

Abstract
Inflammatory processes have been shown to play an important role in the mechanisms involved in the pathogenesis of hypertension. Innate and adaptive immune responses participate in BP elevation and end‐organ damage. Here, we discuss recent studies focusing on novel inflammatory and immune mechanisms that play roles in BP elevation. Different subpopulations of cells involved in innate and adaptive immune responses, such as dendritic cells, monocytes/macrophages and NK cells, on the one hand, and B and T lymphocytes, on the other, contribute to the vascular and kidney injury in hypertension. Unconventional innate‐like T cells such as γδ T cells also participate in hypertensive mechanisms by priming both innate and adaptive immune cells, contributing to trigger vascular inflammation and BP elevation. These cells exert their effects in part via production of various cytokines including pro‐inflammatory IFN‐γ and IL‐17 and anti‐inflammatory IL‐10. The present review summarizes some of these immune mechanisms that participate in the pathophysiology of hypertension.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- Ang
angiotensin
- APCs
antigen presenting cells
- BCR
B cell receptor
- C3a and C5a
complement component 3a and 5a
- C3ar and C5ar
C3a and C5a receptors
- CD
cluster of differentiation
- DAMPs
damage‐associated molecular patterns
- DCs
dendritic cells
- DT
diphtheria toxin
- Foxp3
forkhead box P3
- G‐MDSCs
granulocytic‐myeloid‐derived suppressor cells
- iTreg
inducible T regulatory cells
- ILCs
innate lymphoid cells
- Ifng
IFN‐γ gene
- Ifngr1
IFN‐γ receptor 1 gene
- JT
Jurkat T
- LysM+
lysozyme M‐positive
- MDSCs
myeloid‐derived suppressor cells
- MHC
major histocompatibility complex
- MR1
MHC I related
- Mo‐MDSCs
monocytic‐myeloid‐derived suppressor cells
- PAMPs
pathogen‐associated molecular patterns
- PRRs
pattern recognition receptors
- Tbx21
T‐Box 21
- Tc
cytotoxic T
- TCR
T cell receptor
- TEM
effector memory T
- TLRs
toll‐like receptors
- Treg
T regulatory lymphocytes or cells
- TRM
tissue‐resident memory T
Introduction
High BP or hypertension is the leading risk factor for cardiovascular disease worldwide, accounting for 9.4 million deaths and 7% of global disability‐adjusted life years (Lim et al., 2012). The aetiology of essential hypertension is unclear, and its mechanisms are complex. Perturbations of the sympathetic and parasympathetic nervous systems, the renin‐angiotensin‐aldosterone system and the endothelin system, genetic predisposition, environmental factors such as a high‐salt intake, and ageing, in combination or independently contribute to elevation of BP, and BP and/or the pro‐hypertensive stimuli induce cardiovascular injury. It is widely appreciated that low‐grade inflammation is an important mediator in the initiation and maintenance of BP elevation and could contribute to its occurrence in association with chronic inflammatory diseases (Caillon and Schiffrin, 2016; Rodriguez‐Iturbe et al., 2017; Norlander et al., 2018). Infiltration of innate (monocytes/macrophages) and adaptive immune cells (T lymphocytes) in perivascular fat, kidneys and myocardium, increased expression of adhesion molecules and chemokines, cytokine production and release and ROS generation are consistent features of hypertension. Gain‐ and loss‐of‐function experiments have shown that innate and adaptive immune cells such as monocyte/macrophages, on the one hand, and B and T lymphocytes, on the other hand, are implicated in hypertension and cardiovascular injury, in a ying/yang relationship with anti‐inflammatory cells that include anti‐inflammatory M2 macrophages and T regulatory lymphocytes (Treg). These studies have been reviewed previously (Idris‐Khodja et al., 2014; Mian et al., 2014; Caillon and Schiffrin, 2016). The purpose of the present review is to discuss recent advances in the knowledge of BP control by immune mechanisms from basic and clinical studies in hypertension, illustrated in Figure 1. The immune system has also been implicated in the pathogenesis of pre‐eclampsia, but this is beyond the scope of the current review.
Figure 1.
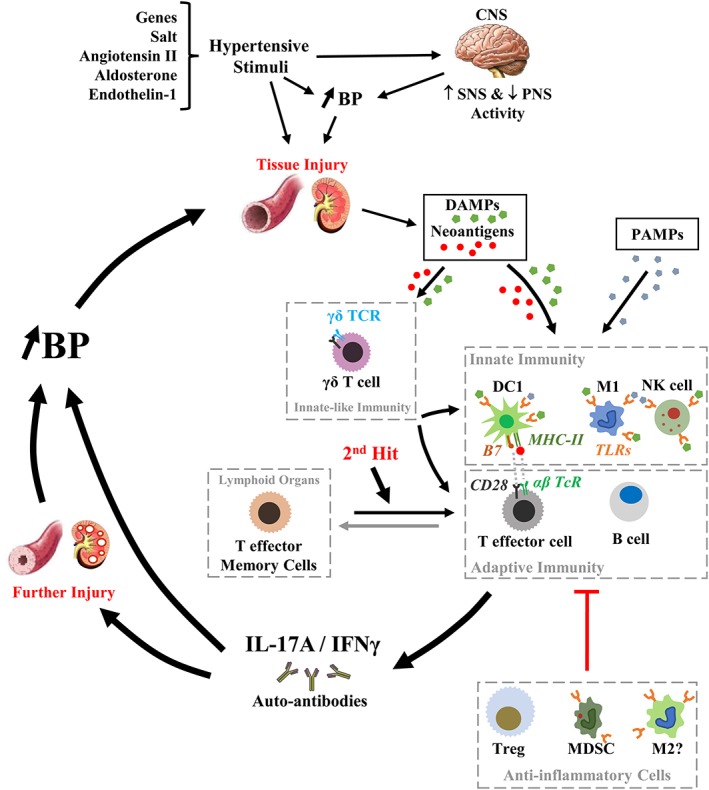
Role of immune cells in inflammation in hypertension. Environmental factors such as salt, in the presence of genetic susceptibility, may lead to small rises of BP, in part, by triggering the activation of the sympathetic nervous system (SNS) and inhibition of the parasympathetic nervous system (PNS) alone or together with other hypertensive stimuli such as Ang II, aldosterone and endothelin‐1. Over time, high BP and/or the pro‐hypertensive stimuli induce tissue injury, which together with oxidative stress caused by vasoactive peptides such as Ang II or endothelin‐1 create favourable conditions for the development of DAMPs and neoantigens, such as isoketal protein adducts. DAMPs activate innate immunity via TLRs on type 1 macrophages (M1), type 1 dendritic cells (DC1) and NK cells, while neoantigens enhance DC immunogenicity and promote DC release of IL‐6, IL‐1β and IL‐23, causing proliferation of T cells and production of IL‐17A, IFN‐γ and TNF‐α. DAMPs and neoantigens could also activate unconventional lymphocytes such as innate‐like γδ T cells, which will activate T lymphocytes. Opportunistic diseases such as periodontitis could also exacerbate the activation of the innate immune system through binding of PAMPs to pattern recognition receptors such as TLRs. Innate immune cells and γδ T cells contribute to inflammation, both directly or via the activation of adaptive immunity, inducing pro‐inflammatory cytokines, such as IL‐17 and IFN‐γ, and the production of autoantibodies, leading to vascular and kidney injury, which closes the pro‐hypertensive circle that is a feed‐forward process resulting in progressive BP elevation. During this process, TEM cells are produced and stored in lymphoid organs including the bone marrow. Upon a second hit such as high‐salt intake, TEM cells could be reactivated and contribute to the vicious cycle to maintain high BP or further increase its level. Throughout this process, anti‐inflammatory cells such as Treg, MDSCs and type 2 macrophages (M2) could provide homeostatic fine‐tuning of the inflammatory process in blood vessels and the kidney. However, this anti‐inflammatory mechanism could be rendered unable to counteract the development of hypertension because of factors such as Ang II that can cause a decrease in the number and function of Treg through activation of the complement system. B7, co‐stimulatory molecule on antigen presenting cell surface (CD80 or CD86).
