided dose tapering group and the anti‐TNF continuation group after 24, 48, 72, and 96 weeks' follow‐up (24 weeks: RR 3.25, 95% CI 0.14 to 76.01; 48 weeks: RR 1.09, 95% CI 0.24 to 4.86; 72 weeks: RR 1.09, 95% CI 0.36 to 3.27; 96 weeks: RR 1.27, 95% CI 0.50 to 3.22). Fautrel 2016 (STRASS) defined flare as DAS28 > 2.6 with an increase in DAS28 of > 0.6. They reported a higher proportion of participants with flare in the anti‐TNF disease activity‐guided dose tapering group compared to the anti‐TNF continuation group after 18 months' follow‐up (RR 1.64, 95% CI 1.24 to 2.18). In van Herwaarden 2015 (DRESS), participants had a flare if DAS28 increased > 1.2, or if DAS28 increased > 0.6 and current DAS28 was ≥ 3.2. The authors reported a higher proportion of participants with flare in the anti‐TNF disease activity‐guided dose tapering group compared to the anti‐TNF continuation group after 9 and 18 months (9 months: RR 2.68, 95% CI 1.58 to 4.56; 18 months: RR 2.68, 95% CI 1.74 to 4.13). van Herwaarden 2015 (DRESS) found no difference in major flares (duration > 3 months) after 9 and 18 months (9 months: RR 1.71, 95% CI 0.37 to 7.96; 18 months: RR 1.22, 95% CI 0.50 to 2.98).
Quality of life: Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), reported on this outcome, with 98 and 180 participants, respectively. Both studies reported mean quality‐adjusted life years (QALYs) of the 18‐month study period. Fautrel 2016 (STRASS) found that the anti‐TNF disease activity‐guided dose tapering group gained fewer QALYs during the 18‐month study period than the anti‐TNF continuation group (MD −0.158). No confidence intervals were available. van Herwaarden 2015 (DRESS) reported no difference between the anti‐TNF disease activity‐guided dose tapering group versus the anti‐TNF continuation group (MD −0.02, 95% percentiles −0.06 to 0.02).
Costs: Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), reported on this outcome, with 98 and 180 participants, respectively. Both studies reported lower costs in the anti‐TNF disease activity‐guided dose tapering group compared to the anti‐TNF continuation group after 18 months' follow‐up (Fautrel 2016 (STRASS): MD EUR −8440. No confidence intervals were available. van Herwaarden 2015 (DRESS): MD EUR −9051, 95% percentiles −10,278 to −7731 (rectification submitted)).
Decremental cost‐effectiveness ratio: Of the three studies included in this comparison (365 participants), two studies, Fautrel 2016 (STRASS) and van Herwaarden 2015 (DRESS), reported on this outcome, with 98 and 180 participants, respectively. Fautrel 2016 (STRASS) reported a decremental cost‐effectiveness ratio (DCER) of EUR 53,417 per QALY loss. van Herwaarden 2015 (DRESS) reported a DCER of EUR 379,433 per QALY loss (rectification submitted).
Time to flare: None of the three included studies reported data on this outcome.
Change in other medication: Of the three studies included in this comparison (365 participants), one study with 180 participants reported on this outcome (van Herwaarden 2015 (DRESS)). No difference was found between the anti‐TNF disease activity‐guided dose tapering group and the anti‐TNF continuation group after 18 months concerning use of intramuscular or intra‐articular glucocorticosteroids (RR 1.50, 95% CI 0.89 to 2.51); use of oral glucocorticosteroids (RR 0.65, 95% CI 0.24 to 1.79); DMARD initiation or dose escalation (RR 3.90, 95% CI 0.93 to 16.41); and use of a DMARD (RR 0.88, 95% CI 0.71 to 1.10). The proportion of participants that reduced the dose of their DMARD or discontinued the DMARD after 18 months' follow‐up was lower in the anti‐TNF disease activity‐guided dose tapering group compared to the anti‐TNF continuation group (RR 0.37, 95% CI 0.19 to 0.72).
Subgroup and sensitivity analyses
We planned to perform a subgroup analysis as described in the Subgroup analysis and investigation of heterogeneity section. However, because of the small number of included studies, analyses were not informative.
We also planned to perform sensitivity analyses as described in the Sensitivity analysis section. However, because of the small number of included studies, analyses were not informative.
Discussion
This is the first update of the original Cochrane Review first published in 2014. We identified eight new studies for inclusion in this update and additional data on old studies, which changed some results. We retrospectively excluded one study that had been included in the original review. Our main conclusions remained largely the same.
Summary of main results
This systematic review summarises evidence from 14 studies (13 RCTs and one CCT) of down‐titration of anti‐TNF agents in people with RA with low disease activity. We considered three down‐titration strategies to be sufficiently different to warrant separate reviewing: (1) anti‐TNF dose reduction, (2) anti‐TNF discontinuation, and (3) anti‐TNF disease activity–guided dose tapering. We presented the available data on these strategies separately.
Anti‐TNF dose reduction compared with anti‐TNF continuation
Six studies (three on etanercept, one on etanercept and adalimumab, one on certolizumab pegol, and one on all anti‐TNF agents) reported data on fixed anti‐TNF dose reduction compared with anti‐TNF continuation. After pooling of data where possible, we can conclude that anti‐TNF dose reduction leads to little or no difference in mean disease activity, the proportion of participants with persistent remission, number of serious adverse events and withdrawals due to adverse events compared to continuation. Also, anti‐TNF dose reduction does not result in an important deterioration in function but probably slightly increases the proportion of participants with minimal radiographic damage. Next to this, anti‐TNF dose reduction may slightly reduce the proportion of participants switched to another biologic. We found no data on important outcomes like cost‐effectiveness.
Anti‐TNF discontinuation compared with anti‐TNF continuation
Eight RCTs reported data on anti‐TNF discontinuation compared with anti‐TNF continuation for all anti‐TNF agents, but mainly adalimumab and etanercept. Different types of outcome measures were reported, and marked heterogeneity was present. The results showed that anti‐TNF discontinuation probably increases the mean disease activity score slightly, and that the risk ratio of persistent remission lies between 0.16 and 0.77 (data not pooled). Anti‐TNF discontinuation increases the proportion participants with minimal radiographic progression of > 0.5 mSvdH point per year and may lead to a slight deterioration in function and probably slightly increases the number of withdrawals due to adverse events. It is uncertain whether anti‐TNF discontinuation influences the number of serious adverse events due to very low‐certainty evidence. Again, we found no data on cost‐effectiveness.
Anti‐TNF disease activity–guided dose tapering compared with anti‐TNF continuation
Three studies (all anti‐TNF agents, but again mostly etanercept and adalimumab) compared disease activity–guided anti‐TNF dose tapering with anti‐TNF continuation. Anti‐TNF disease activity–guided dose tapering may result in little or no difference in mean disease activity score and the proportion of participants switched to another biologic. Furthermore, anti‐TNF disease activity–guided dose tapering results in little or no difference in the proportion of participants with persistent remission. Next to this, tapering may result in a slight increase in the proportion of participants with minimal radiographic progression and probably causes a slight deterioration in function. It is uncertain whether anti‐TNF disease activity‐guided dose tapering influences the number of serious adverse events due to very low‐certainty evidence. No data were available on withdrawals due to adverse events. In the two studies that reported on cost‐effectiveness, costs were significantly lower, and decremental cost‐effectiveness ratios were found to be between EUR 53,000 and EUR 379,000 per QALY lost.
Overall completeness and applicability of evidence
The number of controlled studies on this matter is increasing, although they are mostly limited to adalimumab and etanercept. Data were available on all three strategies, although discontinuation was studied most extensively. We considered the included studies to be quite comparable clinically and decided to accept some clinical heterogeneity in order to obtain more precision. One study was reported as abstract only (Bejerano 2016 (OPTIBIO)). Excluding the results reported in this abstract did not change our conclusions.
An important issue remains that the superiority design of the fixed‐dose reduction or discontinuation studies hampers interpretability and generalisability to clinical practice. With regard to the first issue, these studies do provide point estimates about between‐group differences in important outcomes between the strategies, but they do not compare these point estimates and their confidence intervals with a prespecified relevant non‐inferiority margin. Consequently, independent of whether superiority tests demonstrate a significant difference, the interpretation has to be made post hoc whether this (non‐)significant difference is relevant compared to a non‐inferiority claim, although this issue might become less important in meta‐analyses where sample sizes are large. Next to this, the preferred method of analysis for non‐inferiority trials is per protocol, while an intention‐to‐treat analysis is favoured for superiority trials. This hampers comparison of studies with different approaches. With regard to generalisability, it seems important to mention that for patients and clinicians alike, the outcome of fixed‐dose reduction or discontinuation is valuable to know. However, it would be much more valuable to know whether disease activity‐guided dose reduction and discontinuation is non‐inferior with regard to important outcomes, as this supports shared decision making with patients.
The participants included in these studies vary from early RA with short treatment duration and no prior DMARD treatment to longstanding, established RA patients who have been treated for a long time with several other DMARDs, and also include patients with and without concomitant DMARD. This increases generalisability, although it also has been shown that no clinical patient‐, disease‐, or treatment‐related variable is clearly an effect modifier for the chance of successful dose reduction or discontinuation (Tweehuysen 2017).
With regard to outcome measures, we noted that domains are often missing, such as functioning, radiographic damage progression, or cost (effectiveness). Also, when a domain is included as an outcome, there is marked heterogeneity in the way the outcome is assessed, leaving much room for improvement of outcome standardisation.
Quality of the evidence
Anti‐TNF dose reduction compared with anti‐TNF continuation
Using the GRADE approach, we assessed the overall certainty of evidence as high for two of the seven 'Summary of findings' outcomes in the anti‐TNF dose reduction versus continuation comparison: mean disease activity and function. We assessed the certainty of the evidence for the proportion of participants with minimal radiographic progression as moderate because of imprecision. We assessed the certainty of the evidence for the remaining four main outcomes as low. For the proportion of participants with persistent remission, this was the result of heterogeneity (downgraded two times). For the proportion of participants that switched to another biologic, this was due to concern about risk of bias in the reporting study (downgraded two times). We downgraded the evidence on the number of serious adverse events and the number of withdrawals due to adverse events one level for risk of bias and one level for imprecision.
Anti‐TNF discontinuation compared with anti‐TNF continuation
The overall certainty of the evidence was high for one of the seven 'Summary of findings' outcomes in the anti‐TNF discontinuation versus continuation comparison: proportion of participants with minimal radiographic progression. We assessed the certainty of the evidence for the outcomes mean disease activity and withdrawals due to adverse events as moderate. We performed a subanalysis for the outcome mean disease activity, since heterogeneity was present between the studies that could be explained by the study characteristics. We downgraded the evidence on mean disease activity for discontinuation without restarting or with restarting and with LOCF analysis one level because of imprecision. We downgraded the evidence for mean disease activity with restarting and without LOCF analysis one level due to concerns about risk of bias. We downgraded the evidence on withdrawals one level because of imprecision. We assessed the certainty of the evidence on the proportion of participants with persistent remission and function as low because of substantial heterogeneity (downgraded two levels). Lastly, we assessed the certainty of the evidence on the number of serious adverse events as very low because of (1) concerns about risk of bias, (2) moderate heterogeneity between effect estimates, and (3) imprecision. The included anti‐TNF discontinuation studies did not report the proportion of participants that switched to another biologic.
Anti‐TNF disease activity–guided dose tapering compared with anti‐TNF continuation
The certainty of the evidence for the outcome proportion of participants with persistent remission was high in the anti‐TNF disease activity‐guided dose tapering versus continuation comparison. The data came from one study with a low risk of bias, and there were no other reasons to downgrade the certainty of the evidence. We assessed the certainty of the evidence on function as moderate because of imprecision. We assessed the certainty of the evidence on mean disease activity as low because of heterogeneity (downgraded two levels). We graded the certainty of the evidence on the proportion of participants that switched to another biologic as low because of imprecision (downgraded two levels). We graded the certainty of the evidence on the proportion of participants with minimal radiographic progression as low due to heterogeneity and imprecision. We assessed the certainty of the evidence for serious adverse events as very low because of substantial heterogeneity (downgraded two levels) and imprecision (downgraded two levels). The included studies for this comparison did not report on withdrawals due to adverse events.
Potential biases in the review process
Two review authors independently reviewed all titles and abstracts, extracted data, and performed bias and quality assessment. Consequently, errors in extraction have been minimised. Risk of bias could not be completely assessed for some studies due to restricted information despite efforts to obtain additional information from study authors. Post‐hoc decisions had to be made regarding the presentation and pooling of outcomes (e.g. time of follow‐up, threshold values) which could have had implications on the results. The authors have tried to be as transparent as possible about choices that have been made, although they remain subjective.
Agreements and disagreements with other studies or reviews
A few other systematic reviews have examined anti‐TNF down‐titration, although the focus differed to some extent (Galvao 2016; Kuijper 2015; Navarro‐Millán 2013; Yoshida 2014). Galvao 2016 focused on discontinuation of biological DMARDs, and Kuijper 2015 on discontinuation of biologic and synthetic DMARDs. Navarro‐Millán 2013 investigated discontinuation of anti‐TNF agents specifically. The results of these reviews are comparable to the findings in our review. The systematic review by Yoshida 2014 looked into the design and failure definitions in anti‐TNF discontinuation studies. The review authors concluded that heterogeneity can be seen across studies in both study design and failure definition. This is consistent with the findings reported in our review.
Authors' conclusions
Implications for practice.
This review of the data has several implications for clinical practice with regard to the three different strategies studied: dose reduction, discontinuation, and disease activity‐guided tapering of anti–tumour necrosis factor (anti‐TNF).
Firstly, fixed‐dose reduction of anti‐TNF (especially etanercept) in people with rheumatoid arthritis (RA) with at least three to 12 months of low disease activity is comparable with continuing the standard dose with regard to mean disease activity, the proportion of participants remaining in remission, and mean function. Consequently, an attempt to reduce the dose (or increase the dosage interval) in people with low disease activity with RA on full‐dose anti‐TNF seems sensible in clinical practice. It should be mentioned, however, that all treatment changes in RA should be done carefully on a background of 'treat to target', that is guided by disease activity. Dose reduction probably slightly increases the proportion of participants with minimal radiographic progression. Anti‐TNF dose reduction may cause little or no difference in number of serious adverse events and withdrawals due to adverse events, although certainty of evidence was low. This review showed that slightly fewer participants undergoing anti‐TNF dose reduction may switch to another biologic disease‐modifying antirheumatic drug (bDMARD) compared to continuation. An explanation for this might be that temporary disease flares (inherent to the disease) are treated in the dose reduction group by increasing the dose, while in the continuation group patients are switched to another biologic.
Secondly, this review shows that anti‐TNF discontinuation (without disease activity–guided restarting of treatment) is an inferior strategy compared with continuation of anti‐TNF in terms of disease control (mean disease activity and the proportion of participants remaining in remission), minimal radiographic damage, function and the number of withdrawals due to adverse events. Although a sizeable proportion of patients can stop the anti‐TNF without deterioration, the large majority of patients who cannot discontinue the drug are harmed if the treatment is not reinstated. The effect of discontinuation on the number of serious adverse events remains uncertain. However, given the current evidence, discontinuation should not be attempted without regular assessment of disease activity, setting a treatment goal, and reinstatement of treatment when necessary.
The abovementioned findings converge finally in the evidence on the disease activity‐guided dose tapering strategy. This review shows that disease activity‐guided tapering is comparable to continuation with regard to mean disease activity, the proportion of participants remaining in remission and the proportion of participants switched to another biologic. Tapering may result in a slight increase in the proportion of participants with radiographic progression and probably leads to a slight deterioration in function. The effect of disease activity‐guided dose tapering of anti‐TNF on the number of serious adverse events and withdrawals due to adverse events could not be determined with certainty. This evidence is similar to that for fixed‐dose reduction. Because disease activity‐guided dose tapering provides the opportunity to find the lowest effective dose for each individual patient and to discontinue treatment as the final step of the tapering process, this may be the most cost‐effective and feasible approach in clinical practice. Since uncertainty remains on several important outcome measures, more data on, for example, radiographic damage progression, function, (serious) adverse events, and costs are warranted.
With respect to interpretation, it should be noted that the burden of proof in this case does not lie solely with dose reduction or stopping compared with continued use. To our knowledge, no controlled data are available on anti‐TNF continued use after week 52, including all registration studies. Consequently, there remains equipoise on what is the best strategy after one year of treatment with anti‐TNF.
Implications for research.
Our review highlights what is already known about anti‐TNF down‐titration in people with low RA disease activity, and on the other hand identifies gaps in our knowledge. Here we would like to mention a number of aspects that could be targeted in future studies. Of note, most of these points are currently being addressed in several ongoing studies.
The design selected for studies comparing an anti‐TNF down‐titration strategy versus an anti‐TNF continuation strategy should include a non‐inferiority approach instead of the classical superiority analyses, as the aim is to maintain and not improve clinical outcomes, while minimising the amount of treatment that is needed. Superiority analyses can be reserved for domains were superiority can be expected, such as drug use, infections, and costs. As guidelines for performing a systematic review on non‐inferiority studies are absent, development of such guidelines would be helpful.
The intervention should include disease activity–guided dose tapering or stopping of the anti‐TNF agent using tight control/treat to target instead of fixed‐dose adaptation or stopping, as the former is more compatible with clinical practice.
The domain in which an intervention should be non‐inferior is long‐term RA disease control. Although temporary flaring will inevitably be seen more often in the trial‐and‐error dose‐tapering arm, both the incidence of more severe or prolonged flaring and mean disease activity at study end should be comparable.
Consequently, in addition to mean disease activity at study end, cumulative incidence of a validated RA flare criterion could be used. Use of (one of) validated Outcome Measures in Rheumatology Clinical Trials (OMERACT) disease activity score in 28 joints (DAS28)‐based flare criteria should be considered (van der Maas 2013 Flare). Use of a validated flare criterion also increases standardisation for future meta‐analyses.
