

Abstract
The inability of the yeast Saccharomyces cerevisiae to produce ethanol from xylose has hampered the biofuel production from lignocellulosic biomass. However, prior studies reveal that functional expression of xylose isomerase (XI) from Burkholderia cenocepacia (XylABc) in S. cerevisiae has remarkably improved xylose consumption and ethanol productivity. Yet, little is known about kinetic and structural properties of this enzyme. Hereby, a purified recombinant XylA was assayed in vitro, showing optimal enzyme activity at 37 °C and pH 7.2. The Km of XylA for d-xylose was at least threefold lower than the Km results for any XI published to date (e.g. XylA from Piromyces sp.). In addition, oligomerization behavior as a tetramer was observed for XylA in solution. Functional and structural comparative analyses amongst three microbial XIs were further performed as theoretical models, showing that xylose orientation at the active site was highly conserved among the XIs. Mg2+ ions anchor the sugar and guide its pyranoside oxygen towards a histidine residue present at the active site, allowing an acid–base reaction, linearizing xylose. Electrostatic surface analyses showed that small variations in the net charge distribution and dipole moment could directly affect the way the substrate interacts with the protein, thus altering its kinetic properties. Accordingly, in silico modeling suggested the tetramer may be the major functional form. These analyses and the resulting model promote a better understanding of this protein family and pave the way to further protein engineering and application of XylA in the ethanol industry.
Electronic supplementary material
The online version of this article (10.1186/s13568-019-0795-4) contains supplementary material, which is available to authorized users.
Keywords: Xylose isomerase, XylA, Burkholderia cenocepacia, Ethanol, Xylose fermentation, Enzyme kinetics
Introduction
The increasing concern about climate changes and its effects on human health has created a global effort for the development of new technologies based on renewable energy matrixes (Tibbetts 2015; Perera 2016; Ko and Lee 2018). In this scenario, ethanol is the preferred biofuel produced worldwide and has proven to have the lowest impact on current economies based on fossil fuels (Nielsen and Keasling 2016). Furthermore, the growing population creates an increasing demand for energy dedicated to transports, thus raising the pressure for fuel production expansion. However, the ethanol fermentation process based on sugarcane and starch as feedstock is already close to theoretical maximum efficiency (Carlos et al. 2011). Moreover, the expansion of croplands suffers social and political embargo. Then, the use of residual lignocellulosic biomass from sugarcane bagasse has gained great attention. After the pretreatment and hydrolysis steps, it is possible to retrieve large amounts of hexoses and pentoses from cellulose and hemicellulose polymers (Mosier et al. 2005). Xylose is the second most common sugar present on lignocellulosic hydrolysates. However, the inability of the yeast Saccharomyces cerevisiae to produce ethanol from xylose is one of the problems associated with the direct application of this technology (Stambuk et al. 2008).
Xylose metabolism in S. cerevisiae requires the uptake of the sugar by non-specific hexose transporters of the HXT family (Kruckeberg 1996). Then, xylose is reduced to xylitol by xylose reductase (XR; E.C. 1.1.1.307) and xylitol is further oxidized into xylulose by xylitol dehydrogenase (XDH; E.C. 1.1.1.B19). These reactions are NAD(P)H and NAD+-dependent, respectively. D-xylulose has to be phosphorylated into 5P-xylulose by xylulokinase (XK) in order to enter the pentose phosphate pathway (PPP). PPP mainly performs NADP+ reduction and the production of glyceraldehyde-3-phosphate (G3P) and fructose-6P that can be oxidized into pyruvate via glycolysis and further converted into ethanol.
The redox imbalance generated by XR/XDH pathway cannot be overcome due to the lack of transhydrogenase activity in S. cerevisiae, responsible for converting NADH to NAD(P)H, leading to cytosolic NADH accumulation (Bruinenberg et al. 1985). Attempts to clone fungi recombinant enzymes that use the same cofactors for conversion of d-xylose into d-xylulose have failed in producing an efficient strain capable of performing alcoholic fermentation of glucose and xylose simultaneously, without accumulation of by-products (xylitol and glycerol) (Matsushika et al. 2009; Gonc et al. 2014).
On the other hand, xylose Isomerase (XI; E.C. 5.3.1.5) pathway, used by some bacterial species to metabolize xylose (Hahn-Hägerdal et al. 2007), directly converts d-xylose into d-xylulose commonly using divalent metals as cofactors (Asboth and Naray-Szabo 2000; Hahn-Hägerdal et al. 2007). As reviewed in Moysés et al. (2016), the first functional recombinant XI expressed in S. cerevisiae was cloned from Thermus thermophiles and showed low activity at fermentation conditions. Another strain was successfully obtained by Kuyper et al. (2003), where the fungal XI from Piromyces sp. showed high activity but slow growth in xylose. Moreover, the same strategy using xylA from Orpinomyces was tested and likewise, high activities were observed but also had a slow growth in xylose (Madhavan et al. 2009). Nevertheless, XI from anaerobic bacterium Clostridium phytofermentans was expressed in an industrial S. cerevisiae strain with a similar Km compared to Piromyces xylA and approximately three times smaller Ki for xylitol. However, the strain could not efficiently produce ethanol without xylitol accumulation.
Similarly, Vilela et al. (2015) were able to successfully express the recombinant XI from Burkholderia cenocepacia (XylABc) with high activity in S. cerevisiae. After adaptive engineering in xylose as a sole carbon source, the recombinant yeast presented one of the best fermentation results so far (Moysés et al. 2016). Under anaerobic conditions, glucose and xylose were simultaneously consumed while no xylitol was accumulated. Those results show the great potential of this strategy, considering that no further genetic modifications were required, as occurred in other constructions (Cai et al. 2012). For a better understanding of the model proposed for S. cerevisiae expressing XylABc, detailed analyses of enzymatic functionality and structure was still necessary.
