

Abstract
The immune system plays a prominent role in the initiation and maintenance of hypertension. The innate immune system, via toll‐like receptors (TLRs), identifies distinct signatures of invading microbes and damage‐associated molecular patterns and triggers a chain of downstream signalling cascades, leading to secretion of pro‐inflammatory cytokines and shaping the adaptive immune response. Over the past decade, a dysfunctional TLR‐mediated response, particularly via TLR4, has been suggested to support a chronic inflammatory state in hypertension, inducing deleterious local and systemic effects in host cells and tissues and contributing to disease progression. While the underlying mechanisms triggering TLR4 need further research, evidence suggests that sustained elevations in BP disrupt homeostasis, releasing endogenous TLR4 ligands in hypertension. In this review, we discuss the emerging role of TLR4 in the pathogenesis of hypertension and whether targeting this receptor and its signalling pathways could offer a therapeutic strategy for management of this multifaceted disease.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- AngII
angiotensin II
- AT1 receptor
angiotensin II type 1 receptor
- DAMPs
damage‐associated molecular patterns
- ECs
endothelial cells
- HMGB1
high‐mobility group box 1
- HSPs
heat shock proteins
- IKK
IκB kinase
- iNOS
inducible NOS
- IRAK
IL‐1 receptor‐associated kinase
- IRF
IFN regulatory factor
- MAL
myeloid differentiation primary response 88 adaptor‐like protein
- MAP
mean arterial pressure
- MCP‐1
monocyte chemoattractant protein 1
- MD2
myeloid differentiation factor 2
- MyD88
myeloid differentiation primary response 88
- PA
pulmonary artery
- PAH
pulmonary arterial hypertension
- PASMCs
pulmonary arterial smooth muscle cells
- PE
pre‐eclampsia
- PH
pulmonary hypertension
- PRRs
pattern recognition receptors
- PVN
paraventricular nucleus of the hypothalamus
- RAS
renin–angiotensin system
- RIPK1
receptor‐interacting protein 1
- SAPKs
stress‐activated protein kinases
- SHR
spontaneously hypertensive rat
- TAK1
TGFβ‐activated kinase 1
- TIR
toll–IL‐1 receptor
- TIRAP
toll–IL‐1 receptor domain‐containing adaptor protein
- TLR
toll‐like receptor
- TRAF
TNF receptor‐associated factor
- TRAM/TICAM‐2
toll–IL‐1 receptor domain‐containing adaptor‐inducing IFN‐β‐related adaptor molecule
- TRIF/TICAM‐1
toll–IL‐1 receptor domain‐containing adaptor‐inducing IFN‐β
- VSMCs
vascular smooth muscle cells
Introduction
Hypertension, one of the most prevalent cardiovascular risk factors, can exhibit numerous pathophysiologies and is typically attributed to dysregulation of the cardiovascular and renal systems, and the CNS (Dominiczak and Kuo, 2018). Such dysregulation has been known to be associated with abnormal immune system activity for more than a half century. A proper immune response requires precise coordination of the innate and adaptive elements of immunity. The contributions of aberrant activation of adaptive immunity to high BP have been well documented in various models of hypertension (Harrison et al., 2010; Idris‐Khodja et al., 2014; Lopez Gelston and Mitchell, 2017; Norlander et al., 2018). However, the molecular mechanisms that activate the innate immune system and lead to priming/activation of the adaptive immune response in hypertension are still not fully understood.
Recent work reveals a core and pathogenic role for innate immune system activity in hypertension (Bomfim et al., 2017; Lopez Gelston and Mitchell, 2017; Norlander et al., 2018). Evidence suggests that the receptors of innate immune cells act as gateways in hypertension, promoting end‐organ damage through the propagation of chronic inflammation, oxidative stress and vascular remodelling (Abais‐Battad et al., 2017; Bomfim et al., 2017; Nosalski et al., 2017). The initial response to potential pathogens and tissue damage is determined by the innate immune cells through pattern recognition receptors (PRRs), which include the toll‐like receptors (TLRs) (Kawai and Akira, 2010). In the absence of stimulation, TLRs are primarily expressed at either the cell membrane (TLR1, TLR2, TLR4–TLR6 and TLR9–TLR12) or within endosomes (TLR3, TLR7, TLR8 and TLR9) (McGettrick and O'Neill, 2010). TLR activation occurs in response to both exogenous pathogen‐associated molecular patterns and endogenous molecules released by cells following tissue damage, termed damage‐associated molecular patterns (DAMPs) (Matzinger, 2002; Kawai and Akira, 2010). DAMP‐stimulated TLR activation has gained much attention as a central component of hypertension pathogenesis (McCarthy et al., 2014). In particular, persistent activation of TLR4, resulting in low‐grade chronic inflammation, has been linked to significant kidney, cardiovascular and CNS tissue damage within the context of hypertension (Figure 1) (McCarthy et al., 2014; Biancardi et al., 2017). Thus, the TLR4 signalling pathway may allow pharmaceutical targeting of the innate immune system through direct TLR4 modulation and/or signal transduction inhibitors. In this review, we will focus on recent findings regarding the TLR4 signalling pathways, discussing the putative role for their various components in hypertension as well as advancements in potential target discovery within TLR4‐mediated hypertension.
Figure 1.