Other outcomes besides disease activity that should be included are cost, quality of life, cost‐effectiveness, and (long‐term) safety, because these constitute the reason why down‐titration is contemplated in the first place.
The drugs that are studied should preferably also include other anti‐TNF agents like certolizumab pegol, golimumab, and infliximab.
Prediction of (un)successful dose tapering would perhaps further improve outcomes of individualised disease activity–guided dose tapering, and prediction modelling should be considered, using, for example, genetics, imaging, biomarkers, and drug levels. Possible gains when using a good prediction rule include (1) prevention of unnecessary flaring in patients that cannot be dose reduced; and (2) prevention of months of slow dose tapering in patients that can be stopped directly.
Finally, although outside the scope of this review, efforts should be (and already are) directed toward other non–anti‐TNF biologicals (abatacept, tocilizumab) and toward other inflammatory diseases in which biologicals are used, both within rheumatology (ankylosing spondylitis, psoriatic arthritis) and within other medical specialties (gastroenterology, dermatology).
Feedback
Comments ‐ Lo and Tejani, 30 March 2015
Summary
Comment: Written by:
Elaine Lo, MSc. (Clin Pharm), BCPS, PharmD Student Aaron M Tejani, BSc.Pharm, PharmD
We read with interest the review on down‐titration and discontinuation strategies of tumor necrosis factor (TNF)‐blocking agents for rheumatoid arthritis (RA) in patients with low disease activity and there are a few points we wish to address.
The authors concluded that dose reduction of etanercept after at least 3 to 12 months of low disease activity, seems as effective as continuing the standard dose with respect to disease activity and functional outcomes; while discontinuation of adalimumab and etanercept is inferior to continuation of treatment. The conclusion is driven mainly by the results of the PRESERVE trial2, which weighs 59.7% and 30% in the meta‐analyses of the dose reduction and discontinuation endpoint respectively. We feel that the conclusion should be rephrased as “in patients who were put on anti‐TNF agents (with methotrexate) and improved from moderate to low disease activity for at least 3 to 12 months” to reflect the patient population studied in the PRESERVE trial. This would prevent unintentional extrapolation of the conclusion to patients who start out with severe disease activity or whose disease activity remains unchanged on anti‐TNF agents.
The PRESERVE trial is rated as having a low risk of bias for all parameters except “incomplete outcome data” and that differs from our evaluation. PRESERVE is the largest study (n=604) included in the review and the only trial identified for many endpoints. Thus an accurate evaluation of its risk of bias is important in the synthesis of data. Though not reported in PRESERVE, injection site reaction is a significant adverse event with etanercept. In the Cochrane review by Lethaby et al3, more patients receiving etanercept plus DMARD developed injection site reactions than those taking DMARD alone (at six months: 25.6% versus [vs] 3.8%, RR 6.9; 95% CI 2.2 to 21.3). For patients who were given etanercept for 9 months in the open‐label phase, the sudden lack of injection site reaction might be a trigger for unblinding in the placebo group despite the double blind design and identical drug package. By the same token, assessors/ study investigators could have been unblinded by the lack of injection site reaction in the placebo group. Potentially compromised blinding for a subjective outcome like Disease activity score‐28 (DAS28) meets the criteria for judgment of high risk of performance bias.4 Inadequate blinding combined with blocked randomization might increase the risk of selection bias. With a block size of 3, when assignments are revealed because of the characteristic injection site reaction, it might be possible to predict future assignments, thus undermining allocation concealment. The chance of seeing a pattern and hence being able to predict assignment is arguably small in a trial that involves 80 centres recruiting 834 patients. Yet we have no information regarding the distribution of recruitment among centres and how many centres were recruiting at the same time. Selection bias should be unclear rather than low.4
We agree with the authors’ assessment that the attrition risk is high in PRESERVE. There were significantly more treatment discontinuation due to unsatisfactory responses in the placebo group compared to the 25mg etanercept and 50mg etanercept group (43 (21.5%) vs 27 (13.4%) vs 4 (2%)). Patients were assumed to be non‐responders if they discontinued early because of poor efficacy. This might exaggerate the number of non‐responders, especially in the placebo group. There is no objective criteria for discontinuation due to inefficacy described in the trial – patient may not have reached the point of being considered a non‐responder when they left the study. Though the benefit of etanercept continuation withstands the test of sensitivity analysis for the outcome of DAS28<3.2, other endpoints with a smaller effect size or lower incidence e.g. normal HAQ‐DI and ACR70 may have become insignificant should a different analysis be used. Having mentioned that the risk of unblinding is high, the threshold of discontinuation for a “perceived” lack of drug effect might be lower in PRESERVE than an adequately blinded study. As for patients who discontinued for reasons other than unsatisfactory response, missing data were imputed with last observation carried forward (LOCF) method. There is no information about the number of patients that were affected by LOCF. A patient may be categorized as having low disease activity when he was lost to follow up and imputed as a responder but indeed would be rated as having high disease activity if assessment was done at week 88. It appears that the review authors took the data straight from Table 3 of the PRESERVE trial which do not account for the uncertainty around imputation. We suggest communicating with the authors of PRESERVE about the extent of LOCF/ non‐responder imputation and performing sensitivity analyses for outcomes presented in the review (e.g. DAS28<2.6, HAQ‐DI etc) like those performed for DAS28<3.2 in the supplementary appendix of PRESERVE.2
We also feel that the reporting for assessment of adverse events (AE) and co‐intervention (e.g. methotrexate and glucocorticoid dose) was inadequate in PRESERVE. Besides cost, another reason to attempt dose reduction or discontinuation is to avoid unnecessary exposure to drug toxicity. PRESERVE is the only trial included for the evaluation of serious adverse events (SAE). However, the trial authors did not describe how SAE were assessed or adjudicated. Indeed, PRESERVE captured significantly less SAE and AE compared to other trials comparing etanercept plus methotrexate vs methotrexate alone (e.g. Emery, 20085: SAE 12%, any AE 91%; Weinblatt, 19996: infection 51‐63%; Klareskog, 20047: infection AE 81%, 67%‐64% PRESERVE2: SAE 3‐8%, any AE 53‐61%, infection 1‐2%). The dose of methotrexate and glucocorticoid is not reported but is deemed pertinent as there seemed to be no restriction on adjustment of these drugs for lack of efficacy. The review authors should consider explaining the risk explicitly to the readers and incorporating this as part of the quality assessment.
The terms “low disease activity” and “remission” were used loosely in the review. Uninformed readers might find this misleading and confusing. For instance, in the plain language summary, the author stated the impact of stopping or lowering the dose of anti‐TNF drugs on “RA remission”. However, this is referred to as “persistent low disease activity” in the main text. Under this umbrella of “persistent low disease activity”, the authors pooled outcomes on DAS28 remission, non‐failure and DAS28‐CRP<2.7 – not remission alone. We suggest the authors revising the wording in the plain language summary from “RA remission” to “persistent low disease activity” for the sake of consistency.
We noted with regret that the authors of the review changed the major outcome from “proportion of patients with a flare” to “proportion of patients with persistent low disease activity”, claiming that the two are highly comparable. Despite the understanding that only 2 of the included studies reported the former outcome, we still want to acknowledge its merit as an endpoint. As revealed in a study by the OMERACT group8, the validated flare criteria (i.e. an increase in DAS28>1.2 or >0.6 if DAS28>3.2) was found to be more discriminating and more valid than a threshold criteria (i.e. DAS28>2.6 or 3.2) (see Table 1) It gives a good balance of specificity/ sensitivity in the transition scale where patient/ physician were asked whether disease activity had changed compared with the last visit on a 7‐point Likert scale (criterion validity), and is well associated with DMARD/ corticosteroid and CRP change. As pointed out by the review authors, in addition to mean disease activity, a validated RA flare criterion like the OMERACT DAS28‐based flare criteria should be considered. We look forward to seeing studies with standardized outcomes for future meta‐analyses.
We hope this provides some constructive feedbacks for the next review. We look forward to hearing from you.
1. van Herwaarden N, den Broeder AA, Jacobs W, van der Maas A, Bijlsma JW, van Vollenhoven RF. Down‐titration and discontinuation strategies of tumor necrosis factor–blocking agents for rheumatoid arthritis in patients with low disease activity. The Cochrane Library 2013.
2. Smolen JS, Nash P, Durez P, et al. Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): a randomised controlled trial. Lancet 2013;381(9870):918‐29. 10.1016/s0140‐6736(12)61811‐x
3. Lethaby A, Lopez‐Olivo MA, Maxwell LJ, Burls A, Tugwell, P., Wells GA. Etanercept for the treatment of rheumatoid arthritis. The Cochrane Library 2013.
4. Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
5. Emery P, Breedveld FC, Hall S, et al. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): a randomised, double‐blind, parallel treatment trial. Lancet 2008;372(9636):375‐82. 10.1016/s0140‐6736(08)61000‐4
6. Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. The New England journal of medicine 1999;340(4):253‐9. 10.1056/NEJM199901283400401
7. Klareskog L, van der Heijde D, de Jager JP, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double‐blind randomised controlled trial. Lancet 2004;363(9410):675‐81. 10.1016/S0140‐6736(04)15640‐7
8. van der Maas A, Lie E, Christensen R, et al. Construct and criterion validity of several proposed DAS28‐based rheumatoid arthritis flare criteria: an OMERACT cohort validation study. Annals of the rheumatic diseases 2013;72(11):1800‐5. 10.1136/annrheumdis‐2012‐202281
I agree with the conflict of interest statement below:
I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Reply
Hereby our response:
We read the comment by Lo et al on our review 'Down‐titration and discontinuation strategies of tumor necrosis factor–blocking agents for rheumatoid arthritis in patients with low disease activity' with interest. Herewith we thank them for taking the time and effort to do this, and provide a point to point response:
First it should be noted that we plan to update the review within the next year already. This because in the original review the majority of studies had not yet been published as a full text article. Since then, some important trials have been published in full, and also we expect some strategy studies to be published within the next months. In this update, we will incorporate the comments by Lo et all were applicable.
With respect to our conclusion that "dose reduction of etanercept after at least 3 to 12 months of low disease activity, seems as effective as continuing the standard dose with respect to disease activity and functional outcomes; while discontinuation of adalimumab and etanercept is inferior to continuation of treatment." We think that this statement is correct. Although PRESERVE study weights in heavily, both Botsios 2007 and van Vollenhoven 2012 did not include only patients that had only moderate disease activity at study start. We therefore think that our conclusion is still valid. In the update we will hopefully be able to include STRASS and DRESS strategy studies, that also did not limit patient inclusion to patients with moderate disease activity at study start
With regard to risk of bias being high due to possible unblinding and blocked randomisation, we disagree. Injection site reaction can occur, but are firstly for the most part limited to the first 6 months of treatment. Also, patients with severe ISR stop treatment. Secondly, low dose etanercept patients could not have been unblinded by sudden, as ISR do not seem to depend on dose. Also, even when unblinding occurred in some patients, it could only give bias in the conservative direction, ie patients and physicians would expect a flare more so in these patients. Estimates for disease activity would not he lower, but higher in these patients. Finally, for radiographic outcome, any unblinding would not play a role. Of course classifying a certain characteristic of a study as having a risk of bias remains a judgement call, but we do not think that it is fair to expect a large risk of bias in the estimates of this study. In the update of the review, we will address these issues more in depth.
Adverse event reporting is indeed overall suboptimal, and we have mentioned this in our conclusion. The same holds true for absence of any cost effectiveness analyses. In the upcoming update, we expect to have more data on this.
"We suggest the authors revising the wording in the plain language summary from “RA remission” to “persistent low disease activity” for the sake of consistency. ". Thanks for the suggestion, we will do this in the next update.
Finally, we agree wholeheartedly with the suggestion to add % patients with flare as outcome. We are the authors by the way of the OMERACT flare criteria paper that is referred to, so we are quite familiar with the upside of using this outcome. Unfortunately, this outcome was not used in the trials we have identified. Furthermore, in strategy studies, the outcome of prolonged flare is probably better reflecting (non) inferiority of a certain tapering strategy, see for ample discussion about this in our recent BMJ paper. www.bmj.com/content/350/bmj.h1389‐0.long
Thank again for taking the time to review our systematic review.
Best regards,
Contributors
Dr A.A. (Alfons) den Broeder, on behalf of all co‐authors.
What's new
| Date | Event | Description |
|---|---|---|
| 3 June 2019 | Amended | Correction to article metadata; no impact on article content. |
History
Protocol first published: Issue 4, 2013 Review first published: Issue 9, 2014
| Date | Event | Description |
|---|---|---|
| 22 May 2019 | New citation required and conclusions have changed | One previously included trial was excluded in this update. We included eight additional trials, for a total of 14 studies |
| 29 March 2018 | New search has been performed | Conclusions have changed. We have now more evidence on other tumour necrosis factor blocking agents, and disease activity tapering is comparable to continuation of treatment with respect to the proportion of participants with persistent remission and may be comparable regarding disease activity |
| 20 May 2015 | Feedback has been incorporated | Incorporated feedback from Lo and Tejani |
Acknowledgements
We thank Tamara Rader for her help with modifying the search strategies and performing the searches for the first version of this review.
Appendices
Appendix 1. The Cochrane Library search strategy
#1 MeSH descriptor: [Arthritis, Rheumatoid] explode all trees
#2 caplan* near/2 syndrome
#3 felty* near/2 syndrome
#4 "rheumatoid nodule*"
#5 "rheumatoid vasculitis"
#6 sjogren* near/2 syndrome
#7 "still* disease"
#8 arthritis near/2 rheumat*
#9 (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8)
#10 MeSH descriptor: [Tumor Necrosis Factor‐alpha] this term only and with qualifier(s) [Antagonists & Inhibitors – Al]
#11 MeSH descriptor: [Adalimumab] this term only
#12 MeSH descriptor: [Certolizumab Pegol] this term only
#13 MeSH descriptor: [Etanercept] this term only
#14 MeSH descriptor: [Infliximab] this term only
#15 "anti‐tumor necrosis factor"
#16 "anti‐tumour necrosis factor"
#17 anti‐tnf
#18 "tumor necrosis factor" near/3 inhibit*
#19 "tumour necrosis factor" near/3 inhibit*
#20 tnf near/3 inhibit*
#21 tnfi
#22 tnf near/3 block*
#23 bDMARD*
#24 "biologic* DMARD*"
#25 adalimumab
#26 humira
#27 etanercept
#28 enbrel
#29 benepali
#30 infliximab
#31 remicade
#32 remsima
#33 inflectra
#34 golimumab
#35 simponi
#36 certolizumab near/2 pegol
#37 cimzia
#38 (#10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37)
#39 MeSH descriptor: [Dose‐Response Relationship, Drug] this term only
#40 MeSH descriptor: [Drug Administration Schedule] this term only
#41 MeSH descriptor: [Remission Induction] this term only
#42 MeSH descriptor: [Withholding Treatment] this term only
#43 down near/3 titrat*
#44 dose near/3 reduc*
#45 dose near/3 de‐escalat*
#46 discontinu*
#47 dose near/3 taper*
#48 spac*
#49 cessat*
#50 stop*
#51interval near/3 widen*
#52 biologic near/2 free
#53 withdraw*
#54 dose near/3 titrat*
#55 (#39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54)
#56 (#9 and #38 and #55)
Appendix 2. MEDLINE search strategy
1 exp arthritis, rheumatoid/
2 (arthritis adj2 rheumat$).tw.
3 (felty$ adj2 syndrome).tw.
4 (caplan$ adj2 syndrome).tw.
5 rheumatoid nodule$.tw.
6 rheumatoid vasculitis.tw.
7 (sjogren$ adj2 syndrome).tw.
8 still$ disease.tw.
9 or/1‐8
10 Adalimumab/
11 Certolizumab Pegol/
12 Etanercept/
13 Infliximab/
14 Infliximab.tw.
15 remicade.tw.
16 remsima.tw.
17 inflectra.tw.
18 adalimumab.tw.
19 humira.tw.
20 (Certolizumab adj2 pegol).tw.
21 cimzia.tw.
22 Etanercept.tw.
23 enbrel.tw.
24 benepali.tw.
25 Golimumab.tw.
26 simponi.tw.
27 Tumor Necrosis Factor‐alpha/ai [Antagonists & Inhibitors]
28 anti‐tnf.tw.
29 anti‐tumor necrosis factor.tw.
30 anti‐tumour necrosis factor.tw.
31 (Tumor Necrosis Factor adj3 inhibit$).tw.
32 (tumour necrosis factor adj3 inhibit$).tw.
33 (tnf adj3 inhibit$).tw.
34 Tnfi.tw.
35 (tnf adj3 block$).tw.
36 bDMARD$.tw.
37 biologic$ DMARD$.tw.
38 or/10‐37
39 Dose‐Response Relationship, Drug/
40 (down adj3 titrat$).tw.
41 (dose adj3 titrat$).tw.
42 (dose adj3 reduc$).tw.
43 (dose adj3 de‐escalat$).tw.
44 withdraw$.tw.
45 discontinu$.tw.
46 (dose adj3 taper$).tw.
47 (biologic adj2 free).tw.
48 spac$.tw.
49 cessat$.tw.
50 stop$.tw.
51 (interval adj3 widen$).tw.
52 Drug Administration Schedule/
53 remission induction/
54 Withholding Treatment/
55 or/39‐54
56 randomized controlled trial.pt.
57 controlled clinical trial.pt.
58 randomized.ab.
59 randomised.ab
60 placebo.ab.
61 clinical trials as topic.sh.
62 randomly.ab.
63 trial.ti.
64 or/56‐63
65 exp animals/ not humans.sh.
