

Abstract
Cannabinoid CB1 and CB2 receptors are activated by Δ9-tetrahydrocannabinol, a psychoactive component of marijuana. The cannabinoid CB1 receptor is primarily located in the brain and is responsible for the psychoactive side effects, whereas the cannabinoid CB2 receptor is located in immune cells and is an attractive target for immune-related maladies. We identify small molecules that selectively bind to the cannabinoid CB2 receptor and can be further developed into therapeutics. The affinity of three molecules, ABK5, ABK6, and ABK7, to the cannabinoid CB2 receptor was determined with radioligand competition binding. The potency of G-protein coupling was determined with GTPγS binding. The three compounds bound selectively to the cannabinoid CB2 receptor, and no binding to the cannabinoid CB1 receptor was detected up to 10 μM. Immunoblotting studies show that the amount of ERK1/2 and MEK phosphorylation increased in a Gi/o-dependent manner. Furthermore, an immune cell line (Jurkat cells) was treated with ABK5, and as a result, inhibited cell proliferation. These three compounds are novel cannabinoid CB2 receptor agonists and hold promise to be further developed to treat inflammation and the often-associated pain.
Keywords: cannabinoids, cannabinoid CB2 receptor agonists, drug discovery, receptors
1. Introduction
Δ9-tetrahydrocannabinol (THC) in marijuana binds the cannabinoid receptors (Gaoni and Mechoulam, 1964), of which there are two subtypes. The cannabinoid CB1 receptor is located primarily in the central nervous system (CNS) (Matsuda et al., 1990) and is responsible for the psychoactive side effects of THC. The cannabinoid CB2 receptor is located in immune cells and the peripheral nervous system (Galiègue et al., 1995; Munro et al., 1993). Cannabinoid CB1 and CB2 receptors are G protein-coupled receptors (GPCRs), which primarily couple to Gi upon activation (Glass and Northup, 1999) resulting in inhibition of adenylyl cyclase with subsequent inhibition of cAMP production. (Howlett and Fleming, 1984). When activated, cannabinoid CB2 receptor produces analgesic effects without the psychoactive side effects of cannabinoid CB1 receptor activation, making it an attractive target for pain medication and that associated with inflammation in particular (Malan et al., 2001; Malan et al., 2003).
T lymphocytes play an important role in immune disorders and prior studies have shown that cannabinoids such as the endogenous anandamide (AEA) and 2 arachidonoylglycerol (2-AG), and the plant-derived THC inhibit the proliferation of T lymphocytes (Cancioni et al., 2019; Robinson et al., 2013; Robinson et al 2015). Nociceptive pain and inflammation are linked. When injury occurs, inflammatory mediators are released and exacerbate pain. Furthermore, the experimental evidence suggests that cannabinoid CB2 receptors may have a beneficial role in regulating the immune response, inflammation, and the associated pain. Cannabinoid CB2 receptor agonists attenuate inflammatory and neuropathic pain without the psychoactive effects caused by activation of cannabinoid CB1 receptor or opioid receptors (Ibrahim et al., 2006; Malan et al., 2003; Yamamoto et al., 2008). Over half of American adults experience pain, and as many as one in ten suffer from chronic pain, or daily pain for at least three months (Nahin, 2015). There are multiple types of pain medications available, but they come with serious side effects. Some cannabinoids, such as THC in marijuana, have analgesic properties, but are not optimal due to psychoactive side effects of sedation and impaired memory.
Compounds were identified as potential cannabinoid CB2 receptor agonists from a high throughput screen that showed that these compounds effectively inhibited the production of cAMP as a result of Gi coupling to the cannabinoid CB2 receptor (Ogawa et al., 2017). These agonists have very different chemical structures from known cannabinoid agonists such as THC and its derivative, CP55,940, as shown in Fig. 1. THC, CP55,940, and many other related agonists bind significantly to the cannabinoid CB2 receptor yet also the cannabinoid CB1 receptor (McPartland et al., 2007; Ross et al., 1999), leading to the undesirable psychoactive side effects. The compounds reported here, however, bind cannabinoid CB2 receptor with no detectable cannabinoid CB1 receptor binding.
Fig. 1.

Schematic illustrations of chemical structures of known CB2 agonists (THC and CP55,940) and CB2 agonists examined in this study: ABK5, ABK6, and ABK7. The * denotes the chiral center in the racemic mixture of ABK7.
Here, we describe the impact of agonists on both the cannabinoid CB2 receptor level and the mammalian cellular level. These are: ethyl 2-(2-(N-(2,3-dimethylphenyl)phenylsulfonamido)acetamido)benzoate (ABK5), benzoic acid, 2-[[2-[(2-methoxyphenyl)[(4-methylphenyl)sulfonyl]amino]acetyl]amino]-, methyl ester (ABK6), and 2-(2-phenylbutanamido)-4,5,6,7,8,9-hexahydrocycloocta[b]thiophene-3-carboxamide (ABK7). All ligands are available from ChemBridge Corporation (San Diego, CA). To date, there are no known drugs on the market that interact selectively and with such high affinity for cannabinoid CB2 receptor indicating the value of these compounds as potential scaffolds to be later developed into analgesics and the often-associated inflammation.
2. Materials and Methods
2.1. Cell culture
HEK293T and HEK293 cells were maintained in Dulbecco’s Modified Eagle Medium with 4.5 mg/ml of D-glucose (DMEM) (Thermo Fisher Scientific; Pittsburgh, PA USA) and 10% fetal bovine serum (FBS) at 37°C and 5% carbon dioxide saturation. Cells between passage four to twelve were used in experiments. Jurkat cells, a T-lymphoblastic leukemia cell line endogenously expressing cannabinoid CB2 receptor, were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and 1% antibiotic-antimycotic (Life Technologies, Carlsbad, CA USA) and were maintained at 37°C and 5% CO2 saturation. Jurkat cells between passage four to fifteen were used in experiments.