In this work, the XylABc was expressed in Escherichia coli and purified. In vitro assays with purified recombinant XylABc exhibited a similar performance to that observed in vivo with yeast strains that were submitted to extensive metabolic and adaptive engineering (Cai et al. 2012). Functional analyses in different pH and temperature conditions were performed. Enzyme activity was measured in optimal conditions and kinetic parameters such as Km, Vmax and Kcat were determined. The conditions used on in vitro assays were used to guide the creation of theoretical models by comparative modeling, in order to evaluate the overall structure, metal binding sites and xylose interaction at the putative active site. Molecular dynamics techniques were used to refine the models in conditions similar to those of enzymatic assays, aiming to determine any difference regarding the active site geometry, surface electrical charges, dipole moment and substrate interaction. The XIs from Burkholderia cenocepacia, Piromyces sp and Clostridium phytofermentans were analyzed, since those three microbial species present some of the best results published to date.
Materials and methods
A synthetic DNA fragment containing the full-length XylA (GenBank accession no: AM747722) which encodes the enzyme Xylose Isomerase from Burkholderia cenocepacia J2315 strain (de Figueiredo et al. 2013) was obtained from Epoch Life Science (TX, USA). The gene was cloned into HindIII and BglII digested pTrcHis-B plasmid (Thermo Scientific, USA) and transformed in E. coli TOP10 [F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZ ΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu) 7697 galU galK rpsL (StrR) endA1 nupGλ] cells. Transformant cells were selected in LB broth with 200 μg/mL ampicillin. Protein expression was performed in 1-liter flasks containing 500 mL of LB broth and inoculated at 0.1 OD600nm starting cell density. Cells were incubated at 37 °C under 160 rpm agitation until OD600nm reached 0.6. Then, the culture was refrigerated to 18 °C and 500 μM IPTG was added to the medium for induction of protein expression for 16 h.
Protein purification
IPTG-induced cells were washed with washing buffer (50 mM sodium phosphate, 300 mm sodium chloride, pH 7) and centrifuged at 2900×g for 30 min at 4 °C. Cell pellet was frozen at − 80 °C. In the day of use, thawed samples were disrupted with 10 sonication cycles of 1 min with 1 min intervals on ice-water bath using a 4 mm titanium micro-point (QR 300, Ultronique). The cell lysate suspension was centrifuged at 9100×g for 30 min at 4 °C and supernatant was collected. A Nickel (Ni2+) resin (Novagen®, Merck, USA) was equilibrated with 30 bed volumes of washing buffer. The cell lysate supernatant was incubated with the nickel resin for 30 min under gentle agitation on ice-cold water bath for His-tagged protein trapping. Then, the resin was added into a disposable Bio-Rad column and washed twice with 10 bed volumes of washing buffer. His-tagged proteins were eluted in a stepwise manner, with elution buffer (50 mM sodium phosphate, 300 mm sodium chloride, pH 7.5) supplemented with 100, 200 or 300 mM imidazole.
Polyacrylamide gel electrophoresis (PAGE), protein concentration and determination
Protein expression and purification were initially analyzed in SDS-PAGE (5% stacking, 12% resolving gel). Sample loading dye contained 100 mM Tris–HCl pH 6.8, 8 M urea, 20% glycerol, 4% SDS, 0.2% bromophenol blue and 100 mM β-mercaptoethanol. Electrophoresis was performed at room temperature at a constant current of 0.06 A. Gel content was evaluated with Coomassie Brilliant Blue R-250 staining (BioRad). Eluted protein samples from the Ni2+ -resin were concentrated with Amicon Ultra 30K (Merck) by centrifuging at 4000×g at 4 °C for 10 min. The concentrated sample was fractionated into the Superdex 200 (GE Healthcare) resin with 1 mL/min flux of buffer (100 mM Tris–HCl, 20 mM NaCl, pH 7.5). The protein elution was followed by absorbance at 280 nm (ÄKTA Prime PLUS, GE Healthcare). Protein concentration after purification was assessed with BCA assay (Smith et al. 1985). The standard curve was made using BSA concentrations varying from 25 to 2000 μg/μL.
The molecular size and oligomerization of native His6-XylA was determined with a non-denaturing gradient PAGE (4–20% Mini-PROTEAN® TGX™ Precast Protein Gel, BIO-RAD). Electrophoresis was performed as described above, with SDS-free sample and running buffers. A mixture of 10 recombinant proteins ranging from 10 a 250 kDa was used as standard (Precision Plus Protein™, BIO-RAD).
Mass spectrometry
Protein bands were extracted from either native or SDS-PAGE and unstained on a solution of 30% methanol (v/v) and 10% acetic acid (v/v). Then, the sliced gel was incubated on a washing solution containing 50% acetonitrile (v/v) and 25 mM ammonium bicarbonate at pH 8.0 for 15 min. Protein samples were reduced on 8 M urea, 10 mM DTT and 25 mM ammonium bicarbonate solution at room temperature for 30 min. Then, the samples were incubated in a dark chamber in a 55 mM iodoacetamide and 25 mM ammonium bicarbonate solution for 30 min at room temperature. Trypsin (Promega) was added to the solution for proteolysis at 37 °C overnight. The digestion reaction was stopped with the addition of 1% formic acid (v/v). The peptides were cleaned using homemade C-18 stage tips (Rappsilber et al. 2007), dried, and resuspended in 0.1% formic acid (v/v) solution.
Peptides were fractionated using a nanoHPLC system Easy-nLC II (Proxeon) system on an in-house packed 2 cm × 150 μm i.d. pre-column (Reprosil-Pur C18-AQ, 5 μm, 120 Å, Dr. Maisch), and 15 cm × 75 μm i.d. analytical column (Reprosil-Pur C18-AQ, 3 μm, 120 Å, Dr. Maisch) coupled to an LTQ Velos Orbitrap (Thermo Fisher Scientific). Chromatography was performed at 300 nL/min. flow rate with 95% water, 5% Acetonitrile (ACN) and 0.1% formic acid (FA) as mobile phase A and 85% ACN, 15% water, and 0.1% FA as phase B. Two technical replicates of each sample were performed in optimized gradient of 55 min. Mass spectra were acquired by Tune and Xcalibur software operating in data-dependent acquisition (DDA) mode, switching between full scan MS1 (60,000 resolution at 200 m/z, 100 ms accumulation time, AGC 1 × 106 ions, range 375–1800 m/z) and MS2 (15,000 resolution at 200 m/z, 200 ms accumulation time, AGC 105 ions, range 100–2000 m/z). MS2 spectra were obtained by HCD fragmentation of the 10 most intense ions using 30 normalized collision energy.