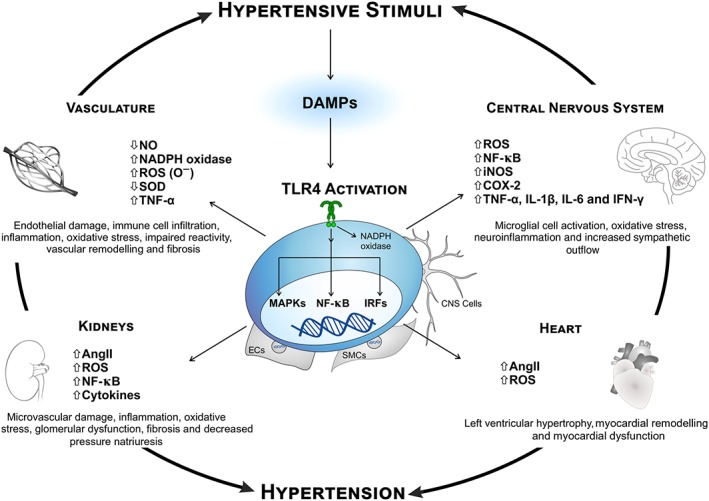
Summary of effect of TLR4 overactivation on the vasculature, CNS, kidneys and heart. An up‐regulation of TLR4 in the systems depicted increases pro‐inflammatory mediator levels, leading to deleterious effects in hypertension. SMCs, smooth muscle cells.
DAMP‐mediated TLR4 activation in hypertension
When homeostatic disturbances inflict tissue insult, whether via cellular stress or direct damage, DAMPs are released from the site of insult and act as PRR ligands. Among the molecules classified as DAMPs are cell‐derived nucleic acids, fatty acids, heat shock proteins (HSPs) and high‐mobility group box 1 (HMGB1), as well as components of the extracellular matrix (ECM), such as proteoglycans, hyaluronic acid and fibronectin (Kawai and Akira, 2011). The activation of TLRs by DAMPs plays an inherently protective role, alerting cells to damage for its resolution and repair. However, excessive or prolonged DAMP‐mediated stimulation of these innate immune system receptors provokes a chronic inflammatory state that contributes to the maintenance of hypertension (McCarthy et al., 2014). Through intracellular adaptor protein‐dependent signal cascades, DAMP‐induced TLR activation increases the expression of pro‐inflammatory genes (Akira and Takeda, 2004). TLR4 is unique in this sense, being the only TLR known to recruit four adaptor molecules and signal through two distinct pathways to produce pro‐inflammatory cytokines and chemokines (Kawai and Akira, 2010). Of particular interest, a variety of cell types that have long‐been associated with cardiovascular diseases have been found to express TLR4, such as macrophages, renal epithelial cells, cardiomyocytes, vascular smooth muscle cells (VSMCs), endothelial cells (ECs), glial cells and neurons (Vaure and Liu, 2014). Indeed, a myriad of studies show that abnormal activation of TLR4, primarily by DAMPs, contributes to cardiovascular dysfunction and remodelling, kidney disease and CNS dysregulation. These studies form the basis of the suggestion that DAMP‐induced TLR4 stimulation may be the missing link between inflammation and hypertension.
In this review, we will examine a variety of organs and systems known to be impacted by DAMP‐mediated TLR4 activation during hypertension. Of note, it is not the purpose of this review to provide an in‐depth discussion of TLR4 endogenous ligands in hypertension but rather to concisely list those ligands specifically related to induction of TLR4's downstream pathways. Table 1 contains a summary of hypertension‐related DAMPs that are proposed to modulate TLR4.
Table 1.
DAMPs that can activate TLR4 in hypertension
| DAMP | Cell type/tissue (reference) |
|---|---|
| AngII | Arteries (Bomfim et al., 2012; De Batista et al., 2014; Hernanz et al., 2015), VSMCs (De Batista et al., 2014), PVN (Dange et al., 2014; Dange et al., 2015; Li et al., 2016a), corpus cavernosum (Nunes et al., 2017), mesangial cells (Wolf et al., 2006), tubular epithelial cells (Nair et al., 2015) and cardiomyocytes (Eißler et al., 2011) |
| ADMA | Adipocytes (Yang et al., 2009b) |
| C‐reactive protein | VSMCs (Liu et al., 2010b; Liu et al., 2011) |
| Fibrinogen | Cardiomyocytes (Li et al., 2009) and monocytes (Smiley et al., 2001) |
| Fibronectin‐EDA | Aorta (Doddapattar et al., 2015) |
| HMGB1 | ECs (Szasz et al., 2016) and macrophages (Park et al., 2004) |
| HSPs | VSMCs (Zhao et al., 2015) and cardiomyocytes (Kim et al., 2009) |
| Hyaluronan | ECs (Taylor et al., 2004) |
| Oxidized LDL | Macrophages (Miller et al., 2003) |
| Uric acid | Macrophages (Liu‐Bryan et al., 2005) |
ADMA, asymmetric dimethylarginine.
TLR4 signal transduction in hypertension
TLR4 contains an extracellular domain of leucine‐rich repeats and an intracellular toll–IL‐1 receptor (TIR) domain that is responsible for signal transmission. Once activated, TLR4 signal transduction occurs through both myeloid differentiation primary response 88 (MyD88)‐dependent and MyD88‐independent (TRIF‐dependent) pathways (Akira and Takeda, 2004). Together, the two arms of TLR4's signalling cascade induce the production and release of pro‐inflammatory cytokines, chemokines and costimulatory factors (pro‐inflammatory profile). For an overview of TLR4 signalling in hypertension, please refer to Figure 2.
Figure 2.