66 64 not 65
67 9 and 38 and 55
68 66 and 67
Appendix 3. Embase search strategy
1 exp arthritis, rheumatoid/
2 (arthritis adj2 rheumat$).tw.
3 (felty$ adj2 syndrome).tw.
4 (caplan$ adj2 syndrome).tw.
5 rheumatoid nodule$.tw.
6 rheumatoid vasculitis.tw.
7 (sjogren$ adj2 syndrome).tw.
8 still$ disease.tw.
9 or/1‐8
10 infliximab.tw.
11 infliximab/
12 remicade.tw.
13 remsima.tw.
14 inflectra.tw.
15 humira.tw.
16 adalimumab/
17 adalimumab.tw.
18 cimzia.tw.
19 certolizumab pegol/
20 (certolizumab adj2 pegol).tw.
21 enbrel.tw.
22 etanercept/
23 etanercept.tw.
24 benepali.tw.
25 simponi.tw.
26 golimumab/
27 golimumab.tw.
28 tumor necrosis factor antibody/
29 anti‐tnf.tw.
30 anti‐tumor necrosis factor.tw.
31 anti‐tumour necrosis factor.tw.
32 (Tumor Necrosis Factor adj3 inhibit$).tw.
33 (Tumour Necrosis Factor adj3 inhibit$).tw.
34 (tnf adj3 inhibit$).tw.
35 Tnfi.tw.
36 (tnf adj3 block$).tw.
37 bDMARD$.tw.
38 biologic$ DMARD$.tw.
39 or/10‐38
40 (down adj3 titrat$).tw.
41 (biologic adj2 free).tw.
42 drug dose reduction/
43 (dose adj3 reduc$).tw.
44 (dose adj3 de‐escalat$).tw.
45 discontinu$.tw.
46 (dose adj3 taper$).tw.
47 spac$.tw.
48 cessat$.tw.
49 stop$.tw.
50 (interval adj3 widen$).tw.
51 withdraw$.tw.
52 (dose adj3 titrat$).tw.
53 remission/
54 treatment withdrawal/
55 dose response/
56 or/40‐55
57 9 and 39 and 56
58 random$.tw.
59 factorial$.tw.
60 crossover$.tw.
61 cross over.tw.
62 cross‐over.tw.
63 placebo$.tw.
64 (doubl$ adj blind$).tw.
65 (singl$ adj blind$).tw.
66 assign$.tw.
67 allocat$.tw.
68 volunteer$.tw.
69 crossover procedure/
70 double blind procedure/
71 randomised controlled trial/
72 single blind procedure/
73 or/58‐72
74 57 and 73
Appendix 4. Web of Science search strategy
#1 rheumat* NEAR/2 arthritis or caplan* NEAR/2 syndrome or felty* NEAR/2 syndrome or "rheumatoid nodule*" or "rheumatoid vasculitis" or sjogren* NEAR/2 syndrome or "still* disease"
#2 "anti‐tumor necrosis factor" or "anti‐tumour necrosis factor" or "tumor necrosis factor" NEAR/3 inhibit* or "tumour necrosis factor" NEAR/3 inhibit* or anti‐tnf or tnf NEAR/3 inhibit* or tnf NEAR/3 block* or tnfi or adalimumab or humira or etanercept or enbrel or benepali or infliximab or remicade or remsima or inflectra or golimumab or simponi or certolizumab NEAR/2 pegol or cimzia or bDMARD* or "biologic* DMARD*"
#3 down NEAR/3 titrat* or dose NEAR/3 reduc* or dose NEAR/3 de‐escalat* or discontinu* or dose NEAR/3 taper* or spac* or cessat* or stop* or interval NEAR/3 widen* or dose NEAR/3 titrat* or withdraw* or biologic NEAR/2 free
#4 trial* or random* or control*
#5 #1 AND #2 AND #3 AND #4
Appendix 5. Search strategies trial registries
Registry: US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov/)
Date of search: 11 April 2018
Search terms and results:
| Date | Terms | Hits |
| 11‐4‐2018 | Rheumatoid arthritis AND biologics | 470 |
| 11‐4‐2018 | Rheumatoid arthritis AND anti TNF | 291 |
| 11‐4‐2018 | Rheumatoid arthritis AND etanercept | 168 |
| 11‐4‐2018 | Rheumatoid arthritis AND adalimumab | 201 |
| 11‐4‐2018 | Rheumatoid arthritis AND infliximab | 119 |
| 11‐4‐2018 | Rheumatoid arthritis AND golimumab | 38 |
| 11‐4‐2018 | Rheumatoid arthritis AND certolizumab | 71 |
| 11‐4‐2018 | Rheumatoid arthritis AND discontinuing | 14 |
| 11‐4‐2018 | Rheumatoid arthritis AND reducing | 250 |
| 11‐4‐2018 | Rheumatoid arthritis AND withdrawal | 96 |
| 11‐4‐2018 | Rheumatoid arthritis AND “dose reduction” | 25 |
Registry: EU Clinical Trials Register (www.clinicaltrialsregister.eu/)
Date of search: 11 April 2018
Search terms and results:
| Date | Terms | Hits |
| 11‐4‐2018 | Rheumatoid arthritis AND biologics | 66 |
| 11‐4‐2018 | Rheumatoid arthritis AND anti TNF | 144 |
| 11‐4‐2018 | Rheumatoid arthritis AND etanercept | 141 |
| 11‐4‐2018 | Rheumatoid arthritis AND adalimumab | 159 |
| 11‐4‐2018 | Rheumatoid arthritis AND infliximab | 119 |
| 11‐4‐2018 | Rheumatoid arthritis AND golimumab | 53 |
| 11‐4‐2018 | Rheumatoid arthritis AND certolizumab | 56 |
| 11‐4‐2018 | Rheumatoid arthritis AND discontinuing | 4 |
| 11‐4‐2018 | Rheumatoid arthritis AND reducing | 25 |
| 11‐4‐2018 | Rheumatoid arthritis AND withdrawal | 62 |
| 11‐4‐2018 | Rheumatoid arthritis AND “dose reduction” | 12 |
Registry: Dutch trial register (www.trialregister.nl)
Date of search: 11‐4‐2018
Search terms and results:
| Date | Terms | Hits |
| 11‐4‐2018 | Rheumatoid arthritis | 59 |
| 11‐4‐2018 | TNF‐blockers | 1 |
| 11‐4‐2018 | Anti‐TNF | 11 |
| 11‐4‐2018 | TNF | 19 |
Registry: World Health Organization International Clinical Trials Registry Platform (www.who.int/ictrp/en/)
Date of search: 11 April 2018
Search terms and results:
| Date | Terms | Hits |
| 11‐4‐2018 | Anti‐TNF OR biologic OR adalimumab OR etanercept OR certolizumab OR infliximab OR golimumab AND Rheumatoid arthritis AND dose OR taper OR withdrawal OR discontinuation OR reduce OR titration (recruitment status: ALL) |
131 |
Appendix 6. Risk of bias
Article nr:
Reviewer: BvdB/LV
Date:
| Domain | Support for judgement |
Judgement low risk of bias, high risk of bias, or unclear risk of bias |
| Selection bias | ||
| Random sequence generation | ||
| Allocation concealment | ||
| Performance bias | ||
| Blinding of participants and personnel | |
|
| Detection bias | ||
| Blinding of outcome assessment (subjective outcomes) |
|
|
| Blinding of outcome assessment (objective outcomes) |
||
| Attrition bias | ||
| Incomplete outcome data | |
|
| Reporting bias | ||
| Selective reporting | ||
| Other bias | ||
| Other sources of bias | |
|
Data and analyses
Comparison 1. Anti‐TNF dose reduction versus anti‐TNF continuation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean disease activity score (DAS28) | 2 | 501 | Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.11, 0.24] |
| 2 Proportion persistent remission (DAS28) | 2 | 612 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.80, 1.28] |
| 3 Proportion switched to another biologic | 1 | 323 | Risk Ratio (IV, Random, 95% CI) | 0.40 [0.17, 0.93] |
| 4 Proportion radiographic progression (mSvdH > 0.5) | 2 | 553 | Risk Ratio (IV, Random, 95% CI) | 1.22 [0.76, 1.95] |
| 5 Function (Health Assessment Questionnaire) | 2 | 501 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.00, 0.19] |
| 6 Number of serious adverse events | 5 | 1084 | Risk Ratio (IV, Random, 95% CI) | 1.09 [0.65, 1.82] |
| 7 Withdrawals due to adverse events | 3 | 937 | Risk Ratio (IV, Random, 95% CI) | 1.07 [0.51, 2.24] |
| 8 Proportion of participants with a flare | 3 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 9 Quality of life | 2 | 501 | Mean Difference (IV, Random, 95% CI) | ‐0.00 [‐0.04, 0.03] |
1.8. Analysis.
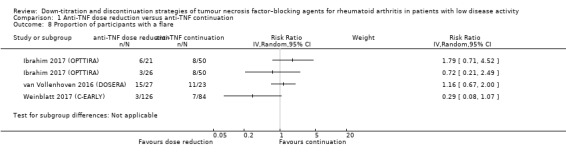
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 8 Proportion of participants with a flare.
1.9. Analysis.
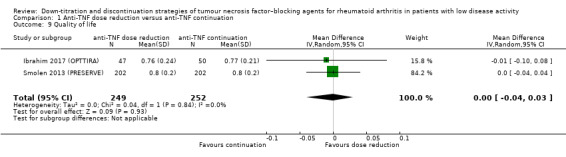
Comparison 1 Anti‐TNF dose reduction versus anti‐TNF continuation, Outcome 9 Quality of life.
Comparison 2. Anti‐TNF discontinuation versus anti‐TNF continuation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean disease activity score (DAS28) | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Discontinuation without restarting, or with restarting and LOCF analysis | 2 | 733 | Mean Difference (IV, Random, 95% CI) | 0.96 [0.67, 1.25] |
| 1.2 Discontinuation with restarting without LOCF analysis | 1 | 692 | Mean Difference (IV, Random, 95% CI) | 0.29 [0.14, 0.44] |
| 2 Proportion persistent remission (DAS28) | 6 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 3 Proportion radiographic progression (mSvdH > 0.5) | 3 | 549 | Risk Ratio (IV, Random, 95% CI) | 1.69 [1.10, 2.59] |
| 4 Function (Health Assessment Questionnaire) | 4 | 1498 | Mean Difference (IV, Random, 95% CI) | 0.18 [0.05, 0.31] |
| 5 Number of serious adverse events | 8 | 2095 | Risk Ratio (IV, Random, 95% CI) | 1.29 [0.82, 2.03] |
| 6 Withdrawals due to adverse events | 4 | 1116 | Risk Ratio (IV, Random, 95% CI) | 1.46 [0.75, 2.84] |
| 7 Proportion flare | 5 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 7.1 DAS28 ≥ 3.2 and ΔDAS ≥ 0.6 (24 to 52 weeks' follow‐up) | 3 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 DAS28 ≥ 3.2 OR ΔDAS ≥ 0.6 (follow‐up 52 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 DAS28 ≥ 2.6 OR ΔDAS > 1.2 (follow‐up 28 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Proportion failure (follow‐up 48 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Quality of life | 2 | 733 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.13, ‐0.07] |
2.7. Analysis.
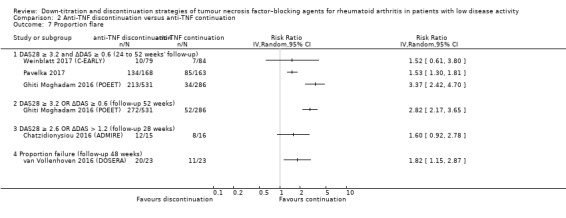
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 7 Proportion flare.
2.8. Analysis.
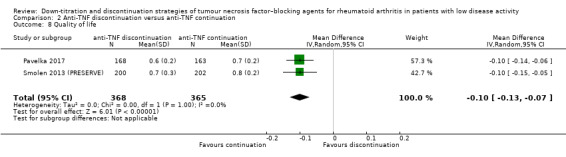
Comparison 2 Anti‐TNF discontinuation versus anti‐TNF continuation, Outcome 8 Quality of life.
Comparison 3. Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean disease activity score (DAS28) | 3 | 357 | Mean Difference (IV, Random, 95% CI) | 0.25 [‐0.17, 0.67] |
| 2 Proportion persistent remission (DAS28) | 1 | 180 | Risk Ratio (IV, Random, 95% CI) | 0.89 [0.75, 1.06] |
| 3 Proportion switched to another biologic | 2 | 317 | Risk Ratio (IV, Random, 95% CI) | 0.62 [0.25, 1.54] |
| 4 Proportion radiographic progression (mSvdH > 0.5 or > 1.0) | 2 | 312 | Risk Ratio (IV, Random, 95% CI) | 1.45 [0.77, 2.73] |
| 5 Function (Health Assessment Questionnaire) | 1 | 123 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.02, 0.42] |
| 6 Number of serious adverse events | 2 | 317 | Risk Ratio (IV, Random, 95% CI) | 1.24 [0.42, 3.70] |
| 7 Proportion flare | 3 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 7.1 DAS28‐ESR >2.6 with ∆DAS28‐ESR > 0.6 after 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 ΔDAS28‐CRP > 1.2 OR ΔDAS28‐CRP > 0.6 and current DAS28‐CRP ≥ 3.2 for > 3 months at 9 months' follow‐up | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 ΔDAS28‐CRP > 1.2 OR ΔDAS28‐CRP > 0.6 and current DAS28‐CRP ≥ 3.2 for > 3 months at 18 months' follow‐up | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 ΔDAS28‐CRP > 1.2 OR ΔDAS28‐CRP > 0.6 and current DAS28‐CRP ≥ 3.2 for < 3 months at 9 months' follow‐up | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 ΔDAS28‐CRP > 1.2 OR ΔDAS28‐CRP > 0.6 and current DAS28‐CRP ≥ 3.2 for < 3 months at 18 months' follow‐up | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.6 DAS28 > 2.6, SDAI > 5 or ACR/EULAR criteria not fulfilled (follow‐up 24 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.7 DAS28 > 2.6, SDAI > 5 or ACR/EULAR criteria not fulfilled (follow‐up 48 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.8 DAS28 > 2.6, SDAI > 5 or ACR/EULAR criteria not fulfilled (follow‐up 72 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.9 DAS28 > 2.6, SDAI > 5 or ACR/EULAR criteria not fulfilled (follow‐up 96 weeks) | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Change in other medication | 1 | Risk Ratio (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Use of intramuscular or intra‐articular glucocorticoid injections at 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Use of oral glucocorticoids at 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 DMARDs reduction or discontinuation after 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 DMARD initiation or dose escalation after 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Use of a DMARD at 18 months | 1 | Risk Ratio (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
3.7. Analysis.
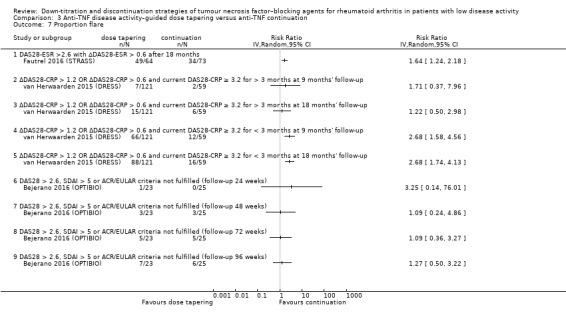
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 7 Proportion flare.