Peptide spectrum match (PSM) search was performed using Comet search engine from PatternLab for Proteomics v4.0 (Carvalho et al. 2016) against forward and reverse protein sequences from E. coli and B cenocepacia, downloaded from UniProtKB on January 31, 2017, and containing 34,214 entries. Carboxamidomethylation of cysteines was set as a fixed modification and oxidation of methionine was set as a variable modification. Semi-tryptic peptides were considered with a maximum of 3 missed cleavages and mass tolerance for MS1 was set at 40 ppm. Resultant peptides were processed and evaluated by Search Engine Processor (SEPro) from using the following filter parameters: 10 ppm deviation from theoretical peptide precursor, peptides longer than six amino acid residues, minimum number of peptides per protein 3, Delta CN of 0.001, and a 1% estimated protein-level FDR.
Enzymatic assays
XI activity was measured according to Brat et al. (2009). A solution containing 195 μg/mL of purified XI was incubated at the desired working temperature until the moment of use. Assays were performed in a quartz cuvette with 1 cm optical path. A reaction mixture containing 0.23 mM NADH, 10 mM MgCl2, 2 U/mL sorbitol dehydrogenase and 29.25 μg of XI in 100 mM Tris–HCl (pH 7.2) was fresh made for a final volume of 1 mL and incubated for 5 min at temperatures varying from 25 to 37 °C. The assay was performed in a quartz cuvette with 1 cm optical path. The reaction started with the addition of d-xylose and then followed for 30 min by measuring the oxidation of NADH at 340 nm until its complete oxidation to NAD+. Variations of d-xylose concentration from 10 to 500 mM were used to determine kinetic parameters. The protein theoretical isoelectric point (pI) was calculated using ProtParam Tool (http://web.expasy.org/protparam/) in order to determine the pH interval for activity measurement. From that, optimal pH was found by varying the buffer pH from 5.8 to 7.6. For pH values between 5.8 and 7.0, it was used a 100 mM Bis–Tris + 70 mM NaCl buffer. The ionic strength of each buffer was calculated for each condition to remain constant at 87 mM. Xylitol inhibition assay was done by measuring enzyme activity in the presence of 125 mM xylose and 10–50 mM xylitol at pH 7.2, 37 °C.
Protein tertiary structure prediction
Templates were chosen over the Protein Databank (PDB) based on their % identities and coverage to sequences using the BLAST tool (NCBI). For homology modeling, a multiple sequence alignment between target and templates were generated using MUSCLE (Edgar 2004) and MODELLER (Martí-Renom et al. 2000; Webb and Sali 2014) salign script. Alignments were manually checked for overall quality and edited when necessary. Modeling of the tertiary structures was performed by MODELLER v9.17 with 500 models generated. In order to choose the best model, Modeller’s objective function, DOPE score and Z-score (normalized DOPE) were used to pick the best models from the pool. Each model quality was assessed with PROCHECK for overall structure geometry, ERRAT for non-bonded interactions statistics and VERIFY 3D for compatibility of the atomic model of tertiary structure with its primary structure (Bowie et al. 1991; Lüthy et al. 1992; Laskowski et al. 1993). Identified anomalies in loop regions were fixed by running MODELLER’s Loop refining protocol. RMSD was calculated with PyMol v1.8.4.
Molecular dynamics and energy minimization
Structural models for B. cenocepacia, C. phytofermentans and Piromyces sp. were positioned to 1a0c, 1a0d, 1s5 m and 1s5n crystallographic structures. Cyclical d-xylose structure was obtained by modifying the glucose (removal of the methyl group and addition of H) from 1s5 m and transferring the coordinates to the constructed models. Water molecules from 1a0c crystal structure were preserved, except for HOH 494, 495, 496, 639 (chain A), 527, 528, 529, 671 (chain B), 525, 526, 527, 670 (chain C), 558, 559, 560, 702 (chain D) that were overlapping xylose atoms at the active site.
Amino acid side chain protonation state was determined according to PROPKA algorithm (Olsson et al. 2011) for pH 7.0. Structures were prepared with AMBER 14 program package (Simmerling 2015). Topology parameters for protein construction were obtained from AMBER 14SB force field. Xylose topology parameters were obtained from GLYCAM 06j-1 force field (Kirschner et al. 2008). The system was involved in an octahedral water box with 15 Å distance from sides and neutralized with Na+ or Cl− when necessary.
The model structures were relaxed before molecular dynamics simulation with the following protocol: (i) structural minimization with position restrictions at 100 kcal/mol for heavy atoms present on the protein and xylose; (ii) structural minimization with position restrictions at 10 kcal/mol for protein backbone and xylose heavy atoms; (iii) whole system minimization with weak positioned restrictions at 1 kcal/mol for the complex heavy atoms. Then, the system temperature was increased from 0 to 100 K with the canonical ensemble (NVT) for 20 ps. Next, it was used the isothermal-isobaric (NPT) ensemble from 100 to 29,815 K for 20 ps. Both were controlled with Berendsen temperature protocol (Berendsen et al. 1984). After the system heating, the system went through 250 ps at 1 kcal/mol for restriction density equilibration. Finally, the system went through another cycle of 250 ps for heavy atoms restriction equilibration at 0.1 kcal/mol. All production cycles were performed by approximately 20 ns.
Electrostatic surface and electric field vector
The file preparation for electric field calculations was done using the online software PDB2PQR (Dolinsky et al. 2004). The protein topology parameters were calculated using PARSE force field. Amino acid side chain protonation state was designated with PROPKA software at the following pH: 7.2 (B. cenocepacia) and 7.5 (C. phytofermentans and Piromyces sp.). Electrostatic field calculation and visualization were done on VMD v1.9.3 with APBS v1.4.1 plugin. Grid boxes were automatically calculated and kept at 298.15 K for C. phytofermentans and Piromyces sp. and 310.15 K for B. cenocepacia.
Statistical analysis
All values obtained during enzymatic assays were analyzed on GraphPad Prism 6 and were considered statistically different on a 95% confidence interval using ANOVA test. All assays were performed as two independent replicates with three technical replicates. Results presented are the mean values of the assay replicates with standard error values.