2.2. Cannabinoid receptor expression and membrane preparation
After four-twelve passages, cells were seeded to a density of 5×105 cell/100 mm plate. The calcium phosphate method (Chen and Okayama, 1987) was used for transfection. 24 h after transfection, the cells were prepared as described previously (Abadji et al., 1999) with some alterations. The cells were suspended in phosphate buffered saline (PBS) and centrifuged at 500 g for 5 min at 4°C. This process was repeated, and the pellet was resuspended in PBS and 1% (vol/vol) protease-inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) consisting of 104 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 80 μM aprotinin, 4 mM bestatin, 1.64 mM E-64, 2 mM leupeptin, and 1.5 mM pepstatin A. Following rupture, the cellular suspension underwent nitrogen cavitation for 5 min at 750 psi using a Parr cell disruption bomb. The resulting lysate was centrifuged for 10 min at 500 g and 4°C, and the resulting supernatant was centrifuged for 45 min at 116,000 g and 4°C. The pellet consisting of cell membranes was suspended in TME buffer (25 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM EDTA) with 7% (vol/vol) sucrose, and the suspension was aliquoted at a protein concentration of 1–2 mg/ml, which was determined with the Bradford assay (Bradford, 1976). The aliquots were stored at −80°C.
2.3. Radioligand binding
Radioligand competition binding was conducted as described previously (Abadji et al., 1994; Abadji et al., 1999; Murphy and Kendall, 2003) with some adjustments. Typically, 5 μg of protein membrane was incubated in a shaking water bath for 1 h at 30°C in 200 μl consisting of 1.5 nM [3H]CP55,940 (150.2 Ci/mmol; PerkinElmer Life Sciences, Waltham, MA USA), TME buffer and 0.2% (vol/vol) bovine serum albumin (BSA), TME buffer and 7% (w/v) sucrose, and 2 μl of the unlabeled ligand in nine concentrations ranging from 0.1 μM to 1 mM. Non-specific binding was determined using 10 μM CP55,940 in DMSO, in place of the radiolabeled ligand. The reactions are terminated with the addition of 300 μl of TME and 5% BSA (w/v) then washed with TME buffer and filtered with a Brandel cell harvester using Whatman GF/C filter paper. The filter paper samples are placed in scintillation vials with 4 ml Ultima Gold XR liquid scintillation cocktail (PerkinElmer Life Sciences, Waltham, MA USA) and counted with a Beckman Coulter 6500 liquid scintillation counter.
2.4. GTPγS binding
The guanosine 5’−3-O-(thio)triphosphate (GTPγS) binding assays were done as described previously (Abadji et al., 1994; Abadji et al., 1999; Murphy and Kendall, 2003) with some modifications. Typically, 8 μg of the protein membrane was incubated in a shaking water bath for 1 h at 30°C in 200 μl containing TME buffer and 0.1% fatty acid free BSA (vol/vol), 10 μM guanosine diphosphate (GDP), 0.1 nM [35S]GTPγS (1250 Ci/mmol; PerkinElmer Life Sciences, Waltham, MA USA), TME buffer and 7% (vol/vol) sucrose, GTPγS binding assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl), and 2 μl of nine concentrations of unlabeled ligand. Non-specific binding was determined in the presence of unlabeled 10 μM GTPγS. Reactions were terminated by filtration with the Brandel cell harvester, and bound radiolabeled compounds were separated on Whatman GF/C filter paper. Preparation of the scintillation vials and the liquid scintillation counting was conducted as described above.
2.5. Immunoblotting studies
The procedures below are based on those described previously (Ahn et al., 2012; Delgado-Peraza et al., 2016) with some modifications. HEK293 cells were seeded into 12-well plates to a density of 1×105 cells/ml. Subsequently, the cells were transfected with cannabinoid CB2 receptor DNA using Lipofectamine® 2000 (Invitrogen; Carlsbad, CA USA). 24 h after transfection, the cells were incubated in DMEM and pretreated with 10 ng/ml of pertussis toxin (PTX) (MilliporeSigma; Burlington, MA USA) for 16 h to abrogate Gi binding. The cells were treated for 5 min at 37°C and 5% CO2 saturation with ABK5, ABK6, ABK7, or CP55,940 at a concentration of 1.0 μM in DMEM. Untransfected cells and the cannabinoid CB2 receptor-vehicle were treated with DMEM and DMSO, both at 0.1%. Cell lysates were obtained by harvesting the cells in lysis buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 7.5 containing protease-inhibitor cocktail) (MilliporeSigma; Burlington, MA USA) and incubating on ice for 30 min. The resulting lysates were treated with β-mercaptoethanol. SDS-PAGE gel electrophoresis was used to resolve the 13 μg of protein lysate in 10% gels, and then transferred to polyvinylidene fluoride (PVDF) membrane (MilliporeSigma; Burlington, MA USA). After incubating the PVDF membrane overnight with Superblock T20 (PBS) blocking reagent (Fisher Scientific; Pittsburgh, PA USA), the membranes were incubated with the respective primary antibodies (1:3000 phospho-p44/42 and phospho-MEK antibodies; Cell Signaling Technology, Danvers, MA USA) for 2 h followed by 1 h of washing with Tris-buffered saline, 0.1% Tween 20 (20 mM Tris, 137 mM NaCl, 0.1% Tween 20) buffer, 1 h with goat anti-rabbit peroxidase-conjugated secondary antibodies (1:6000, Cell Signaling Technology, Danvers, MA USA), and 1 h of washing in buffer all at room temperature. The specific binding to the immunoreactive proteins was visualized with the SuperSignal West Femto Chemiluminescent Substrate System (Thermo Fisher Scientific; Rockford, IL USA) according to the manufacturer’s protocol.