3.8. Analysis.
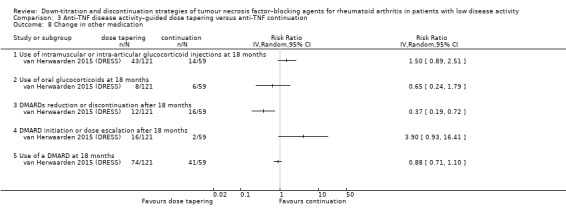
Comparison 3 Anti‐TNF disease activity–guided dose tapering versus anti‐TNF continuation, Outcome 8 Change in other medication.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bejerano 2016 (OPTIBIO).
| Methods | Open, randomised and controlled study in a hospital in Spain | |
| Participants | Inclusion criteria:
Exclusion criteria: none described Baseline characteristics: not reported |
|
| Interventions | Participants are assigned to 2 groups randomly:
|
|
| Outcomes | Primary endpoint is to evaluate the proportion of patients that after 1 year are maintained in clinical remission with a dose reduction treatment regimen of biological therapy in people with RA, and to evaluate if the proportion of participants in remission with new regimen dose of treatment is not inferior to participants in remission with standard dose regimen. Analyses: ITT |
|
| Notes | Acronym: OPTIBIO At time of review, study presented in abstract form only (preliminary results). EudraCT: 2012‐004482‐40 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described in this abstract |
| Allocation concealment (selection bias) | Unclear risk | Not described in this abstract |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open‐label study" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Open‐label study" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient reporting of attrition/exclusions to permit judgement of ‘low risk’ or ‘high risk’ |
| Selective reporting (reporting bias) | Unclear risk | Study protocol available. Not all outcome measures described in this abstract. |
| Other bias | Unclear risk | Insufficient information to assess whether an important risk of bias exists |
Chatzidionysiou 2016 (ADMIRE).
| Methods | 52‐week, multicentre, randomised, controlled, open‐label pilot study in Sweden | |
| Participants | Inclusion criteria:
Baseline characteristics: median age 61 (IQR 53 to 65) years, 65% female, median disease duration 8 (IQR 5 to 16) years, median DAS28 1.9 (IQR 1.55 to 2.39), median number of previous DMARDs 2 (IQR 1 to 3), median number of previous bDMARDs 0 (IQR 0 to 1) |
|
| Interventions | People fulfilling the inclusion criteria were randomised in a 1:1 ratio to arm AM (continue with adalimumab and methotrexate) or to arm M (discontinue adalimumab and continue with methotrexate monotherapy) for 52 weeks. Any participant experiencing disease "flare" at any visit could continue in the rescue arm, where adalimumab would be re‐instituted. Disease flare was defined as DAS28 ≥ 2.6 or a change in DAS28 (ΔDAS28) > 1.2 from baseline at any time.
|
|
| Outcomes | Primary outcome: proportion of participants in remission (DAS28 < 2.6) at week 28 in both arms Secondary outcomes:
Analyses: ITT; non‐responder imputation (i.e. "flare" imputed) was performed for participants with no available DAS28 score at week 28 (this included most participants who had a flare in the M arm and who restarted treatment with adalimumab) |
|
| Notes | Acronym: ADMIRE Pilot study Funding: AbbVie Disclosures: KC has received consultancy fees and/or speaker honoraria from Pfizer and Roche. CT has received consultancy fees and/or speaker honoraria from AbbVie, Bristol‐Myers Squibb, Janssen, MSD, Pfizer, Roche, Novartis, and UCB, and has received unrestricted research grants from AbbVie, Pfizer, and Roche. KF has received consultancy fees and/or speaker honoraria from AbbVie, Bristol‐Myers Squibb, MSD, and Pfizer. RvV has received research support and/or honoraria from AbbVie, Biotest, BMS, GSK, Lilly, Merck, Pfizer, Roche, UCB, and Vertex. MH is an employee of AbbVie. EudraCT: 2008‐004398‐16 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described; "The randomization was 4 patients in each block" |
| Allocation concealment (selection bias) | Low risk | "Use of an independent person and a sealed, opaque envelope system" |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open‐label study" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Open‐label study" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
El Miedany 2016.
| Methods | 12‐month randomised controlled trial | |
| Participants | Inclusion criteria:
Exclusion criteria:
Baseline characteristics: mean (SD) DAS28 group 1: 1.97 (0.4), group 5: 2.2 (0.5); RF+ group 1: 55%, group 5: 56%; ACPA+ group 1: 61%, group 5: 59% |
|
| Interventions | Participants were randomised 1:1:1:1:1 to 5 groups (group 1 and 5 of interest for this review):
|
|
| Outcomes | Primary endpoint was the maintenance of remission for 12 months. Secondary endpoint was identifying the potential predictors, whether ultrasonographic, clinical, or lab measures, for relapses in patients tapering and/or stopping DMARD(s) and/or biologic therapy. Analyses: ITT |
|
| Notes | This study included participants using anti‐TNF as well as non‐anti‐TNF. The authors provided data for the participants using anti‐TNF only for this review. Disclosures: none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Blinding not described, however very unlikely due to the absence of a description of placebo treatment |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcome measure assessment was unblinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 participant lost to follow‐up, in group 1. |
| Selective reporting (reporting bias) | Unclear risk | No study protocol present, therefore insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Fautrel 2016 (STRASS).
| Methods | 18‐month, multicentre randomised controlled equivalence trial in France | |
| Participants | Inclusion criteria:
Exclusion criteria
Baseline characteristics: age (mean, SD) 55.6 (11.2) years, female 78%, disease duration 9.5 (8.0) years, 69% rheumatoid factor positive, 78% anti‐CCP positive, DAS28 1.8 (0.6), 54% etanercept, 46% adalimumab, number of previous DMARDs 2.7 (1.7), percentage of participants with previous bDMARD treatment 24% |
|
| Interventions | Participants were randomised into 1 of 2 arms: maintenance of the subcutaneous injections at the standard full regimen (M‐arm) or injections spacing by 50% every 3 months up to complete stop (S‐arm). Spacing was reversed to the previous interval in case of relapse, and eventually reattempted after remission was re‐achieved.
|
|
| Outcomes | Primary outcome: evolution in RA inflammatory activity over 18 months as measured by the DAS28 every 3 months Secondary outcomes:
Analyses: primary analysis PP, supplemented by an ITT analysis |
|
| Notes | Acronym: STRASS Funding: the trial was conducted under the auspices of the CRI‐IMIDIATE clinical research FCRIN network. Institutional support by a grant from the Ministry of Health (PHRC national 2007, AOM 07 127/P 070120), France. The sponsor was the Département à la Recherche Clinique et au Développement, Assistance Publique–Hôpitaux de Paris. Disclosures: none declared EudraCT: 2007‐004483‐41 ClinicalTrials.gov: NCT00780793 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was generated by use of a computer‐generated sequence with blocks of variable (2–6) and undisclosed size stratified by centre and TNF‐blocker (ie, ETA or ADA)" |
| Allocation concealment (selection bias) | Low risk | "Treatment allocation was concealed to patients, research staff and clinical staff until randomisation by use of an internet‐based randomisation module (Cleanweb, Telemedecine technologies S.A.S, Boulogne‐Billancourt, France)." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Open study |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Most important outcome assessments (DAS28 and X‐ray) blinded. Other outcomes likely to be influenced. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing outcome data unlikely to be related to true outcome |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | High risk | Number of included participants was lower than anticipated in the sample size calculation. |
Ghiti Moghadam 2016 (POEET).
| Methods | 12‐month, open‐label randomised controlled study in the Netherlands | |
| Participants | Inclusion criteria:
There were no exclusion criteria. Participant characteristics: mean (SD): age intervention 60.0 (11.8), control 59.7 (10.6); female: intervention 68%, control 66%; disease duration: intervention 12.0 (8.8), control 11.1 (8.4); previous bDMARD: intervention 13.4%, control 15% |
|
| Interventions | Participants were randomised 2:1 to either stop or continue their anti‐TNF. All other medications, including csDMARDs, glucocorticoids, and NSAIDs, were left at the discretion of the treating rheumatologist and were continued unchanged as much as possible. In case of flare, defined as a DAS28 ≥ 3.2 plus an increase ≥ 0.6 compared to the baseline DAS28, anti‐TNF treatment could be restarted in the stop group or switched in the continuation group.
|
|
| Outcomes | Primary outcome: percentage of participants who experienced a flare (DAS28 ≥ 3.2 and DAS28 increase ≥ 0.6) of RA during the first year Secondary outcomes:
Analyses: ITT with participants that were correctly included |
|
| Notes | Acronym: POEET Dutch trial register: NTR3112 Funding: The Netherlands Organization for Health Research and Development (ZonMw)/Government of the Netherlands, Ministry of Health, Welfare and Sport (VWS) Disclosures: none |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer block randomization was used to achieve balance in allocation per center." |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open‐label study" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Open‐label study" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | High risk | The flare criterion differs between the trial register and the final publication, which might have introduced risk of bias. |
Ibrahim 2017 (OPTTIRA).
| Methods | Open‐label, 6‐month, multicentre randomised controlled proof‐of‐principle trial in the United Kingdom | |
| Participants | 103 RA patients receiving etanercept or adalimumab plus a DMARD who achieved sustained good responses with DAS28 scores of ≤ 3.2 without increases of > 0.6 during the previous 3 months Baseline characteristics: mean (SD): age 57 (11), female 74%, median disease duration 11.3 (IQR 7.3 to 16.7) |
|
| Interventions | Participants were randomised 1:1:2 to:
Note: in months 6 to 12, controls tapered anti‐TNF and experimental groups discontinued anti‐TNF (data not included in review). |
|
| Outcomes | Primary outcome: time to first flare, defined as an increase in DAS28 scores ≥ 0.6 resulting in a DAS28 > 3.2 together with an increase in the swollen joint count; both had to be present on 2 occasions at least 1 week apart. An increase in DAS28 score ≥ 1.2 resulting in DAS28 > 3.2 was defined as flare irrespective of changes in swollen joints. Secondary outcomes:
Analyses: ITT, participants without flares who withdrew or were lost to follow‐up were censored at the time of their last visit. |
|
| Notes | Acronym: OPTTIRA The trial was funded by Arthritis Research UK (grant reference number 18813). Disclosures: JBG has received honoraria for speaking from UCB, Pfizer, Celgene, and Bristol‐Myers Squibb. All other authors declared no conflicts of interest. EudraCT: 2010‐020738‐24 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomisation by online electronic data capture system using minimization protocol with randomly permuted blocks" |
| Allocation concealment (selection bias) | Low risk | "Trial team were blinded to allocation process and sequences" |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | Open‐label study |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study, and most outcomes likely to be influenced. "X‐ray reading was blinded" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Numbers of participants included in different analysis sets clearly described. 100% of participants included in ITT analyses. |
| Selective reporting (reporting bias) | Low risk | Study protocol available; similar outcome measures in the paper compared to the protocol |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Pavelka 2017.
| Methods | 52‐week, double‐blind, multicentre randomised controlled study in locations around the world Period 1: 24 weeks open‐label; period 2: 28 weeks double‐blind |
|
| Participants | Inclusion criteria (period 1):
Exclusion criteria (period 1):
Inclusion criteria (period 2):
Baseline characteristics: mean (SD): age: intervention 47.2 (11.8), control 46.1 (12.9); female: intervention 85%, control 83%; symptom duration in years: intervention 8.3 (6.8), control 8.0 (7.4); prior csDMARD (not MTX): intervention 34%, control 38% |
|
| Interventions | Period 1:
Period 2:
|
|
| Outcomes | Primary outcome measure: proportion of participants who remained in LDA (DAS28 < 3.2) at week 52 without rescue medication Secondary outcome measures:
Analyses: ITT with LOCF approach on the full analysis set, which included participants who had taken at least 1 dose of study medication and had at least 1 DAS28‐ESR evaluation |
|
| Notes | EudraCT: 2011‐005448‐87 ClinicalTrials.gov: NCT01578850 Funding: Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Screening, enrollment, and randomization were accomplished using an automated internet/telephone randomization system (i.e., the Interactive Web Response System)." |
| Allocation concealment (selection bias) | Low risk | "Screening, enrollment, and randomization were accomplished using an automated internet/telephone randomization system (i.e., the Interactive Web Response System)." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | The study was participant‐, investigator‐, and sponsor‐blinded. Prefilled syringes were labelled in such a way that participants’ treatment assignment could not be determined. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Double‐blind trial" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data < 10% and balanced in numbers across intervention groups, with similar reasons for missing data across groups. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Raffeiner 2015.
| Methods | Open‐label quasi‐randomised controlled study in a hospital in Italy | |
| Participants | Inclusion criteria:
Baseline characteristics: mean (SD): age: intervention 55.7 (13.5), control 55.6 (12.8); female: intervention 85%, control 81%; disease duration: intervention 14.3 (9), control 13.4 (5.9); number of previous DMARDs: intervention 2.4 (1.1), control 2.4 (1.3) |
|
| Interventions | Participants were randomised to 1 of the following 2 subcutaneous dose regimens: etanercept 25 mg weekly (group A) or etanercept 25 mg biweekly (group B). The randomisation was done in a consecutive manner, 1:1, and treatment was continued until disease flare‐up.
|
|
| Outcomes | Primary outcome: maintained DAS28 remission (DAS28 < 2.6) Secondary outcomes:
Analyses: ITT |
|
| Notes | Funding: This study was supported in part by AIFA (Agenzia Italiana del Farmaco). Disclosures: none declared |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "The randomisation was done according to the order of the recruitment. The first patient recruited was allocated to group A, the second one to group B, and so on." Comment: semi‐randomisation |
| Allocation concealment (selection bias) | High risk | Predictability of allocation due to consecutive randomisation |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Patients, physicians and study nurses were not blinded." |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study, and most outcomes likely to be influenced; "the radiologist was blinded" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing outcome data unlikely to be related to true outcome |
| Selective reporting (reporting bias) | Unclear risk | No study protocol present, therefore insufficient information to permit judgement of ‘low risk’ or ‘high risk’ |
| Other bias | High risk | No study protocol present, but information from earlier abstract indicates that inclusion criteria, outcome measures, and duration of follow‐up have changed over time. |
Smolen 2013 (PRESERVE).
| Methods | 88‐week, randomised, placebo‐controlled trial in 80 centres in Europe, Latin America, Asia, and Australia Open‐label period: weeks 0 through 36. Randomisation at week 36. Double‐blind period: weeks 36 through 88 |
|
| Participants | Open‐label period:
Double‐blind period:
Baseline characteristics:
|
|
| Interventions | Open‐label period:
Double‐blind period:
|
|
| Outcomes | Primary outcome (double‐blind period): proportion of participants with DAS28 ≤ 3.2 in the etanercept 50 mg/week versus placebo group at week 88 (52 weeks after randomisation) In case of significantly more low disease activity in the etanercept 50 mg/week group compared with the placebo group, the conditional primary endpoint was proportion of participants receiving etanercept 25 mg/week who achieved DAS28 ≤ 3.2. Secondary outcomes:
Analyses: modified ITT population made up of participants who had received at least 1 dose of study drug and had 1 or more DAS28 evaluations. A modified non‐responder analysis was done in which participants who discontinued early because of poor efficacy were imputed as non‐responders for all time points; all other participants were analysed with the LOCF method. |
|
| Notes | Acronym: PRESERVE Funding: "PRESERVE was sponsored by Wyeth, which was acquired by Pfizer in October 2009. Pfizer was responsible for data collection and analyses. The academic authors and sponsors representatives were involved in the study design, data analyses, data interpretation, writing of the report, and the final decision to submit for publication. Biostatisticians at Pfizer did and verified all data analyses. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication." Disclosures: JSS has received honoraria for consultations or speaking engagements, or grant support, or both, from Abbott, Amgen, AstraZeneca, Bristol‐Myers Squibb, Celgene, GlaxoSmithKline, MSD, Novo Nordisk, Pfizer, Roche, Sandoz, Sanofi, and UCB. PN has received grant support and honoraria for consultations from Pfizer. FI‐P has received grant support and honoraria for consultations or speaking engagements from Bristol‐Myers Squibb, Janssen, Pfizer, and Roche. PM has received grant support from Abbott, Bristol‐Myers Squibb, Celltrion, Centocor, GlaxoSmithKline, Human Genome Sciences, Merck, Neovacs, and Pfizer. KP has received honoraria for lectures from Abbott, Fidia, MSD, Pfizer, Roche, and UCB. RP, CH, ASK, and BV are employees of Pfizer and own Pfizer stock. AS is an employee of Inventive Health, who are paid contractors for Pfizer, providing statistical support for the PRESERVE study and the development of this report. The other authors declare that they have no conflicts of interest. EudraCT: 2007‐000896‐41 ClinicalTrials.gov: NCT00565409 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomly assigned by a centralised system" |
| Allocation concealment (selection bias) | Low risk | "allocation of patients to treatment groups was done with the ICOPhone interactive voice response system on the basis of information supplied by the investigator or the study staff" |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "patients, investigators, data‐analysts and study staff were all masked to treatment allocation" "etanercept packages for each patient were identical" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "patients, investigators, data‐analysts and study staff were all masked to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Patients who discontinued early because of poor efficacy were imputed as non‐responders for all time points" "Significantly more patients discontinued in group given placebo than in 50 mg and 25 mg groups" |
| Selective reporting (reporting bias) | Low risk | Study protocol available; similar outcome measures in the paper compared to the protocol |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Smolen 2014 (OPTIMA).
| Methods | 78‐week, multicentre, randomised, double‐blind, placebo‐controlled trial in 161 sites across Europe (n = 71), North America (n = 73), South America (n = 5), Africa (n = 6), Australia (n = 3), and New Zealand (n = 3) Period 1: weeks 0 through 26, re‐randomisation at week 26; period 2: weeks 26 through 78 |
|
| Participants | Period 1:
Period 2:
Baseline characteristics: mean (SD): age: intervention 50.1 (14.9), control 49.5 (15.3); female: intervention 73%, control 73%; RF+: intervention 95%, control 89%; ACPA+: intervention 90%, control 90%; disease duration in months: intervention 3.9 (3.3), control 3.9 (2.9); percentage with ≥ 1 previous DMARDs: intervention 8.8%, control 9.5% |
|
| Interventions | Period 1: participants were randomised in a ratio 1:1 to:
Period 2: participants in the adalimumab plus methotrexate group reaching DAS28‐CRP < 3.2 at weeks 22 and 26 were re‐randomised in a 1:1 ratio to:
|
|
| Outcomes | Primary outcome (period 2): not described Outcomes:
Analyses: ITT including all participants who had received at least 1 dose of study drug in period 2. The primary endpoint was assessed using non‐responder imputation (NRI). NRI or LOCF, or both, was used for additional clinical outcomes; LOCF was used for functional outcomes. Markov chain Monte Carlo method was used to impute missing radiographic data 10 times (multiple imputation). |
|
| Notes | Acronym: OPTIMA Funding: AbbVie Disclosures: JSS has received grant fees, research fees, consulting fees, or other remuneration from AbbVie, Amgen, AstraZeneca, Bristol‐Myers Squibb, Celgene, Centocor‐Janssen, GlaxoSmithKline, Lilly, Pfizer (Wyeth), MSD (Schering‐Plough), Novo Nordisk, Roche, Sandoz, and UCB. PE has provided paid expert advice and has done trials for AbbVie, Merck, Pfizer, UCB, Roche, and Bristol‐Myers Squibb. RF has received research grants and consulting fees or other remuneration from AbbVie, Pfizer, Merck, Roche, UCB, Celgene, Centocor‐Janssen, Amgen, AstraZeneca, Bristol‐Myers Squibb, Lilly, and Novartis. RFvV has served as a consultant for, or received grant or research support from, AbbVie, GlaxoSmithKline, Merck, Pfizer, Roche, and UCB. KP has received consulting fees or other remuneration and speaker honoraria from AbbVie, Pfizer, MSD, Roche, Amgen, and Bristol‐Myers Squibb. PD has served on speaker’s bureaus for BMS. BG, HK, and VA are shareholders and employees of AbbVie. LR is a former employee of AbbVie. AK has received grant fees, research fees, or provided paid expert advice to AbbVie, Amgen, AstraZeneca, Bristol‐Myers Squibb, Celgene, Centocor‐Janssen, Pfizer, Roche, and UCB. EudraCT: 2006‐004139‐31 ClinicalTrials.gov: NCT00420927 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised trial, method not described |
| Allocation concealment (selection bias) | Low risk | "Patients were centrally randomised in blocks of four by interactive voice response system on the basis of information supplied by the investigator" |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "During period 2, treatment reallocation of patients who achieved the target was also masked to patients and investigators" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "During period 2, treatment reallocation of patients who achieved the target was also masked to patients and investigators" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for missing outcome data unlikely to be related to true outcome |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
van Herwaarden 2015 (DRESS).
| Methods | Pragmatic, multicentre, open‐label, randomised, controlled, cost‐effectiveness non‐inferiority strategy trial, stratified for anti‐TNF agent in 2 hospitals in the Netherlands | |
| Participants | Inclusion criteria: we enrolled consenting patients with rheumatoid arthritis (based on 2010 or 1987 ACR criteria, or clinical diagnosis by the treating rheumatologist) using adalimumab or etanercept at any stable dose and interval for at least 6 months, with stable low disease activity at 2 subsequent visits Exclusion criteria: none described Baseline characteristics: mean (SD): age: intervention 59 (10.5), control 58 (9.3); female: intervention 62%, control 69%; median disease duration: intervention 10 (IQR 6 to 17), control 10 (IQR 6 to 16), median number of previous DMARD treatment: intervention 2 (IQR 1 to 3), control 2 (IQR 1 to 3); median number of previous TNFi treatment: intervention 0 (IQR 0 to 1), control 0 (IQR 0 to 1) |
|
| Interventions | Participants were randomised (stratified by anti‐TNF) in a ratio of 2:1.