Results
Purification and oligomerization analysis of XylABc, a XI from Burkholderia cenocepacia
To assess the functional properties of XylABc, the gene was cloned into pTrcHis plasmid under the control of PTrc promoter, generating pTrcHis-XylABc vector. E. coli TOP10 cells were transformed with the pTrcHis-XylABc vector and the induction of protein expression was performed in Lysogen broth supplemented with 0.5 mM IPTG, at 18 °C for 16 h. The induction of XylA at a low temperature provided high yields of soluble protein and was capable of preventing the protein wrong folding and consequent precipitation. Expression of the His6-XylA fusion protein, with a theoretical mass of 53,598.67 Da, was analyzed on SDS-PAGE after cell lysis and sample enrichment on a Ni+2-resin (Additional file 1: Figure S1). The protein presented a band of the expected size (~ 54 kDa) after His-trap resin enrichment. The fractions containing His-tagged XylA were pooled and loaded onto a Superdex 200 resin column for size-exclusion chromatography. The His-tagged XylABc eluted in a major peak, at 70 mL (Fig. 1a), as identified by digestion with trypsin. The resulting peptides were submitted to LC–MS/MS and eight proteins with maximum parsimony were identified. The best-scored protein (12,795.258 Xcorr) with 100% coverage was XylABc (B4ENA5) and all the other 7 were identified with lowers scores, spectrum counts, and coverage (see additional files). Hence, XylABc-containing samples were analyzed on SDS-PAGE for approximate size estimative. Interestingly, even under denaturing conditions, XylA appeared as two band sizes of approximately 54 kDa and 108 kDa (Fig. 1b). Then, the most intense peak was loaded on a 4–20% gradient native-PAGE and ran as a single band of ca. 208 kDa, therefore compatible with a homotetramer (Fig. 1c). In order to confirm if the observed protein bands corresponded to the XyIA monomer, dimer, and tetramer, they were excised from the respective gels (Fig. 1b, c), digested with trypsin, and submitted to LC–MS/MS analysis. In all the three bands, the top score, coverage, spectrum count, and unique peptides pointed towards XI (B4ENA5) as the top1 identified protein, the same identified in the purified sample from His-trap enrichment step.
Fig. 1.

Purification and oligomerization behavior of a recombinant XylA from B. cenocepacia. a Size exclusion chromatogram obtained from Superdex 200 revealed a major peak. b Two major bands are observed on SDS-PAGE after size exclusion chromatography, revealing molecular sizes consistent with the monomer (ca. 54 kDa) and dimer (ca. 108 kDa) of His6XylA fusion. c Native 4–20% gradient PAGE gel shows a single protein band of ca. 216 kDa, when all His6XylA-containing fractions were pooled in a single sample. mAu, milli absorbance unit. *XylA containing fraction
Functional analysis of XylABc
The XI specific enzymatic activity of the purified XylABc samples was investigated. In order to find the optimal kinetics conditions, xylose isomerization was measured in a range of pH and temperatures. To find the best working temperature, buffers were kept at pH 7.5 and temperature varied from 25 to 37 °C. The temperatures used in the assays were chosen since they are generally the default temperatures used in sugarcane fermenters (Carlos et al. 2011). It was found that temperature variation has a great impact on enzymatic activity, as expected (Fig. 2a). The highest activity was 22.47 μmol min−1 mg ptn−1 at 37 °C. On each subsequent lower temperature, protein activity was lowered and reached 8.42 μmol min−1 mg ptn−1 at 30 °C and 3.53 μmol min−1 mg ptn−1 at 25 °C. Further assays used the optimum temperature (37 °C). To evaluate the influence of pH on XI activity, reaction mixes were buffered on a pH range from 5.8 to 7.6, varying − 1 pH below and +0.8 pH above the predicted isoelectric point (pI) and keeping all protein content soluble. Enzyme activity revealed to be substantially altered by pH variations as small as 0.2. The highest conversion rate was found at pH 7.2 being 51.61 μmol min−1 mg ptn−1 (Fig. 2b). Then, with the optimal conditions established, the Km, Vmax and Kcat were calculated from the non-linear fit of a Michaelis–Menten equation. The apparent Km for d-xylose is 17.08 ± 5.45 mM and Vmax is 44.97 ± 2.06 μmol min−1 mg ptn−1 (Fig. 2c). The calculated Kcat for XylABc was 19.02 ± 0.87 min−1. In addition, xylitol inhibition on this protein was evaluated since this by-product is one of the biggest concerns on using the XI strategy for ethanol production. The impact of the xylitol on XylABc activity was tested on minimal xylose concentration of 7 times the Km (125 mM), a guarantee that the Vo–Vmax. Xylitol concentration varied from 10 to 50 mM. The smaller concentration of xylitol tested was capable of reducing the enzyme activity by 67% while 30 mM and 50 mM have reduced the enzymatic activity by 85% and 93%, respectively (Fig. 2d). The calculated IC50 for xylitol was 3.75 mM.
Fig. 2.

Functional analysis of XI from B. cenocepacia. a XI activity at different temperatures. b XI activity at different pH values. c Initial velocity dependence on the substrate concentration. d Xylitol inhibition with 125 mM d-xylose. Data shown are representative of the average of two independent replicates. Column bars represent the assay standard error of a technical duplicate
In silico structural analysis of XylA
The structural model of XylABc was investigated in order to determine whether environmental or structural factors could have a major impact on the protein activity in vivo (Fig. 3a). Furthermore, for comparison reasons, theoretical models for C. phytofermentas and Piromyces sp. XIs were also created (Additional file 1: Figures S2, S3). Then, the structures were assessed for overall folding quality. The Ramachandran plot analysis of the models presented a majority of atoms in allowed regions (approximate average of all models 94%) and less than 0.5% in disallowed regions (Additional file 1: Figures S4–S6). Further RMSD calculation between models and templates were performed and showed a distance < 0.6 Å to all comparisons. Likewise, all three models were very similar among themselves (RMSD < 0.5 Å) (Additional file 2: Table S1). These results show that the models have a high conservation of their tridimensional predicted structures despite they have small variations in the geometry of the active site. This somehow could be related to the differences observed on their performance in vivo. Then, molecular dynamics simulations were performed in order to allow the energy minimization of the protein-substrate complex structures.