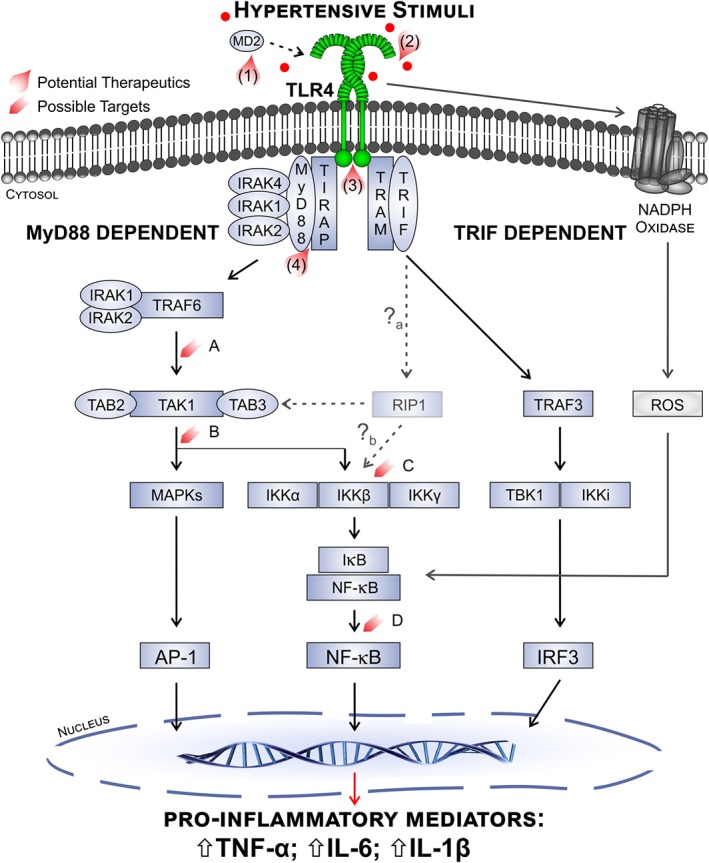
Overview of TLR4 signalling in hypertension and potential therapeutic targets. Activation of TLR4 by hypertensive stimuli, such as DAMPs, may involve a primary interaction with MD2. Stimulated TLR4 initiates the early MyD88‐dependent phase and late TRIF‐dependent phase cascades. In the MyD88‐dependent pathway, TIRAP associates with the receptor TIR domain, facilitating MyD88 association. MyD88 then recruits and activates IRAKs, leading to sequential stimulation of TRAF6 and TAK1. The downstream MAPK and IKK pathways ultimately result in the nuclear translocation of activator protein 1 (AP‐1) and NF‐κB transcription factors and production of pro‐inflammatory mediators. In the delayed TRIF‐dependent phase, recruitment of TRAM to the TIR domain allows TRIF binding, which up‐regulates IRF3 expression through TRAF3. TRAM recruitment also leads to NF‐κB activation through RIPK1 (RIP1). Although NF‐κB expression is shown to be increased in animal models of hypertension, it has not yet been determined whether RIPK1 activation is involved in hypertension (a), nor is the exact mechanism of RIPK1‐mediated NF‐κB activation fully understood (b). TLR4 stimulation also activates NADPH oxidase, increasing ROS production and thus ROS‐induced NF‐κB translocation. As a result of these signalling pathways, TLR4 stimulation causes the up‐regulation of pro‐inflammatory mediators, such as TNFα, IL‐6 and IL‐1β. Several molecules have been developed that inhibit TLR4 signalling and may be beneficial in hypertension management. Eritoran, tested in sepsis patients, binds to the pocket of the MD2 adaptor protein (1). NI‐0101 inhibits both exogenous and endogenous bindings and is currently in a phase III clinical trial for rheumatoid arthritis (2). TAK‐242 binds to the TIR domain of TLR4 and has been tested in clinical trial for sepsis (3). ST2825 inhibits the dimerization of MyD88, thus halting the MyD88‐dependent pathway (4). Other drugs (not shown here) have been developed that inhibit TLR4 binding of AngII, while several decrease TLR4 signalling through unknown mechanisms. Suggested therapeutic targets within the TLR4 signalling cascades are highlighted above (A–D) and discussed throughout the review.
Following activation, TLR4 recruits TIR domain‐containing adaptor/MyD88 adaptor‐like protein (TIRAP/MAL), which connects MyD88 to the TIR domain and initiates the MyD88‐dependent pathway (Yamamoto et al., 2002). MyD88 then recruits and activates IL‐1 receptor‐associated kinase (IRAK) 4, forming the myddosome (Suzuki et al., 2002). IRAK4 participates in the recruitment, phosphorylation and degradation of IRAK1 and IRAK2, after which these IRAKs dissociate from MyD88 to bind TNF receptor‐associated factor (TRAF) 6 (Kawagoe et al., 2007). TRAF6 is crucial for signal transduction downstream of IRAK4 and IRAK1/2 (Li et al., 2002). In combination with TGF‐β‐activated kinase 1 (TAK1) binding proteins 2 and 3, TAK1 triggers activation of the IκB kinase (IKK) complex and MAPK pathways (Kawai and Akira, 2011). Within the IKK pathway, two catalytic subunits, IKKα and IKKβ, along with one regulatory subunit (IKKγ/NEMO) degrade IκB proteins through phosphorylation and ubiquitination (Israël, 2010). This TLR4‐mediated activation of the IKK pathway results in early nuclear translocation and activation of NF‐κB (Oeckinghaus and Ghosh, 2009).
