

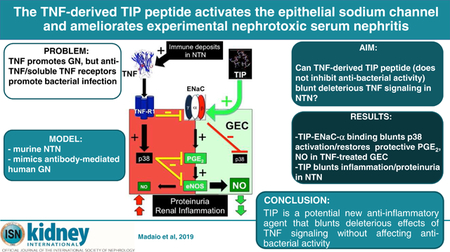
Abstract
In mice, the initial stage of nephrotoxic serum-induced nephritis (NTN) mimics antibody-mediated human glomerulonephritis. Local immune deposits generate tumor necrosis factor (TNF), which activates pro-inflammatory pathways in glomerular endothelial cells (GEC) and podocytes. Because TNF receptors mediate antibacterial defense, existing anti-TNF therapies can promote infection; however, we have previously demonstrated that different functional domains of TNF may have opposing effects. The TIP peptide mimics the lectin-like domain of TNF, and has been shown to blunt inflammation in acute lung injury without impairing TNF receptor-mediated antibacterial activity. We evaluated the impact of TIP peptide in NTN. Intraperitoneal administration of TIP peptide reduced inflammation, proteinuria, and blood urea nitrogen. The protective effect was blocked by the cyclooxygenase inhibitor indomethacin, indicating involvement of prostaglandins. Targeted glomerular delivery of TIP peptide improved pathology in moderate NTN and reduced mortality in severe NTN, indicating a local protective effect. We show that TIP peptide activates the epithelial sodium channel (ENaC), which is expressed by GEC, upon binding to the channel’s α subunit. In vitro, TNF treatment of GEC activated pro-inflammatory pathways and decreased the generation of prostaglandin E2 and nitric oxide, which promote recovery from NTN. TIP peptide counteracted these effects. Despite the capacity of TIP peptide to activate ENaC, it did not increase mean arterial blood pressure in mice. In the later autologous phase of NTN, TIP peptide blunted the infiltration of Th17 cells. By countering the deleterious effects of TNF through direct actions in GEC, TIP peptide could provide a novel strategy to treat glomerular inflammation.
Keywords: cytokines, glomerulus, endothelium, proteinuria, prostaglandins
Graphical Abstract
INTRODUCTION.
Acute glomerulonephritis (GN) is an inflammatory condition, which accounts for up to 20% of end stage renal disease (ESRD). Current therapies aimed to limit disease progression, to retard underlying mechanisms of renal injury, or to reverse the primary underlying disease are inadequate, lack specificity and are often associated with systemic toxicity. Understanding how renal tissue recovers or regenerates after insult is therefore especially relevant in developing plans to enhance the process1.
The initial stage of nephrotoxic serum (NTS)-induced nephritis (NTN) in mice is a valid model system for antibody-mediated GN2. Both infiltrating cells and intrinsic renal cells play crucial roles in nephritis, as they participate in leukocytes and macrophage recruitment and their activation and dysfunction contributes to proteinuria and renal failure1,3. During the course of GN, the pro-inflammatory cytokine TNF is increased in infiltrating monocytes and resident renal cells following immune deposit formation3–5. TNF mediates glomerular inflammation and cellular injury by activating the pro-inflammatory p38 MAP kinase pathway in glomerular endothelial cells (GEC) and podocytes (POD) during experimental and human GN, which in turn increases MCP-1-dependent renal monocyte infiltration. Pharmacological inhibition of the p38 MAP kinase pathway has been shown to reduce pathology6–8. TNF receptor 1-deficient mice subjected to immune GN develop significantly less renal activation of the p38 MAP kinase pathway, with fewer leukocyte infiltrates and reduced proteinuria9,10. Based on these observations, our goal was to pursue inhibition of leukocyte infiltration and GEC activation, mediated by NTS-induced TNF, by interfering with TNF receptor 1-mediated activation of the p38 MAP kinase pathway.
A critical limitation of more general modulation of TNF activity during disease is impairment of host response to infection, as well as development of solid organ tumors, resulting from chronic inhibition of TNF receptor signaling, observed with TNF antibodies (Ab) or soluble receptor constructs11–15. We have shown that different functional domains of TNF, i.e. the receptor binding region versus the lectin-like domain16,17, can have opposing effects on tissue injury and barrier dysfunction18–23. Mutations in the lectin-like domain did not impair TNF’s anti-bacterial activities in a murine model of septic peritonitis24. This finding provided a unique opportunity to evaluate a TNF-derived circular peptide mimicking the lectin-like domain of TNF, the TIP peptide17 (sequence: CGQRETPEGAEAKPWYC), to resolve ongoing inflammation during the course of NTN, without interfering with the cytokine’s role in immune defense. We demonstrated that the TIP peptide binds to the α subunit of the epithelial sodium channel (ENaC)20,22, which can be expressed in both epithelial and endothelial cells25,26.
Support for this experimental direction is provided by the finding that inhaled TIP peptide (a.k.a. AP301 and Solnatide) was recently found to be safe in a phase 1 clinical trial in volunteers27 and displayed promising activities on lung function in two phase 2a clinical trials in patients with acute lung injury28 and following lung transplantation29.
Initially, we assessed whether TIP peptide treatment could blunt pathology and restore renal function during the course of acute nephritis in a murine NTN model and whether this was primarily mediated by renal or systemic activities of the TIP peptide. Our results indicate that, during the course of NTN, TIP peptide, given either systemically, or targeted to glomeruli by conjugation of the peptide with a human monoclonal antibody against the type IV collagen α3NC1 domain30–33, significantly reduced pathology, diminished leukocyte renal infiltration and improved kidney function, without increasing mean arterial blood pressure. These protective activities were blunted upon co-treating mice with the cyclooxygenase inhibitor indomethacin, indicating a role for prostaglandins in recovery. We subsequently found that TIP peptide reduced TNF-mediated activation of the pro-inflammatory p38 MAP kinase and NF-κB pathways in GEC. Consistent with the results obtained with indomethacin, TIP peptide increased the generation of PGE2 and eNOS-mediated NO in hTNF-treated GEC, two mediators shown to reduce pathology in NTN32,34.
Taken together, these results support the therapeutic potential of the TIP peptide in NTN, and they indicate that this effect is at least in part mediated through increased PGE2 generation in GEC. They also provide the potential to delivery TIP peptide to glomeruli during established disease to restore pathology and function.