Fig. 3.

Xylose isomerase from B. cenocepacia predicted structure. a Overall structure of XylABc as a tetramer. b Monomer structure of XylABc. c Active site representation
Molecular dynamics and energy minimization of XI protein-substrate complexes
In order to obtain models likely to happen in vivo, enzymatic assay conditions such as temperature and pH were used to calculate the protonation states of amino acid residues during the simulation. These variables can heavily contribute to the enzymatic activity since it can alter the interaction between the protein and the substrate. The PDB crystallographic structure 1s5 m was chosen as template to obtain metallic ions and substrate coordinates. The reason for the use of this template was the appropriate cyclic form of glucose (β-d-glucofuranose) presented at the active site. The glucose structure present at 1s5 m was edited into a xylose (β-d-xylopyranose) prior to coordinates transposing to the models. This form of xylose is believed to be preferred for isomerization reaction (Asboth and Naray-Szabo 2000). The protein-ion-substrate complexes were finally created by the transposition of the metallic ions and xylose coordinates from 1s5 m-xylose structure into the XI constructed models for each species. Then, simulations were performed in a water box with approximately 120 Å3 kept at 295.15 K and 1 atm for 20 ns. The results were analyzed in two steps: (i) overall and secondary structure stability, and general thermodynamic properties: (ii) active site geometry, substrate interaction and orientation.
All simulation models were visually inspected and no significant conformational transition was observed. The global RMSD and RMSF calculations were used as a metric for structural stability. The overall RMSD analysis revealed a similar relaxation for all structures of approximately 2 Å from its initial position (Additional file 1: Figure S7). The overall protein RMSF analysis showed that the larger fluctuations are concentrated at the amino acid residues closer to the surface that are directly exposed to the solvent (Additional file 1: Figure S8). The substrate RMSF result shows the fluctuation of each atom’s position along the structure (Additional file 1: Figure S10). The wide range of the torsion angles allowed by the primary and secondary C–OH ligations can explain this phenomenon. It is noteworthy that there is little movement of the atoms on the pyranoside ring of the sugar. However, the hydroxyl radicals at positions 0, 15, 17 and 19 considerably vary their rotation during simulations. In addition, the simulation indicates that the protonated nitrogen from the imidazole HIS residue side chains is likely to act as a catalyst by donating a proton to the oxygen in the xylose pyranoside ring (Fig. 3c).
Since all models from B. cenocepacia, C. phytofermentans and Piromyces sp. had similar tridimensional structures even after relaxation, it was thought that small variations in the active site could explain the results obtained in vitro. Then, the average volume of the pocket that contains the xylose binding site was calculated. The results have shown that there is no significant difference between the pocket volume of each chain in a same protein or among each predicted XI model (Additional file 1: Figure S11). Therefore, this suggests that the pocket size do not limit the active site availability for substrate binding and that cannot explain the results obtained in the functional analysis.
Yet no significant difference has been observed in the active site geometry of the theoretical models created, XIs from each species can vary its primary sequence to some degree and therefore, its specific activity. Therefore, a thorough investigation on the physical and chemical properties of these proteins was still necessary.
Electrostatic surface and dipole moment analysis
Although the tridimensional structures are very similar, the sequence alignment only shows an identity about 50% (Additional file 1: Figure S12). Therefore, it is expected that residues substitutions could substantially alter the enzyme activity by altering electrostatic net charges along the protein tridimensional structure. Thus, the electrostatic surface of each protein was predicted for the models obtained after the molecular dynamics simulation. The three proteins analyzed showed a negative net charge distributed along the structure’s surface (Fig. 4). However, the charges can have different intensities for one protein, which directly correlates with the dipole moment of each protein. Then, variations in the electrical field vector intensity and direction were determined with all models after the RMSF stabilization in the molecular dynamics. It was revealed that XylABc has the smallest value between the three species analyzed, followed by Piromyces sp. and C. phytofermentans, respectively (Fig. 5). This result corroborates the data in Table 1, where the XI from C. phytofermentans has the highest Km for xylose and the highest dipole moment intensity.
Fig. 4.

Electric field representation. A B. cenocepacia. B C. phytofermentans. C Piromyces sp. Red lines represent a negative charges. Blue lines represent positive charges. Grey lines represent neutral charges. Black arrows show the active site domains
Fig. 5.
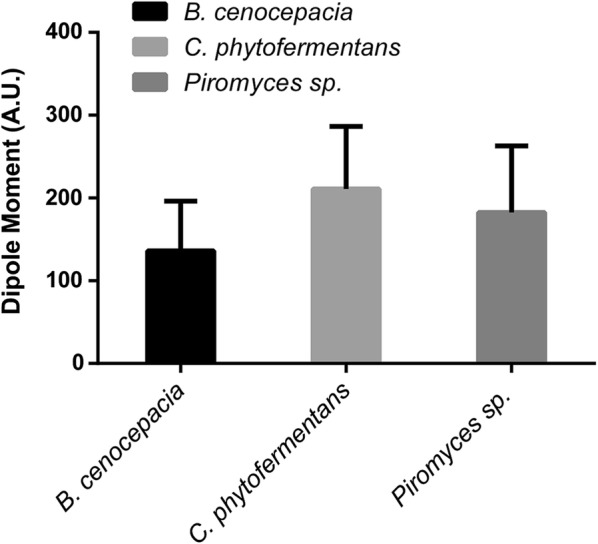
Dipole moment representing the net protein charge. All proteins atoms charge and topology were used to calculate the protein electric vector under similar conditions to the enzymatic assay. Statistical analyses of the data shown were performed with one-way ANOVA test and have shown a p value < 0.001. n = 11,407 models analyzed
Table 1.