The innate immune system in hypertension
Innate immune system
Innate immune cells play a role in the initiation and subsequent sustained response of adaptive immunity, key mechanisms in hypertension and vascular injury. Innate immune cells are characterized by their rapid response as a first line of defence. They can sense pathogen‐associated molecular patterns (PAMPs) or damage molecules (including neoantigens) from stressed or injured tissue (damage‐associated molecular patterns, DAMPs) via their pattern recognition receptors (PRRs) such as toll‐like receptors (TLRs). Innate responses are mediated by specialized effector cells that can initiate host defence responses and inflammation by either direct mechanisms (e.g. cytokine and chemokine production, activation of proteins of the complement system and phagocytosis) or by activating the adaptive immune system via antigen presentation. Innate immune effector cells include the professional phagocytes [macrophages and dendritic cells (DCs)], which can perform phagocytosis and present antigens (antigen presenting cells or APCs). Macrophages and DCs can be distinguished into pro‐inflammatory cells like M1 macrophages and DC1 or anti‐inflammatory cells such as M2 macrophages and DC2 (Shortman and Liu, 2002). M1 and DC1 release pro‐inflammatory cytokines such as IL‐1β, IL‐6, IL‐12, IL‐23, TNF‐α and ROS. On the other hand, M2 macrophages and DC2 release anti‐inflammatory cytokine IL‐10. Macrophages and DCs express on their surface pro‐inflammatory or anti‐inflammatory co‐stimulator molecules belonging to the B7 family (Ceeraz et al., 2013). DCs are able to present antigens to activate both CD4+ and CD8+ T lymphocytes, and macrophages (Leibowitz and Schiffrin, 2011). Other cells can present antigens but are less susceptible of activating the adaptive immune system and are called non‐professional antigen presenting cells, such as neutrophils (see below). NK cells are innate lymphoid cells (ILCs), which express a repertoire of activating and inhibitory receptors implicated in self‐tolerance while being capable of rapid responses against pathogens (Vivier et al., 2011). NK cells can secrete pro‐ or anti‐inflammatory cytokines such as IFN‐γ or IL‐10. The cytokines released and co‐stimulation molecular pattern participate together with the antigen presenting process in T cell or B cell activation.
Monocyte/macrophages in hypertension
The role of monocyte/macrophages in hypertension was demonstrated using genetic manipulation and/or adoptive transfer experiments (De Ciuceis et al., 2005; Wenzel et al. 2011). The role of monocyte/macrophages was first demonstrated by showing that mice with a deficiency in monocyte/macrophage function due to a mutation in the colony stimulating factor 1 gene presented blunted angiotensin (Ang) II‐ or deoxycorticosterone acetate (DOCA) plus salt‐induced BP elevation and vascular injury (De Ciuceis et al., 2005; Ko et al., 2007). Wenzel et al. (2011) elegantly extended these findings by selective ablation of lysozyme M‐positive (LysM+) myelomonocytic cells by low‐dose diphtheria toxin (DT) in mice with inducible expression of the DT receptor, which prevented Ang II‐induced BP elevation and vascular dysfunction. This was reversed by adoptive transfer of monocytes but not neutrophils.
Antigen presenting cells in hypertension
Very recently, Hevia et al. (2018) demonstrated that myeloid CD11c+ APCs are important for the development and maintenance of hypertension in response to Ang II infusion plus a high‐salt diet. This was demonstrated using ablation/reconstitution protocols in transgenic mice expressing the DT receptor under the control of the Cd11c gene promoter (CD11c.DOG). CD11c is an integrin α‐X chain protein that is highly expressed in DCs, macrophages and NK cells. Ang II infusion plus a high‐salt diet induced BP elevation in wild‐type mice associated with an increase in the fraction of pro‐inflammatory CD11c+ APCs in the spleen (CD86 +) and the kidneys (CD86+, MHC I+ and MHC II+) (Hevia et al., 2018).
NK cells in hypertension
NK cells play a role in vascular injury in hypertension (Kossmann et al., 2013). Ang II caused NK cell recruitment in the aortic wall, which was blunted in mice lacking T‐Box 21 (Tbx21) or IFN‐γ. NK cell depletion using anti‐NK1.1 antibody reduced aortic endothelium‐dependent and independent dysfunction induced by Ang II infusion. Although not directly tested, NK cells do not seem to play a role in Ang II‐induced BP elevation as BP was unaffected in mice lacking Tbx21, which is highly expressed in NK cells and controls the expression of IFN‐γ.
Neutrophils in hypertension
Neutrophils are another type of innate cell that could play a role in hypertension and cardiovascular injury. The reader is directed to excellent reviews on neutrophils for detailed information on these cells (Hampton and Chtanova, 2016; Rosales, 2018). In brief, neutrophils or polymorphonuclear leukocytes, which are the most abundant cell type in the human blood, are the first line of defence of the innate immune system. They are involved in eliminating microbial infection through phagocytosis, degranulation, and the release of nuclear material in the form of neutrophil extracellular traps. However, it has been revealed recently that neutrophils have a more diverse function and could regulate the adaptive immune system. Neutrophils induce somatic hypermutation and antibody production by marginal B lymphocytes through cytokine secretion, to activate T lymphocytes by antigen presentation in lymph nodes and to inhibit T lymphocytes by direct cell contact and hydrogen peroxide generation. Wenzel et al. (2011) showed using flow cytometry that a 7‐day Ang II infusion increased the number of neutrophils in the aorta. However, as indicated above, adoptive transfer of neutrophils did not restore the inhibition of Ang II‐induced BP elevation and vascular dysfunction caused by selective ablation of LysM+ myelomonocytic cells using low‐dose DT. This observation is not unexpected since this experiment did not really examine the role of neutrophils in hypertension, as the selective ablation of LysM+ myelomonocytic cells reduced the number of circulating and infiltrating CD11b+GR‐1+ monocytes but not the number of CD11b+GR‐1+ neutrophils. More recently, it was shown in humans that the neutrophil to lymphocyte ratio and neutrophil count correlate with an increasing risk of developing hypertension (Liu et al., 2015).