RESULTS
TIP peptide reduces clinical features of nephritis in NTN.
As shown in Fig. 1, NTN induced by injection of 13.5 μg/g NTS increases BUN levels and proteinuria. TIP peptide17,20, but not mutant TIP peptide (sequence: CGQREAPAGAAAKPWYC), which has lost ENaC-α20 binding activity (both at 2.5 mg/kg), significantly reduced BUN levels, proteinuria and body weight, when applied ip on day 2 post NTS (Fig. 1A,B,C). In a previous study, using the same preparation and dose of NTS, we have shown that both proteinuria and BUN levels were already significantly elevated at day 2, as compared to controls. As such, pathology was already established when TIP peptide treatment was initiated32.
Figure 1 |. TIP peptide restores renal function and pathology during nephrotoxic nephritis.

(a) Body weight gain (g), (b) blood urea nitrogen (BUN) levels (mg/dl), and (c) urinary albumin (mg/d) on day 7 in control, nephrotoxic serum-induced nephritis (NTN) (13.5 μl/g nephrotoxic serum [NTS]), TIP+NTN, and mutant TIP+NTN mice (peptides were injected i.p. on days 2, 4, and 6 of NTN at 2.5 mg/kg); n = 5 per group, *P < 0.05 versus ctrl; #P < 0.05 versus NTN. (d) Representative images of synaptopodin expression in isolated glomeruli from control, NTN, and TIP+NTN mice (scale bar: 10 μm). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Moreover, TIP peptide treatment restored expression of the actin-binding protein synaptopodin35 in podocytes (Fig. 1D). In control mice a fine linear staining with intervals between the lines can be observed, corresponding to healthy foot processes. In NTN mice the linear pattern is more diffuse, presumably reflecting foot process effacement. In glomeruli from NTS/TIP mice the normal linear pattern is restored. As such, this indicates that TIP peptide restored expression of synaptopodin, consistent with its anti-proteinuric effect.
There was an accompanying reduction in both glomerular and tubulo-interstitial inflammation (assessed as glomerular and tubulo-interstitial injury score36,37), associated with TIP peptide therapy (Fig. 2A,B,C). As also shown by others38 the cyclooxygenase inhibitor, indomethacin, given in drinking water (approximately 2.2 mg/kg/day), aggravated NTN (Fig. 3A,B), Furthermore, indomethacin blocked the protective actions of TIP peptide (Fig. 3A,B). This finding is consistent with the anti-proteinuric effects of PGE2 in NTN32 and suggests a potential link between actions of the peptide and PGE2 generation.
Figure 2 |. TIP peptide reduces kidney injury score in nephrotoxic serum-induced nephritis (NTN) mice.

(a) Light microscopy (hematoxylin and eosin [H&E]) images were investigated on day 7 (scale bar: 50 μm). NTN mice displayed proliferative glomerulonephritis with interstitial nephritis and proteinuria. Limited signs of nephritis are evident in TIP peptide-treated mice. Quantification of H&E images done by scoring of (b) glomerular and (c) tubulointerstitial damage36,37 (n = 4, *P < 0.05 vs. Ctrl; #P < 0.05 vs. nephrotoxic serum [NTS]). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Figure 3 |. Indomethacin blunts the protective effect of TIP peptide in nephrotoxic serum-induced nephritis (NTN).

Effect of indomethacin, applied in the drinking water (2.2 mg/kg per day) from day 0 on, on (a) body weight gain and (b) blood urea nitrogen (BUN) levels on day 7 in NTN (13.5 μl/g nephrotoxic serum [NTS]) and NTN+TIP mice. (TIP peptide injected i.p. on days 2, 4, and 6 after NTS [2.5 mg/kg];n = 5 per group;*P < 0.05 vs. ctrl; #P < 0.05 vs. NTN.)
TIP peptide reduces NTS-induced renal inflammation.
Renal macrophage and neutrophil infiltration, assessed by F4–80/CD11b and GR-1 staining, respectively, was significantly increased in NTN and this was significantly blunted in mice treated with TIP peptide (Fig. 4A–D). F4/80 is an exclusive macrophage marker, while CD11b, apart of macrophages, also stains other innate immune cells, such as monocytes, neutrophils and macrophages and is important for adhesion of those cells to activated endothelium. We observed in this study an increase in leukocytes in the whole kidney and, to lesser extent, in glomeruli in the NTN group. NTN mice have significantly increased plasma levels of the pro-inflammatory cytokines TNF, IL-1 β and IL-6, and chemokines KC (neutrophil attractant) and MCP-1 (macrophage attractant) and of the anti-inflammatory cytokine IL-10 at 7 days after injection of NTS. After treatment with TIP peptide (but not with mutant TIP peptide20), pro-inflammatory cytokines were significantly reduced (Table 1). TIP peptide did not significantly affect the basal cytokine and chemokine profiles in control animals (data not shown).
Figure 4 |. TIP peptide blunts renal leukocyte infiltration in nephrotoxic serum-induced nephritis (NTN).

Assessment of leukocyte infiltration in kidney slices. (a) Representative immunohistochemistry images of CD11b+, F4/80+, and GR-1+ cells in frozen kidney slices from either control, NTN (13.5 μl/g nephrotoxic serum [NTS]), or TIP+NTN mice on day 7. (b-d) Quantification of the number of CD11b-, F4/80-, and GR1-positive cells per 20× field (4 fields were checked per condition). *P < 0.05 versus ctrl; #P < 0.05 versus NTN. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Table 1.
Cytokine and chemokine concentrations in plasma at day 7 post NTS (pg/ml).
| TNF | IL-1beta | IL-6 | IL-10 | MCP-1 | KC | |
|---|---|---|---|---|---|---|
| ctrl | 3.4±3.4 | 3.2±4.3 | 1.2±1.8 | 7.8±6.4 | 56.1±9.8 | 111.1±53.6 |
| NTS | 15.7±8.2* | 14.8±8.6* | 17.4±10.6* | 61.0±13.4* | 117±13.4* | 336.2±65.1* |
| TIP/NTS | 9.1 ±5 | 4.1±2.9# | 5.3±3# | 14.3±13.3# | 66.9±20.1# | 197.6±53.7*# |
| mutTIP/NTS | 19.9±1.2* | 24.0±3.7* | 24.5±7.6* | 63.5±12.7* | 82.8±2.8* | 355.5±67.1* |
p<0.05 vs. ctrl;
p<0.05 vs. NTS
NTN mice had significantly decreased levels of soluble receptors for IL-6 and IL-1, whereas soluble TNF receptor 1 was increased (but not soluble TNF receptor 2). TIP peptide treatment significantly increased plasma levels of IL-1 and IL-6 soluble receptors during NTN, whereas it did not affect levels of soluble TNF receptors 1 or 2 (Table 2). Levels of the glycoprotein gp-130, crucial for IL-6 signaling39,40 did not change significantly.