2.6. RNA extraction
Jurkat cells were seeded at 2×105 cells/well in 12-well plates. Total RNA was extracted from cells at 48 h using TRIzol Reagent (Life Technologies, Carlsbad, CA USA) followed by reverse transcription by High-Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA USA) according to manufacturer’s instructions. Quantitative real-time PCR was performed using Applied Biosystems 7500 Fast Real-Time PCR System in a 10 μl reaction volume containing 2 μl diluted cDNA and 0.5 μM each of forward and reverse primers, Fast SYBR Green PCR Master Mix (Life Technologies, Carlsbad, CA USA). Primers used in the reaction are as follows: human cannabinoid CB2 receptor forward, 5’-CATGGAGGAATGCTGGGTGAC-3’, and human cannabinoid CB2 receptor reverse, 5’-GAGGAAGGCGATGAACAGGAG-3’ (Roth et al., 2015), human GAPDH forward, 5’-AGCCACATCGCTCAGACAC-3’, and human GAPDH reverse, 5’-AATGAAGGGGTCATTGATGG-3’ (Hartwell et al., 2006). The PCR cycles are as follows: 95°C for 20 s, 40 cycles of 95°C for 3 s and 60°C for 30 s.
2.7. Cell toxicity
HEK293T cells were seeded in 96-well plate at 0.75 × 104 cells/well and cultured for 24 h before compound treatment. Cells were incubated as the vehicle alone (DMSO) or 0.1 or 10 μM ABK5 for 72 h, and the viable cell number was measured by CellTiter 96®AQ ueous One Cell Proliferation Assay (Promega; Madison, WI USA) following manufacturer’s instructions.
2.8. Cell proliferation
After four-twelve passages, the Jurkat cells were cultured in media in a 96-well plate at a density of 105 cells/ml. For 70 h, the 50 μl of the cells were treated with 50 μl solutions containing the RPMI 1640 media with the compound ABK5, CP55,940, or DMSO alone. After the compound treatment, 20 μl of CellTiter 96® AQueous One Solution was added to each well for 2 h of incubation. Plate readings were conducted at an absorbance of 490 nm on a PowerWaveX Microplate Spectrophometer (BioTek Instruments, Inc.; Winooski, VT USA) with KC4 version 3.4 Data Analysis software.
2.9. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The ligand (Fig. 2) and GTPγS (Fig. 3) binding assays were carried out in duplicate for three independent experiments. Data are presented as the mean of three assays ± standard error mean (S.E.M.). The Ki values were calculated using Prism software (GraphPad Software Inc.; San Diego, CA USA) and non-linear regression fitted to the one-site binding model. The EC50 values for the GTPγS binding assays are calculated also using this software. The “zero percentage” is defined as the percentage of GTPγS binding in the presence of 100 pM of the compound, where no stimulation was observed. The increase in GTPγS binding with higher concentrations of the compound is shown relative to that point. The immunoreactive bands from the Western blots were quantified with the ImageJ program (https://imagej.nih.gov/ij/index.html). Data are expressed as a fold increase above the basal level of phosphorylation (Fig. 4), which is denoted as the vehicle and represented by cannabinoid CB2 receptor-transfected HEK293 cells treated with DMSO alone. For mRNA extraction (Fig. 5A), to ensure equal amounts of mRNA loading, the cannabinoid CB2 receptor mRNA levels were analyzed by the ΔΔCt method and normalized by the housekeeping gene GAPDH (Barber et al., 2005). For cell proliferation (Fig. 5A), experiments were completed in triplicate. Cells treated with DMSO alone were used as a control, and the percent inhibition was normalized with respect to this. Results are expressed as the mean ± S.E.M. of the experiment performed in triplicate. Significance was assessed by one-way ANOVA followed by Tukey’s post-hoc test.
Fig. 2.
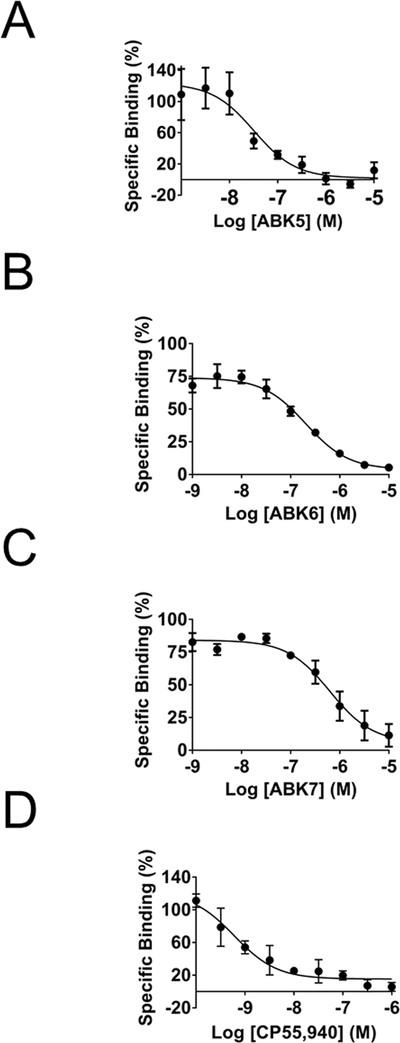
Specific binding of [3H]CP55,940 to CB2 in the presence of increasing concentrations of (A) ABK5, (B) ABK6, (C) ABK7, and (D) CP55,940. Membrane preparations of CB2-expressing HEK293T cells and their evaluation for binding is described in Materials and Methods. Each data point represents the mean ± S.E.M. of three independent assays performed in duplicate.