Myeloid‐derived suppressor cells in hypertension
Myeloid‐derived suppressor cells (MDSCs) are anti‐inflammatory immune cells characterized by expression of CD11b and Gr‐1 (Gabrilovich and Nagaraj, 2009; Rosales, 2018). The anti‐Gr‐1 antibodies bind to two epitopes, Ly6G and Ly6C. The use of epitope‐specific antibodies has permitted the identification of two subsets of MDSCs, granulocytic (G‐MDSCs, CD11b+Ly6G+Ly6Clow) and monocytic (Mo‐MDSCs, CD11b+Ly6G−Ly6Chi). G‐MDSCs resemble neutrophils and may represent a neutrophil subset with immunosuppressive capacity (Rosales, 2018). MDSCs have been shown to suppress CD8+ T cell responses in a MHC I‐dependent manner. However, whether MDSCs could suppress CD4 + T cell responses via a similar mechanism was not examined. MDSCs can counteract the development of hypertension (Shah et al., 2015). CD11b+Gr‐1+ cells were increased in the blood, spleen and kidneys of Ang II‐infused mice. An increase in the blood CD11b+Gr‐1+ cells was also observed in L‐NAME‐ and L‐NAME/washout/high‐salt diet‐induced hypertension. Furthermore, flow cytometry analysis revealed that mouse blood contains a small population of Mo‐MDSCs (~2% of leukocytes) and a larger population of G‐MDSCs (~11% of leukocytes). Ang II increased both populations of MDSCs, but G‐MDSCs were predominant. MDSCs from hypertensive mice had increased capacity to suppress T cells in vitro and CD8+ T cells in vivo. Gemcitabine‐ and anti‐Gr‐1 antibody‐induced MDSC depletion exaggerated Ang II‐induced BP elevation and increased preferentially CD8+ T cells expressing IL‐17A and both IL‐17A and IFN‐γ. Adoptive transfer of MDSCs from hypertensive but not normotensive mice reduced or reversed Ang II‐induced BP elevation.
Role of adaptive immune system in hypertension
Lymphocytes
T and B lymphocytes belong to the adaptive immune system and could be considered as commanders of the immune response, orientating pro‐ or anti‐inflammatory processes (Janeway et al., 2001). These lymphocytes help combat specific pathogens like invading bacteria, viruses and parasites by producing cytokines and/or cytotoxic molecules or antibodies. In the absence of infection, or during sterile inflammation, T and B cells could be involved in enhancing responses, thus contributing to the development of chronic or auto‐immune diseases.
T lymphocytes
T cells are characterized by their cell surface markers, which define their function. T cells typically express the T cell receptor (TCR). This complex contains two TCR chains αβ, surrounded by CD3 complex formed by CD3γ, δ, ε, ζ‐chains to transduce the TCR signal. In the thymus, the site of T cell maturation, CD3+ T lymphocytes become either CD4+ or CD8+ single‐positive cells. A few cells remain CD4−CD8− double negative or become CD4+CD8+ double positive cells. After leaving the thymus, CD4+ and CD8+ T cells acquire immune competence. T lymphocytes require two signals for activation: one, through TCR activation, due to an antigenic peptide presented by APCs via their MHC molecules. The second is a co‐stimulatory signal, which is usually the interaction between the receptors of B7 family ligands on APCs and the T cell surface co‐stimulatory molecule family. CD4 and CD8 are co‐receptors of the TCR that bind MHC class II and I molecules respectively. In response to combined stimulation with antigen, co‐stimulators and particular cytokines, naïve CD4+ T helper cells differentiate into Th1, Th2 and Th17 effector cells, or Treg, each producing its own panel of cytokines and mediating separate functions. Th1 cells secrete IFN‐γ, IL‐2 and TNF‐α and were described originally to play roles in cell‐mediated defence against intracellular microorganisms and protozoa and are involved in chronic and autoimmune diseases. Th2 lymphocytes produce IL‐4, IL‐5, IL‐9 and IL‐13, known to influence B cell activation and antibody production and function in the defence against extracellular parasites including helminths. Th17 cells secrete IL‐17, IL‐21 and IL‐22 and participate in the defence against extracellular bacteria and fungi and in autoimmune diseases in similar ways to Th1 cells. Treg are important in self‐tolerance and immune homeostasis and they have the capacity to suppress innate and adaptive immune responses to autoantigens, alloantigens, tumour antigens and infectious agents (Vignali et al., 2008). They express the IL‐2 receptor α‐subunit (CD25) and the transcription factor forkhead box P3 (Foxp3). The suppressive actions of Treg are mediated by cell–cell contact mechanisms and/or via the release of anti‐inflammatory cytokines IL‐10, IL‐35 and TGF‐β (Schiffrin, 2014). Treg include natural Treg, which develop in the thymus and constitute the majority of circulating Treg, and inducible Treg (also known as adaptive, peripheral or iTreg). Upon activation with a MHC I‐restricted antigen, naïve CD8+ T cells mature into cytotoxic T (Tc) cells that secrete IFN‐γ and TNF‐α, perforin and granzyme B. Perforin creates pores within the target cell membranes, through which granzymes can enter and induce apoptosis, leading to increased circulating apoptotic bodies, another manner of inducing inflammation (Zhang and Bevan, 2011).
Memory T lymphocytes
Some activated T cells become memory T cells that could be reactivated upon subsequent interaction with previously encountered antigen (Devarajan and Chen, 2013). Memory lymphocytes express CD44 in mice (CD45RO in humans). Two memory T cell subsets have been classically identified based on CD62L and CCR7 homing receptors that permit entry into lymphoid tissues. Central memory T cells are CD62L+CCR7+ and are found in lymphoid organs, whereas effector memory T (TEM) cells are CD62L−CCR7− and are able to recirculate between the lymphoid tissues, blood and peripheral organs. There is another subset of memory T cells that reside permanently in peripheral tissues, tissue‐resident memory T (TRM) cells. TRM are CD62L−CCR7− like TEM but CD69+CD109+. TEM cells have been shown to produce the pro‐inflammatory cytokines IFN‐γ and IL‐17A in autoimmune diseases.
B lymphocytes
B lymphocytes, like T cells, express a specific receptor on the cell surface, the B cell receptor (BCR). They also express the B cell co‐receptor (protein complex consisting of CD19, CD21 and CD81). However, B cells differ from T cells in that they recognize antigens in their native forms. The binding of an antigen on the BCR engages B cells to differentiate into short‐lived plasmablasts for immediate production of antibodies (also known as Ig) or in long‐lived plasma cells and memory B cells for persistent protection. B cells could be activated with the help of T cells (T cell‐dependent) or in their absence (T cell‐independent).
T and B lymphocytes in hypertension
Svendsen (1976) made the first demonstration of a role of T cells in hypertension. DOCA‐salt treatment had no effect on BP in nude mice, an athymic animal with no T cells. A graft of thymus from wild‐type into nude mice restored the DOCA‐salt‐induced BP elevation response (Svendsen, 1976). In 2007, Guzik et al. demonstrated a role for T cells in hypertension using mice lacking the recombination activating gene‐1 that plays an important role in the rearrangement and recombination of the genes of Igs and the TCR, and as a result are deficient in T and B cells (Guzik et al., 2007). Ang II‐induced BP elevation, endothelial dysfunction and vascular remodelling, and superoxide production were reduced in Rag1 −/− mice, but could be restored by adoptive transfer of T but not B cells. However, a role of B cells in hypertension was recently suggested (Chan et al., 2015). Twenty‐eight‐day Ang II infusion caused an increase in activated B cells (CD19+CD86+), plasmablasts and plasma cells in the spleen and IgG in circulation and aorta. BP elevation was attenuated, and the increase in aortic macrophage infiltration, collagen deposition and stiffening were prevented in Ang II‐infused B‐cell activating factor knockout mice that are deficient in B but not T cells. Altogether, these results suggest that activation of B cells in hypertension is T cell‐dependent (Guzik et al., 2007; Chan et al., 2015). B cells could contribute to the elevation in BP and generate a pro‐inflammatory environment leading to vascular injury.