Table 2.
Soluble Cytokine receptor concentrations in plasma at day 7 post NTS (pg/ml).
| sol IL-1R | sol IL-6R | sol gp130 | sol TNF-R1 | sol TNF-R2 | |
|---|---|---|---|---|---|
| ctrl | 1123±630 | 9170±2478 | 1687±434 | 2616±547 | 7393±1323 |
| NTS | 344.4±33* | 6120±1169* | 1029±589 | 7548.3±1382* | 8448±1967 |
| TIP/NTS | 757.3±246# | 9843±1863# | 1268±623 | 6997.5±2237* | 9546±3927 |
p<0.05 vs. ctrl;
p<0.05 vs. NTS
Glomerular delivery of TIP peptide protects from NTN.
To investigate whether there were direct effects of TIP peptide in glomeruli during NTN, we employed a glomerular delivery strategy shown to be effective with other drugs33. TIP peptide was chemically linked to the human mAb, F1.1, directed against α3(IV) collagen30,31. The conjugate was administered to mice on day 2 and day 4 of NTN (10 mg/kg body weight). Mice were either treated with a moderate (13.5 μl/g) or a high dose (14.5 μl/g) of NTS, and they subsequently received native or F1.1-mAb-complexed TIP peptide every other day, starting on day 2 of NTS-induced nephritis.
As shown by histology (Fig 5A), mice who received a moderate NTS dose developed glomerulonephritis and proteinuria, whereas these parameters were significantly reduced in NTS animals treated with F1.1 mAb-bound TIP peptide. Proteinaceous material is indicated by stars and glomerular hypercellularity and diminution of capillary lumina is indicated by arrows (scale bar: 50 μm). In moderate NTN, the F1.1-TIP complex significantly reduced kidney injury, to the same extent as native TIP peptide (glomerular injury and tubulo-interstitial injury scores36,37 shown in Fig. 5B,C).
Figure 5 |. Glomerular delivery of TIP peptide reduces nephritis and mortality in nephrotoxic serum-induced nephritis (NTN).

(a) Representative histology of kidney sections of control, NTN (13.5 μl/g nephrotoxic serum [NTS]), NTN+native TIP peptide, and NTN+F1.1 mAb-conjugated TIP peptide. TIP peptide (2.5 mg/kg) and F1.1-TIP conjugate (10 mg/kg) were injected i.p. on days 2, 4, and 6 after NTS. (b) Quantification of glomerular injury score (n = 4 per group). (c) Quantification of tubulointerstitial injury score (n = 4 per group), (d) Survival curve of control, high-dose NTN (14.5 μl/g NTS), and high-dose NTN+F1.1 mAb-TIP mice (n = 5 per group). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
NTN mice had significantly elevated levels of the pro-inflammatory cytokines TNF, IL-1 β and IL-6, and chemokines KC (neutrophil attractant) and MCP-1 (macrophage attractant) and of the anti-inflammatory cytokine IL-10 in kidney homogenates (assessed in pg per mg of protein) at 7 days after injection of NTS. Treatment with the F1.1-TIP peptide complex significantly reduced the cytokines TNF, IL-1 β, IL-6, IL-10 and the chemokines KC and MCP-1 (Table 3).
Table 3.
Cytokine and chemokine concentrations in kidney homogenates at day 7 post NTS (pg per mg protein).
| TNF | IL-1beta | IL-6 | IL-10 | MCP-1 | KC | |
|---|---|---|---|---|---|---|
| ctrl | 13.2±0.7 | 95.0±2.0 | 48.3±17.2 | 55.3±10.9 | 95.3±21.7 | 150.6±35.4 |
| NTS | 16.7+2.2* | 126.5±11.3* | 80.8±9.2* | 98.9±6.6* | 140.1±18.1* | 588.8±94.6* |
| F1.1-TIP/NTS | 13.1+1.2# | 64.6±24.8# | 48.2±7.6# | 56.7±25.6# | 106.4±5.7# | 124.4±19.9# |
p<0.05 vs. ctrl;
p<0.05 vs. NTS
High dose NTS caused mortality in 100% of mice on day 6, whereas 80% of mice treated with the F1.1-TIP peptide complex survived (n=5) (Fig 5C). NTS mice treated with unconjugated F1.1 had the same severity of nephritis as NTS mice, indicating that antibody alone was not responsible for the reduction in disease (data not shown and33).
TIP peptide induces ENaC activation in glomerular endothelial cells.
Since our results above indicated that TIP peptide, which binds to ENaC-α20,22, reduced local glomerular inflammation in NTN, we asked whether there was an ENaC-mediated effect in intrinsic GEC. As shown in Fig. 6A, both human and mouse GEC express all three ENaC subunits: α, β and γ. Bands around 95–100kD represent uncleaved glycosylated subunits and bands around 65–70kD represent cleaved matured subunits. In single channel recordings, we detected channels with conductances typical for ENaC (4–5 pico-Siemens) in 22 out of 38 hGEC (data not shown). As shown in a representative single channel recording (Fig. 6B,C), hGEC express ENaC channels and their open probability (Po) can be significantly increased by TIP peptide (50 μg/ml; before TIP (mean ± SEM): Po=0.117±0.0267, after TIP: Po=0.222±0.0313, n=8 per group, *:p≤0.003 versus untreated).
Figure 6 |. Human and murine glomerular endothelial cells (GEC) express functional epithelial sodium channel (ENaC).