Increase in interval for adalimumab: start 14, 21, 28 days and stop, etanercept start 7, 10, 14 days and stop |
|
| Outcomes | Primary outcome is to assess whether the difference in cumulative incidence in persistent RA flares (DAS28 increase > 1.2 or DAS28 increase > 0.6 with a current DAS28 ≥ 3.2) and a duration of > 3 months between the intervention group and the usual care group does not exceed the non‐inferiority margin of 20% after 18 months' follow‐up. Secondary outcomes include cost‐effectiveness ratio between intervention and usual care groups. Other secondary outcomes are predictive factors for successful dose reduction and progression of radiological joint damage. Analyses: primary analyses were done PP by including only participants who (1) completed follow‐up, (2) actually started dose reduction of TNF inhibitors in the dose reduction arm, and (3) had not stopped or reduced TNF inhibitor use at 18 months’ follow‐up in the usual care arm. Additional ITT analyses were also done. No multiple imputation was deemed necessary since almost no data were missing. |
|
| Notes | Dutch trial register: NTR3216 Acronym: DRESS Funding: The study received no external funding. Disclosures: JB received grants and personal fees from Pfizer and AbbVie during the conduct of the study, and grants and personal fees from Roche, Bristol‐Myers Squibb, and Union Chimique Belge outside the submitted work. RvV received grants from AbbVie, BMS, GlaxoSmithKline, Pfizer, Roche, and UCB, and personal fees from AbbVie, Biotest, BMS, GlaxoSmithKline, Janssen, Lilly, Merck, Pfizer, Roche, UCB, and Vertex outside the submitted work. The other authors declare no conflicts of interest. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A randomisation list generated by computer" |
| Allocation concealment (selection bias) | Low risk | "To conceal the sequence until treatment strategy was assigned, sequentially numbered sealed opaque envelopes that contained the randomly assigned allocations were used." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open‐label study" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study, and most outcomes likely to be influenced. X‐ray reading was blinded: "Radiographs of hands and feet (at baseline and 18 months) were assessed in chronological order by two blinded, trained readers" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data < 10% and balanced in numbers across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | Lack of DMARD cotreatment and a higher level of radiological damage at baseline were more prevalent in the dose reduction group than in the usual care group, but there was a low chance of inducing bias. |
van Vollenhoven 2016 (DOSERA).
| Methods | 48‐week, double‐blind, randomised, placebo‐controlled trial in 16 rheumatology units in Sweden (n = 5), Denmark (n = 2), Finland (n = 2), Norway (n = 3), Hungary (n = 3), and Iceland (n = 1) Period 1: 8‐week run‐in period; period 2: RCT period |
|
| Participants | Inclusion criteria for period 1:
Exclusion criteria for period 1: key exclusions were: prior therapy with biologics except anti‐TNFs and a prior attempt at etanercept discontinuation or dose reduction for the purpose of maintaining a good clinical result (i.e. for the same purpose to be assessed in this study) Inclusion criteria for period 2:
Baseline characteristics: mean (SD): age 56.7 (11.0), female 70%, disease duration 13.6 (8.8), RF+ 69%, prior treatment with DMARDs other than MTX 66% |
|
| Interventions | Period 1: run‐in period with etanercept + methotrexate. Methotrexate dose was kept unchanged throughout the study, etanercept was provided in a once‐weekly 50 mg dose
in the form of the lyophilised product. (n = 91) Period 2: participants were randomised in a ratio 1:1:1
If a flare occurred during period 2, the participant was withdrawn from this phase. Participants who discontinued period 2 were designated as failures in the primary analysis, transferred to the third phase (period 3), and received etanercept 50 mg weekly plus methotrexate. |
|
| Outcomes | Primary outcome: proportion of non‐failures for ETN50 versus PBO Secondary outcomes:
Analyses: modified ITT consisting of the participants who had received a randomised treatment assignment and who had at least 1 available evaluation after the first dose of study medication at randomisation. For dichotomous clinical outcomes, a non‐responder imputation was applied, designating a participant as a ‘failure’ if he/she had discontinued double‐blinded treatment for any reason. The primary analysis was performed on LOCF data. |
|
| Notes | Acronym: DOSERA Funding: This study was sponsored by Wyeth, which was acquired by Pfizer in October 2009. Disclosures: RFvV has received research support and honoraria from AbbVie (Abbott), BMS, GSK, Lilly, MSD, Pfizer, Roche, and UCB Pharma. MØ has received research support or honoraria, or both from AbbVie (Abbott), BMS, Centocor, GSK, Janssen, Merck, Mundipharma, Novo, Pfizer, Schering‐Plough, Roche, UCB, and Wyeth. TU has received research support and honoraria from AbbVie (Abbott), BMS, MSD, Pfizer, Roche, and UCB Pharma. ML‐R has been a consultant for Abbott, Pfizer, MSD, Roche, and BMS. EL and MJ were employees of Pfizer Sweden at the time of the study; EL is currently an employee of Eli Lilly. FB is an employee of Quanticate, who were paid consultants to Pfizer in connection with statistical support for the development of this manuscript. KF‐L was an employee of Wyeth/Pfizer at the time of the conduct of the study and is currently employed by the Swedish Medical Products Agency. EudraCT: 2007‐006657‐63 ClinicalTrials.gov: NCT00858780 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Pfizer generated a randomization schedule. Allocation of subjects to treatment groups proceeded through the use of the Clinical Operations Randomization Environment II (CORE II) system /Impala system that was accessible 24 hours a day, 365 days a year." |
| Allocation concealment (selection bias) | Low risk | "Pfizer generated a randomization schedule. Allocation of subjects to treatment groups proceeded through the use of the Clinical Operations Randomization Environment II (CORE II) system /Impala system that was accessible 24 hours a day, 365 days a year." |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "Double‐blind trial" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double‐blind trial" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Double‐blind trial" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | Low risk | The study appears to be free of other sources of bias. |
Weinblatt 2017 (C‐EARLY).
| Methods | Multicentre, double‐blind randomised controlled trial conducted at 103 study centres in Europe, Australia, North America, and Latin America (of the 181 that participated in C‐EARLY period 1) Period 1: 52 weeks, double‐blind. Period 2: week 52 to 104, double‐blind (period of interest) |
|
| Participants | Inclusion for period 1:
Exclusion period 1:
Inclusion for period 2:
Exclusion period 2:
Baseline characteristics: CZP standard dose: mean (SD): age 49.1 (13.1), female 79%, RF+ 98%, ACPA+ 92%, time since diagnosis in months 2.5 (2.5) CZP reduced dose: age 49.2 (12.5), female 68%, RF+ 95%, ACPA+ 89%, time since diagnosis in months 2.6 (2.8) CZP stopped: age 47.6 (14.0), female 73%, RF+ 100%, ACPA+ 86%, time since diagnosis in months 2.9 (3.1) |
|
| Interventions | Period 2: participants were randomised into 1 of 3 groups in ratio 2:3:2.
In the event of a confirmed disease flare, participants received a loading dose of certolizumab pegol (400 mg at 3 subsequent visits) followed by the standard dose (200 mg every 2 weeks) until the end of the study. |
|
| Outcomes | Primary: proportion of participants with sustained low disease activity (DAS28‐ESR of ≤ 3.2 at both week 40 and week 52) who maintained low disease activity (DAS28‐ESR of ≤ 3.2) for all 5 consecutive study visits to week 104 without flares Secondary outcome measures:
Analyses: ITT. Missing data from participants who entered period 2 but withdrew before the end of the study were imputed using non‐responder imputation for the primary and key secondary endpoints. Radiographic analyses used linear extrapolation. In post hoc analyses, LOCF imputation was used for the proportions of participants achieving low disease activity, remission, and normative physical function. |
|
| Notes | Acronym: C‐EARLY period 2 Funding: supported by UCB Pharma Disclosures: Dr Weinblatt has received consulting fees, speaking fees, and/or honoraria from AbbVie, Amgen, Bristol‐Myers Squibb, Roche (less than USD 10,000 each), Lilly, Pfizer, UCB Pharma, AstraZeneca, Merck, Novartis, Crescendo Bioscience, and MedImmune (more than USD 10,000 each). Dr Bingham has received consulting fees, speaking fees, and/or honoraria from UCB Pharma (less than USD 10,000). Dr Burmester has received consulting fees, speaking fees, and/or honoraria from AbbVie, Bristol‐Myers Squibb, Celgene, MSD, UCB Pharma, Roche, Pfizer, and Lilly (less than USD 10,000 each). Dr Bykerk has received consulting fees, speaking fees, and/or honoraria from Pfizer, AbbVie, Bristol‐Myers Squibb, Sanofi, and UCB Pharma (less than USD 10,000 each). Dr Furst has received consulting fees, speaking fees, and/or honoraria from Abbott, AbbVie, Amgen, Biogen, Bristol‐Myers Squibb, Cytori, Gilead, GlaxoSmithKline, Janssen, NIH, Novartis, Pfizer, Roche/Genentech, and UCB Pharma (less than USD 10,000 each) and research grants from Abbott, Actelion, Amgen, Biogen, Bristol‐Myers Squibb, GlaxoSmithKline, Novartis, Pfizer, Roche/Genentech, and UCB Pharma. Dr Mariette has received consulting fees, speaking fees, and/or honoraria from Bristol‐Myers Squibb, GlaxoSmithKline, Pfizer, and UCB Pharma (less than USD 10,000 each). Dr van der Heijde has received consulting fees, speaking fees, and/or honoraria from AbbVie, Amgen, Astellas, AstraZeneca, Bristol‐Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi, Eli Lilly and Company, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, and UCB Pharma (less than USD 10,000 each). Dr van Vollenhoven has received consulting fees and/or honoraria from AbbVie, Biotest, Bristol‐Myers Squibb, Celgene, Crescendo Bioscience, GlaxoSmithKline, Janssen, Lilly, Merck, Novartis, Pfizer, Roche, UCB Pharma, and Vertex (less than USD 10,000 each) and research grants from AbbVie, Amgen, Bristol‐Myers Squibb, GlaxoSmithKline, Pfizer, Roche, and UCB Pharma. Ms VanLunen and Drs Ecoffet and Cioffi own stock or stock options in UCB Pharma. Dr Emery has received consulting fees and/or honoraria from Pfizer, MSD, AbbVie, Bristol‐Myers Squibb, UCB Pharma, Roche, Novartis, Samsung, Sandoz, and Lilly (more than USD 10,000 each). EudraCT: 2011‐001729‐25 ClinicalTrials.gov: NCT01521923 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Re‐randomisation at week 52 was performed centrally using an interactive voice/web response system (IXRS). |
| Allocation concealment (selection bias) | Low risk | Re‐randomisation at week 52 was performed centrally using an interactive voice/web response system (IXRS). |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | Double‐blind study. Placebo was supplied as 0.9% saline, and certolizumab pegol was supplied as a 200 mg solution. Both were in prefilled syringes for subcutaneous injection and were administered up to week 102. |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All study personnel were blinded with respect to treatment, except for a separate group who supervised and administered the study medication and determined ESR but who otherwise had no involvement in the study. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | High risk | The number of included participants was lower than anticipated in the sample size calculation. “The primary end point of the present study was not achieved. Fewer patients than projected from period 1 were eligible to enter period 2 (i.e., achieved sustained low disease activity), which may have resulted in an underpowered study.” |
Yamanaka 2016 (ENCOURAGE).
| Methods | Multicentre, open‐label randomised controlled study in Japan and Korea Period 1: first year. Period 2: second year (period of interest for review) |
|
| Participants | Period 1:
Period 2:
Baseline characteristics: mean (SD): age: intervention 52.9 (14.9), control 49.8 (13.0); female: intervention 88%, control 88%; RF+: intervention 71%, control 70%; DAS28: intervention 1.8 (0.5), control 1.7 (0.5); disease duration in years: intervention 2.4 (1.4), control 1.9 (1.4) |
|
| Interventions | Period 1: participants were randomised into 1 of 2 groups at a ratio of 1:4
Period 2: participants were randomised at a ratio of 1:1
|
|
| Outcomes | Primary outcome period 2: maintenance of remission rates, including clinical remission, structural remission, and functional remission rates at 12 months after the second randomisation Secondary outcomes:
Analyses: ITT and PP. Missing data were imputed using the LOCF strategy. |
|
| Notes | Acronym: ENCOURAGE Funding: This investigator‐initiated study was supported by a grant from Pfizer Inc through the nonprofit corporation Advanced Clinical Research Organization (www.npo‐acro.jp/), due to an international multicentre co‐operative study led by the Institute of Rheumatology, Tokyo Women’s Medical University. Disclosures: HY has received research grants from AbbVie, Asahi Kasei Pharma, Astellas, Bristol‐Myers Squibb, Chugai, Daiichi‐Sankyo, Eisai, GlaxoSmithKline, Janssen, Mitsubishi‐Tanabe, MSD, Nippon Kayaku, Pfizer, Santen, Taisho‐Toyama, Takeda, and Teijin Pharma, and has received honorarium for lectures or consultancy from Teijin Pharma, Chugai, Astellas, Bristol‐Myers Squibb, AbbVie, Daiichi‐Sankyo, Nippon Kayaku, Mitsubishi‐Tanabe, Pfizer, Takeda, and UCB. SN has received research grants from AbbVie, Bristol‐Myers Squibb, and Pfizer, and honorarium for lectures or consultancy from Bristol‐Myers Squibb and Pfizer. TK has received research grants from AbbVie, Astellas, Bristol‐Myers Squibb, Chugai, Eisai, Mitsubishi‐Tanabe, Pfizer, and Takeda, and honorarium for lectures from Chugai, Astellas, AbbVie, and Mitsubishi‐Tanabe. YU has received speaking fees from Pfizer, Mitsubishi‐Tanabe, Chugai, Bristol‐Myers Squibb, and Astellas. YS has received speaking fees from AbbVie and Pfizer. NT has received research grant or speaking fee, or both from Mitsubishi‐Tanabe Pharma, Chugai, Takeda, Astellas, and Eisai. KS has received speaking fees from Mitsubishi‐Tanabe, Chugai, Eisai, AbbVie, Bristol‐Myers Squibb, Takeda, and Astellas. YI has received speaking fees from Eisai and Pfizer. YM has received research grants from Mitsubishi‐Tanabe, Astellas, Pfizer, and Eisai. YT has received consulting fees, speaking fees, and/or honoraria from AbbVie, Chugai, Astellas, Takeda, Santen, Mitsubishi‐Tanabe, Pfizer, Janssen, Eisai, Daiichi‐Sankyo, UCB, GlaxoSmithKline, and Bristol‐Myers Squibb, and research grants from Mitsubishi‐Tanabe, Chugai, MSD, Astellas, and Novartis. TTa has received research grants from Astellas, Bristol–Myers Squibb, Chugai, Daiichi‐Sankyo, Eisai, Mitsubishi‐Tanabe, Pfizer, Santen, Takeda, Teijin Pharma, AbbVie, Asahikasei Pharma, Taisho‐Toyama, and SymBio Pharmaceuticals; speaking fees from AbbVie, Bristol–Myers Squibb, Chugai, Eisai, Janssen, Mitsubishi‐Tanabe, Pfizer, Takeda, Astellas, and Daiichi‐Sankyo; and consultant fees from AstraZeneca, Eli Lilly, Novartis, Mitsubishi‐Tanabe, Asahi Kasei Pharma, AbbVie, Daiichi‐Sankyo, and Bristol–Myers Squibb. UMIN Clinical Trials Registry Japan: UMIN000002687 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgement of ‘low risk’ or ‘high risk’ |
| Allocation concealment (selection bias) | Low risk | "Randomization of ENCOURAGE study was conducted by the sealed envelope method in central study secretariat" |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open‐label study" |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | Open‐label study, but outcome not likely to be influenced |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label study, and most outcomes likely to be influenced. X‐ray reading was blinded: "Assessment of outcome was mostly conducted by the investigators without blinding, but only radiological scores (Total Sharp score) was assessed centrally by experts of evaluating scores, thus radiological score was assessed by blinding manner." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Proportion of missing data is likely to induce clinically relevant bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol is available, and all of the study’s prespecified (primary and secondary) outcomes that are of interest in the review have been reported in the prespecified way. |
| Other bias | High risk | The number of included participants was lower than anticipated in the sample size calculation. |
ACPA: Anti‐Citrullinated Protein Antibody ACR: American College of Rheumatology bDMARD: biological DMARD CCP: cyclic citrullinated peptide CDAI: Clinical Disease Activity Index CRP: C‐reactive protein csDMARD: conventional synthetic DMARD CZP: certolizumab pegol DAS28: disease activity score in 28 joints DAS28‐ESR: disease activity score in 28 joints using erythrocyte sedimentation rate DAS44: disease activity score in 44 joints DMARD: disease‐modifying antirheumatic drug ESR: erythrocyte sedimentation rate ETN: etanercept EULAR: European League Against Rheumatism EQ‐5D: European Quality of Life‐5 Dimensions (a standardised measure of health‐related quality of life) FACIT: Functional Assessment of Chronic Illness Therapy GH: growth hormone HAQ‐DI: Health Assessment Questionnaire‐Disability Index IA: intra‐articular IM: intramuscular IV: intravenous IQR: interquartile range ITT: intention‐to‐treat LDA: low disease activity LOCF: last observation carried forward mSvdH score: modified Sharp‐van der Heijde score mTSS: modified total Sharp score MTX: methotrexate NSAID: non‐steroidal anti‐inflammatory drug PP: per protocol RA: rheumatoid arthritis RCT: randomised controlled trial RF: rheumatoid factor SC: subcutaneous SD: standard deviation SDAI: Simplified Disease Activity Index SF‐36: 36‐item short‐form health survey SJ: swollen joint TJ: tender joint TNF: tumour necrosis factor
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aletaha 2010 | Overview article |
| Awan 2011 | No anti‐TNF continuation comparison group. No separate RA data |
| Bejarano 2010 | After 1 year, all participants receiving infliximab plus methotrexate discontinued infliximab. No comparison with infliximab continuation arm |
| CADTH Report 2014 | Literature study |
| Detert 2013 (HIT‐HARD) | HIT‐HARD study After 24 weeks, all participants receiving adalimumab plus methotrexate discontinued adalimumab. No comparison with adalimumab continuation arm |
| Emery 2013 (PRIZE) | PRIZE study Etanercept reduced or discontinued, no continuation comparison arm. |
| Greenberg 2014 | Cohort study |
| Haraoui 2014 | No certolizumab pegol dose below standard dose |
| Harigai 2012 (BRIGHT) | BRIGHT study Not an RCT or CCT according to the Cochrane definition; randomisation based on physician preference |
| Haschka 2016 (RETRO) | RETRO study Dose reduction of bDMARDs and sDMARDs at the same time |
| Heimans 2016 (IMPROVED) | IMPROVED study No comparison with an adalimumab continuation arm |
| Ichikawa 2007 | Overview article |
| Keystone 2003 | Overview article |
| Klarenbeek 2011 | BeSt study In the combination therapy with infliximab arm, infliximab dose could be reduced and stopped. No comparison with infliximab continuation arm |
| Kobelt 2011 | No real participant data. Markov model |
| Kobelt 2014 | No real participant data. Markov model |
| Oba 2017 (RRRR study) | RRRR study No comparison with a continuation arm |
| Quinn 2005 | After 1 year, all participants receiving infliximab plus methotrexate discontinued infliximab. No comparison with infliximab continuation arm |
| Rakieh 2013 | Not an RCT or CCT according to the Cochrane definition; allocation based on patient decision |
| Ramírez‐Herráiz 2013 | Retrospective study |
| Seddighzadeh 2014 (NORD‐STAR) | NORD‐STAR study No comparison with a continuation arm |
| Smolen 2012 (CERTAIN) | CERTAIN study No certolizumab pegol continuation comparison arm |
| Tada 2012 (PRECEPT) | PRECEPT study Participants who required biological therapy were randomly assigned to receive low‐dose versus standard‐dose etanercept. Participants had not used etanercept before study start. No dose reduction protocol |
| Tanaka 2013 (HONOR) | HONOR study Not an RCT or CCT according to the Cochrane definition |
| Tanaka 2014 (HOPEFUL‐2) | HOPEFUL‐2 Not an RCT or CCT according to the Cochrane definition; allocation based on patient/investigator decision |
| van den Broek 2011 | BeSt study In the combination therapy with infliximab arm, infliximab dose could be reduced and stopped. No comparison with infliximab continuation arm |
| van der Kooij 2009 | BeSt study In the combination therapy with infliximab arm, infliximab dose could be reduced and stopped. No comparison with infliximab continuation arm |
| Villeneuve 2012 | No etanercept continuation control arm, and no data after etanercept withdrawal |
| Wiland 2016 (PRIZE) | PRIZE study No comparison with etanercept continuation arm |
bDMARD: biological DMARD CCT: controlled clinical trial DMARD: disease‐modifying antirheumatic drug RA: rheumatoid arthritis RCT: randomised controlled trial sDMARD: synthetic DMARD TNF: tumour necrosis factor
Characteristics of ongoing studies [ordered by study ID]
2012‐004631‐22.