Memory T lymphocytes in hypertension
Immunological memory has been shown to play a role in hypertension. Finn Olsen reported BP elevation in control rats after transfer of splenocytes from DOCA‐salt hypertensive rats and from rats rendered hypertensive by unilateral nephrectomy and ligation of a branch of the renal artery of the remaining kidney (Olsen, 1980). Recently, memory T cells were shown to be increased in the aorta and kidney at a pre‐hypertensive stage and in the kidney after development of hypertension and renal injury caused by Ang II infusion in mice (Carnevale et al., 2014; Xiao et al., 2015). Also, TEM cells developed during an initial hypertensive episode sensitizing mice to develop hypertension to a second mild hypertensive challenge (Itani et al., 2016b). This was demonstrated in mice subjected to an initial exposure to L‐NAME or a hypertensive dose of Ang II, followed by a washout period and subsequent high‐salt diet or sub‐pressor dose of Ang II respectively. Repeated hypertensive challenges caused CD4+ and CD8+ TEM cell accumulation in the kidney and bone marrow and increased the production of IL‐17A and IFN‐γ in kidney‐infiltrating CD4+ and CD8+ TEM cells. Inhibition of TEM cell development by destruction of the APC CD70 ligand–T cell CD27 receptor interaction blocked the hypertensive responses to second mild hypertensive challenges. Finally, bone marrow transplantation experiments demonstrated that hypertension‐induced CD4+ and CD8+ TEM cells reside in the bone marrow and expand upon a second hypertensive challenge. TEM cells could have a role in human hypertension, as Ang II infusion caused BP elevation and TEM cell accumulation in the aorta, kidney and lymph nodes of humanized mice (that have a human immune system) (Itani et al., 2016a). As well, uncontrolled hypertensive patients with high plasma renin and aldosterone levels presented greater frequency of CD4+ and CD8+ TEM cells in the blood compared to normotensive subjects.
CD4+ TEM lymphocytes could play an important role in cholinergic anti‐inflammatory pathways and BP homoeostasis (Okusa et al., 2017). Sensory afferent vagal nerve endings detect PAMPs, Igs, pro‐inflammatory cytokines and ATP. The danger signals are transmitted to activate neurons of the brainstem nucleus tractus solitarius, which causes activation of efferent vagal nerve fibres, generating signals that suppress monocyte/macrophage production of inflammatory cytokines in the reticuloendothelial system. The spleen is a major component of this system and a source of TNF‐α. However, the efferent vagal nerve fibres do not innervate the spleen directly but end in the celiac ganglion. Efferent vagal signals are transmitted to the spleen by noradrenergic nerves that end in the white pulp around splenic T cells. Rosas‐Ballina et al. (2011) discovered that a subpopulation of CD4+ TEM cells expressing choline acetyltransferase (ChAT) mediate the cholinergic anti‐inflammatory pathway in the spleen. Upon activation by noradrenaline via β2‐adrenoceptors, CD4+ ChAT+ TEM cells release acetylcholine, which binds to α7‐nicotinic acetylcholine receptors on macrophages and suppresses LPS‐induced TNF‐α in the serum and spleen. This response was blocked by deletion of the corresponding Chrna7 gene. Recently, Olofsson et al. (2016) show that BP was higher in mice lacking ChAT in CD4+ T cells than in control mice, and infusion of Jurkat T cells overexpressing ChAT (JTChAT) decreased BP. They postulated that CD4+ ChAT+ TEM cells could decrease BP through an ACh‐induced vasodilatory mechanism. This was supported by experiments showing that co‐culture of JTChAT cells with endothelial cells increased endothelial NO synthase phosphorylation and NO production. However, these authors did not determine whether CD4+ ChAT+ TEM cells lower BP by decreasing pro‐inflammatory cytokine levels.
T cell receptor subtypes in hypertension
Study of TCR subtypes demonstrated a role for CD8+ but not CD4+ T cells in hypertension. Trott et al. discovered using TCR Vβ spectratyping analysis that Ang II infusion resulted in the clonal amplification of CD8+ TCR Vβ3, 8.1, and 17 variants in the kidneys but not in the spleen and mesenteric vascular arcade. Mice lacking CD8+ but not CD4+ T cells were protected against Ang II‐ and DOCA‐salt‐induced BP elevation, and Ang II‐induced BP elevation in Rag1 −/− mice previously injected with CD8+ T cells but not CD4+CD25− T cells. CD8+ T cells could also play a role in human hypertension. Newly diagnosed hypertensive patients presented greater frequency of circulating senescent (CD28 −CD57+) CD8+ but not CD4+ T cells compared to age‐ and sex‐matched normotensive controls (Youn et al., 2013). An increased frequency of CD8+ T cells expressing IFN‐γ, TNF‐α, perforin and granzyme B was also observed. However, it was recently reported that the frequency of circulating Th1 and Th17 cells was increased in uncontrolled hypertensive patients compared to normotensive subjects, which could implicate CD4+ T cells in human hypertension (Ji et al., 2017).
T regulatory cells in hypertension
Decreased immune suppressive activity could play a role in hypertension and cardiovascular injury (Idris‐Khodja et al., 2014). Treg are decreased in the kidney of animal models of hypertension (Barhoumi et al., 2011; Kasal et al., 2012). We and others have shown that adoptive transfer of Treg (CD4+CD25+ T cells) reduced Ang II‐ or aldosterone/salt‐induced BP elevation, including vascular and cardiac injury (Kvakan et al., 2009; Barhoumi et al., 2011; Matrougui et al., 2011; Kasal et al., 2012). Ang II‐induced vascular injury was exaggerated in Rag1 −/− mice reconstituted with T cells isolated from Scurfy mice that are devoid of Treg due to a mutation in Foxp3 (Mian et al., 2016). Activation of the complement system in hypertension could mediate the decrease in Foxp3+ Treg number and function. The complement system is an important component of the innate immune system that also plays an important role in the induction of adaptive immune responses (Cravedi et al., 2013; Killick et al., 2018). Complement components 3a (C3a) and 5a (C5a) are increased and play a role in end‐organ damage in different models of hypertension (Shagdarsuren et al., 2005; Zhang et al., 2014; Ruan et al., 2015). Recently, Chen et al. (2018) demonstrated using double knockout of the C3a (C3ar1) and C5a (C5ar1) receptors, depletion of Treg with anti‐CD25 antibodies and adoptive transfer of C3ar1 and C5ar1 deficient Treg that Ang II decreases Treg function via the binding of C3a and C5a to their cognate receptor on Treg, which causes a decrease in Foxp3 expression. C3ar1 C5ar1 double knockout blunted Ang II‐induced BP elevation, whereas a single injection of C3ar1 C5ar1 deficient Treg was sufficient to reduce BP elevation. Decrease in Treg could play a role in human hypertension. An inverse correlation was observed between the frequency of circulating CD4+CD25High CD127 Low Treg and vascular remodelling evaluated by the media/lumen ratio of small subcutaneous resistance arteries and also retinal arterioles in human essential hypertension (De Ciuceis et al., 2017). Altogether, these studies suggest that a loss of Treg contributes to the development of hypertension and end‐organ damage.