Fig. 3.

Increase in the specific binding of [35S]GTPγS to CB2 in the presence of increasing concentrations: (A) ABK5, (B) ABK6, (C) ABK7, and (D) CP55,940. Stimulation of GTPγS binding in response to compounds treatment of membranes prepared from HEK293T cells transfected with the CB2 receptor as described in Materials and Methods. Each data point represents the mean ± S.E.M. of three independent assays performed in duplicate.
Fig. 4.

CB2-induced (A) ERK1/2 and (B) MEK phosphorylation in HEK293 cells in response to ABK5, ABK6, and ABK7. Western blots (left) and their respective quantifications (right) are shown. The compounds were tested at a concentration of 1.0 μM, and treatment with 10 ng/ml of PTX to preclude G protein binding is indicated. One-way ANOVA plus Tukey’s post-hoc test was used and *P < 0.05 versus vehicle alone and #P < 0.05 versus compound alone (without PTX.).
Fig. 5.

Inhibition of Jurkat-cell proliferation in response to ABK5. (A) mRNA levels of CB2 in Jurkat and HEK293T cells. As indicated, Jurkat cells have substantial levels of endogenous CB2 while HEK293T cells have none and must be transfected. The bars indicate that the CB2 levels were done on cells in 3–4 wells. (B) Toxicity analysis as percentage of viable HEK293T cells in the presence of the vehicle alone (DMSO), 0.1 μM, and 10 μM ABK5. (C) Percentage of Jurkat-cell proliferation inhibition with relative to vehicle alone (DMSO, indicated as 0 μM) by the concentration of ABK5 given. CP55,940 is given for comparison. Results are expressed as the mean ± S.E.M. (n=3) for each concentration. One-way ANOVA plus Tukey’s post-hoc test were used and *P < 0.05 versus vehicle alone.
3. Results
We determined that the compounds bound to the cannabinoid CB2 receptor using competitive radiolabeled ligand binding of membranes from HEK293T cells transfected with the cannabinoid CB2 receptor. Fig. 2 (A–C) shows how the concentration of the respective compounds affects the specific binding of the radiolabeled tracer (agonist CP55,940). In each case, all three compounds bound the cannabinoid CB2 receptor. ABK5 has the strongest binding affinity, Ki=16±8 nM, followed by ABK6, Ki=102±7 nM. ABK7, a racemic compound with the chiral center shown in Fig. 1, has the weakest binding affinity of the three compounds with a Ki=317± 117 nM. To ensure that these compounds bind selectively to the cannabinoid CB2 receptor and not to the cannabinoid CB1 receptor, we also measured their respective binding affinities for the cannabinoid CB1 receptor in membranes of HEK293T cells transfected with the cannabinoid CB1 receptor. All three compounds showed no specific binding to the cannabinoid CB1 receptor up to 10 μM of the test compounds. Thus, these compounds are highly selective for the cannabinoid CB2 receptor over cannabinoid CB1 receptor. CP55,940 is shown (Fig. 2D) for comparison. The binding parameters are summarized in Table 1. As noted in the table, we determined a Ki=0.3±0.2 nM for CP55,940 binding to the cannabinoid CB2 receptor and a Ki = 1.0±0.2 nM for CP55,940 binding to the cannabinoid CB1 receptor. This is within the range of reported values (Pertwee et al., 2010; Ross et al., 1999).
Table 1.
Binding and G protein-stimulation properties of ABK5, ABK6, ABK7, and CP55,940.
| CP55,940 | GTPγS | ||
|---|---|---|---|
| Ki (nM)a | EC50 (nM)b | ||
| Compound | CB2 | CB1 | CB2 |
| ABK5 | 16±8 | N.B.c | 4±3 |
| ABK6 | 102±7 | N.B.c | 13±4 |
| ABK7 | 317±117 | N.B.c | 31±14 |
| CP55,940 | 0.3±0.2 | 1±0.2 | 36±26 |
Ki values were determined from competition binding assays using [3H]CP55,940.
EC values were determined from stimulation of [35S]GTPγS binding.
N.B.: No specific binding detected up to 10 μM of test compounds.
Each data point represents the mean ± S.E.M. of three independent assays performed in duplicate.
To ensure that these compounds activate the cannabinoid CB2 receptor in a G protein-coupled manner, we measured their potency at inducing GTPγS binding. Fig. 3 (A–C) shows the increase in the percentage of specific GTPγS binding with respect to increasing concentration of the test compound. ABK5 is the most potent of the three compounds with an EC50 of 4±2 nM. ABK6 has an EC50 of 13±4 nM, and the racemic mix of ABK7 is the least potent with an EC50 of 31±14 nM. However, all of the compounds have a potency less than 100 nM. The compounds are effective at inducing G-protein coupling to the cannabinoid CB2 receptor, which is indicative of receptor activation and suggests downstream signaling is G-protein dependent. CP55,940 is shown (Fig. 3D) for comparison. These parameters are summarized in Table 1.