Km values of xylose isomerase from three different microbial species
| Species | Km (mM)a | References |
|---|---|---|
| B. cenocepacia | 17.08 ± 5.45 | This work |
| C. phytofermentans | 61.85 ± 3.41 | Brat et al. (2009) |
| Piromyces sp. | 49.85 ± 2.82 | Kuyper et al. (2003) |
aKm values were obtained for d-xylose and are expressed with standard error calculated from a duplicate of independent assays
Discussion
The purified XylABc samples have shown multiple band sizes on SDS-Page denaturing conditions, which was a strong indication that the protein could have different oligomerization states, including a monomer and an SDS-resistant dimer. However, under native conditions, the protein has shown a single band of approximately 208 kDa. Taken together, these results strongly suggest that XylABc is able to form homodimers and homotetramers, corroborating with previous structural studies of other bacterial XI (Peng et al. 2015). Considering that the protein sample of ca. 208 kDa appears as the only higher-order structure of XylA and was highly active in the XI enzymatic assays, we suggest that XylA may be able to assemble itself as homotetramer in vivo. Furthermore, the protein concentration used in the functional assays is consistent with protein concentration commonly found in yeasts (Milo 2013), showing that it is likely that XylABc would present itself mostly as a homotetramer and that the homotetramer self-assembly observed in vitro is not a concentration dependent effect. The strong reduction power required to denature this protein shows its potential for industrial fermentation, where temperature variations can go from 30 to 42 °C depending on environmental conditions (Basso et al. 2008; Carlos et al. 2011).
To this date, most attempts to measure xylose isomerase (XI) activity have employed the use of crude cell extracts, through a coupled reaction with sorbitol dehydrogenase (SDH) to measure XI activity by monitoring and quantifying the oxidation of NADH (Kersters-Hilderson et al. 1987). Notwithstanding, it is difficult to accurately estimate the functional characteristics of the protein within the conditions of a complex biological extract. In the most used assay, the target protein is diluted in the cytoplasmic content within other NADH and NAD+ -dependent proteins that can affect the results of XI activity measurements. Therefore, it has been of great interest to evaluate the dynamics of this protein on both in vivo and in vitro conditions in order to determine the enzymatic activity, functionality and productivity. The XylA from B. cenocepacia was already proven to be functional in fermentation conditions and has shown to be one of the most promising XI performances described so far (Vilela et al. 2015). The functional analysis of this enzyme is necessary to understand the reasons why the sole expression of this protein was able to achieve such results, when other researchers had to perform multiple modification in strains to observe similar results (Cai et al. 2012). The XylA from B. cenocepacia showed a threefold lower Km for xylose than the XI from C. phytofermentans and Piromyces sp xylA, one of the best microbial XI results published to date (Kuyper et al. 2003; Brat et al. 2009; Moysés et al. 2016). Those are remarkable results considering that a lower Km can have a striking effect on the yeast xylose metabolism during fermentation. As XylA requires a smaller concentration of sugar to reach ½ Vmax, it is expected that it can compete for xylose with the yeast’s endogenous XR. It has been reported that S. cerevisiae endogenous XR has an approximate Km for xylose of 27.9 mM (Jeong et al. 2001). Considering the results obtained in this work, it is possible to assert that the recombinant XylABc is capable of diverting the metabolic flow towards XI pathway instead of the two-steps XR/XDH pathway. Therefore, it corroborates previous observation by our group (Vilela et al. 2015) showing that XylABc-engineered S. cerevisiae does not significantly accumulate by-products such as xylitol and glycerol, improving ethanol production.
Many researchers have found that xylitol is a potent inhibitor of XI, thus it is one of the biggest concerns of researchers using the XI strategy (Walfridsson et al. 1996; Klimacek et al. 2010; Zhang et al. 2012). On the other hand, Ha et al. (2011) brought up to light that xylitol inhibition may not be as severe in vivo as it is in vitro. Furthermore, Vilela et al. (2015) demonstrated that accumulation of xylitol is negligible during co-fermentation of glucose and xylose, thus it should not be a major concern for our model.
Further analyses revealed that the XI from B. cenocepacia Kcat for d-xylose is one of the highest ever reported and the specificity constant Kcat/Km is approximately 1056 s−1 M−1. It has been recently reported that mutations in the Ca+2/Mn+2 ATPase gene (PMR1) can alter the XI activity in vivo by increasing the overall concentration of divalent ions in the cell (Verhoeven et al. 2017). In this case, XylA from Piromyces sp. E2 was able to reach a Km for d-xylose as low as 3.9 ± 0.2 mM and a Kcat of 7.8 ± 0.1 s−1, while the calculated Kcat/Km for XylA was 2000 s−1 M−1. Thus, the XylA from B. cenocepacia seems to have a lower specificity for the same substrate in comparison with XylA from Piromyces sp. However, the Kcat values indicate that XylA catalysis is more efficient in native conditions. It is possible that further modifications in our evolved strain could improve the XI activity even further as observed by Verhoeven et al. (2017).
Although recombinant XIs enzymatic activity greatly differ upon expression in S. cerevisiae, it was observed that all three XIs studied have similar tridimensional structures. It is commonly accepted that the primary sequence of a protein is responsible for the structure folding upon exposure to water in the cytoplasm. Therefore, it is expected that proteins with a large difference on their amino acid sequences should have different tridimensional structures as well. The XIs of B. cenocepacia, C. phytofermentans and Piromyces sp. showed amino acid identities of approximately 50% when compared to PDB 1s5 m sequence and the same can be observed in the multiple alignment of the three XIs (Additional file 1: Figure S12). The generated 3D models revealed a striking similarity among them with RMSD values smaller than 0.5 Å (Additional file 2: Table S1), despite the low average-value sequence identity. Molecular dynamics was used to minimize the energy and relax the protein-ion-substrate complex, in order to observe if small differences in the active site domain could explain the results observed.