The influence of TLR4 upon downstream MAPKs may be a critical component of cytokine and chemokine production. The highly conserved MAPK family is typically divided into three subfamilies: (i) ERKs, (ii) JNKs and (iii) p38/stress‐activated protein kinases (SAPKs) (Morrison, 2012). Downstream of TLR4, the primary MAPKs appear to be JNKs and p38/SAPKs (Küper et al., 2012). MAPK‐stimulated transcriptional factors, activator protein 1 and cAMP‐responsive element protein, are intrinsically involved in cytokine and chemokine production, and TLR4‐induced transcriptional activation of COX‐2 is shown to depend upon MAPK signalling in cells of the renal collecting ducts (Küper et al., 2012).
Conversely, signal transduction through MyD88‐independent pathway involves recruitment of TIR domain‐containing adaptor‐inducing IFN‐β (TRIF/TICAM‐1) via TRIF‐related adaptor molecule (TRAM/TICAM‐2) (Yamamoto et al., 2003). TRIF interacts with TRAF3 to activate IKK pathways, resulting in translocation of NF‐κB, via adapter kinase receptor‐interacting protein (RIPK) 1 and IFN regulatory factor (IRF) 3 stimulation (Kawai and Akira, 2010). Phosphorylated IRF3 translocates to the nucleus and modulates the expression of type I IFNs, which are crucial to viral defence (Kumar et al., 2011; Chen et al., 2017).
Understanding TLR4 in BP regulation through hypertensive animal models
Animal models are an essential tool for understanding the pathophysiology of diseased states and for exploring new pharmacological interventions. Different rodent models have been used to study the role of TLR4 and its downstream signalling mechanisms in inflammation, oxidative stress, vascular remodelling, sympathetic overactivity and renal injury in the context of hypertension. The studies discussed herein were primarily conducted using animal models, particularly rodents, and, as such, care is required in translating their results to human applications. However, such animal‐based research continues to allow for the development of new pharmacological approaches to prevent and/or manage hypertension. With regard to TLR4, rodents and humans share approximately 60–70% similarity in the amino acid sequence of TLR4's extracellular and transmembrane domains (Vaure and Liu, 2014).
While the influence of TLR4 on BP control is not completely elucidated, TLR4 up‐regulation is suggested to contribute to the pathogenesis of hypertension in animal models. Spontaneously hypertensive rats (SHRs), a genetically hypertensive model, have proved crucial to evaluating TLR4 expression and cytokine profiles. SHRs are characterized by age‐dependent elevations in BP, reaching approximately 175–200 mmHg during the established phase of hypertension at 10–15 weeks of age (Okamoto and Aoki, 1963). SHRs are also susceptible to multiple types of organ damage, including cardiac hypertrophy and failure, impaired endothelium‐dependent vascular relaxation, increased sympathetic drive and renal dysfunction (Leong et al., 2015). In these animals, in vivo treatment against TLR4 has shown attenuation of some of the organ damage observed. For instance, systemic long‐term treatment with a TLR4 antibody lowered BP and decreased both cardiac hypercontractility and remodelling in SHRs (Bomfim et al., 2012; Bomfim et al., 2015). Furthermore, a study investigating TLR4 modulation of hypertension via CNS cardioregulatory centres showed that TLR4 inhibition in the paraventricular nucleus of the hypothalamus (PVN) decreases BP in SHRs (Dange et al., 2015; Wang et al., 2018).
Conversely, systemic TLR4 blockade in the angiotensin II (AngII) infusion model of hypertension results in less pronounced BP changes as compared with SHR. Systemic infusion of AngII progressively enhances BP, mainly due to increases in oxidative stress and vascular remodelling (Lerman et al., 2005). However, in AngII‐infused mice with a TLR4 deficiency, despite significant inhibition of vascular remodelling through reduced levels of ROS, TLR4 deficiency did not impact AngII's effects on BP (Nakashima et al., 2015). In corroboration with these results, AngII‐infused mice treated with a TLR4 antibody had superior vascular function but no difference in BP compared with untreated animals (Nunes et al., 2017). Based on the literature, there is a consensus that, in response to higher levels of AngII, TLR4 is an important contributor to vascular dysfunction and oxidative stress, both hallmarks of hypertension. Interestingly, systemic blockade of TLR4 does not affect BP in AngII‐infused animals in the same manner as observed in SHR. Zhang et al. (2015) showed that after 4 weeks of treatment with TAK‐242, a specific TLR4 inhibitor, not only was a reversal in high BP observed in aldosterone‐induced hypertensive animals, but renal and cardiac inflammation were also inhibited. Aldosterone, the primary human mineralocorticoid, is significantly involved in cardiovascular morbidity and hypertension (Freel and Connell, 2004). The main regulators of aldosterone production are plasma levels of potassium and AngII. Thus, in hypertension, plasma levels of aldosterone and AngII positively correlate. However, in animals where hypertension is induced by external aldosterone administration, the plasma levels of AngII are not modified. Because the levels of AngII remain within normal ranges, the aldosterone model of hypertension might be considered an AngII‐independent model. This could explain the controversial results regarding BP changes in hypertensive animals with higher circulating levels of AngII.
In order to target TLR4, however, the following question remains to be elucidated: why do the BP‐lowering effects of TLR4 blockade differ between animal models while appearing to modulate similar events during the progression of hypertension? At this point, we can speculate that blockade of TLR4 may minimize the end‐organ damage triggered by hypertension; however, it is not yet clear whether such blockade would be enough to prevent this pathology.