| Trial name or title | TapERA: Maintaining remission in RA while tapering Etanercept |
| Methods | Open‐label randomised controlled trial |
| Participants | People with established RA in remission for ≥ 6 months, treated with etanercept ≥ 1 year |
| Interventions |
|
| Outcomes | Primary outcome: the proportion of participants maintaining remission 6 months after decreasing the dose of etanercept to 50 mg every 2 weeks compared to the proportion of participants maintaining remission while continuing the established dose of 50 mg weekly Secondary outcomes: baseline predictors, maintenance of remission, regaining remission after retreatment, FLARE questionnaire, adverse events |
| Starting date | 2012 |
| Contact information | Rene Westhovens, University Hospitals Leuven |
| Notes | EudraCT Number: 2012‐004631‐22 |
2017‐001970‐41.
| Trial name or title | The BIODOPT trial (BIOlogical Dose OPTimisation). Dose reduction and discontinuation of biological therapy in patients with rheumatoid arthritis, psoriatic arthritis and axial spondyloarthritis: protocol for a 18 months randomised, open label, parallel‐group, multi‐centre trial |
| Methods | Randomised, open‐label, parallel group, multicentre |
| Participants | People with RA, PsA, or SpA treated with a bDMARD in stable dose in sustained clinical remission |
| Interventions | Dose optimisation tapering strategy for biological therapy |
| Outcomes | The co‐primary endpoint is: 1A Superiority: the proportion of participants who at 18 months are reduced to 50% or less of their inclusion dose of biological therapy. 1B Equivalence: disease activity assessed 18 months from baseline |
| Starting date | December 2017 |
| Contact information | MD Line Uhrenholt, The Department of Rheumatology, Aalborg University Hospital |
| Notes | EudraCT: 2017‐001970‐41 |
NCT01793519.
| Trial name or title | Stopping tumor necrosis factor‐alpha inhibitors in rheumatoid arthritis |
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled, non‐inferiority trial |
| Participants | People with RA, remission for > 6 months while taking etanercept, adalimumab, or infliximab and at least 1 DMARD |
| Interventions | Randomisation 2:1 Matching placebo OR current anti‐TNF agent (etanercept/adalimumab/infliximab) |
| Outcomes | Primary outcome: 48‐week relapse‐free status Secondary outcomes: difference in progression of joint damage on radiographs, differences in physical function and predictors of relapse |
| Starting date | January 2013 |
| Contact information | Arthur Weinstein, MD/Michael M Ward, MD |
| Notes | ClinicalTrials.gov: NCT01793519 |
NCT01881308.
| Trial name or title | Remission in rheumatoid arthritis ‐ assessing withdrawal of disease‐modifying antirheumatic drugs in a non‐inferiority design |
| Methods | Randomised, open, controlled, parallel‐group, multicentre, phase 4, non‐inferiority strategy study in Norway |
| Participants | People with RA with disease duration < 5 years, stable DAS28 remission > 12 months and unchanged treatment with anti‐TNF and/or sDMARDs > 12 months |
| Interventions | Stable dose anti‐TNF OR stepdown and withdrawal of anti‐TNF (half‐dose anti‐TNF first 4 months, thereafter withdrawal) OR stable dose sDMARD OR sDMARD dose reduction (half‐dose sDMARD for first 12 months, after 12 months re‐randomisation, continue half‐dose or withdraw DMARD) OR ARCTIC follow‐up |
| Outcomes | Primary endpoint is the proportion of participants who are non‐failures (have not experienced a flare) at 12 months' follow‐up. Secondary endpoints include composite disease activity scores and remission criteria, joint damage and inflammation assessed by various imaging modalities, work participation, healthcare resource use, and health‐related quality of life. |
| Starting date | June 2013 |
| Contact information | Siri Lillegraven, MD, MPH/Espen A Haavardsholm, MD, PhD |
| Notes | ClinicalTrials.gov: NCT01881308 |
NCT02198651.
| Trial name or title | PREDICTRA: A Phase 4 Trial Assessing the ImPact of Residual Inflammation Detected Via Imaging TEchniques, Drug Levels and Patient Characteristics on the Outcome of Dose TaperIng of Adalimumab in Clinical Remission Rheumatoid ArThritis (RA) Subjects |
| Methods | Randomised, double‐blind, phase 4 trial |
| Participants | People with RA treated with adalimumab and methotrexate who are in sustained clinical remission |
| Interventions |
|
| Outcomes | Primary outcome measures: hand and wrist synovitis RAMRIS score, bone marrow oedema RAMRIS score, flare occurrence Secondary outcome measures: time to flare, flare severity, DAS28, CDAI, SDAI, HAQ‐DI, RAPID‐3, TSQM, WPAI, SF‐36 |
| Starting date | December 2014 |
| Contact information | AbbVie |
| Notes | ClinicalTrials.gov: NCT02198651 |
NCT02373813.
| Trial name or title | Study of etanercept monotherapy vs methotrexate monotherapy for maintenance of rheumatoid arthritis remission |
| Methods | Multicentre, double‐blind randomised controlled study |
| Participants | People with RA on etanercept plus methotrexate therapy in very good disease control for 6 months prior to study entry |
| Interventions |
|
| Outcomes | Primary outcome measure: SDAI remission at week 48 Secondary outcome measures: DAS28, CDAI, adverse events |
| Starting date | February 2015 |
| Contact information | Amgen |
| Notes | ClinicalTrials.gov: NCT02373813 |
NTR3903.
| Trial name or title | Dose‐to‐target of etanercept treatment: a dose‐tapering randomised controlled trial in patients with rheumatoid arthritis, ankylosing spondylitis or psoriatic arthritis |
| Methods | Open randomised controlled study of a dose‐to‐target step‐down treatment strategy in the Netherlands |
| Participants | People with RA, PsA, and AS, etanercept treatment 50 mg/week > 6 months and minimal disease activity |
| Interventions | Continuation of etanercept 50 mg/week OR etanercept 50 mg/2 weeks After 6 months among participants still in a state of minimal disease activity, etanercept 50 mg/2 weeks in the original continuation group and etanercept discontinuation in the original etanercept 50 mg/2 weeks |
| Outcomes | Primary outcome: proportion of participants with RA, AS, PsA maintaining minimal disease activity after dose interval prolongation of etanercept Secondary outcomes: cost‐effectiveness of tapering down etanercept treatment, whether the lowest effective etanercept dose will reduce the risk of adverse events, predictive value of serum etanercept through levels and other participant‐related factors for successful down‐titration |
| Starting date | May 2013 |
| Contact information | Dr GJ Wolbink |
| Notes | Dutch trial register: NTR3903 |
AS: ankylosing spondylitis bDMARD: biological DMARD CDAI: Clinical Disease Activity Index DAS28: disease activity score in 28 joints DMARD: disease‐modifying antirheumatic drug HAQ‐DI: Health Assessment Questionnaire‐Disability Index PsA: psoriatic arthritis RA: rheumatoid arthritis RAMRIS: Rheumatoid Arthritis Magnetic Resonance Imaging Score RAPID‐3: Routine Assessment of Patient Index Data 3 SDAI: Simplified Disease Activity Index sDMARD: synthetic DMARD SF‐36: 36‐item Short Form Health Survey SpA: spondyloarthritis TNF: tumour necrosis factor TSQM: Treatment Satisfaction Questionnaire for Medication WPAI: Work Productivity and Activity Impairment
Differences between protocol and review
We removed the sentence: "The intervention should include the option for a patient to restart the anti‐TNF agent in case of loss of response." from Types of interventions. We did this because the largest study included in this review did not include the option to restart the anti‐TNF agent in case of loss of response. It was not always clear for the other included studies whether participants could restart the anti‐TNF agent. We believe the possibility of restarting an anti‐TNF agent in case of loss of response is important.
Different review authors for selecting studies: BJFvdB replaced AAdB in selecting studies, abstracting data, and assessing risk of bias. AAdB was the referee. This change was made because AAdB had time limitations.
Switch in primary outcome: We made "Proportion of patients with persistent low disease activity" a major outcome and "Proportion of patients with a flare" a minor outcome. We switched these outcomes because the two are highly comparable. Most included studies used the first outcome.
Additional types of studies: Both superiority and non‐inferiority trials were included. One of the studies included in this review was reported to be a non‐inferiority study. Also, some of the identified ongoing trials were reported to be non‐inferiority studies. A non‐inferiority design is the best study design for a down‐titration strategy.
Additional types of participants: standard (or lower) anti‐TNF dose. We added the "or lower dose" because some studies might also include participants who used a lower‐than‐standard dose before entering the study.
Addition to other sources of bias: We added imbalance in prognostic variables as another source of bias, as we believe this is an important addition for the 'Risk of bias' assessment in our review.
The outcome "proportion persistent loss of response, refractory to re‐instalment of the tapered anti‐TNF" was changed to "proportion of participants that switched to another biologic due to persistent loss of response, refractory to re‐instalment of the tapered anti‐TNF in the intervention group". We made this change since the definition was not specific enough and insufficiently distinct from the other outcome measures.
For the outcome "proportion participants with persistent low disease activity" we have chosen to report the proportion of participants in persistent remission to have more data available for this outcome. Since remission is more stringent than low disease activity, we might be more sensitive to differences between continuation and down‐titration of anti‐TNF.
Contributions of authors
Title and protocol: AAdB, NvH.
Review of abstracts and full‐text articles: BJFvdB, NvH, AAdB, LMV.
Data extraction: BJFvdB, NvH, LMV.
Results and analyses: NvH, BJFvdB, WJ, AAdB, AvdM, LMV.
Interpretation of data: NvH, BJFvdB, WJ, AAdB, AvdM, JEV, FHJvdH, MH, LMV.
Draft of the review: NvH, BJFvdB, AAdB, LMV.
Editing of the draft: WJ, AvdM, JEV, FHJvdH, MH.
Sources of support
Internal sources
-
Sint Maartenskliniek, Nijmegen, Netherlands.
Office space and computer access
External sources
No sources of support supplied
Declarations of interest
Disclosures
Lise M Verhoef: none known.
Bart(holomeus) JF van den Bemt: none known.
Aatke van der Maas: none known.
Johanna Vriezekolk: none known
Marlies Hulscher: none known.
Frank van den Hoogen: none known.
Wilco Jacobs: none known.
Noortje van Herwaarden: none known.
Alfons A den Broeder: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Bejerano 2016 (OPTIBIO) {published data only}
- Bejerano C, Oreiro N, Fernandez‐Lopez C, Pinto‐Tasende JA, Atanes A, Aspe B, et al. Clinical evaluation usefulness of standardized protocol strategies of dose reduction in patients with rheumatoid arthritis in clinical remission treated with biologic therapies. The OPTIBIO study [abstract]. Arthritis and Rheumatology 2016;68:(Suppl 10). [Google Scholar]
Chatzidionysiou 2016 (ADMIRE) {published data only}
- Chatzidionysiou K, Turesson C, Teleman A, Knight A, Lindqvist E, Larsson P, et al. A multicentre, randomised, controlled, open‐label pilot study on the feasibility of discontinuation of adalimumab in established patients with rheumatoid arthritis in stable clinical remission. RMD Open 2016;2:e000133. [PUBMED: 26819752 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidionysiou K, Turesson C, Teleman A, Knight A, Lindqvist E, Larsson P, et al. Multicenter, randomised, controlled, open‐label pilot study of the feasibility of discontinuation of adalimumab in rheumatoid arthritis patients is stable clinical remission. Arthritis and Rheumatism 2012;64(Suppl 10):776. [DOI] [PMC free article] [PubMed] [Google Scholar]
El Miedany 2016 {published data only}
- Miedany Y, Gaafary M, Ahmed I, Bahlas S, Hegazi M, Nasr A. Optimizing therapy in inflammatory arthritis: prediction of relapse after tapering or stopping treatment for rheumatoid arthritis patients achieving clinical and radiological remission. Clinical Rheumatology 2016;35:2915‐23. [PUBMED: 27658417] [DOI] [PubMed] [Google Scholar]
Fautrel 2016 (STRASS) {published data only}
- Fautrel B, Gandjbakhch F, Foltz T, Pham T, Morel J, Alfaiate T, et al. Targeting the lowest efficacious dose for rheumatoid arthritis patients in remission: clinical and structural impact of a step‐down strategy trial based on progressive spacing of TNF‐blocker injections (STRASS trial). Annals of Rheumatic Disease 2013;72(Suppl 3):72. [Google Scholar]
- Fautrel B, Pham T, Alfaiate T, Gandjbakhch F, Foltz V, Morel J, et al. Step‐down strategy of spacing TNF‐blocker injections for established rheumatoid arthritis in remission: results of the multicentre non‐inferiority randomised open‐label controlled trial (STRASS: Spacing of TNF‐blocker injections in Rheumatoid ArthritiS Study). Annals of the Rheumatic Diseases 2016;75(1):59‐67. [PUBMED: 26103979 ] [DOI] [PubMed] [Google Scholar]
- Fautrel B, Pham T, Tubach F, Alfaiate T, Morel J, Dernis E, et al. Tapering TNF‐blockers in established rheumatoid arthritis patients in DAS28 remission: results of a DAS28‐driven step‐down strategy randomized controlled trial. Arthritis and Rheumatism 2012;64(12):4169‐70. [Google Scholar]
- Vanier A, Mariette X, Tubach F, Fautrel B. Cost‐Effectiveness of TNF‐Blocker Injection Spacing for Patients with Established Rheumatoid Arthritis in Remission: An Economic Evaluation from the Spacing of TNF‐Blocker Injections in Rheumatoid Arthritis Trial.. Value in Health 2017;20(4):577‐585. [DOI] [PubMed] [Google Scholar]
Ghiti Moghadam 2016 (POEET) {published data only}
- Ghiti Moghadam M, Vonkeman HE, Klooster PM, Tekstra J, Schaardenburg D, Starmans‐Kool M, et al. Stopping tumor necrosis factor‐inhibitors in patients with established rheumatoid arthritis in remission or stable low disease activity: a pragmatic randomized multicenter open‐label controlled trial. Arthritis and Rheumatology 2016;68(8):1810‐7. [DOI] [PubMed] [Google Scholar]
Ibrahim 2017 (OPTTIRA) {published data only}
- Galloway JB, Kingsley G, Ma M, Lorente‐Canovas B, Cope A, Ibrahim F, et al. Optimising treatment with TNF inhibitors in rheumatoid arthritis with different dose tapering strategies: the OPTTIRA trial. Annals of the Rheumatic Diseases 2015;74 (Suppl 2):706. [Google Scholar]
- Ibrahim F, Lorente‐Cánovas B, Doré CJ, Bosworth A, Ma MH, Galloway JB, et al. Optimizing treatment with tumour necrosis factor inhibitors in rheumatoid arthritis ‐ a proof of principle and exploratory trial: is dose tapering practical in good responders?. Rheumatology 2017;56:2004‐14. [PUBMED: 28968858] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pavelka 2017 {published data only}
- NCT01578850. Study conducted in subjects with rheumatoid arthritis who have moderate to severe disease activity despite methotrexate therapy with or without other non biologic disease modifying antirheumatic drugs (DMARDs) for at least 12 weeks prior to screening. https://clinicaltrials.gov/ct2/show/results/NCT01578850 First posted April 17, 2012.