The molecular dynamics simulations were able to show that regions exposed to the solvent have a greater degree of flexibility while regions closer to the active site undergo few structural modifications (Additional file 1: Figure S8). Furthermore, the xylose binding site residues are highly conserved among the analyzed species, as well as their tridimensional geometry. In either case, it is clear that the Mg2+ ion and the water molecules present at the active site have an important role on the substrate orientation within the active site domain. The presence of the ion and water molecules creates an anchoring platform caused by the anion–cation–anion charge distribution, similar to those observed in lysozymes and serine proteases (Asboth and Naray-Szabo 2000). Moreover, the oxygen present at the pyranoside ring of xylose is always aligned towards the HIS residue sidechains. The alignment of the pyranosidic oxygen creates a higher resonant movement in the molecule, reducing the energy barrier required for linearization of the substrate. Adding to this, the simulations have shown that the imidazolic hydroxyl group donates a proton for the oxygen and allows the linearization of the sugar. This kind of interaction has been previously described by Fenn et al. (2004) and Toteva et al. (2011) but with no clarity whether the HIS residue participated in the catalysis. We believe that our results corroborate with previous founds reported in the literature.
As no significant difference in the overall protein tridimensional structure was observed and all modeled proteins present the same type of interaction with the substrate, we decided to assess the electrostatic surface net charge and the protein dipole moment. Since the only reasonable difference between XIs was their primary amino acid sequence, any residues substitutions could alter the net charge of the protein electric field. The protein electrical surface directly influences the accessibility of the substrate to the active site. The results presented herein show a clear correlation between the dipole moment resultant from the electric field vector and the Km shown by each XI. As the electric field gets more negatively intense, the Km values are also increased. However, there is no canonical link stablished between the dipole moment of a protein structure related to its function. It is known that in some cases the electrostatic properties of a protein family are highly specific and can help directing the substrate towards the active site (Felder et al. 2007). We hypothesize that this may be one of the mechanisms behind the results observed in the enzymatic assays, since the compared structures are very similar. Further studies on phylogeny, amino acid conservation, structure and net charges are necessary in order to fully explain the correlation between these characteristics and the enzyme activity for this protein family.
Additional files
Acknowledgements
The authors thank Pedro Geraldo Pascutti, Anderson de Sá Pinheiro and Carolina Oliveira Matos for their contributions to this project.
Authors’ contributions
BCN conceived this study, with input from IPVV and ECAE. IPVV, GTC, DEBG, and LFV performed experiments and analyzed data. IPVV drafted the manuscript. IPVV, BCN, ECAE, DEBG, GBD, and RDM were involved in manuscript editing and/or revisions. All authors read and approved the final manuscript.
Funding
This study was supported by the Brazilian National Council of Research and Development (CNPq), Grant No. 482880/2013-9 awarded to B.C.N and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Model PDB structures, mass spectrometry datasheet and dynamics simulation raw data are available and free of charge.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Asboth B, Naray-Szabo G. Mechanism of action of d-xylose isomerase. Curr Protein Pept Sci. 2000;1:237–254. doi: 10.2174/1389203003381333. [DOI] [PubMed] [Google Scholar]
- Basso LC, De Amorim HV, De Oliveira AJ, Lopes ML. Yeast selection for fuel ethanol production in Brazil. FEMS Yeast Res. 2008;8:1155–1163. doi: 10.1111/j.1567-1364.2008.00428.x. [DOI] [PubMed] [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- Bowie JU, Lüthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Brat D, Boles E, Wiedemann B. Functional expression of a bacterial xylose isomerase in Saccharomyces cerevisiae. Appl Environ Microbiol. 2009;75:2304–2311. doi: 10.1128/AEM.02522-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinenberg PM, Jonker R, Van Dijken JP, Scheffers WA. Utilization of formate as an additional energy source by glucose-limited chemostat cultures of Candida utilis CBS 621 and Saccharomyces cerevisiae CBS 8066. Arch Microbiol. 1985;1442:302–306. doi: 10.1007/BF00693408. [DOI] [Google Scholar]
- Cai Z, Zhang B, Li Y. Engineering Saccharomyces cerevisiae for efficient anaerobic xylose fermentation: reflections and perspectives. Biotechnol J. 2012;7:34–46. doi: 10.1002/biot.201100053. [DOI] [PubMed] [Google Scholar]
- Carlos L, Olitta T, Nitsche S. Ethanol production in Brazil: the industrial process and its impact on yeast fermentation. In: Bernardes MA, editor. Biofuel production-recent developments and prospects. InTech: São Paulo; 2011. pp. 85–100. [Google Scholar]
- Carvalho PC, Lima DB, Leprevost FV, Santos MDM, Fischer JSG, Aquino PF, Moresco JJ, Yates JR, Barbosa VC. Integrated analysis of shotgun proteomic data with PatternLab for proteomics 4.0. Nat Protoc. 2016;11:102–117. doi: 10.1038/nprot.2015.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Figueiredo Vilela L, de Mello VM, Reis VCB, Bon EPDS, Gonçalves Torres FA, Neves BC, Eleutherio ECA. Functional expression of Burkholderia cenocepacia xylose isomerase in yeast increases ethanol production from a glucose–xylose blend. Bioresour Technol. 2013;128:792–796. doi: 10.1016/j.biortech.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CE, Prilusky J, Silman I, Sussman JL. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007;35:512–521. doi: 10.1093/nar/gkm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn TD, Ringe D, Petsko GA. Xylose isomerase in substrate and inhibitor michaelis states: atomic resolution studies of a metal-mediated hydride shift. Biochemistry. 2004;43(21):6464–74. doi: 10.1021/bi049812o. [DOI] [PubMed] [Google Scholar]