TLR4 effects in specific organs and systems during hypertension
Kidneys
The kidneys, in combination with the renin–angiotensin system (RAS), play central roles in BP regulation (Yim and Yoo, 2008). AngII has powerful control of sodium uptake in proximal tubules and affects glomerular filtration rate (GFR), which increases water reabsorption in a process responsible for maintaining homeostatic BP levels. However, prolonged increases in circulating AngII levels contribute to the aetiology of hypertension (Crowley et al., 2006). Likewise, there is substantial evidence supporting the hypothesis that increased natriuresis, with a rightward shift in the sodium retention curve, contributes to the maintenance of a hypertensive state. In the kidneys, AngII, via its angiotensin type 1 receptor (AT1 receptor), has the potential to damage the renal microvasculature and is posited to be associated with fibrosis, vascular rarefaction and glomerular dysfunction (Xu et al., 2017). Additionally, AngII has been demonstrated to be a strong inflammatory mediator and is suggested to act in concert with TLR4 pathways to promote inflammation (Phillips and Kagiyama, 2002; Biancardi et al., 2017; Xu et al., 2017). In fact, AngII is among the most commonly investigated possible endogenous ligands of TLR4 during hypertension. Still, while we have compiled extensive knowledge regarding AngII's actions in the kidneys as a long‐term mechanism of BP regulation, the implications of AngII‐mediated TLR4 activation in the renal system is not completely understood.
TLR4 is expressed in renal epithelial cells, and its overactivation is implicated in the nephropathy associated with various diseases (Zhang et al., 2008; Souza et al., 2015). In the context of the hypertensive kidney, current literature points to AngII as the main mediator of TLR4 activation. In corroboration with this statement, it has been reported that myeloid differentiation factor 2 (MD2)‐deficient mice were protected from renal inflammatory injury and fibrosis (Xu et al., 2017). The adaptor protein MD2 is known to play a role in LPS recognition by promoting TLR4–LPS–MD2 complex dimerization (Park et al., 2009). More specifically, Han et al. (2017) have demonstrated that direct hydrogen bond interactions may occur between MD2 and AngII in a manner similar to that of the MD2 and LPS interaction. Based on this evidence, it is reasonable to speculate that AngII could mediate the activation of TLR4 within the kidneys, thereby contributing to inflammation and oxidative stress, both intrinsic factors for the development of end‐organ damage in hypertensive patients.
TLR4‐mediated renal damage in hypertension plays a key role in the development and progression of microvascular complications and may represent a new treatment target. Diuretics are one of the main antihypertensive drug classes targeting the kidneys, favouring water and sodium excretion to help relieve tubular pressure and lower BP. However, as AngII is produced systemically, it may continue to activate TLR4 in the face of diuretic therapy, thereby propagating renal microvascular damage. Supporting this notion, combining diuretics with angiotensin converting enzyme (ACE) inhibitors appears to produce better outcomes in hypertensive patients (Ruoff, 1989). A plausible explanation is that inhibiting AngII production may decrease the extent of TLR4 activation, ameliorating renal oxidative stress and inflammation.
Another cause of renal damage in hypertension comes from the high BP itself, which injures the endothelial layer and contributes to elevated ROS generation. While the precise molecular mechanisms underlying the damage‐induced ROS generation are not completely elucidated, recent findings showing an association between increased renal TLR4 activation and worsened outcomes in hypertensive model point to DAMP‐mediated TLR4 activation (Pushpakumar et al., 2017). In the kidneys, increased levels of ROS are associated with dysfunctional glomerular and tubular cells (Araujo and Wilcox, 2014). Importantly, as NF‐κB is mediated downstream of TLR4 activation and is described as a source of pro‐inflammatory cytokines, this may be one mechanism by which TLR4 contributes to renal dysfunction and end‐organ damage in hypertension. Renal parenchymal TLR4 was shown to mediate inflammation and tissue damage following cisplatin exposure in a murine model of nephrotoxicity (Zhang et al., 2008). In a mouse model of hypertension, TLR4 deficiency protected against renal oxidative damage and was further found to increase antioxidant capacity (Pushpakumar et al., 2017). Taken together, the aforementioned data implicate TLR4 activation in tying inflammation to kidney dysfunction in hypertension.
CNS
As in other systems, CNS alterations in TLR4 expression, ROS and the pro‐inflammatory cytokine profile are linked to the pathogenesis of hypertension. In neural tissue, innate pro‐inflammatory mediators are produced primarily by the resident immune cells, microglia and astrocytes (Ransohoff and Brown, 2012). TLR4 is constitutively expressed by microglia, while the nature of its expression in other CNS cells, including astrocytes and neurons, remains somewhat controversial (Olson and Miller, 2004; for review, see Lehnardt, 2010; Hanke and Kielian, 2011). Despite low TLR4 surface expression detection, particularly in astrocytes and neurons, stimulation with LPS is shown to trigger innate immune activity in non‐microglia cells through various processes.
A complex communication system exists among microglia, astrocytes and neurons that allow the innate immune cells to sense environmental perturbations and subsequently influence neuronal activity. A primary mechanism by which such communication is achieved is through the secretion and detection of pro‐inflammatory mediators. The neuroimmune communication has been implicated in the pathogenesis of hypertension, wherein DAMP/danger signal recognition by CNS cells propagates a pro‐inflammatory CNS milieu, resulting in inflammation, elevated sympathetic outflow and increased BP.