Innate lymphoid cells in hypertension
ILCs have been shown to play a role in initiation, regulation and resolution of inflammation (Juelke and Romagnani, 2015). ILCs are innate lymphocytes lacking rearranged antigen receptors. ILCs can be grouped according to surface markers, transcription factors and effector cytokines into three groups (ILC1, 2 and 3). IFN‐γ is produced by ILC1, IL‐5 and IL‐13 by ILC2, and IL‐17A and/or IL‐22 by ILC3. NK cells belong to the ILC1 and are the only ILCs for which a role in hypertension has been described (see above).
Unconventional T cells in hypertension
Unconventional T cells or non‐MHC restricted T cells include αβ TCR T cells such as CD1‐restricted T cells, MHC I related (MR1)‐restricted mucosal‐associated invariant T cells and MHC class Ib–reactive T cells, and γδ T cells that have an γδ TCR (Godfrey et al., 2015). These cells are unconventional T cells because they do not recognize peptide antigens and present a more limited TCR diversity and therefore a higher precursor frequency, they have a tendency to reside in non‐lymphoid tissues and a faster response after activation including proliferation or cytokine release, and a faster response after a recall stimulation with an antigen compared to MHC‐restricted T cells, although their memory response is controversial. Unconventional T cells recognize non‐polymorphic antigen‐presenting molecules, some of which are encoded by genes outside of the MHC locus (CD1a, CD1b, CD1c, CD1d and MR1) or within the MHC locus (MHC I‐E, homeostatic iron regulator, H2‐M3 and H2‐Q9). Unconventional αβ TCR T cells will not be further discussed as no role in hypertension has been demonstrated for these cells as yet.
γδ T cells represent ~0.5–10% of circulating lymphocytes in humans and mice, are mostly CD4 and CD8 double negative and are activated by non‐protein phosphoantigens, isoprenoid pyrophosphates, alkylamines, non‐classical MHC class I molecules, MHC class I chain‐related proteins A and B, as well as heat shock protein‐derived peptides, without antigen processing and MHC presentation. γδ T cells are sequestered in many peripheral tissues, with specific homing and retention (Vantourout and Hayday, 2013; Chien et al., 2014; Krebs et al., 2014; Godfrey et al., 2015; Paul et al., 2015). For example, γδ T cells localized in the skin are known as dendritic epidermal T cells. These cells can perform professional antigen‐presenting functions similar to DCs and can at the same time produce IFN‐γ. γδ T cells are unique because, in addition to effector functions shared with αβ T cells, they are ‘innate‐like’ T lymphocytes with functions that span across the biology of innate and adaptive immune cells. γδ T cells can produce IL‐17A in response to the pro‐inflammatory cytokines IL‐1β and IL‐23 like Th17 cells, and both express CCR6. γδ T cells may produce IFN‐γ and typically express CD27.
Recently, we demonstrated that γδ T cells play a critical role in hypertension and vascular injury (Caillon et al., 2017). Ang II caused an increase in number and activation of γδ T cells after 7 days of Ang II infusion. Deficiency in γδ T cells due to Tcrδ knockout or injection of γδ T cell‐depleting antibodies prevented Ang II‐induced BP elevation, small artery endothelial dysfunction and spleen and mesenteric artery perivascular adipose tissue CD4+ and CD8+ T cell activation. This suggests that γδ T cells participate in the initiation of inflammatory responses involved in the development of hypertension. Moreover, using multiple linear regression, we found a correlation between whole blood expression of the TCR‐γ chain constant regions 1 and 2 that was used as an estimate of the frequency of blood γδ T cells, and systolic BP in a human cohort with and without coronary artery disease and a full range of BPs, from low to high (Caillon et al., 2017). γδ T cells are a major source of IL‐17A in the aorta, kidneys and the heart of Ang II‐infused mice (Li et al., 2014; Saleh et al., 2016). Li et al. (2014) demonstrated that depletion of γδ T cells using anti‐γδ TCR antibodies did not affect 7‐day Ang II infusion‐induced BP elevation but it reduced markedly the ventricular fibrosis induced by 7‐day and 4‐week Ang II infusions. The lack of effect on BP elevation might be due to the high dose of Ang II (1500 ng·kg−1·min−1) used in this study. Thus, γδ T cells could play a key pathophysiological role in hypertension and end‐organ damage.
Cytokines produced in hypertension by T cells
Some cytokines are thought to play a role in hypertension (McMaster et al., 2015), and here, we will focus on two cytokines produced by T cells, IL‐17A and IFN‐γ. Covering cytokines produced by cell lineages other than T cells is beyond the scope of the current review.
IL‐17A belongs to a group of six isoforms (A–F), of which IL‐17A and IL‐17F are the most closely related and generally produced by the same cells (Gaffen, 2009). IL‐17A is produced by Th17 cells, γδ T cells, Tc cells, NK cells, NK T cells and lymphoid tissue inducer cells. Madhur et al. (2010) demonstrated that Ang II infusion increased the production of IL‐17A in circulating T cells and aortic IL‐17A protein levels. Il17a knockout did not affect the initial BP elevation but reduced the level of BP elevation during the third and fourth weeks of Ang II infusion. IL‐17A also plays a role in end‐organ damage as lack of IL‐17A prevented Ang II‐induced aortic endothelial dysfunction, oxidative stress and infiltration of leukocytes and T cells. Similarly, Li et al. (2014) reported, in Ang II‐infused mice, a greater T‐cell production of IL‐17A mostly by infiltrating γδ T cells and that Il17a knockout affected high BP maintenance and reduced cardiac fibrosis. Recently, Saleh et al. (2016) demonstrated in Ang II‐infused mice that both IL‐17A and IL‐17F production was increased in aortic and renal infiltrating γδ T cells, CD4+ T cells, CD8+ T cells and CD4−CD8− T cells, cells in which γδ T cells were the main source of IL‐17A and IL‐17F. Furthermore, they demonstrated in Ang II‐infused mice that depleting IL‐17A or blocking its receptor with monoclonal antibodies affected the maintenance of high BP and reduced aortic and renal inflammation and renal injury, whereas depleting IL‐17F had no effect. Paradoxically, Krebs et al. (2014) have shown that Il17a knockout did not affect maintenance of high BP and exaggerated renal injury in a severe model of hypertensive end‐organ damage induced by a combination of DOCA‐salt and high dose Ang II infusion. The lack of effect of Il17a knockout on the maintenance of high BP could be due to the severity of the hypertensive model used to induce renal injury. Interestingly, this severe model of hypertensive end‐organ damage revealed a renal protective role of IL‐17A. Finally, IL‐17A could play a role in human hypertension since hypertensive diabetic patients presented greater IL‐17A plasma levels than normotensive diabetic patients (Madhur et al., 2010).