- Pavelka K, Akkoç N, Al‐Maini M, Zerbini CAF, Karateev DE, Nasonov EL, et al. Maintenance of remission with combination etanercept–DMARD therapy versus DMARDs alone in active rheumatoid arthritis: results of an international treat‑to‑target study conducted in regions with limited biologic access. Rheumatology International 2017;37:1469‐79. [PUBMED: 28597306] [DOI] [PubMed] [Google Scholar]
Raffeiner 2015 {published data only}
- Botsios C, Furlan A, Ostuni P, Sfriso P, Todesco S, Punzi L. Effects of low‐dose etanercept in maintaining DAS‐remission previously achieved with standard‐dose in patients with rheumatoid arthritis. Annals of the Rheumatic Diseases 2007;66(Suppl II):54. [Google Scholar]
- Raffeiner B, Botsios C, Ometto F, Bernardi L, Stramare R, Todesco S, et al. Effects of half dose etanercept (25 mg once a week) on clinical remission and radiographic progression in patients with rheumatoid arthritis in clinical remission achieved with standard dose. Clinical and Experimental Rheumatology 2015;33:63‐8. [PUBMED: 25535985] [PubMed] [Google Scholar]
Smolen 2013 (PRESERVE) {published data only}
- Smolen JS, Nash P, Durez P, Hall S, Ilivanova E, Irazoque‐Palazuelos F, et al. Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): a randomised controlled trial. Lancet 2013;381:918‐29. [DOI] [PubMed] [Google Scholar]
Smolen 2014 (OPTIMA) {published data only}
- Emery P, Smolen JS, Kavanaugh A, Vollenhoven R, Pavelka K, Durez P, et al. Maintenance of biologic‐free disease control in early rheumatoid arthritis patients after induction of low disease activity with adalimumab plus methotrexate. Annals of the Rheumatic Diseases 2011;70 (Suppl 3):262. [Google Scholar]
- Smolen JS, Emery P, Fleischmann R, Vollenhoven R, Florentius S, Santra S, et al. Biologic free disease control (BFDC) in early RA: predictors of successful withdrawal of adalimumab (ADA) after achieving stable low disease activity with ADA plus methotrexate (MTX) ‐ data from the OPTIMA study. Annals of the Rheumatic Diseases 2012;71 (Suppl 3):516. [Google Scholar]
- Smolen JS, Emery P, Fleischmann R, Vollenhoven RF, Pavelka K, Durez P, et al. Adjustment of therapy in rheumatoid arthritis on the basis of achievement of stable low disease activity with adalimumab plus methotrexate or methotrexate alone: the randomised controlled OPTIMA trial. Lancet 2014;383:321‐32. [PUBMED: 24168956] [DOI] [PubMed] [Google Scholar]
van Herwaarden 2015 (DRESS) {published data only}
- Kievit W, Herwaarden N, Hoogen FHJ, Vollenhoven RF, Bijlsma JWJ, Bemt BJF, et al. Disease activity‐guided dose optimisation of adalimumab and etanercept is a cost‐effective strategy compared with non‐tapering tight control rheumatoid arthritis care: analyses of the DRESS study. Annals of the Rheumatic Diseases 2016;75(11):1939‐1944. [PUBMED: 26764260] [DOI] [PubMed] [Google Scholar]
- Herwaarden N, Maas A, Minten MJM, Hoogen FHJ, Kievit W, Vollenhoven RF, et al. Disease activity guided dose reduction and withdrawal of adalimumab or etanercept compared with usual care in rheumatoid arthritis: open label, randomised controlled, non‐inferiority trial. BMJ 2015;350:h1389. [PUBMED: 25858265] [DOI] [PMC free article] [PubMed] [Google Scholar]
van Vollenhoven 2016 (DOSERA) {published data only}
- Vollenhoven R, Østergaard M, Leirisalo‐Repo M, Uhlig T, Jansson M, Klackenberg A, et al. Rheumatoid arthritis patients with stable low disease activity on methotrexate plus etanercept, continuation of etanercept 50 mg weekly or 25 mg weekly are both clinically superior to discontinuation: results from a randomized, 3‐armed, double‐blind clinical trial. Arthritis and Rheumatism 2012;64(12):4171. [Google Scholar]
- Vollenhoven RF, Østergaard M, Leirisalo‐Repo M, Uhlig T, Jansson M, Larsson E, et al. Full dose, reduced dose or discontinuation of etanercept in rheumatoid arthritis. Annals of the Rheumatic Diseases 2016;75(1):52‐8. [PUBMED: 25873634] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weinblatt 2017 (C‐EARLY) {published data only}
- Weinblatt ME, Bingham CO 3rd, Burmester G, Bykerk VP, Furst DE, Mariette X, et al. A phase III study evaluating continuation, tapering, and withdrawal of certolizumab pegol after one year of therapy in patients with early rheumatoid arthritis. Arthritis & Rheumatology 2017;69:1937‐48. [PUBMED: 28666080] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yamanaka 2016 (ENCOURAGE) {published data only}
- Yamanaka H, Nagaoka S, Lee S, Bae S, Kasama T, Kobayashi H, et al. Discontinuation of etanercept after achievement of sustained remission in patients with rheumatoid arthritis who initially had moderate disease activity ‐ results from the ENCOURAGE study, a prospective, international, multicenter randomized study. Modern Rheumatology 2016;26(5):651‐61. [PUBMED: 26698929] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aletaha 2010 {published data only}
- Aletaha D. Challenging the course of RA: time for drug‐free remission?. Nature Reviews Rheumatology 2010;6:442‐3. [DOI] [PubMed] [Google Scholar]
Awan 2011 {published data only}
- Awan S, Bannon C, O'Sullivan, Duffy M, Murphy E, Barry M. Sustained remission at 6 months, despite reduction in anti‐TNF dosing frequency in inflammatory arthritis. Irish Journal of Medical Science 2011;181(Suppl 2):54. [Google Scholar]
Bejarano 2010 {published data only}
- Bejarano V, Conoghan P, Quinn MA, Saleem B, Emery P. Benefits 8 years after a remission induction regime with an infliximab and methotrexate combination in early rheumatoid arthritis. Rheumatology 2010;49:1971‐4. [DOI] [PubMed] [Google Scholar]
CADTH Report 2014 {published data only}
- Unknown. Long‐term sustained clinical remission after stopping first‐line anti‐TNF agents in patients with rheumatoid arthritis, psoriatic arthritis, or ankylosing spondylitis: clinical effectiveness. Canadian Agency for Drugs and Technologies in Health 2014.
Detert 2013 (HIT‐HARD) {published data only}
- Detert J, Bastion H, Listing J, Weiss A, Wassenberg S, Liebhaber A, et al. Induction therapy with adalimumab plus methotrexate for 24 weeks followed by methotrexate monotherapy up to week 48 versus methotrexate therapy alone for DMARD‐naïve patients with early rheumatoid arthritis: HIT HARD, an investigator‐initiated study. Annals of the Rheumatic Diseases 2013;72:844‐50. [DOI] [PubMed] [Google Scholar]
Emery 2013 (PRIZE) {published data only}
- Emery P, Hammoudeh M, FitzGerald O, Combe B, Martin Mola E, Bukowski J, et al. Assessing maintenance of remission with reduced dose etanercept plus methotrexate, methotrexate alone or placebo in patients with early rheumatoid arthritis who achieved remission with etanercept and methotrexate: the PRIZE study. Annals of the Rheumatic Diseases 2013;72 (Suppl 3):399. [Google Scholar]
Greenberg 2014 {published data only}
- Greenberg JD, Shan Y, Reed GW, Bitman B, Collier D. Comparison of switching to reduced dose vs continuation of standard dose etanercept for rheumatoid arthritis patients in the CORRONA registry. Annals of the Rheumatic Diseases 2014;73 (Suppl 2):241. [Google Scholar]
Haraoui 2014 {published data only}
- Haraoui B, Bykerk VP, Vollenhoven R, Longueville M, Luijtens K, Ralston P, et al. Analysis of pooled data from two randomized controlled trials and their open‐label extensions: long‐term safety in rheumatoid arthritis before and after certolizumab pegol dose increase/decrease. Arthritis & Rheumatology 2014;66:S199. [Google Scholar]
Harigai 2012 (BRIGHT) {published data only}
- Harigai M, Takeuchi T, Tanaka, Y, Matsubara T, Yamanaka H, Miyasaka N. Discontinuation of adalimumab treatment in rheumatoid arthritis patients after achieving low disease activity. Modern Rheumatology 2012;22:814‐22. [DOI] [PubMed] [Google Scholar]
Haschka 2016 (RETRO) {published data only}
- Haschka J, Englbrecht M, Hueber AJ, Manger B, Kleyer A, Reiser M, et al. Relapse rates in patients with rheumatoid arthritis in stable remission tapering or stopping antirheumatic therapy: interim results from the prospective randomised controlled RETRO study. Annals of the Rheumatic Diseases 2016;75:45‐51. [PUBMED: 25660991] [DOI] [PubMed] [Google Scholar]
Heimans 2016 (IMPROVED) {published data only}
- Heimans L, Akdemir G, Wevers‐de Boer KVC, Goekoop‐Ruiterman YP, Molenaar ET, Groenendael JHLM, et al. Two‐year results of disease activity score (DAS)‐remission‐steered treatment strategies aiming at drug‐free remission in early arthritis patients (the IMPROVED‐study). Arthritis Research and Therapy 2016;28:23. [26794605] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ichikawa 2007 {published data only}
- Ichikawa N, Yamanaka H. Maintenance therapy for rheumatoid arthritis after remission following successful treatment using biologics. Nihon Rinsho 2007;65(7):1293‐8. [PubMed] [Google Scholar]
Keystone 2003 {published data only}
- Keystone EC, Haraoui B, Bykerk VP. Role of infliximab in the treatment of early rheumatoid arthritis. Clinical and Experimental Rheumatology 2003;21 (Suppl 31):S200‐2. [PubMed] [Google Scholar]
Klarenbeek 2011 {published data only}
- Klarenbeek NB, Kooij SM, Güler‐Yüksel M, Groenendael JHLM, Han KH, Kerstens PJSM, et al. Discontinuing treatment in patients with rheumatoid arthritis in sustained clinical remission: exploratory analyses from the BeSt study. Annals of the Rheumatic Diseases 2011;70:315‐9. [DOI] [PubMed] [Google Scholar]
Kobelt 2011 {published data only}
- Kobelt G, Lekander I, Lang A, Raffeiner B, Botsios C, Geborek P. Cost‐effectiveness of etanercept treatment in early active rheumatoid arthritis followed by dose adjustment. International Journal of Technology Assessment in Health Care 2011;27(3):193‐200. [DOI] [PubMed] [Google Scholar]
Kobelt 2014 {published data only}
- Kobelt G. Treating to target with etanercept in rheumatoid arthritis: cost‐effectiveness of dose reductions when remission is achieved. Value in Health 2014;17:537‐44. [PUBMED: 25128046] [DOI] [PubMed] [Google Scholar]
Oba 2017 (RRRR study) {published data only}
- Oba K, Norie N, Sato N, Saito K, Takeuchi T, Mimori T, et al. Remission induction by Raising the dose of Remicade in RA (RRRR) study: rationale and study protocol for a randomized controlled trial comparing for sustained clinical remission after discontinuation of infliximab in patients with rheumatoid arthritis. Contemporary Clinical Trials Communications 2017;8:49‐54. [PUBMED: 29696196 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Quinn 2005 {published data only}
- Quinn MA, Conaghan PG, O'Connor PJ, Karim Z, Greenstein A, Brown A, et al. Very early treatment with infliximab in addition to methotrexate in early, poor‐prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve‐month randomized, double‐blind, placebo‐controlled trial. Arthritis and Rheumatism 2005;52(1):27‐35. [DOI] [PubMed] [Google Scholar]
Rakieh 2013 {published data only}
- Rakieh C, Saleem B, Takase K, Nam JL, Keen H, Wakefield RJ, et al. Long term outcomes of stopping tumour necrosis factor inhibitors (TNFi) in patients with established rheumatoid arthritis (RA) who are in sustained remission: is it worth the risk?. Annals of the Rheumatic Diseases 2013;72 (Suppl 3):208‐9. [Google Scholar]
Ramírez‐Herráiz 2013 {published data only}
- Ramírez‐Herráiz E, Escudero‐Vilaplana V, Alañón‐Plaza E, Trovato‐López N, Herranz‐Alonso A, Morell‐Baladrón A, et al. Efficiency of adalimumab, etanercept and infliximab in rheumatoid arthritis patients: dosing patterns and effectiveness in daily clinical practice. Clinical and Experimental Rheumatology 2013;31(4):559‐65. [PubMed] [Google Scholar]
Seddighzadeh 2014 (NORD‐STAR) {published data only}
- Seddighzadeh M. NORD‐STAR: a multicentre, randomized, open‐label, blinded‐assessor, phase 4 study in patients with early rheumatoid arthritis. Scandinavian Journal of Rheumatology 2014;43 (Suppl 127):31‐2. [Google Scholar]
Smolen 2012 (CERTAIN) {published data only}
- Smolen JS, Emery P, Ferraccioli G, Samborski W, Berenbaum F, Davies O, et al. Maintenance of remission in rheumatoid arthritis patients with low‐moderate disease activity following withdrawal of certolizumab pegol treatment: week 52 results from the CERTAIN study. Annals of the Rheumatic Diseases 2012;71 (Suppl 3):25. [Google Scholar]
Tada 2012 (PRECEPT) {published data only}
- Tada M, Koiki T, Okano T, Sugioka Y, Wakitani S, Fukushima K, et al. Comparison of joint destruction between standard‐ and low‐dose etanercept in rheumatoid arthritis from the Prevention of Cartilage Destruction by Etanercept (PRECEPT) study. Rheumatology 2012;51:2164‐9. [DOI] [PubMed] [Google Scholar]
Tanaka 2013 (HONOR) {published data only}
- Tanaka Y, Hirata S, Kubo S, Fukuyo S, Hanami K, Sawamukai N, et al. Discontinuation of adalimumab after achieving remission in patients with established rheumatoid arthritis: 1‐year outcome of the HONOR study. Annals of the Rheumatic Diseases 2015;74:389‐95. [PUBMED: 24288014] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanaka 2014 (HOPEFUL‐2) {published data only}
- Tanaka Y, Yamanaka H, Ishiguro N, Miyasaka N, Kawana K, Hiramatsu K, et al. Attainment of low disease activity is predictive of maintenance of disease control upon adalimumab discontinuation for two years following combination therapy in Japanese patients with early rheumatoid arthritis. Arthritis & Rheumatology 2014;66:S1059. [Abstract number: 2427] [Google Scholar]
van den Broek 2011 {published data only}
- Broek M, Klarenbeek NB, Dirven L, Schaardenburg D, Hulsmans HMJ, Kerstens PJSM, et al. Discontinuation of infliximab and potential predictors of persistent low disease activity in patients with early rheumatoid arthritis and disease activity score‐steered therapy: subanalysis of the BeSt study. Annals of the Rheumatic Diseases 2011;70:1389‐94. [DOI] [PubMed] [Google Scholar]
van der Kooij 2009 {published data only}
- Kooij SM, Goekoop‐Ruiterman YPM, Vries‐Bouwstra JK, Guler‐Yuksel M, Zwinderman AH, Kerstens PJSM, et al. Drug‐free remission, functioning and radiographic damage after 4 years of response‐driven treatment in patients with recent‐onset rheumatoid arthritis. Annals of the Rheumatic Diseases 2009;68:914‐21. [DOI] [PubMed] [Google Scholar]
Villeneuve 2012 {published data only}
- Villeneuve E, Nam E, Hensor JL, Wakefield E, Conoghan RJ, Green PG, et al. Preliminary results of a multicentre randomised controlled trial of etanercept and methotrexate to induce remission in patients with newly diagnosed inflammatory arthritis. Arthritis and Rheumatism 2011;63 (Suppl 10):2465. [Google Scholar]
Wiland 2016 (PRIZE) {published data only}
- Wiland P, Dudler J, Veale D, Tahir H, Pedersen R, Bukowski J, et al. The effect of reduced or withdrawn etanercept‐methotrexate therapy on patient‐reported outcomes in patients with early rheumatoid arthritis. Journal of Rheumatology 2016;43:1268‐77. [PUBMED: 27252426] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
2012‐004631‐22 {published data only}
- 2012‐004631‐22. TapERA: Maintaining remission in RA while tapering Etanercept.. https://www.clinicaltrialsregister.eu/ctr‐search/trial/2012‐004631‐22/BE Date of first entry 11‐12‐2012.