Numerous studies have characterized, at least partially, the responses of various CNS cell types to stimulation by LPS. Surface expression of TLR4 is particularly abundant in microglia, and it is well accepted that LPS stimulation activates quiescent microglia, up‐regulating the innate immune response and increasing cytokine and chemokine secretion (Olson and Miller, 2004). While direct activation of the MyD88‐dependent pathway is shown in astrocytes upon LPS exposure, they are also shown to have an alternative response, propagating the LPS‐mediated microglial inflammatory response and furthering neurotoxic factor production (Saijo et al., 2009; Gorina et al., 2011). In neuronal cultures, LPS stimulation induces transendothelial migration of neutrophils and activation of cerebral ECs, the hallmarks of neuroinflammatory response (Leow‐Dyke et al., 2012). Together, these studies point to TLR4 dysregulation as a key candidate for modulating neuroinflammation in hypertension. Most of the evidence implicating neural TLR4 dysregulation within the CNS in the pathogenesis of hypertension is derived from animal models, which either have elevated levels of circulating AngII or are exposed to exogenous AngII (in vivo or in vitro). Within the CNS, AngII is intrinsic to the inflammatory process, acting as a pro‐hypertensive neurotransmitter and promoting innate immune activation through AT1 receptors (Ando et al., 2004; Zhou et al., 2006; Benicky et al., 2009; Benicky et al., 2011; Harrison et al., 2011; Zubcevic et al., 2011; Young and Davisson, 2015).
Whether endogenous to the hypertensive model or exogenously applied, chronic AngII elevations aggravate TLR4, activate microglia and up‐regulate pro‐inflammatory cytokine production (Benicky et al., 2009; Shi et al., 2010; Benicky et al., 2011; Zubcevic et al., 2011; Biancardi et al., 2016). Benicky et al. (2009; 2011) found LPS‐induced neuroinflammation in normotensive animals to be blocked by AT1 receptor antagonists. This AT1 receptor blockade decreased production of TLR4‐regulated pro‐inflammatory mediators and reduced microglia activation, both in vitro and in vivo. These alterations were observed in multiple nuclei, including, of note, several associated with autonomic control such as the PVN and the subfornical organ (SFO) (Benicky et al., 2009; Benicky et al., 2011). In TLR4‐competent mice, exogenous AngII applied to PVN‐containing hypothalamic slices caused microglial activation and ROS production (Biancardi et al., 2016). These response were shown to be attenuated in the PVN of TLR4‐deficient mice, demonstrating the contribution of TLR4 to these predecessors of hypertensive autonomic dysfunction (Biancardi et al., 2016).
TLR4 protein and mRNA expression within the PVN are elevated in both AngII infusion models and SHR, and chronic i.c.v. infusion of a viral TLR4 inhibitory peptide normalized these parameters in AngII‐infused animals (Dange et al., 2014; Dange et al., 2015; Li et al., 2016a). Furthermore, this chronic TLR4 blockade ameliorated cardiac function, decreased the cardiac inflammatory profile and reduced mean arterial pressure (MAP) (Dange et al., 2014). In SHRs, inhibition of PVN TLR4 attenuates the pro‐inflammatory cytokine profile as well as elevations in inducible NOS (iNOS) and NF‐κB levels, which are linked to the elevated BP and circulating plasma noradrenaline characteristic of this model (Dange et al., 2014; Dange et al., 2015). Li et al. (2016a) showed reduced MAP in SHR via chronic bilateral PVN infusion of the AT1 receptor inhibitor, telmisartan. PVN AT1 receptor inhibition also down‐regulated the MyD88‐dependent pathway, resulting in decreased CNS IL‐1β and IL‐6 levels (Li et al., 2016a).
Compellingly, these studies show that abolishment or attenuation of altered microglial and TLR4 signalling activity occurs upon blockade of TLR4 or the AT1 receptor, suggesting a role for neural TLR4, particularly through AT1 receptor–TLR4 crosstalk, in hypertension. The findings discussed above highlight the involvement of hypothalamic TLR4 and pro‐inflammatory signal transduction in driving AngII‐mediated hypertension and demonstrate the powerful cardiovascular effects of TLR4 activity in the CNS. As such, the TLR4 signalling pathways represent potential antihypertensive therapeutic targets. Importantly, CNS inflammation is a known component in numerous pathologies beyond hypertension, including neurodegenerative disorders (Appel et al., 2010; Perry et al., 2010; Lopes Pinheiro et al., 2016). Thus, elucidating the extent of TLR4's role in the hypertensive CNS may allow for an expansion of pharmaceutical targets in many neuroinflammatory diseases. On the basis of this commonality, there arises the potential that therapeutics currently employed in the management of other CNS‐associated diseases may represent novel antihypertensive therapies.