Type II IFN, IFN‐γ, is produced by Th1 cells, Tc cells, γδ T cells and NK T cells. The role of IFN‐γ in hypertension and end‐organ damage differs in function in hypertensive models and different mouse strains. INF‐γ was elevated in plasma and in kidney infiltrating CD8+ T cells but not CD4+ T cells of Ang II‐infused C57BL/6J mice (Saleh et al., 2015). Deletion of IFN‐γ receptors (Ifngr1) reduced cardiac hypertrophy, macrophage and T cell infiltration and fibrosis without affecting BP elevation caused by Ang II in 129SV mice (Marko et al., 2012). It had, however, divergent effects on the kidney. It reduced tubulointerstitial injury and improved GFR but worsened glomerular injury. However, Garcia et al. (2012) observed that IFN‐γ (Ifng) knockout slightly reduced BP elevation and exaggerated cardiac hypertrophy and diastolic dysfunction in uninephrectomized BALB/c mice treated with aldosterone and salt. Other studies have shown that Ifng knockout reduced Ang II‐induced BP elevation in C57BL/6J mice (Kamat et al., 2015; Saleh et al., 2015). As well, Ifng knockout prevented Ang II‐induced proximal tubule sodium reabsorption (Kamat et al., 2015). Finally, IFN‐γ plays a role in immunological memory as shown using repeated hypertensive challenges (Itani et al., 2016b). CD4+ and CD8+ TEM cells that expressed IFN‐γ were increased in the kidney following treatment with L‐NAME/washout/high‐salt diet. Ifng knockout did not affect BP elevation during the first hypertensive challenge (L‐NAME treatment) but prevented the subsequent rise in BP caused by the second hypertensive challenge (high‐salt diet) and prevented kidney accumulation of CD4+ and CD8+ TEM cells.
Role of chemokines on immune cell infiltration in hypertension
Infiltration of immune cells in perivascular adipose tissue and the kidney plays an important role in the development of hypertension and end‐organ damage. Immune cells expressing appropriate chemokine receptors are attracted to injury sites by increasing gradients of chemokines (Olson and Ley, 2002). Forty‐five chemokine ligands (L) and 15 chemokine receptors (R) have been identified and are divided into four subgroups based on the spacing of conserved cysteine residues: CC, CXC, C and CX3C. The chemokine ligand–receptor pairings are complex as most chemokines bind to more than one receptor and vice versa. Few chemokine ligands and receptors have been studied in hypertension (Rudemiller and Crowley, 2017).
The chemokine CCL2 guides neutrophils, monocytes and T cells to inflammation sites via CCR2. In hypertension, CCL2 and CCR2 expression is increased in cardiovascular cells and in circulating monocytes respectively (Rudemiller and Crowley, 2017). Ccr2 knockout experiments have shown that CCR2 plays a role in the development of vascular remodelling and kidney injury in hypertension but not in BP elevation (Bush et al., 2000; Ishibashi et al., 2004; Liao et al., 2008). However, the study of CCR2 might be complicated by the fact that it also binds CCL7, CCL8, CCL12 and CCL13 (Olson and Ley, 2002).
CCL5 attracts monocytes and T cells to inflammation sites. Increased CCL5 expression and infiltration of T cells expressing CCL5 receptors CCR1, CCR3 and CCR5 in perivascular adipose tissue has been reported in Ang II‐infused mice (Mikolajczyk et al., 2016). Ccl5 knockout reduced Ang II‐induced vascular injury and perivascular infiltration of IFN‐γ‐expressing CD8+ and CD3+CD4−CD8− T cells without altering BP elevation. Surprisingly, Rudemiller et al. (2016) showed that although Ccl5 knockout did not affect BP elevation, it exaggerated renal macrophage infiltration and kidney injury after 4 weeks of Ang II infusion in uninephrectomized mice. CCL5 could have different roles in different tissues and be protective in the kidney.
CXCR3 binds CXCL9, CXCL10 and CXCL11, which guide T cells, B cells and NK cells to inflammation sites (Olson and Ley, 2002). Hsu et al. demonstrated that Cxcr3 null mice present greater basal BP, which was further increased by an acute sodium load or when fed a 0.8% sodium diet. Increased contractile responses to Ang II and decreased relaxation responses to ACh were found in mesenteric arteries of Cxcr3 −/− mice. Whether CXCR3 plays a role in hypertension is unclear. This study was done using C57BL/6 mice, a strain resistant to salt‐induced hypertension. It could have been more instructive to use a more appropriate hypertensive model. As CXCR3 is also expressed on endothelial cells and vascular smooth muscle cells, Cxcr3 knockout could have affected BP homoeostasis.
Recently, Wang et al. (2016) demonstrated an important role for the CXCL1‐CXCR2 axis in hypertension and vascular injury, as they reported that Cxcl1 was the most highly expressed chemokine gene in mouse aorta among 13 examined after 1 day of Ang II infusion. However, CXCL1 producing cells were not characterized. Furthermore, Ang II infusion increased infiltration of leukocytes expressing the CXCL1 receptor CXCR2. Cxcr2 knockout, or selective CXCR2 inhibition with SB265610, blunted Ang II‐induced BP elevation, aortic injury and macrophage and T cell infiltration. Cxcr2 knockout was also effective in preventing DOCA/salt hypertension and vascular injury. Bone marrow transplantation experiments demonstrated that it is CXCR2 expressed on immune cells that plays a role in hypertension and vascular injury. CXCR2 may be an interesting therapeutic target since these authors observed that numbers of monocytes, neutrophils and macrophages expressing CXCR2 were increased in the blood of hypertensive patients compared to normotensive subjects. Using the same experimental design, Wang et al. (2018) extended the role of CXCL1‐CXCR2 axis to hypertension, and cardiac hypertrophy and remodelling.
Mechanisms of immune system activation in hypertension
The mechanism of activation of the immune system, i.e., what actually triggers its involvement in hypertension, remains unclear. However, it has been postulated that the initial BP elevation and/or pro‐hypertensive stimuli induce cardiovascular injury will contribute to the production of DAMPs and neoantigens. In 2015, Harrison's group proposed a mechanism of formation of neoantigens (Kirabo et al., 2014; McMaster et al., 2015). Ang II causes ROS generation in DCs, leading to formation of γ‐ketoaldehydes or isoketals, which react rapidly with the lysine residues of proteins in injured tissue to form isoketal protein adducts that are recognized as non‐self. These isoketal protein adducts enhance DC immunogenicity and promote DC release of IL‐6, IL‐1β and IL‐23, causing proliferation of T cells and production of IL‐17A, IFN‐γ and TNF‐α. Interestingly, treatment of mice with the isoketal scavenger 2‐hydroxybenzylamine prevented BP elevation induced by Ang II infusion and DOCA‐salt. These authors also reported that isoketal protein adducts were elevated in circulating monocytes and DCs from hypertensive humans. More recently, it was demonstrated in vitro that like Ang II, increased extracellular sodium through amiloride‐sensitive sodium channels can cause ROS generation in DCs, formation of isoketal protein adducts and production of IL‐1β (Barbaro et al., 2017). Furthermore, these authors showed that high extracellular sodium‐stimulated DCs could activate T cells isolated from mice in which salt‐sensitive hypertension was induced, but not T cells from untreated mice. Finally, adoptive transfer of high extracellular sodium‐activated DCs sensitized mice to develop hypertension to a subpressor dose of Ang II. However, whether high sodium intake increases BP through activation of DCs in vivo remains to be demonstrated.