(a) Representative Western blot of ENaC-α, -β, and -γ expression in human and mouse GEC (isolated from female C57BL6 mice, Taconic). Bands around 95–100 kD represent uncleaved glycosylated subunits and bands around 65–70 kD represent cleaved matured subunits. (b) Representative current trace of a single-channel patch clamp recording demonstrating that TIP peptide increases the open probability Po of ENaC in hGEC. (c) Quantification of Po before and after TIP peptide treatment in hGEC (n = 8). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
TIP peptide does not increase mean arterial pressure in mice on standard or high salt diet.
Since TIP peptide activates ENaC by binding to its α subunit and since ENaC-mediated Na+ reabsorption in the collecting duct can be involved in the development of hypertension41, we investigated whether it can alter mean arterial pressure (MAP). TIP peptide did not increase MAP in normal mice on standard chow (0.4% sodium, Harlan-Teklad) (Fig. 7A). When administered 24h after NTS administration, TIP peptide did not further increase MAP under these conditions (Fig 7A).
Figure 7 |. TIP peptide does not increase mean arterial pressure (MAP) in nephrotoxic serum-induced nephritis (NTN) in mice under standard chow or high salt diet.

(a) MAP in TIP-treated, NTN and NTN+TIP treated mice, as measured in radio-telemetry during day (D) and night (N) cycles over a total of 10 days (n = 4 per group). (b-d) Effect of TIP peptide on MAP in mice that were under standard chow diet for 3 days, and which were then switched for 10 days to high salt diet (n = 3 per group). (b) TIP peptide (2.5 mg/kg) was injected on days 10, 12, 14, and 16 of high salt diet. Mice were euthanized on day 19. (c) Nephrotoxic serum (NtS) (13.5 μl/g) was injected i.p. on day 10 of high salt diet, followed by vehicle (0.9% saline, i.p.) (c) or TIP peptide (2.5 mg/kg, i.p.) (d) on days 1,3,5, and 7 after NTS injection (red cross indicates death).
We moreover investigated whether the TIP peptide, in mice on high salt diet (4% sodium, Harlan-Teklad), would affect hypertension. As shown in Fig. 7B, TIP peptide (2.5 mg/kg) did not further increase MAP when given on days 10, 12, 14 and 16 of high salt diet. Injection of NTS (13.5 μl/g) at day 10 of high salt diet induced a significant increase in MAP (Fig. 7C) and all three mice succumbed on day 4 and 6 post NTS. By contrast, NTN mice treated with TIP peptide on days 1, 3, 5 and 7 post NTS did not show an increased MAP (Fig. 7D), and whereas one mouse died on day 8 post NTS, two mice survived up to 14 days after NTS, when they were euthanized in order to recuperate transmitters. Taken together, these data demonstrate that TIP peptide does not increase MAP in mice on regular chow or on high salt diet. In NTN mice on high salt diet, the peptide rather blunts the NTS-induced increase in MAP.
TIP peptide reduces TNF-induced p38 and NF-κB activation and restores PGE2 and NO generation in human GEC.
TNF actions on renal intrinsic cells play a crucial role in nephritis, since they mediate recruitment of leukocytes, proteinuria and renal failure. We assessed whether TIP peptide counteracted TNF actions in GEC. The pro-inflammatory p38 MAP kinase pathway in GEC was shown to be crucially involved in MCP-1-dependent renal monocyte infiltration7,8, so we investigated whether TIP peptide blunted human TNF-induced p38 MAP kinase, assessed as the ratio of phosphorylated over total p38 ratio in human GEC. As shown in Fig. 8A,B pretreatment of hGEC with TIP peptide (50 μg/ml) blunted hTNF (1 ng/ml)-induced p38 MAP kinase activation. Apart from blunting TNF-induced p38 activation, TIP peptide also partially inhibited hTNF-induced NF-κB activation in hGEC (Fig. 8C,D).
Figure 8 |. TIP peptide blunts tumor necrosis factor (TNF)-mediated p38 and nuclear factor κB (NF-κB) activation.

Representative Western blot and quantification (duplicates in 2 independent experiments) of phospho- and total p38 (a,b) and NF-κB (c,d) in ctrl, human tumor necrosis factor (hTNF)-treated (30 minutes, 1 ng/ml), or TIP/hTNF-treated human glomerular endothelial cells. *P < 0.05 versus ctrl; #P < 0.05 versus hTNF. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Since we have previously shown that glomerular-targeted PGE2 ameliorates nephritis32,33, we investigated a role for PGE2 in TIP-peptide-mediated restoration. PGE2 generation was measured in supernatants from hTNF-treated hGEC, in the presence or absence of TIP peptide. hTNF significantly decreased PGE2 generation within 30 min. (Fig. 9A). This decrease was partially blunted by pretreating the cells with either TIP peptide (50 μg/ml) or with the p38 MAP kinase inhibitor, SB203580 (10 μM). Depletion of ENaC-α by specific siRNA (Fig. 9B) eliminated TIP-induced increase in PGE2 generation and the decrease in p38 MAP kinase activity in hTNF-treated cells (transfection with scrambled siRNA had no effect) (Fig. 9C,D). Taken together, these results show that TIP peptide in an ENaC-α-dependent manner restores PGE2 generation and reduces p38 activity in hTNF-treated hGEC.
Figure 9|. TIP peptide restores prostaglandin E2 (PGE2) generation in human tumor necrosis factor (hTNF)-treated glomerular endothelial cells (GEC).

(a) PGE2 generation (pg/ml) in supernatants from human GEC left untreated (Ctrl), or treated for 30 minutes with hTNF (1 ng/ml), in the presence or absence of TIP peptide (50 μg/ml) or p38 inhibitor SB203580 (10 μM) (n = 4). (b) Representative Western blot of epithelial sodium channel-α (ENaC-α) depletion efficacy. (c) PGE2 generation (n = 5) and (d) p38 activation (n = 4) in hTNF-treated GEC transfected with scrambled or ENaC-α-specific small, interfering RNA (siRNA) in the presence or absence of TIP peptide. *P < 0.05 versus respective ctrl;#P < 0.05 versus hTNF. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Since endothelial nitric oxide synthase (eNOS) reduces proteinuria and renal injury in murine models of glomerular injury, at least partially by preserving endothelial integrity34,42–44, and since PGE2 activates eNOS in endothelial cells45, we investigated whether TIP peptide affects NO generation in hTNF-treated GEC. As shown in Fig. 10, hTNF significantly reduced NO generation in hGEC, and this was restored by TIP peptide. These results indicate that TIP peptide protects from TNF-induced glomerular injury in NTN, in large part by inducing the generation of mediators in GEC that are protective, including PGE2 and NO. These protective effects of TIP peptide in TNF-treated GEC are at least partially dependent on the expression of ENaC-α.