2017‐001970‐41 {published data only}
- 2017‐001970‐41. The BIODOPT trial (BIOlogical Dose OPTimisation). https://www.clinicaltrialsregister.eu/ctr‐search/trial/2017‐001970‐41/DK Date first received 15‐09‐2017.
NCT01793519 {published data only}
- NCT01793519. Stopping TNF Alpha Inhibitors in Rheumatoid Arthritis (STARA). https://clinicaltrials.gov/ct2/show/NCT01793519 First posted on February 15, 2013.
NCT01881308 {published data only}
- NCT01881308. Assessing Withdrawal of Disease‐Modifying Antirheumatic Drugs in Rheumatoid Arthritis (ARCTIC REWIND). https://clinicaltrials.gov/ct2/show/NCT01881308 First posted on June 19, 2013.
NCT02198651 {published data only}
- NCT02198651. A Phase 4 Trial Assessing the ImPact of Residual Inflammation Detected Via Imaging TEchniques, Drug Levels and Patient Characteristics on the Outcome of Dose TaperIng of Adalimumab in Clinical Remission Rheumatoid ArThritis (RA) Subjects (PREDICTRA). https://clinicaltrials.gov/ct2/show/NCT02198651 First posted on July 24, 2014. [DOI] [PMC free article] [PubMed]
NCT02373813 {published data only}
- NCT02373813. Study of Etanercept Monotherapy vs Methotrexate Monotherapy for Maintenance of Rheumatoid Arthritis Remission. https://clinicaltrials.gov/ct2/show/NCT02373813 First posted on February 27, 2015.
NTR3903 {published data only}
- NTR3903. Dose‐to‐target of etanercept treatment in rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis.. https://www.trialregister.nl/trial/3705 Register date 2013‐03‐14.
Additional references
Aletaha 2005
- Aletaha D, Smolen J. The Simplified Disease Activity Index (SDAI) and the Clinical Disease Activity Index (CDAI): a review of their usefulness and validity in rheumatoid arthritis. Clinical and Experimental Rheumatology 2005;23(Suppl 39):S100‐8. [PubMed] [Google Scholar]
Aletaha 2010 RA criteria
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis and Rheumatism 2010;62(9):2569‐81. [DOI] [PubMed] [Google Scholar]
Arnett 1988
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis and Rheumatism 1988;31(3):315‐24. [DOI] [PubMed] [Google Scholar]
Bartelds 2007
- Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti‐adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Annals of the Rheumatic Diseases 2007;66(7):921‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Blumenauer 2002
- Blumenauer BBTB, Judd M, Wells GA, Burls A, Cranney A, Hochberg MC, et al. Infliximab for the treatment of rheumatoid arthritis. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD003785] [DOI] [PMC free article] [PubMed] [Google Scholar]
Blumenauer 2003
- Blumenauer BBTB, Cranney A, Burls A, Coyle D, Hochberg MC, Tugwell P, et al. Etanercept for the treatment of rheumatoid arthritis. Cochrane Database of Systematic Reviews 2003, Issue 3. [DOI: 10.1002/14651858.CD004525] [DOI] [PubMed] [Google Scholar]
Bongartz 2006
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti‐TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta‐analysis of rare harmful effects in randomised controlled trials. JAMA 2006;295(19):2275‐85. [DOI] [PubMed] [Google Scholar]
Brocq 2009
- Brocq O, Millasseau E, Albert C, Grisot C, Flory P, Roux CH, et al. Effect of discontinuing TNFa antagonist therapy in patients with remission of rheumatoid arthritis. Joint Bone Spine 2009;76(4):350‐5. [DOI] [PubMed] [Google Scholar]
den Broeder 2002
- Broeder AA, Creemers MCW, Gestel AM, Riel PLCM. Dose titration using the Disease Activity Score (DAS28) in rheumatoid arthritis patients treated with anti‐TNF‐α. Rheumatology 2002;41(6):638‐42. [DOI] [PubMed] [Google Scholar]
den Broeder 2010
- Broeder AA, Maas A, Bemt BJF. Dose de‐escalation strategies and role of therapeutic drug monitoring of biologics in RA. Rheumatology 2010;49(10):1801‐3. [DOI] [PubMed] [Google Scholar]
Doherty 2009
- Doherty M, Dieppe P. The ‘placebo’ response in osteoarthritis and its implications for clinical practice. Osteoarthritis Cartilage 2009;17(10):1255‐62. [DOI] [PubMed] [Google Scholar]
Fautrel 2015
- Fautrel B, Broeder AA. De‐intensifying treatment in established rheumatoid arthritis (RA): why, how, when and in whom can DMARDs be tapered?. Best Practice & Research: Clinical Rheumatology 2015;29(4‐5):550‐65. [PUBMED: 26697766] [DOI] [PubMed] [Google Scholar]
Felson 2011
- Felson DT, Smolen JS, Wells G, Zhang B, Tuyl LHD, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis and Rheumatism 2011;63(3):573‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fransen 2005
- Fransen J, Riel PLCM. The Disease Activity Score and the EULAR response criteria. Clinical Experimental Rheumatology 2005;23(5 Suppl 39):S93‐9. [PubMed] [Google Scholar]
Galvao 2016
- Galvao TF, Zimmerman IR, Mota LMH, Silva MT, Pereira MG. Withdrawal of biologic agents in rheumatoid arthritis: a systematic review and meta‐analysis. Clinical Rheumatology 2016;35:1659‐68. [DOI] [PubMed] [Google Scholar]
Genovese 2002
- Genovese MC, Bathon JM, Martin RW, Fleischmann RM, Tesser JR, Schiff MH, et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: two‐year radiographic and clinical outcomes. Arthritis & Rheumatology 2002;46(6):1443‐50. [PUBMED: 12115173] [DOI] [PubMed] [Google Scholar]
GRADEpro 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed on 28 June 2018. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Kavanaugh 2012
- Kavanaugh A, Emery P, Vollenhoven R, Cifaldi M, Shaw J, Chen N, et al. Effect of adalimumab discontinuation on patient‐reported outcomes and work productivity in early rheumatoid arthritis patients who achieved low disease activity following 26 weeks of treatment: data from the OPTIMA study. Annals of the Rheumatic Diseases 2012;71 (Suppl 3):663. [Google Scholar]
Klareskog 2011
- Klareskog L, Gaubitz M, Rodríguez‐Valverde V, Malaise M, Dougados M, Wajdula J. Assessment of long‐term safety and efficacy of etanercept in a 5‐year extension study in patients with rheumatoid arthritis. Clinical Experimental Rheumatology 2011;29(2):238‐47. [PubMed] [Google Scholar]
Kuijper 2015
- Kuijper TM, Lamers‐Karnebeek FBG, Jacobs JWG, Hazes JMW, Luime JJ. Flare rate in patients with rheumatoid arthritis in low disease activity or remission when tapering or stopping synthetic or biologic DMARD: a systematic review. Journal of Rheumatology 2015;42(11):2012‐22. [PUBMED: 26428204] [DOI] [PubMed] [Google Scholar]
Larsen 1973
- Larsen A. A radiological method for grading the severity of rheumatoid arthritis. Scandinavian Journal of Rheumatology 1973;4(4):225‐33. [DOI] [PubMed] [Google Scholar]
Maini 1998
- Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, et al. Therapeutic efficacy of multiple intravenous infusions of anti‐tumor necrosis factor alpha monoclonal antibody combined with low‐dose weekly methotrexate in rheumatoid arthritis. Arthritis & Rheumatology 1998;41(9):1552‐63. [PUBMED: 9751087] [DOI] [PubMed] [Google Scholar]
Navarro‐Millán 2013
- Navarro‐Millán I, Sattui SE, Curtis JR. Systematic review of tumor necrosis factor inhibitor discontinuation studies in rheumatoid arthritis. Clinical Therapeutics 2013;35(11):1850‐61. [PUBMED: 24156821] [DOI] [PMC free article] [PubMed] [Google Scholar]
Navarro‐Sarabia 2005
- Navarro‐Sarabia F, Ariza‐Ariza R, Hernandez‐Cruz B, Villanueva I. Adalimumab for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005113.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nawata 2008
- Nawata M, Saito K, Nakayamada S, Tanaka Y. Discontinuation of infliximab in rheumatoid arthritis patients in clinical remission. Modern Rheumatology 2008;18:460‐4. [DOI] [PubMed] [Google Scholar]
Prevoo 1995
- Prevoo ML, 't Hof MA, Kuper HH, Leeuwen MA, Putte LB, Riel PL. Modified disease activity scores that include twenty‐eight‐joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis and Rheumatism 1995;38(1):44‐8. [DOI] [PubMed] [Google Scholar]
Ramiro 2017
- Ramiro S, Sepriano A, Chatzidionysiou K, Nam JL, Smolen JS, Heijden D, et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2016 update of the EULAR recommendations for management of rheumatoid arthritis. Annals of the Rheumatic Diseases 2017;76(6):1101‐1136. [PUBMED: 28298374] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ruiz Garcia 2014
- Ruiz Garcia V, Jobanputra P, Burls A, Cabello JB, Gálvez Muñoz JG, Saiz Cuenca ESC, et al. Certolizumab pegol (CDP870) for rheumatoid arthritis in adults. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD007649.pub3] [DOI] [PubMed] [Google Scholar]
Saleem 2010
- Saleem B, Keen H, Goeb V, Parmar R, Nizam S, Hensor EMA. Patients with RA in remission on TNF blockers: when and in whom can TNF blocker therapy be stopped?. Annals of the Rheumatic Diseases 2010;69:1636‐42. [DOI] [PubMed] [Google Scholar]
Schipper 2010
- Schipper LG, Hulst LTC, Grol R, Riel PLCM, Hulscher MEJL, Fransen J. Meta‐analysis of tight control strategies in rheumatoid arthritis: protocolized treatment has additional value with respect to the clinical outcome. Rheumatology 2010;49:2154‐64. [PUBMED: 20671022] [DOI] [PubMed] [Google Scholar]
Sharp 1971
- Sharp JT, Lidsky MD, Collins LC, Moreland J. Methods of scoring the progression of radiologic changes in rheumatoid arthritis. Correlation of radiologic, clinical and laboratory abnormalities. Arthritis and Rheumatism 1971;14(6):706‐20. [DOI] [PubMed] [Google Scholar]
Singh 2009
- Singh JA, Christensen R, Wells GA, Suarez‐Almazor ME, Buchbinder R, Lopes‐Olivo MA, et al. Biologics for rheumatoid arthritis: an overview of Cochrane Reviews. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD007848.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2010
- Singh JA, Noorbaloochi S, Singh JA. Golimumab for rheumatoid arthritis. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD008341] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2011
- Singh JA, Wells GA, Christensen R, Tanjong Ghogomu E, Maxwell L, Macdonald JK, et al. Adverse effects of biologics: a network meta‐analysis and Cochrane overview. Cochrane Database of Systematic Reviews 2011, Issue 16. [DOI: 10.1002/14651858.CD008794.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2016
- Singh JA, Saag KG, Bridges SL, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care & Research 2016;68(1):1‐25. [PUBMED: 26545825] [DOI] [PubMed] [Google Scholar]
Smolen 2003
- Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A simplified disease activity index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford) 2003;42(2):244‐57. [DOI] [PubMed] [Google Scholar]
Smolen 2017
- Smolen JS, Landewé R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Annals of the Rheumatic Diseases 2017;76(6):960‐977. [PUBMED: 28264816] [DOI] [PubMed] [Google Scholar]
St Clair 2004
- Clair EW, Heijde DM, Smolen JS, Maini RN, Bathon JM, Emery P, et al. Active‐Controlled Study of Patients Receiving Infliximab for the Treatment of Rheumatoid Arthritis of Early Onset Study Group. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: a randomized, controlled trial. Arthritis and Rheumatism 2004;50(11):3432‐43. [DOI] [PubMed] [Google Scholar]
Tanaka 2010
- Tanaka Y, Takeuchi T, Mimori T, Saito K, Nawata M, Kameda H, et al. Discontinuation of infliximab after attaining low disease activity in patients with rheumatoid arthritis: RRR (Remission induction by Remicade in RA) study. Annals of the Rheumatic Diseases 2010;69(7):1286‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanaka 2012
- Tanaka Y, Hirata S, Fukuyo S, Nawata M, Kubo S, Yamaoka K, et al. Discontinuation of adalimumab without functional and radiographic damage progression after achieving sustained remission in patients with rheumatoid arthritis (the HONOR study): 1‐year results. Arthritis and Rheumatism 2012;64(Suppl 10):771. [Google Scholar]
Tweehuysen 2017
- Tweehuysen L, Ende CH, Beeren FMM, Been EMJ, Hoogen FHJ, Broeder AA. Little evidence for usefulness of biomarkers for predicting successful dose reduction or discontinuation of a biologic agent in rheumatoid arthritis. Arthritis & Rheumatology 2017;69(2):301‐8. [PUBMED: 27696778] [DOI] [PMC free article] [PubMed] [Google Scholar]
van den Bemt 2008
- Bemt BJF, Broeder AA, Snijders GF, Hekster YA, Riel PLCM, Benraad B, et al. Sustained effect after lowering high‐dose infliximab in patients with rheumatoid arthritis: a prospective dose titration study. Annals of the Rheumatic Diseases 2008;67(12):1697‐701. [DOI] [PubMed] [Google Scholar]
van der Bijl 2007
- Bijl AE, Goekoop‐Ruiterman YPM, Vries‐Bouwstra JK, Wolde S, Han KH, Krugten MV, et al. Infliximab and methotrexate as induction therapy in patients with early rheumatoid arthritis. Arthritis and Rheumatism 2007;56(7):2129‐34. [DOI] [PubMed] [Google Scholar]
van der Heijde 1990
- Heijde DM, 't Hof MA, Riel PL, Theunisse LA, Lubberts EW, Leeuwen MA, et al. Judging disease activity in clinical practice in rheumatoid arthritis: first step in the development of a disease activity score. Annals of the Rheumatic Diseases 1990;49(11):916‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
van der Heijde 2000
- Heijde D. How to read radiographs according to the Sharp/van der Heijde method. Journal of Rheumatology 2000;27(1):261‐3. [PubMed] [Google Scholar]
van der Maas 2012
- Maas A, Kievit W, Bemt BJF, Hoogen FHJ, Riel PL, Broeder AA. Down‐titration and discontinuation of infliximab in rheumatoid arthritis patients with stable low disease activity and stable treatment: an observational cohort study. Annals of the Rheumatic Diseases 2012;71(11):1849‐54. [DOI] [PubMed] [Google Scholar]
van der Maas 2013 Flare
- Maas A, Lie E, Christensen R, Choy E, Man Y, Riel P, et al. Construct and criterion validity of several proposed DAS28‐based rheumatoid arthritis flare criteria: an OMERACT cohort validation study. Annals of the Rheumatic Diseases 2013;72:1800‐5. [DOI] [PubMed] [Google Scholar]
van Vollenhoven 2004
- Vollenhoven RF, Brannemark S, Klareskog L. Dose escalation of infliximab in clinical practise: improvements seen may be explained by a regression‐like effect. Annals of the Rheumatic Diseases 2004;63(4):426‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Vollenhoven 2009
- Vollenhoven RF. How to dose infliximab in rheumatoid arthritis: new data on a serious issue. Annals of the Rheumatic Diseases 2009;48(11):1237‐9. [DOI] [PubMed] [Google Scholar]
Verhoef 2017
- Verhoef LM, Tweehuysen L, Hulscher ME, Fautrel B, Broeder AA. bDMARD dose reduction in rheumatoid arthritis: a narrative review with systematic literature search. Rheumatology and Therapy 2017;4(1):1‐24. [PUBMED: 28255897] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weinblatt 2003
- Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti‐tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis & Rheumatology 2003;48(3):855. [PUBMED: 12528101] [DOI] [PubMed] [Google Scholar]
Wolbink 2006
- Wolbink GJ, Vis M, Lems W, Voskuyl AE, Groot E, Nurmohamed MT. Development of anti‐infliximab antibodies and relationship to clinical response in patients with rheumatoid arthritis. Arthritis and Rheumatism 2006;54(3):711‐5. [DOI] [PubMed] [Google Scholar]
Yoshida 2014
- Yoshida K, Sung Y‐K, Kavanaugh A, Bae S‐C, Weinblatt ME, Kishimoto M, et al. Biologic discontinuation studies: a systematic review of methods. Annals of the Rheumatic Diseases 2014;73:595‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
van Herwaarden 2014
- Herwaarden N, Broeder AA, Jacobs W, Maas A, Bijlsma JWJ, Vollenhoven RF, et al. Down‐titration and discontinuation strategies of tumor necrosis factor–blocking agents for rheumatoid arthritis in patients with low disease activity. Cochrane Database of Systematic Reviews 2014, Issue 9. [DOI: 10.1002/14651858.CD010455.pub2; PUBMED: 25264908] [DOI] [PubMed] [Google Scholar]