Conclusion
Pro‐inflammatory cytokines play an important role in the different phases of the low‐grade inflammation found in cardiovascular disease and hypertension. Interfering with some of these stages in the evolution of hypertension blunts the inflammatory process and prevents vascular injury in hypertensive mouse models and could also be beneficial in human cardiovascular disease. This process may be visualized in terms of inflammatory vicious cycles (Figure 1). Genetic predisposition, or environmental influences such as salt, which could modify gene expression epigenetically or directly, influence pro‐hypertensive mechanisms, such as the renin‐angiotensin system, and acting on the brain may increase sympathetic activity and decrease parasympathetic activity, thus producing minor BP elevations. Over time, high BP and/or the pro‐hypertensive stimuli induce cardiovascular injury, which will contribute to the production of DAMPs that favour activation of immune antigen recognition by PRRs such as TLRs. Furthermore, cardiovascular injury, together with increased ROS production caused by vasoactive peptides such as Ang II or endothelin‐1, induce pro‐inflammatory signalling pathways by protein oxidation and favour the production of neoantigens, such as those resulting from isoketal‐induced protein modification, which enhances DC immunogenicity and promotes DC release of IL‐6, IL‐1β and IL‐23, causing proliferation of T cells and production of IL‐17A, IFN‐γ and TNF‐α. The first part of the loop could correspond to the production of pro‐inflammatory cytokines by APCs and activation of MHC‐restricted T cells. This step would appear to be critical in the development of hypertension. In parallel, activation of γδ T cells could directly prime innate and adaptive immune cells bypassing canonical pathways, stimulated by generic ‘public’ antigens, acting as a bridge between the innate and adaptive immune systems. Opportunistic diseases such as periodontitis could also exacerbate the activation of the innate immune system through binding of PAMPs to PRR such as TLRs. Once Th1 and Th17 lymphocytes are activated, the inflammatory cascade has been triggered and the immune‐mediated injury of the kidney and vasculature closes the pro‐hypertensive circle that is a feed‐forward process, resulting in progressive BP elevation. During this process, TEM cells are generated and stored in lymphoid organs including the bone marrow. Upon a second hit such as high‐salt intake, TEM cells could be reactivated and contribute to the vicious cycle. Activation of B cells could also participate in the vicious circle through production of auto‐antibodies that could participate in the development of additional cardiovascular injury. Throughout this process, anti‐inflammatory cells such as Treg, MDSCs and M2 could provide homeostatic fine‐tuning of the inflammatory process in the blood vessels and the kidney. However, this anti‐inflammatory mechanism could be rendered unable to counteract the development of hypertension by factors such as Ang II that can cause a decrease in number and function of Treg through activation of the complement system.
The future lies in the identification of subsets of these different cellular types and mechanisms of activation, their differential effects and mediators, which could allow the development of immune‐based therapies that are devoid of adverse side‐effects but contribute to the healing of vascular and kidney injury in hypertension and cardiovascular disease, leading to improved patient outcomes.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d, 2017e).
Acknowledgements
The authors' work was supported by Canadian Institutes of Health Research (CIHR) grants 102606 and 123465, CIHR First Pilot Foundation grant 143348, a Canada Research Chair (CRC) on Hypertension and Vascular Research by the CRC Government of Canada/CIHR Program, and by the Canada Fund for Innovation (all to ELS), and by a fellowship to A.C. (Canadian Vascular Network).
Conflict of interest
The authors declare no conflicts of interest.
Caillon A., Paradis P., and Schiffrin E. L. (2019) Role of immune cells in hypertension, British Journal of Pharmacology, 176, 1818–1828. 10.1111/bph.14427.
References
- Alexander SP, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The concise guide to pharmacology 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The concise guide to pharmacology 2017/18: Nuclear hormone receptors. Br J Pharmacol 174 (Suppl 1): S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The concise guide to pharmacology 2017/18: Catalytic receptors. Br J Pharmacol 174 (Suppl 1): S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The concise guide to pharmacology 2017/18: Enzymes. Br J Pharmacol 174 (Suppl 1): S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017e). The concise guide to pharmacology 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174 (Suppl 1): S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L et al (2017). Dendritic cell amiloride‐sensitive channels mediate sodium‐induced inflammation and hypertension. Cell Rep 21: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF et al (2011). T regulatory lymphocytes prevent angiotensin II‐induced hypertension and vascular injury. Hypertension 57: 469–476. [DOI] [PubMed] [Google Scholar]
- Bush E, Maeda N, Kuziel WA, Dawson TC, Wilcox JN, DeLeon H et al (2000). CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II‐induced hypertension. Hypertension 36: 360–363. [DOI] [PubMed] [Google Scholar]
- Caillon A, Mian MOR, Fraulob‐Aquino JC, Huo KG, Barhoumi T, Ouerd S et al (2017). γδ T cells mediate angiotensin II‐induced hypertension and vascular injury. Circulation 135: 2155–2162. [DOI] [PubMed] [Google Scholar]
- Caillon A, Schiffrin EL (2016). Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep 18: 21. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Pallante F, Fardella V, Fardella S, Iacobucci R, Federici M et al (2014). The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 41: 737–752. [DOI] [PubMed] [Google Scholar]
- Ceeraz S, Nowak EC, Noelle RJ (2013). B7 family checkpoint regulators in immune regulation and disease. Trends Immunol 34: 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H et al (2015). Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB et al (2018). Deficiency of complement C3a and C5a receptors prevents angiotensin II‐induced hypertension via regulatory T cells. Circ Res 122: 970–983. [DOI] [PubMed] [Google Scholar]
- Chien YH, Meyer C, Bonneville M (2014). γδ T cells: first line of defense and beyond. Annu Rev Immunol 32: 121–155. [DOI] [PubMed] [Google Scholar]
- Cravedi P, van der Touw W, Heeger PS (2013). Complement regulation of T‐cell alloimmunity. Semin Nephrol 33: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL (2005). Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II‐infused macrophage colony‐stimulating factor‐deficient mice: evidence for a role in inflammation in angiotensin‐induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113. [DOI] [PubMed] [Google Scholar]
- De Ciuceis C, Rossini C, Airo P, Scarsi M, Tincani A, Tiberio GA et al (2017). Relationship between different subpopulations of circulating CD4+ T‐lymphocytes and microvascular structural alterations in humans. Am J Hypertens 30: 51–60. [DOI] [PubMed] [Google Scholar]
- Devarajan P, Chen Z (2013). Autoimmune effector memory T cells: the bad and the good. Immunol Res 57: 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S (2009). Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL (2009). Structure and signalling in the IL‐17 receptor family. Nat Rev Immunol 9: 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N et al (2012). Interferon‐γ ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol 303: H587–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB (2015). The burgeoning family of unconventional T cells. Nat Immunol 16: 1114–1123. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton HR, Chtanova T (2016). The lymph node neutrophil. Semin Immunol 28: 129–136. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R et al (2018). Myeloid CD11c(+) antigen‐presenting cells ablation prevents hypertension in response to angiotensin II plus high‐salt diet. Hypertension 71: 709–718. [DOI] [PubMed] [Google Scholar]
- Idris‐Khodja N, Mian MO, Paradis P, Schiffrin EL (2014). Dual opposing roles of adaptive immunity in hypertension. Eur Heart J 35: 1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S et al (2004). Critical role of monocyte chemoattractant protein‐1 receptor CCR2 on monocytes in hypertension‐induced vascular inflammation and remodeling. Circ Res 94: 1203–1210. [DOI] [PubMed] [Google Scholar]
- Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM et al (2016a). Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 68: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL et al (2016b). CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res 118: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CAJ, Travers P, Walport M, Shlomchik MJ (2001). Immunobiology, 5th edn. Garland Science: New York and London. [Google Scholar]
- Ji Q, Cheng G, Ma N, Huang Y, Lin Y, Zhou Q et al (2017). Circulating Th1, Th2, and Th17 levels in hypertensive patients. Dis Markers 2017: 7146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juelke K, Romagnani C (2015). Differentiation of human innate lymphoid cells (ILCs). Curr Opin Immunol 38: 75–85. [DOI] [PubMed] [Google Scholar]
- Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS et al (2015). Renal transporter activation during angiotensin‐II hypertension is blunted in interferon‐gamma−/− and interleukin‐17A−/− mice. Hypertension 65: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A et al (2012). T regulatory lymphocytes prevent aldosterone‐induced vascular injury. Hypertension 59: 324–330. [DOI] [PubMed] [Google Scholar]
- Killick J, Morisse G, Sieger D, Astier AL (2018). Complement as a regulator of adaptive immunity. Semin Immunopathol 40: 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J et al (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM et al (2007). Resistance artery remodeling in deoxycorticosterone acetate‐salt hypertension is dependent on vascular inflammation: evidence from m‐CSF‐deficient mice. Am J Physiol Heart Circ Physiol 292: H1789–H1795. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M et al (2013). Angiotensin II‐induced vascular dysfunction depends on interferon‐γ‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 33: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer‐Schwesinger C et al (2014). Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin II‐induced hypertension. Hypertension 63: 565–571. [DOI] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al (2009). Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 119: 2904–2912. [DOI] [PubMed] [Google Scholar]
- Leibowitz A, Schiffrin EL (2011). Immune mechanisms in hypertension. Curr Hypertens Rep 13: 465–472. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J et al (2014). γδ T cell‐derived interleukin‐17A via an interleukin‐1β‐dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension 64: 305–314. [DOI] [PubMed] [Google Scholar]
- Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA et al (2008). Role of inflammation in the development of renal damage and dysfunction in angiotensin II‐induced hypertension. Hypertension 52: 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair‐Rohani H et al (2012). A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Q, Wu H, Du H, Liu L, Shi H et al (2015). Blood neutrophil to lymphocyte ratio as a predictor of hypertension. Am J Hypertens 28: 1339–1346. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al (2012). Interferon‐γ signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez‐Villalobos RA et al (2011). Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol 178: 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMaster WG, Kirabo A, Madhur MS, Harrison DG (2015). Inflammation, immunity, and hypertensive end‐organ damage. Circ Res 116: 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL (2016). Deficiency of T‐regulatory cells exaggerates angiotensin II‐induced microvascular injury by enhancing immune responses. J Hypertens 34: 97–108. [DOI] [PubMed] [Google Scholar]
- Mian MO, Paradis P, Schiffrin EL (2014). Innate immunity in hypertension. Curr Hypertens Rep 16: 413. [DOI] [PubMed] [Google Scholar]
- Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D et al (2016). Role of chemokine RANTES in the regulation of perivascular inflammation, T‐cell accumulation, and vascular dysfunction in hypertension. FASEB J 30: 1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018). The immunology of hypertension. J Exp Med 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okusa MD, Rosin DL, Tracey KJ (2017). Targeting neural reflex circuits in immunity to treat kidney disease. Nat Rev Nephrol 13: 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson PS, Steinberg BE, Sobbi R, Cox MA, Ahmed MN, Oswald M et al (2016). Blood pressure regulation by CD4(+) lymphocytes expressing choline acetyltransferase. Nat Biotechnol 34: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen F (1980). Transfer of arterial hypertension by splenic cells from DOCA‐salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C 88: 1–5. [DOI] [PubMed] [Google Scholar]
- Olson TS, Ley K (2002). Chemokines and chemokine receptors in leukocyte trafficking. Am J Physiol Regul Integr Comp Physiol 283: R7–R28. [DOI] [PubMed] [Google Scholar]
- Paul S, Shilpi, Lal G (2015). Role of γ‐δ (γδ) T cells in autoimmunity. J Leukoc Biol 97: 259–271. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Pons H, Johnson RJ (2017). Role of the immune system in hypertension. Physiol Rev 97: 1127–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales C (2018). Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol 9: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas‐Ballina M, Olofsson PS, Ochani M, Valdes‐Ferrer SI, Levine YA, Reardon C et al (2011). Acetylcholine‐synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan CC, Ge Q, Li Y, Li XD, Chen DR, Ji KD et al (2015). Complement‐mediated macrophage polarization in perivascular adipose tissue contributes to vascular injury in deoxycorticosterone acetate‐salt mice. Arterioscler Thromb Vasc Biol 35: 598–606. [DOI] [PubMed] [Google Scholar]
- Rudemiller NP, Crowley SD (2017). The role of chemokines in hypertension and consequent target organ damage. Pharmacol Res 119: 404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudemiller NP, Patel MB, Zhang JD, Jeffs AD, Karlovich NS, Griffiths R et al (2016). C–C motif chemokine 5 attenuates angiotensin ii‐dependent kidney injury by limiting renal macrophage infiltration. Am J Pathol 186: 2846–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR et al (2015). Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end‐organ inflammation. J Clin Invest 125: 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Norlander AE, Madhur MS (2016). Inhibition of interleukin 17‐A but not interleukin‐17F signaling lowers blood pressure and reduces end‐organ inflammation in angiotensin II‐induced hypertension. JACC Basic Transl Sci 1: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffrin EL (2014). Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 126: 267–274. [DOI] [PubMed] [Google Scholar]
- Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N et al (2005). Complement activation in angiotensin II‐induced organ damage. Circ Res 97: 716–724. [DOI] [PubMed] [Google Scholar]
- Shah KH, Shi P, Giani JF, Janjulia T, Bernstein EA, Li Y et al (2015). Myeloid suppressor cells accumulate and regulate blood pressure in hypertension. Circ Res 117: 858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Liu YJ (2002). Mouse and human dendritic cell subtypes. Nat Rev Immunol 2: 151–161. [DOI] [PubMed] [Google Scholar]
- Svendsen UG (1976). Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A 84: 523–528. [DOI] [PubMed] [Google Scholar]
- Vantourout P, Hayday A (2013). Six‐of‐the‐best: unique contributions of γδ T cells to immunology. Nat Rev Immunol 13: 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali DA, Collison LW, Workman CJ (2008). How regulatory T cells work. Nat Rev Immunol 8: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL et al (2011). Innate or adaptive immunity? The example of natural killer cells. Science 331: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL et al (2018). CXCL1‐CXCR2 axis mediates angiotensin II‐induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur Heart J 39: 1818–1831. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhao XC, Cui W, Ma YQ, Ren HL, Zhou X et al (2016). Genetic and pharmacologic inhibition of the chemokine receptor CXCR2 prevents experimental hypertension and vascular dysfunction. Circulation 134: 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F et al (2015). Renal denervation prevents immune cell activation and renal inflammation in angiotensin II‐induced hypertension. Circ Res 117: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY et al (2013). Immunosenescent CD8+ T cells and C‐X‐C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62: 126–133. [DOI] [PubMed] [Google Scholar]
- Zhang C, Li Y, Wang C, Wu Y, Cui W, Miwa T et al (2014). Complement 5a receptor mediates angiotensin II‐induced cardiac inflammation and remodeling. Arterioscler Thromb Vasc Biol 34: 1240–1248. [DOI] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35: 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]