Figure 10 |. TIP peptide restores NO generation in human tumor necrosis factor (hTNF)-treated human glomerular endothelial cells (hGEC).

Nitric oxide generation in supernatants from ctrl, hTNF-treated (1 ng/ml, 30 minutes), TIP-treated, or TIP/hTNF-treated hGEC (n = 5 per group). *P < 0.05 versus ctrl;#P < 0.05 versus hTNF.
TIP peptide blunts T cell infiltration in the autologous phase of NTN
In the autologous, late, phase of NTN, mice produce anti-sheep antibodies, which bind to glomerular bound sheep IgG to perpetuate disease46. TH17 cells, which generate IL-17A, were proposed to play a crucial role in pathology in this stage of the disease47. As shown in Fig. 11B, injection of a low dose of NTS (8 μl/g) induced a small, but significant, increase in BUN at three weeks post NTS, as compared to control mice. Treatment of the mice with TIP peptide (2.5 mg/kg) starting at day 2 post NTS and continuing for every other day thereafter, significantly blunted the increase in BUN (Fig. 11B). As shown in Fig. 11A,C NTS treatment induced a significant renal infiltration of CD3+ T cells, which was abrogated upon treatment of the animals with TIP peptide. Moreover, as shown in Fig 11A,D NTS injection also caused a significant infiltration of IL-17A generating cells, consistent with the TH17 cell population and this was blunted by the TIP peptide.
Figure 11 |. TIP peptide blunts renal T-cell infiltration during the autologous phase of nephrotoxic serum-induced nephritis (NTN).

(a) Representative immunohistochemistry for CD3 expression (left panel) and immunofluorescence for IL-17A generation with 4′,6-diamidino-2-phenylindole (DAPI) counterstaining (right panel). (b) Blood urea nitrogen (BUN) levels in control, NTN (8 μl/g, 3 weeks)and TIP+NTN mice. (c,d) Quantification of infiltrating CD3+ and IL-17A positive T cells in the kidney. *P < 0.05 versus ctrl;#P < 0.05 versus nephrotoxic serum (NTS). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
DISCUSSION.
TNF inhibition is an attractive target for treatment of glomerulonephritis, because of the crucial role of this pro-inflammatory cytokine in renal inflammation and pathology48. A difficulty with this approach is reduction in host defense after inhibition of TNF receptor signaling with neutralizing antibodies or soluble TNF receptor constructs11–15.
The goal of this study was to evaluate the anti-inflammatory action and mechanism of a peptide mimicking the lectin-like domain of TNF17, which is not involved in the cytokine’s anti-bacterial activity24, in a murine model of GN, induced by injection of NTS. The TIP peptide specifically binds to the α subunit of ENaC, but not to the β and γ subunits20,22. Since depletion of ENaC-α abrogated the protective effects of the TIP peptide in barrier function in toxin-treated human lung MVEC25 as well as in TNF-induced p38 activation and PGE2 generation in glomerular endothelial cells (this study), we hypothesize that ENaC- α is the specific receptor for the peptide, although the existence of alternative receptors cannot be fully excluded.
Our focus was reversal of established disease to establish clinical utility of the peptide. The reduction of glomerular inflammation during NTN by TIP peptide was characterized by reduced plasma and local kidney levels of the pro-inflammatory cytokines IL-1 β and IL-6 and by increased plasma levels of soluble receptors for IL-1 and IL-6. Moreover, levels of the macrophage and neutrophil-attracting chemokines MCP-1 and KC were reduced. The number of infiltrating macrophages and neutrophils in kidneys during NTN was also reduced. Functionally, BUN levels, proteinuria and body weight gain in NTN were all blunted by TIP peptide, but not by the mutant TIP peptide.
Targeted glomerular delivery of TIP peptide provided as much protection as systemically delivered native peptide. This strongly suggests that the protective effects of peptide in NTN were mainly due to local renal effects. Noteworthy, based on mass spectrometry, the F1.1 mAb-TIP peptide complex injected into the NTN mice contained about 5 times less peptide, compared to free form (data not shown). Together, our results show that TNF-derived TIP peptide ameliorates immune-mediated nephritis, mainly due to local effects within glomeruli.
To further substantiate that there was a direct renal cell component to the protective action of TIP peptide in NTN, we focused on its interference with TNF-mediated inflammation, particularly p38 MAP kinase activation, in GEC. The rationale was that p38 is a crucial mediator of TNF-mediated glomerular endothelial dysfunction in NTN7,8 and that the receptor for TIP peptide - the α subunit of the epithelial sodium channel (ENaC) - is expressed in GEC. TIP-peptide blunted hTNF-induced activation of p38 MAP kinase and, to a lesser extent, the NF-κB pathway in hGEC. ENaC activation by TIP peptide is consistent with increased Na+ entry into endothelial cells. Sodium entry has been shown to reduce p38 MAP kinase phosphorylation49. Moreover, mice with reduced expression of the β subunit of ENaC, and thus with reduced ENaC activity, had increased renal inflammation50. We observed that TIP peptide increased generation of PGE2 in hTNF-treated GEC, and the same effect occurred when p38 activity was inhibited. Together, these findings suggest that TIP peptide activates ENaC, increases PGE2 generation in hGEC, at least partially, by reducing hTNF-induced p38 MAP kinase activation. ENaC-induced PGE2 generation has previously been shown in endometrial epithelial cells51. Our observation that indomethacin, an inhibitor of cyclooxygenases and PGE2 generation, reduces the protective effect of TIP peptide in NTN, also indicates an important role for PGE2-induction in the protective effect of TIP peptide in vivo. Indomethacin was also shown to inhibit generation of glomerular barrier-protective endothelial nitric oxide in gn34,42,43,44,52,, but separation of hemodynamic from cellular effects were not addressed in these studies. However, PGE2 activated eNOS in endothelial cells45, thereby providing a plausible explanation for the inhibitory effect of indomethacin on eNOS activity. Moreover, p38 MAP kinase negatively affects eNOS function, since it increases the activity of arginase, which competes with eNOS for the same substrate L-arginine53.