To determine the impact on cellular signal transduction, we examined the compound-induced phosphorylation of kinases ERK1/2 and MEK (Alberich Jordà et al., 2004; Herrera et al., 2005; Samson et al., 2003) through the G protein-dependent pathway. To verify this, we used pertussis toxin (PTX) to preclude Gi/o activation (Howlett et al., 1986) and potentially inhibit downstream signaling. Fig. 4A shows the impact the three compounds have on ERK1/2 phosphorylation in the presence and absence of PTX. After 5 min of treatment, ABK5, ABK6, and ABK7 all induce ERK1/2 phosphorylation, which is prevented by the addition of PTX. The corresponding quantification of the Western blot image is shown at the right of Fig. 4A. For each compound, the amount of ERK1/2 is approximately three-fold greater than the vehicle alone. The addition of PTX to ablate Gi coupling, however, absolves the phosphorylation effect, and it decreases to approximately equal that of the vehicle. These overall trends were similar to those observed for phosphorylation of the MEK kinases as shown in Fig. 4B. The Western blots show that the three compounds induce MEK phosphorylation, which is inhibited by PTX. The quantification at right shows that the three compounds induce phosphorylation three to five times greater than the vehicle alone (Fig. 4).
ABK5 was shown above to bind with the strongest affinity to the cannabinoid CB2 receptor and to be the most potent at inducing G-protein coupling. Since this compound seems therapeutically promising, we decided to further explore its physiological impact on Jurkat cells, a human T-lymphocyte cell line that has endogenous cannabinoid CB2 receptor (Fig. 5A). Endogenous expression of the cannabinoid CB2 receptor in Jurkat cells is substantial; however, in HEK293T cells, run as a control for the cannabinoid CB2 receptor primer, it is essentially nonexistent. Also, CB1 endogenous expression in both cell lines was essentially nonexistent. Cytotoxicity of ABK5 was tested by determining viable cell number relative to vehicle alone after 72 h of compound incubation. No significant change in viable cell number was observed up to 10 μM. This result suggests that ABK5 concentrations which we used in the cell proliferation assay were within reasonable range (Fig. 5B). Fig. 5C shows that Jurkat cells treated with either 0.1 μM or 10 μM CP55,940 or ABK5 had less cell proliferation after a 72 h proliferation period than that treated with the vehicle alone. Treatment of CP55,940 resulted in a cell proliferation inhibition of 7±3% for 0.1 μM and 26±8% for 10 μM. This same treatment of ABK5 resulted in a reduction in cell proliferation by 15±2% and 20±1% for concentrations of 0.1 μM and 10 μM, respectively. For both cannabinoid CB2 receptor agonists, the higher concentration caused a greater inhibition of cellular proliferation. Thus, we observe that the addition of a selective cannabinoid CB2 receptor agonist significantly decreases the immune cell population, which is consistent with an antiproliferation effect (Fig. 5C).
4. Discussion
To design effective and safe pain medication associated with inflammation that targets the cannabinoid CB2 receptor, the first steps are to identify compounds that (1) bind to the cannabinoid CB2 receptor and not the cannabinoid CB1 receptor to avoid psychoactive side effects; (2) activate the cannabinoid CB2 receptor via the Gi pathway; and (3) inhibit immune cell proliferation. First, the compounds tested here bind strongly to the cannabinoid CB2 receptor with binding affinities ranging from 16±8 nM (ABK5) to 317±117 (ABK7) (see Fig. 2), which is greater than those of the endocannabinoids (AEA has a Ki=581 nM and 2-AG has a Ki=1400 nM) (Mechoulam et al., 1995). ABK5 and ABK6 have an affinity for the cannabinoid CB2 receptor that is comparable to that of THC (Ki=36 nM) (Showalter et al., 1996). Also of importance, selectivity for the cannabinoid CB2 receptor is established and is especially promising given that existing compounds with the strongest affinities for the cannabinoid CB2 receptor only have an approximately 10- to 100-fold selectivity versus the cannabinoid CB1 receptor. CP55,940, shown here for comparison, binds to the cannabinoid CB2 receptor and the cannabinoid CB1 receptor with similar affinities of 0.3±0.2 nM and 1±0.2 nM, respectfully, whereas the ABK compounds do not show any binding to the cannabinoid CB1 receptor up to 10 μM. Similarly, JWH-133 has a Ki of 3.4 nM for the cannabinoid CB2 receptor, but a Ki of 677 nM for the cannabinoid CB1 receptor (Huffman et al., 1999). Also, JWH-051 has a Ki of 0.032 nM for the cannabinoid CB2 receptor and a Ki of 1.2 nM for the cannabinoid CB1 receptor (Huffman et al., 1996). Even though these three agonists have strong affinities for the cannabinoid CB2 receptor, they still have significant affinities for the cannabinoid CB1 receptor, and can bind to this receptor when they are present in relevant concentrations. Another example is the cannabinoid CB2 receptor agonist GW405833. Its affinity for human the cannabinoid CB2 receptor ranges from 4–12 nM (Gallant et al., 1996; Valenzano et al., 2005), with selectivities for the human cannabinoid CB2 receptor over the human cannabinoid CB1 receptor ranging from 37-to 1217-fold (Valenzano et al., 2005; Yao et al., 2009). However, its anti-inflammatory effects are caused by the cannabinoid CB1 receptor pathway rather than the the cannabinoid CB2 receptor one (Li et al., 2017). Thus, even though this compound preferentially binds to the cannabinoid CB2 receptor, it exerts its physiological effects through the cannabinoid CB1 receptor.
These ABK compounds bind to the cannabinoid CB2 receptor, and are also effective agonists since they stimulate GTPγS binding, which is indicative of cannabinoid CB2 receptor activation and coupling to G protein as a result. Compounds ABK5–7 were identified by a screening process based on cAMP accumulation described previously (Ogawa et al., 2017). In this prior screen, the EC50 values for ABK6 and ABK7 were 2 nM and 32 nM, respectively. These values are similar to those determined in this study with [35S]GTPγS binding, EC50=13±4 nM for ABK6 and EC50=31±14 nM for ABK7. These two sets of values are quite comparable between the different experiments, but the determined potencies for ABK6 are slightly different. This discrepancy could be the result of the differences in experimental procedure as the [35S]GTPγS binding is a direct measurement of the amount of G-protein binding, whereas cAMP accumulation is more indirect as it monitors a biological process that occurs downstream of the G-protein receptor binding event. However, both experimental methods show that ABK6 and ABK7 have a strong potency for the cannabinoid CB2 receptor.