- Gonc DL, Matsushika A, De Sales BB, Goshima T. Enzyme and microbial technology xylose and xylose/glucose co-fermentation by recombinant Saccharomyces cerevisiae strains expressing individual hexose transporters. Enzyme Microbial Technol. 2014;63:13–20. doi: 10.1016/j.enzmictec.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Ha SJ, Kim SR, Choi JH, Park MS, Jin YS. Xylitol does not inhibit xylose fermentation by engineered Saccharomyces cerevisiae expressing xylA as severely as it inhibits xylose isomerase reaction in vitro. Appl Microbiol Biotechnol. 2011;92:77–84. doi: 10.1007/s00253-011-3345-9. [DOI] [PubMed] [Google Scholar]
- Hahn-Hägerdal B, Karhumaa K, Fonseca C, Spencer-Martins I, Gorwa-Grauslund MF. Towards industrial pentose-fermenting yeast strains. Appl Microbiol Biotechnol. 2007;74:937–953. doi: 10.1007/s00253-006-0827-2. [DOI] [PubMed] [Google Scholar]
- Jeong EY, Sopher C, Kim IS, Lee H. Mutational study of the role of tyrosine-49 in the Saccharomyces cerevisiae xylose reductase. Yeast. 2001;18:1081–1089. doi: 10.1002/yea.758. [DOI] [PubMed] [Google Scholar]
- Kersters-Hilderson H, Callens M, Van Opstal O, Vangrysperre W, De Bruyne CK. Kinetic characterization of d-xylose isomerases by enzymatic assays using d-sorbitol dehydrogenase. Enzyme Microb Technol. 1987;9:145–148. doi: 10.1016/0141-0229(87)90067-6. [DOI] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimacek M, Krahulec S, Sauer U, Nidetzky B. Limitations in xylose-fermenting Saccharomyces cerevisiae, made evident through comprehensive metabolite profiling and thermodynamic analysis. Appl Environ Microbiol. 2010;76:7566–7574. doi: 10.1128/AEM.01787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko JK, Lee S. Advances in cellulosic conversion to fuels: engineering yeasts for cellulosic bioethanol and biodiesel production. Curr Opin Biotechnol. 2018;50:72–80. doi: 10.1016/j.copbio.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Kruckeberg AL. The hexose transporter family of Saccharomyces cerevisiae. Arch Microbiol. 1996;166:283–292. doi: 10.1007/s002030050385. [DOI] [PubMed] [Google Scholar]
- Kuyper M, Harhangi H, Stave A, Winkler A, Jetten M, Delaat W, Denridder J, Opdencamp H, Vandijken J, Pronk J. High-level functional expression of a fungal xylose isomerase: the key to efficient ethanolic fermentation of xylose by? FEMS Yeast Res. 2003;4:69–78. doi: 10.1016/S1567-1356(03)00141-7. [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Lüthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- Madhavan A, Tamalampudi S, Srivastava A, Fukuda H, Bisaria VS, Kondo A. Alcoholic fermentation of xylose and mixed sugars using recombinant Saccharomyces cerevisiae engineered for xylose utilization. Appl Microbiol Biotechnol. 2009;82:1037–1047. doi: 10.1007/s00253-008-1818-2. [DOI] [PubMed] [Google Scholar]
- Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Šali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Matsushika A, Inoue H, Kodaki T, Sawayama S. Ethanol production from xylose in engineered Saccharomyces cerevisiae strains: current state and perspectives. Appl Microbiol Biotechnol. 2009;84:37–53. doi: 10.1007/s00253-009-2101-x. [DOI] [PubMed] [Google Scholar]
- Milo R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays. 2013 doi: 10.1002/bies.201300066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosier N, Wyman C, Dale B, Elander R, Lee YY, Holtzapple M, Ladisch M. Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresour Technol. 2005;96:673–686. doi: 10.1016/j.biortech.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Moysés D, Reis V, Almeida J, Moraes L, Torres F. Xylose fermentation by Saccharomyces cerevisiae: challenges and prospects. Int J Mol Sci. 2016;17:207. doi: 10.3390/ijms17030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Keasling JD. Engineering cellular metabolism. Cell. 2016;164:1185–1197. doi: 10.1016/j.cell.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Peng B, Huang S, Liu T, Geng A. Bacterial xylose isomerases from the mammal gut Bacteroidetes cluster function in Saccharomyces cerevisiae for effective xylose fermentation. Microb Cell Fact. 2015;14:70. doi: 10.1186/s12934-015-0253-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera FP. Multiple threats to child health from fossil fuel combustion: impacts of air pollution and climate change. Environ Health Perspect. 2016 doi: 10.1289/ehp299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using stagetips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Simmerling C. ff 14SB: Improving the accuracy of protein side chain and backbone parameters from ff 99SB. J Chem Theory Comput. 2015 doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Stambuk BU, Eleutherio ECA, Florez-Pardo LM, Souto-Maior AM, Bon EPS. Brazilian potential for biomass ethanol: challenge of using hexose and pentose cofermenting yeast strains. J Sci Ind Res (India) 2008;67:918–926. [Google Scholar]
- Tibbetts JH. Air quality and climate change: a delicate balance. Environ Health Perspect. 2015;123:A148–A153. doi: 10.1289/ehp.123-A148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toteva MM, Silvaggi NR, Allen KN, Richard JP. Binding energy and catalysis by D-xylose isomerase: kinetic, product, and X-ray crystallographic analysis of enzyme-catalyzed isomerization of (R)-glyceraldehyde. Biochemistry. 2011;50(46):10170–81. doi: 10.1021/bi201378c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven MD, Lee M, Kamoen L, Van Den Broek M, Janssen DB, Daran JMG, Van Maris AJA, Pronk JT. Mutations in PMR1 stimulate xylose isomerase activity and anaerobic growth on xylose of engineered Saccharomyces cerevisiae by influencing manganese homeostasis. Sci Rep. 2017;7:1–11. doi: 10.1038/srep46155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela LDF, de Araujo VPG, Paredes RDS, Bon EPDS, Torres FAG, Neves BC, Eleutherio ECA. Enhanced xylose fermentation and ethanol production by engineered Saccharomyces cerevisiae strain. AMB Express. 2015 doi: 10.1186/s13568-015-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walfridsson M, Bao X, Anderlund M, Lilius G, Bülow L, Hahn-Hägerdal B. Ethanolic fermentation of xylose with Saccharomyces cerevisiae harboring the Thermus thermophilus xylA gene, which expresses an active xylose (glucose) isomerase. Appl Environ Microbiol. 1996;62:4648–4651. doi: 10.1128/aem.62.12.4648-4651.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform. 2014;47:1–32. doi: 10.1002/0471250953.bi0506s47. [DOI] [PubMed] [Google Scholar]
- Zhang G-C, Liu J-J, Ding W-T. Decreased xylitol formation during xylose fermentation in Saccharomyces cerevisiae due to overexpression of water-forming NADH oxidase. Appl Environ Microbiol. 2012;78:1081–1086. doi: 10.1128/AEM.06635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Model PDB structures, mass spectrometry datasheet and dynamics simulation raw data are available and free of charge.