Besides GEC, TIP peptide also protects podocytes, since it preserves synaptopodin expression in NTN, but this effect may be indirect, since podocytes have not been reported to express ENaC. A potential protective mediator in podocytes could be PGE2, which preserves synaptopodin expression in NTN32 and the generation of which can be stimulated by TIP peptide in neighboring GEC.
TIP peptide’s receptor, ENaC-α, is mainly expressed in kidney epithelial and endothelial cells, but not in infiltrating leukocytes. This may be an advantage of the peptide over classical inhibitors of p38 MAP kinase signaling, which exert significant side effects from off-target effects in the CNS and the immune system54,55.
A potential concern is that ENaC activation in the distal nephron can contribute to hypertension41, but in our study TIP peptide, which activates ENaC20,22, does not increase mean arterial pressure in mice on standard chow or on high salt diet and does not further increase MAP in NTN mice. This suggests that the active peptide does not reach the distal tubules in the inner medullary collecting ducts or that the distal nephron regulatory capacity is sufficient to maintain close to normal ENaC activity. We hypothesize that TIP peptide is either captured by Tamm-Horsfall protein in the loops of Henle16 or proteolytically degraded before it can reach the distal nephron. That TIP peptide reduced inflammation without affecting blood pressure in NTN under standard chow conditions furthermore indicates that it mainly exerts local renal, rather than systemic effects. However, TIP treatment blunted the NTS-induced increase in MAP under high salt conditions, which may be a consequence of the peptide’s inhibitory actions on inflammatory processes, suggested to be increased in hypertension56,57.
We show in this study profound protective activity of the ENaC activator, TIP peptide, in NTN, increasing PGE2 and NO generation in GEC, during inflammation. These results appear to contrast with results of others during physiologic states. Indeed, aldosterone-induced, indirect ENaC activation in vascular endothelial cells causes reduced NO generation and increased stiffening, two hallmarks of endothelial dysfunction58. However, others have shown that in mesenteric arteries ENaC mediates vasodilation and increases NO generation59 and that endothelial ENaC-α knock out mice have decreased vasodilatory capacity26. We have previously shown that TIP peptide increases rather than decreases NO generation in pulmonary microvascular endothelial cells in response to bacterial toxins19. These findings suggest a context-dependent role of ENaC in endothelial function whereby the effects differ during inflammatory and non-inflammatory states.
The TIP peptide not only blunted pathology in the early acute phase of NTN, but also in the later autologous phase, possibly by blunting TH17 cell infiltration. Future studies will investigate whether similar mechanisms are involved in both actions of the peptide.
In conclusion, as summarized in the graphical abstract, our results demonstrate a potent anti-inflammatory, protective, and regenerative activity of the TNF-derived TIP peptide in NTN. This effect requires expression of ENaC-α subunit in GEC and is at least partially mediated by the local stimulation of glomerular PGE2 and possibly NO. These results can foster the development of novel reagents and strategies to target glomeruli and to limit/reverse inflammation during glomerulonephritis.
CONCISE METHODS (detailed methods are in supplemental file)
Single-channel patch-clamp studies.
Experiments were performed at room temperature using the cell-attached patch configuration20,22. Patch pipette and extracellular bath solutions consisted of solutions containing (in mM): 95 NaCl, 3.4 KCl, 0.8 CaCl2, 0.8 MgCl2, and 10 HEPES or 10 Tris, adjusted to pH 7.3–7.4. TIP peptide (50 μg/ml) was added to the hGEC cells cultured on permeable polyester filters (Millipore, Billerica, MA). Open probability (Po) of a single channel was calculated by dividing NPo by the number of channels in a patch. For our experiments, we determined Po for 10 min before TIP peptide and for 20 min after addition.
F1.1-TIP peptide conjugation.
F1.1 human monoclonal antibody, with specificity for Ea and Eb epitopes of α3(IV) collagen, was purified and employed as described33. F1.1-TIP peptide conjugates were prepared by a two-step conjugation procedure, as described33. Precise composition of conjugates was determined by HPLC, using size exclusion columns and by amino acid analysis to calculate number of peptide molecules linked per Ab.
Statistical analysis.
Statistical analysis of experiments was with SigmaPlot 11 (Systat Software, San Jose, CA). Differences between two individual experimental groups were compared using the 2-tailed Student’s t-test. Comparisons of multiple groups were done using the Kruskal-Wallis test, followed by pair-wise Mann-Whitney U tests. Survival curves were compared by log-rank test. Data are means + SD; p<0.05 was considered significant.
Supplementary Material
TRANSLATIONAL STATEMENT.
The pro-inflammatory cytokine TNF is a crucial mediator of glomerulonephritis, but the cytokine is also important in anti-bacterial defense. As such, chronically inhibiting TNF signaling using soluble receptor constructs or neutralizing anti-TNF antibodies can promote infection. We propose an alternative manner of inhibiting deleterious TNF signaling without interfering with the beneficial actions of the cytokine. We demonstrate that the TNF-derived TIP peptide, mimicking the lectin-like domain of the molecule, which does not affect the cytokine’s anti-bacterial effects, significantly blunts renal inflammation and proteinuria in murine nephrotoxic nephritis. This protective activity of the TIP peptide occurs mainly through direct interactions with the α subunit of the epithelial sodium channel, expressed in glomerular endothelial cells. These data can foster the development of novel therapeutic agents for the treatment of glomerulonephritis that mainly affect resident glomerular cells.
Acknowledgments.
This work was supported by grants R01 DK100564 (MPM, RL), DK37963 (DCE), DK099548 (POC) and DK114328 (JKC) from the NIH/NIDDK, as well as grants R01 HL138410 (RL), HL128207 (MB) and HL129843 (MC) and P01 HL134604 (MB, POC) from the NIH/NHLBI and grants 17GRNT33410653 from the AHA (MJR), Innovative Grant 1-INO-2017–459-A-N from the JDRF (MJR) and grant #1–16-IBS-196 from the ADA (RL). Part of these results were published as an abstract for International Kidney Week 2016.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE. The authors have no interests to disclose.
REFERENCES.