Vasculature
Chronic high BP and shear stress damage the vascular endothelium over time and contribute to the migration and accumulation of both innate and adaptive immune cells in blood vessels (Goulopoulou et al., 2016). Augmented TLR4 expression and activation positively correlate with vascular inflammation, remodelling and vasoconstriction. As in other cell types, increased TLR4 activity increases the production of pro‐inflammatory cytokines and ROS. Within the vasculature, TLR4 has been demonstrated to modulate NADPH oxidase activity, enhancing the production of ROS and free radicals (Nakashima et al., 2015). TLR4‐induced ROS production decreases the availability of NO, a vasoprotective molecule imperative to the regulation of blood flow and tissue oxygenation (Schiffrin, 2008; Nunes et al., 2017). Furthermore, it has been suggested that, following TLR4 activation, a crosstalk interaction occurs between ROS and NF‐κB wherein ROS can influence NF‐κB nuclear translocation while NF‐κB may regulate ROS production via gene expression (Morgan and Liu, 2011). Additionally, cytokines secreted from individual cells can diffuse to adjacent tissue where they stimulate ROS production (Mittal et al., 2014). These hypertension‐induced disruptions abolish endothelial and vascular functionality, the latter of which is vital to maintaining vascular homeostasis and depends upon proper VSMC contractile responses (Sanchorawala and Keaney, 1997; De Batista et al., 2014; Biancardi et al., 2017; Nunes et al., 2017). In the hypertensive vasculature, altered TLR4 expression is found in VSMCs and ECs (De Batista et al., 2014; Hernanz et al., 2015). De Batista et al. (2014) reported that TLR4 mRNA levels are up‐regulated in VSMCs and aorta of SHRs and that treatment with a TLR4 antibody reduced heart rate, BP and phenylephrine‐induced contraction. In a separate study, TLR4 blockade with a TLR4 antibody prevented pro‐inflammatory cytokine secretion, decreased vascular structural and mechanical changes, ameliorated vascular reactivity and increased NO production (Hernanz et al., 2015). Additionally, Bomfim et al. (2015) showed TLR4's pro‐inflammatory actions to be ultimately mediated through the MyD88‐dependent pathway in SHRs.
As previously discussed, AngII has regulatory roles in long‐term BP regulation and vascular homeostasis through its interaction with AT1 receptors. TLR4 is implicated as a key mediator of AngII‐induced vascular remodelling in hypertension through MyD88‐dependent ROS generation via JNK/NF‐κB activation (Hernanz et al., 2015; Nakashima et al., 2015). AngII‐induced elevations in ROS have been shown to occur through TLR4‐impairment of SOD in addition to TLR4‐stimulation of NADPH oxidase (Nakashima et al., 2015). Ji et al. (2009) have shown that AngII stimulates TLR4 in VSMCs, triggering the production of TNF‐α and MMP‐9, among others mediators, and contributing to vascular dysfunction. Pharmacological inhibition of TLR4 with CLI‐095 (TAK‐242) attenuated NADPH oxidase activity and superoxide production and decreased both cell migration and proliferation in response to AngII (De Batista et al., 2014). Importantly, antibody inhibition of TLR4 decreases MAP and vascular contractility and TLR4 protein expression in SHR mesenteric resistance arteries (Bomfim et al., 2012). In murine cavernosal smooth muscle, a highly vascularized structure, we have shown that AngII alters TLR4 expression and that chronic TLR4 blockade rescues muscle relaxation, decreases TNF‐α production and improves NO levels (Nunes et al., 2017). As evidenced, TLR4 enhances inflammation and contributes to vascular remodelling in models of hypertension, and its inhibition appears to be protective.
In hypertension, vascular remodelling involves complex interactions between endogenous growth factors, vasoactive substances and haemodynamic alterations (Schiffrin, 2012). As both vascular remodelling and endothelial damage progress, a positive feedback loop forms in which vasoconstriction is constantly favoured over vasodilatation. In this way, the physiological adaptations promoted by TLR4 in response to hypertensive stimuli ultimately turn to pathophysiological consequences. The labyrinth of signalling pathways activated by TLR4 involve numerous overlapping mechanisms and crosstalk interactions that have been highlighted as potential therapeutic targets to combat the pathogenesis of hypertension.
Heart
Although myocardial tissue alterations underlying the transition from a healthy to a hypertensive heart are unclear, the eventual cardiac dysfunction is known to be characterized by myocardial remodelling and low‐grade inflammation (Nadruz, 2015). Chronically elevated BP, coupled with factors such as altered neurohormone and cytokine levels, induces compensatory left ventricular hypertrophy (Drazner, 2011). It has been shown that cardiomyocytes express many TLRs, including TLR4, which trigger signalling pathways leading to local inflammation (Boyd et al., 2006). Consequently, during hypertension, the heart is a significant target for active immune cells.
Indeed, mRNA and protein TLR4 expression levels were found to be up‐regulated in cardiomyocytes of SHR (Eißler et al., 2011). Interestingly, treatment of SHRs with the ACE inhibitor, ramipril, showed a dose‐dependent response: a therapeutic dose (1 mg·kg−1·day−1) was sufficient to lower BP, while a supratherapeutic dose (10 mg·kg−1·day−1) was needed to elicit reductions in the observed up‐regulation of cardiac TLR4 expression (Eißler et al., 2011). This indicates that, while ACE inhibitors can alleviate BP elevations, they may or may not affect the associated TLR4‐driven low‐grade inflammation in cardiac tissue. Thus, it is important to take into consideration that AngII is not the only endogenous ligand for TLR4 in the hypertensive heart. For example, increased circulating levels of HSP60 positively correlate with the development of cardiovascular diseases and have been shown to modulate the TLR4/MyD88/p38/NF‐κB pathway in cardiac cells (Pockley et al., 2000; Tian et al., 2013). Conversely, TLR4 knockout mice have reduced left ventricular hypertrophy after aortic banding compared with wild‐type mice, and animals with a dysfunctional LPS response (TLR4lps‐d mice) show no changes in oxidative stress, ventricular hypertrophy or cardiac dysfunction when infused with AngII (Ha et al., 2005; Matsuda et al., 2015). It has been further demonstrated that these effects are mediated by an essential chemokine, monocyte chemoattractant protein 1 (MCP‐1; also known as CCL2), which regulates macrophage tissue infiltration and is up‐regulated by TLR4 stimulation (Matsuda et al., 2015). These results suggest that TLR4 stimulation, whether via AngII or other ligands, contributes to cardiac damage in hypertension.