Compounds, JWH-133, JWH-015, and O-1966 (Börner et al., 2009; Maresz et al., 2007; Robinson et al., 2013), and other non-selective cannabinoids, such as THC, CP55,940, and AEA (Cencioni et al., 2010; Klein et al., 1991; Yuan et al., 2002), inhibit the proliferation of T cells through interactions with their cannabinoid CB2 receptors. Given the role that T cells play in the immune response, inhibition of T-cell proliferation is a good indicator that some of the effects of inflammation could be prevented. ABK5 inhibits Jurkat-cell proliferation to a similar extent as CP55,940, but since ABK5 is a compound with measurable binding observed only to the cannabinoid CB2 receptor and not the cannabinoid CB1 receptor, it would be a good candidate to be further developed into therapeutics to treat inflammation and the associated pain.
In this work, we show that ABK 5–7 induce downstream signaling through ERK1/2 via Gi in cannabinoid CB2 receptor-transfected cells. Studies of the cannabinoid CB2 receptor agonists acting on cells that endogenously express the cannabinoid CB2 receptor are largely consistent with our results. Cannabinoids 2-AG and JWH-133 induce phosphorylation of ERK1/2 in T lymphocytes through interactions with the cannabinoid CB2 receptor (Coopman et al., 2007) and JWH-133 also causes cannabinoid CB2 receptor-mediated ERK1/2 phosphorylation in retinal pigment epithelium (RPE) cells (Hytti et al., 2017). In microglia, endocannabinoid AEA also activates ERK1/2 as a result of binding to the cannabinoid CB2 receptor (Correa et al., 2009a; Correa et al., 2009b). Furthermore, the cannabinoid CB2 receptor activation in microglia involve inhibition of neuroinflammation (Cabral et al., 2008). In our studies, we show that treatment of Jurkat cells with ABK5 results in the prevention of cell proliferation. In other work, the cannabinoid CB2 receptor-induced ERK1/2 phosphorylation by 2-AG and JWH-133 reduced chemokine CXCL12-induced chemotaxis in T lymphocytes (Coopman et al., 2007). Moreover, AEA increases anti-inflammatory cytokine IL-10 production and downregulates pro-inflammatory cytokine IL-12 and IL-23 production in microglia, which result in antiinflammatory effects (Correa et al., 2009a; Correa et al., 2009b). There is also evidence that the cannabinoid CB2 receptor activation can inhibit neuroinflammation by protecting the blood-brain barrier (Persidsky et al., 2015). Future studies from our work will include examining how ABK5-induced ERK1/2 phosphorylation affects cytokine production and how that causes inhibition of T-cell production.
The compound ABK7 is of interest for future studies given its racemic nature. Racemic mixes of other compounds have shown that the separated isomers often behave very differently. For example, GAT211 is a racemic mix that acts as a cannabinoid CB1 receptor allosteric modulator, but that activity results from the S-(−)-enantiomer (GAT229), whereas the R-(+)-enantiomer (GAT228) acts as an allosteric agonist (Laprairie et al., 2017). More studies are required to show whether the separate isomers of ABK7 both act as cannabinoid CB2 receptor agonists, or if they have differences in binding affinity, for example, one isomer binds while the other does not. We anticipate that the Ki of the binding isomer would be half of the racemic mix (160 nM vs. 317 nM). Such studies are on-going.
Tissue injury often results in inflammation and the associated pain (Ji et al., 2011). This includes arthritis, lower back injury, and surgery. If left unresolved, acute pain can lead to chronic pain. According to the National Center for Health Statistics (2006), approximately 76.2 million, one in every four Americans, have suffered from pain that lasts longer than 24 hours and chronic pain is a common cause of long-term disability. Yet opioid pain medications are over used and can lead to addiction (Leider et al., 2011) and overdose. The molecules described here may be good leads for possible anti-inflammatory pain medications for further study. Future study includes structure-activity analysis to identify improvements due to the use of particular functional groups and how these impact cytokine release, and animal studies of models of inflammatory pain. Since there are many different types of pain, having several types of medications to attenuate each of these is advisable. This is an approach toward that end.
5. Conclusions
Here, we have identified and characterized three compounds that behave as cannabinoid CB2 receptor agonists. These compounds bound strongly with affinities ranging from 16 to 317 nM, which are more favorable than those of endocannabinoids and are comparable to that of the cannabinoid agonist examined here, CP55,940. However, none of the ABK compounds show any binding to the cannabinoid CB1 receptor up to 10 μM, suggesting that they bind selectively to the cannabinoid CB2 receptor over the cannabinoid CB1 receptor and that the likelihood of cannabinoid CB1 receptor-induced psychoactive side effects is small. Furthermore, these compounds induce downstream phosphorylation of ERK1/2 and MEK through Gi. Finally, these compounds inhibit T-cell proliferation. These characteristics indicate that these compounds should be explored and developed further into anti-inflammatory and pain medication. Safe and efficient anti-inflammatory medication that lack psychoactive side effects is especially important, and, here, we have identified some promising candidates.