- 1.Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest. 2014;124(6):2355–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. J Am Soc Nephrol. 2012;23(3):381–399. [DOI] [PubMed] [Google Scholar]
- 3.Timoshanko JR, Sedgwick JD, Holdsworth SR, et al. Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J Am Soc Nephrol. 2003;14(7):1785–1793. [DOI] [PubMed] [Google Scholar]
- 4.Hruby ZW, Shirota K, Jothy S, et al. Antiserum against tumor necrosis factor-alpha and a protease inhibitor reduce immune glomerular injury. Kidney Int. 1991;40(1):43–51. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y, Xie C, Chen J, et al. Innate stimuli accentuate end-organ damage by nephrotoxic antibodies via Fc receptor and TLR stimulation and IL-1/TNF-alpha production. J Immunol. 2006;176(1):632–639. [DOI] [PubMed] [Google Scholar]
- 6.Stambe C, Atkins RC, Hill PA, et al. Activation and cellular localization of the p38 and JNK MAPK pathways in rat crescentic glomerulonephritis. Kidney Int. 2003;64(6):2121–2132. [DOI] [PubMed] [Google Scholar]
- 7.Stambe C, Atkins RC, Tesch GH, et al. Blockade of p38-α MAPK ameliorates acute inflammatory renal injury in rat anti-GBM glomerulonephritis. J Am Soc Nephrol. 2003; 14(2):338–351. [DOI] [PubMed] [Google Scholar]
- 8.Sheryanna A, Bhangal G, McDaid J, et al. Inhibition of p38 mitogen-activated protein kinase is effective in the treatment of experimental crescentic glomerulonephritis and suppresses monocyte chemoattractant protein-1 but not IL-1beta or IL-6. J Am Soc Nephrol. 2007;18(4):1167–1179. [DOI] [PubMed] [Google Scholar]
- 9.Taubitz A, Schwarz M, Eltrich N, et al. Distinct contributions of TNF receptor 1 and 2 to TNF-induced glomerular inflammation in mice. PLoS One. 2013;8(7):e68167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vielhauer V, Mayadas TN. Functions of TNF and its receptors in renal disease: distinct roles in inflammatory tissue injury and immune regulation. Semin Nephrol. 2007;27(3):286–308. [DOI] [PubMed] [Google Scholar]
- 11.Rothe J, Lesslauer W, Lötscher H, et al. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993;364(6440):798–802. [DOI] [PubMed] [Google Scholar]
- 12.Garcia I, Miyazaki Y, Araki K, et al. Transgenic mice expressing high levels of soluble TNF-R1 fusion protein are protected from lethal septic shock and cerebral malaria, and are highly sensitive to Listeria monocytogenes and Leishmania major infections. Eur J Immunol. 1995;25(8):2401–2407. [DOI] [PubMed] [Google Scholar]
- 13.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995;2(6):561–572. [DOI] [PubMed] [Google Scholar]
- 14.Harris J, Keane J. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol. 2010;161(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holdsworth SR, Gan PY, Kitching AR. Biologics for the treatment of autoimmune renal diseases. Nat Rev Nephrol. 2016;12(4):217–231. [DOI] [PubMed] [Google Scholar]
- 16.Hession C, Decker JM, Sherblom AP, et al. Uromodulin (Tamm-Horsfall glycoprotein): a renal ligand for lymphokines. Science. 1987;237(4821):1479–1484. [DOI] [PubMed] [Google Scholar]
- 17.Lucas R, Magez S, De Leys R, et al. Mapping the lectin-like activity of tumor necrosis factor. Science. 1994;263(5148):814–817. [DOI] [PubMed] [Google Scholar]
- 18.Braun C, Hamacher J, Morel D, et al. Dichotomal Role of TNF in Experimental Pulmonary Edema Reabsorption. J. Immunol 2005;175(5):3402–3408. [DOI] [PubMed] [Google Scholar]
- 19.Lucas R, Yang G, Gorshkov BA, et al. Protein kinase C-a and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol 2012;47:445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Czikora I, Alli A, Bao HF, et al. A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am J Respir Crit Care Med. 2014; 190(5):522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sieck GC, Wylam ME. Paradoxical use of tumor necrosis factor in treating pulmonary edema. Editorial. Am J Respir Crit Care Med. 2014;190(6):595–596. [DOI] [PubMed] [Google Scholar]
- 22.Lucas R, Yue Q, Alli A, et al. The lectin-like domain of TNF increases ENaC open probability through a novel site at the interface between the second transmembrane and C-terminal domains of the α-subunit. J. Biol. Chem 2016;291 (45):23440–23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Q, Wang D, Liu Y, et al. Solnatide demonstrates profound therapeutic activity in a rat model of high altitude pulmonary edema induced by acute hypobaric hypoxia and exercise. Chest. 2017;151(3):658–667. [DOI] [PubMed] [Google Scholar]
- 24.Lucas R, Echtenacher B, Sablon E, et al. Generation of a mouse tumor necrosis factor mutant with anti-peritonitis and desensitization activities comparable to those of the wild type but with reduced systemic toxicity. Infect Immun. 1997;65(6):2006–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Czikora I, Alli AA, Sridhar S, et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front Immunol. 2017;8:842. doi: 10.3389/fimmu.2017.00842. eCollection 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sternak M, Bar A, Adamski MG, et al. : The Deletion of Endothelial Sodium Channel a (aENaC) Impairs Endothelium-Dependent Vasodilation and Endothelial Barrier Integrity in Endotoxemia in Vivo. Frontiers in Pharmacol. 2018. doi: 10.3389/fphar.2018.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwameis R, Eder S, Pietschmann H, et al. A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J Clin Pharmacol. 2014;54(3):341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krenn K, Lucas R, Croizé A, et al. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: a phase IIa randomized placebo-controlled trial. Crit Care. 2017;21 (1):194. doi: 10.1186/s13054-017-1795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ware LB. Targeting resolution of pulmonary edema in primary graft dysfunction after lung transplantation: Is inhaled AP301 the answer? J Heart Lung Transplant. 2017;S1053–2498(17)32114–32119. [DOI] [PubMed] [Google Scholar]
- 30.Meyers KE, Christensen M, Madaio MP. Modeling of human anti-GBM antibody-alpha3(IV)NC1 interactions predicts antigenic cross-linking through contact of both heavy chains with repeating epitopes on alpha3(IV)NC1. Am J Nephrol. 2009;30(5):474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaudhary K, Kleven DT, McGaha TL, et al. A human monoclonal antibody against the collagen type IV α3NC1 domain is a non-invasive optical biomarker for glomerular diseases. Kidney Int. 2013;84(2):403–408. [DOI] [PubMed] [Google Scholar]
- 32.Kvirkvelia N, McMenamin M, Chaudhary K, et al. Prostaglandin E2 promotes cellular recovery from established nephrotoxic serum nephritis in mice, prosurvival, and regenerative effects on glomerular cells. Am J Physiol Renal Physiol. 2013;304(5):F463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kvirkvelia N, McMenamin M, Gutierrez VI, et al. Human anti-α3(IV)NC1 antibody drug conjugates target glomeruli to resolve nephritis. Am J Physiol Renal Physiol. 2015;309(8):F680–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilkeson GS, Mashmoushi AK, Ruiz P, et al. Endothelial nitric oxide synthase reduces crescentic and necrotic glomerular lesions, reactive oxygen production, and MCP1 production in murine lupus nephritis. PLoS One. 2013;8(5):e64650. doi: 10.1371/journal.pone.0064650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He FF, Chen S, Su H, et al. Actin-associated Proteins in the Pathogenesis of Podocyte Injury. Curr Genomics. 2013;14(7):477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan O, Madaio MP, Shlomchik MJ. The roles of B cells in MRL/lpr murine lupus. Ann N Y Acad Sci. 1997;815:75–87. [DOI] [PubMed] [Google Scholar]
- 37.Madaio MP, Ahima R, Meade R, et al. Glomerular and tubular epithelial defects in kd/kd mice lead to progressive renal failure. Am J. Kid Dis 2005;6: 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagamatsu T, Pippin J, Schreiner GF, et al. Paradoxical exacerbation of leukocyte-mediated glomerulonephritis with cyclooxygenase inhibition. Am J Physiol. 1992;263(2 Pt 2):F228–236. [DOI] [PubMed] [Google Scholar]
- 39.Karkar AM, Tam FW, Steinkasserer A, et al. Modulation of antibody-mediated glomerular injury in vivo by IL-1 ra, soluble IL-1 receptor, and soluble TNF receptor. Kidney Int. 1995;48(6):1738–1746. [DOI] [PubMed] [Google Scholar]
- 40.Hou T, Tieu BC, Ray S, et al. Roles of IL-6-gp130 Signaling in Vascular Inflammation. Curr Cardiol Rev. 2008;4(3):179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlov TS, Levchenko V, O’Connor PM, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24(7):1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trachtman H Nitric oxide and glomerulonephritis. Semin Nephrol. 2004;24(4):324–332, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Garsen M, Rops AL, Li J, et al. Endothelial Nitric Oxide Synthase Prevents Heparanase Induction and the Development of Proteinuria. PLoS One. 2016; 11(8):e0160894. doi: 10.1371/journal.pone.0160894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao HJ, Wang S, Cheng H, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006; 17(10):2664–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hristovska AM, Rasmussen LE, Hansen PB, et al. Prostaglandin E2 induces vascular relaxation by E-prostanoid 4 receptor-mediated activation of endothelial nitric oxide synthase. Hypertension. 2007;50(3):525–530. [DOI] [PubMed] [Google Scholar]
- 46.Unanue ER, Dixon FJ. Experimental Glomerulonephritis. VI. The autologous phase of nephrotoxic serum nephritis. J Exp Med. 1965;121:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paust HJ, Turner JE, Steinmetz OM, et al. The IL-23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol. 2009;20(5):969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McAdoo SP, Pusey CD. Is there a role for TNFα blockade in ANCA-associated vasculitis and glomerulonephritis? Nephrol Dial Transplant. 2017;32(suppl_1):i80–i88. doi: 10.1093/ndt/gfw361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YB, Leroy V, Maunsbach AB, et al. Sodium transport is modulated by p38 kinase-dependent cross-talk between ENaC and Na,K-ATPase in collecting duct principal cells. J Am Soc Nephrol. 2014;25(2):250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Drummond HA, Grifoni SC, Abu-Zaid A, et al. Renal inflammation and elevated blood pressure in a mouse model of reduced β-ENaC. Am J Physiol Renal Physiol. 2011; 301 (2): F443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruan YC, Guo JH, Liu X, et al. Activation of the epithelial Na+ channel triggers prostaglandin E2 release and production required for embryo implantation. Nat Med. 2012;18(7):1112–1117. [DOI] [PubMed] [Google Scholar]
- 52.Nagappan AS, Varghese J, Pranesh GT, et al. Indomethacin inhibits activation of endothelial nitric oxide synthase in the rat kidney: Possible role of this effect in the pathogenesis of indomethacin-induced renal damage. Chem Biol Interact. 2014;221C:77–87. [DOI] [PubMed] [Google Scholar]
- 53.Shatanawi A, Romero MJ, Iddings JA, et al. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am J Physiol Cell Physiol. 2011; 300(5):C1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chopra P, Kanoje V, Semwal A, et al. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert Opin Investig Drugs. 2008;17(10):1411–1425. [DOI] [PubMed] [Google Scholar]
- 55.Xiao YT, Yan WH, Cao Y, et al. P38 MAPK Pharmacological Inhibitor SB203580 Alleviates Total Parenteral Nutrition-Induced Loss of Intestinal Barrier Function but Promotes Hepatocyte Lipoapoptosis. Cell Physiol Biochem. 2017;41(2):623–634. [DOI] [PubMed] [Google Scholar]
- 56.Foss JD, Kirabo A, Harrison DG. Do high-salt microenvironments drive hypertensive inflammation? Am J Physiol Regul Integr Comp Physiol. 2017;312(1):R1–R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elmarakby AA, Quigley JE, Imig JD, et al. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;294(1):R76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Warnock DG, Kusche-Vihrog K, Tarjus A, et al. Blood pressure and amiloride-sensitive sodium channels in vascular and renal cells. Nat Rev Nephrol. 2014;10(3):146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashley Z, Mugloo S, McDonald FJ, et al. Epithelial Na+ channel (ENaC) differentially contributes to shear stress-mediated vascular responsiveness in carotid and mesenteric arteries from mice. Am J Physiol Heart Circ Physiol. 2018; 314(5):H1022–H1032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.