Acknowledgements
This work was supported in part by a grant from the National Institute of Health (DA 040920). Yu-Hsien Liao is gratefully acknowledged for his contribution to the experiments. The authors also thank Michael Lynes (University of Connecticut, Storrs, CT) for providing Jurkat cells and Charles Giardina (University of Connecticut, Storrs, CT) for providing the CellTiter 96® AQueous One Solution Cell Proliferation Assay and for multiple helpful discussions. We thank José Manautou (University of Connecticut, Storrs, CT) for his plate reader.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST: None
REFERENCES
- Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG, Makriyannis A, 1994. (R)-Methanandamide: A chiral novel anandamide possessing higher potency and metabolic stability. Journal of Medicinal Chemistry 37, 1889–1893. [DOI] [PubMed] [Google Scholar]
- Abadji V, Lucas-Lenard JM, Chin C. n., Kendall DA, 1999. Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid CB1 receptor in constitutive activation of Gs. Journal of Neurochemistry 72, 2032–2038. [DOI] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Kendall DA, 2012. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and G(i) protein-independent ERK1/2 kinase activation. Journal of Biological Chemistry 287, 12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberich Jordà M, Rayman N, Tas M, Verbakel SE, Battista N, van Lom K, Löwenberg B, Maccarrone M, Delwel R, 2004. The peripheral cannabinoid receptor CB2, frequently expressed on AML blasts, either induces a neutrophilic differentiation block or confers abnormal migration properties in a ligand-dependent manner. Blood 104, 526–534. [DOI] [PubMed] [Google Scholar]
- Barber RD, Harmer DW, Coleman RA, Clark BJ, 2005. GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiological Genomics 21, 389–395. [DOI] [PubMed] [Google Scholar]
- Börner C, Smida M, Höllt V, Schraven B, Kraus J, 2009. Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. Journal of Biological Chemistry 284, 35450–35460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM, 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F, 2008. CB2 receptors in the brain: role in central immune function. Br J Pharmacol 153, 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencioni MT, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, Maccarrone M, 2010. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS One 5, e8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H, 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Molecular and Cellular Biology 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman K, Smith LD, Wright KL, Ward SG, 2007. Temporal variation in CB2R levels following T lymphocyte activation: Evidence that cannabinoids modulate CXCL12-induced chemotaxis. International Immunopharmacology 7, 360–371. [DOI] [PubMed] [Google Scholar]
- Correa F, Docagne F, Mestre L, Clemente D, Hernangómez M, Loría F, Guaza C, 2009a. A role for CB2 receptors in anandamide signalling pathways involved in the regulation of IL-12 and IL-23 in microglial cells. Biochemical Pharmacology 77, 86–100. [DOI] [PubMed] [Google Scholar]
- Correa F, Hernangómez M, Mestre L, Loría F, Spagnolo A, Docagne F, Di Marzo V, Guaza C, 2009b. Anandamide enhances IL-10 production in activated microglia by targeting CB2 receptors: Roles of ERK1/2, JNK, and NF-κB. Glia 58, 135–147. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA, Gilchrist A, Hoyer D, Insel P,A, Izzo A,A, Lawrence AJ, MacEwan DJ, Moon LDF, Wonnacott S, Weston AH, McGrath JC, 2015. Experimental design and analysis and their reporting: new guidance for publication in BJP. British Journal of Pharmacology 172, 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Peraza F, Ahn KH, Nogueras-Ortiz C, Mungrue IN, Mackie K, Kendall DA, Yudowski GA, 2016. Mechanisms of Biased β-Arrestin-mediated signaling downstream from the cannabinoid 1 receptor. Molecular Pharmacology 89, 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P, 1995. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. European Journal of Biochemistry 232, 54–61. [DOI] [PubMed] [Google Scholar]
- Gallant M, Dufresne C, Gareau Y, Guay D, Leblanc Y, Prasit P, Rochette C, Sawyer N, Slipetz DM, Tremblay N, Metters KM, Labelle M, 1996. New class of potent ligands for the human peripheral cannabinoid receptor. Bioorganic and medicinal chemistry letters 6, 2263–2268. [Google Scholar]
- Gaoni Y, Mechoulam R, 1964. Isolation, structure, and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society 86, 1646–1647. [Google Scholar]
- Glass M, Northup JK, 1999. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Molecular Pharmacology 56, 1362–1369. [DOI] [PubMed] [Google Scholar]
- Hartwell KA, Muir B, Reinhardt F, Carpenter AE, Sgroi DC, Weinberg RA, 2006. The Spemann organizer gene, Goosecoid, promotes tumor metastasis. Proceedings of the National Academy of Sciences, USA 103, 18969–18974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera B, Carracedo A, Diez-Zaera M, Guzmán M, Velasco G, 2005. p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Letters 579, 5084–5088. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Fleming RM, 1984. Cannabinoid inhibition of adenylate-cyclase. Pharmacology of the response in neuro-blastoma cell-membranes. Molecular Pharmacology 26, 532–538. [PubMed] [Google Scholar]
- Howlett AC, Qualy JM, Khachatrian LL, 1986. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Molecular Pharmacology 29, 307–313. [PubMed] [Google Scholar]
- Huffman JW, Liddle J, Yu S, Aung MM, Abood ME, Wiley JL, Martin BR, 1999. 3-(1’,1’-Dimethylbutyl)-1-deoxy-Δ8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorganic and Medicinal Chemistry 7, 2905–2914. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Yu S, Showalter V, Abood ME, Wiley JL, Compton DR, Martin BR, Bramblett RD, Reggio PH, 1996. Synthesis and pharmacology of a very potent cannabinoid lacking a phenolic hydroxyl with high affinity for the CB2 receptor. Journal of Medicinal Chemistry 39, 3875–3877. [DOI] [PubMed] [Google Scholar]
- Hytti M, Andjelic S, Josifovska N, Piippo N, Korhonen E, Hawlina M, Kaarniranta K, Nevalainen TJ, Petrovski G, Parkkari T, Kauppinen A, 2017. CB2 receptor activation causes an ERK1/2-dependent inflammatory response in human RPE cells. Scientific Reports 7, 16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP Jr., 2006. CB2 cannabinoid receptor mediation of antinociception. Pain 122, 36–42. [DOI] [PubMed] [Google Scholar]
- Ji RR, Xu ZZ, Strichartz G, Serhan CN, 2011. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci 34, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW, Kawakami Y, Newton C, Friedman H, 1991. Marijuana components suppress induction and cytolytic function of murine cytotoxic T cells in vitro and in vivo. Journal of Toxicology and Environmental Health 32, 465–477. [DOI] [PubMed] [Google Scholar]
- Laprairie R, Kulkarni PM, Deschamps JR, Kelly MEM, Janero DR, Cascio MG, Stevenson LA, Pertwee RG, Kenakin TP, Denovan-Wright EM, Thakur GA, 2017. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chemical Neuroscience 8, 1188–1203. [DOI] [PubMed] [Google Scholar]
- Leider HL, Dhaliwal J, Davis EJ, Kulakodlu M, Buikema AR, 2011. Healthcare costs and nonadherence among chronic opioid users. Am J Manag Care 17, 32–40. [PubMed] [Google Scholar]
- Li A-L, Carey LM, Mackie K, Hohmann AG, 2017. Cannabinoid CB2 agonist GW405833 suppresses inflammatory and neuropathic pain through a CB1 mechanism that is independent of CB2 receptors in mice. Journal of Pharmacology and Experimental Therapeutics 362, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan TP, Ibrahim MM, Deng HF, Liu Q, Mata HP, Vanderah T, Porreca F, Makriyannis A, 2001. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain 93, 239–245. [DOI] [PubMed] [Google Scholar]
- Malan TP Jr., Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F, 2003. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol 3, 62–67. [DOI] [PubMed] [Google Scholar]
- Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP, Ledent C, Cheng X, Carrier EJ, Mann MK, Giovannoni G, Pertwee RG, Yamamura T, Buckley NE, Hillard CJ, Lutz B, Baker D, Dittel BN, 2007. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nature Medicine 13, 492–497. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI, 1990. Structure of a cannabinoid receptor and functional expression of the cloned DNA. Nature 346, 561–564. [DOI] [PubMed] [Google Scholar]
- McPartland JM, Glass M, Pertwee RG, 2007. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: Interspecies differences. British Journal of Pharmacology 152, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z, 1995. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical Pharmacology 50, 83–90. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL, Abu-Shaar M, 1993. Molecular characterization of a peripheral receptor for cannabinoids. Nature 365, 61–65. [DOI] [PubMed] [Google Scholar]
- Murphy JW, Kendall DA, 2003. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55,940 but not SR 141716A. Biochemical Pharmacology 65, 1623–1631. [DOI] [PubMed] [Google Scholar]
- Nahin RL, 2015. Estimates of pain prevalence and severity in adults: United States, 2012. Journal of Pain 16, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa LM, Burford NT, Liao Y-H, Scott CE, Hine AM, Dowling C, Chin J, Power M, Hunnicutt J, Edward J, Emerick VL, Banks M, Zhang L, Gerritz SW, Alt A, Kendall DA, 2017. Discovery of selective cannabinoid CB2 receptor agonists by high-throughput screening. SLAS DISCOVERY: Advancing Life Scineces R&D 23, 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Fan S, Dykstra H, Reichenbach NL, Rom S, Ramirez SH, 2015. Activation of Cannabinoid Type Two Receptors (CB2) Diminish Inflammatory Responses in Macrophages and Brain Endothelium. J Neuroimmune Pharmacol 10, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA, 2010. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2). Pharmacol Rev 62, 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RH, Meissler JJ, Breslow-Deckman JM, Gaughan J, Adler MW, Eisenstein TK, 2013. Cannabinoids inhibit T-cells via cannabinoid receptor 2 in an in vitro assay for graft rejection, the mixed lymphocyte reaction. Journal of Neuroimmune Pharmacology 8, 1239–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG, 1999. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. British Journal of Pharmacology 126, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MD, Castaneda JT, Kiertscher SM, 2015. Exposure to Δ9-tetrahydrocannabinol impairs the differentiation of human monocyte-derived dendritic cells and their capacity for T cell activation. Journal of neuroimmune pharmacology 10, 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M-T, Small-Howard A, Shimoda LMN, Koblan-Huberson M, Stokes AJ, Turner H, 2003. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. Journal of Immunology 170, 4953–4962. [DOI] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME, 1996. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther 278, 989–999. [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, Harrison JE, Boulet JM, Gottshall SL, Mark L, Pearson MS, Miller W, Shan S, Rabadi L, Rotshteyn Y, Chaffer SM, Turchin PI, Elsemore DA, Toth M, Koetzner L, Whiteside GT, 2005. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology 48, 658–672. [DOI] [PubMed] [Google Scholar]
- Yamamoto W, Mikami T, Iwamura H, 2008. Involvement of central cannabinoid CB2 receptor in reducing mechanical allodynia in a mouse model of neuropathic pain. Eur J Pharmacol 583, 56–61. [DOI] [PubMed] [Google Scholar]
- Yao BB, Hsieh GC, Frost JM, Fan Y, Garrison TR, Daza AV, Grayson GK, Zhu CZ, Pai M, Chandran P, Salyers AK, Wensink EJ, Honore P, Sullivan JP, Dart MJ, Meyer MD, 2009. In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. British Journal of Pharmacology 153, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Kiertscher SM, Cheng Q, Zoumalan R, Tashkin DP, Roth MD, 2002. Δ9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. Journal of Neuroimmunology 133, 124–131. [DOI] [PubMed] [Google Scholar]