It should be noted that neural TLR4 is reported to partially mediate physiological alterations of the myocardium in AngII‐infused animals. In this model, specific blockade of TLR4 in the CNS down‐regulates myocardial inflammation (Dange et al., 2014). Lastly, reduced myocardial hypertrophy and remodelling were observed in MD2−/− mice infused with AngII and in cardiomyocyte‐like H9c2 cells incubated with AngII in the presence of L6H21 (an inhibitor of MD2) (Han et al., 2017). These data indicate that MD2 may directly bind to AngII in the heart, causing dysfunction of the myocardium via TLR4 activation (Han et al., 2017). As there remains a paucity of information regarding the contribution of TLR4 to myocardial injury in response to high BP, it is unclear if blockade of TLR4 is cardioprotective in this condition. Thus, future experiments are needed to elucidate whether the onset and development of hypertension‐associated heart damage, including complications such as heart failure, would be minimized by targeting TLR4.
Translational potential of TLR4 targets in hypertension
Considering the ability of TLR4 to initiate and boost inflammation, and the literature that links TLR4 to hypertension, there is significant interest in developing novel pharmacological drugs that target either TLR4 itself or its downstream pathways in diseases associated with abnormal innate immune system overactivity. Although many mechanisms underlying disease mediation by TLR4 have yet to be fully clarified, insights into TLR4's signal transduction have recently opened the doors to development of effective modulators. Even now, there is preclinical evidence of the therapeutic potential for targeting TLR4 in inflammatory diseases. Table 2 summarizes pharmacological compounds that are currently being used in basic research to target TLR4 and/or its downstream signalling pathways.
Table 2.
Pharmacological therapies currently in use that target TLR4 and its downstream signalling
| Compound | Description | Target | Mechanism of action | Disease model or cell type tested | Main outcome (reference) |
|---|---|---|---|---|---|
| Eritoran (E5564) | Synthetic LPS lipid A analogue | MD2 | Competitively binds a large pocket of MD2 | Rat model of kidney ischaemia/reperfusion | Ameliorated kidney ischaemia/reperfusion‐related inflammatory responses (Liu et al., 2010a) |
| SPA4 | Peptide | TLR4 | Binds to surfactant A and blocks TLR4 activation | HEK293 cells | Decreased secretion of pro‐inflammatory cytokines (Ramani et al., 2013) |
| TAK‐242 | Small molecule/cyclohexene inhibitor | TLR4 | Binds TIR domain and affects the recruitment of adapters | Rat model of hyperaldosteronism | Inhibited hypertension and cardiac and renal fibrosis and attenuates aldosterone‐induced epithelial–mesenchymal transition (Zhang et al., 2015) |
| Rat VSMCs | Decreased NADPH oxidase activity, superoxide anion production and cell migration and proliferation (De Batista et al., 2014) | ||||
| Mouse model of hypertension (AngII) | Reduced AngII‐induced increase in phospho‐JNK1/2 and p65 NF‐κB subunit nuclear protein expression (Hernanz et al., 2015) | ||||
| NI‐0101 | Monoclonal antibody | TLR4 | Antagonist | Synovial explant culture model | Decreased pro‐inflammatory cytokine secretion (TNF‐α and IL‐6) (Page et al., 2011) |
| Valsartan | AT1 – AngII receptor blocker | TLR4 | Unknown | Rat model of myocardial ischaemia/reperfusion | Improved myocardial injury, such as smaller infarct size, and decreased release of myocardial enzymes and pro‐inflammatory mediators (Yang et al., 2009a) |
| Candesartan | AngII receptor blocker | TLR4 | AngII receptor independent | Rat mesangial cells | Decreased oxidative stress and exerted anti‐apoptotic effects (Lv et al., 2009) |
| Fluvastatin | HMG‐CoA reductase inhibitor | TLR4 | Inhibits NF‐κB activation | Rat model of myocardial ischaemia/reperfusion | Decreased ischaemic injury and inhibited the expression levels of TLR4, TNF‐α and NF‐κB (Yang et al., 2011) |
| Atorvastatin | HMG‐CoA reductase inhibitor | TLR4 | Impairs TLR4 recruitment of lipid raft and inhibits NF‐κB activation | Rabbit model of atherosclerosis | Impaired TLR4/NF‐κB activation in atherosclerotic plaques that decreased inflammation (Fang et al., 2014) |
| ST2825 | Peptidomimetic | MyD88 | Inhibits homodimerization of MyD88 | Mouse model of hypertension (AngII) | Decreased NADPH oxidase activity (Hernanz et al., 2015) |
| dnMyD88 | Mutated form of MyD88 | MyD88 | Inhibits homodimerization of MyD88 | Rat model of myocardial ischaemia/reperfusion | Prevented ischaemia/reperfusion via inhibition of NF‐κB (Ha et al., 2006) |
HMG‐CoA, hydroxymethylglutaryl CoA.
Eritoran (E5564) is a synthetic TLR4 antagonist that has been well studied in inflammatory disease models and has been used in four clinical trials in the last decade (NCT00334828, NCT00756912, NCT02267317 and NCT02321111). Unfortunately, the promising results observed in animal models of inflammatory diseases were not translated to human subjects. As an example, in a phase III randomized control clinical trial that compared the efficacy of eritoran in preventing mortality in patients with severe sepsis, human subjects receiving the drug did not have an enhanced chance of survival compared with those who received the placebo (NCT00334828). However, in the context of evaluating drug efficacy, it is important to take into consideration the aetiological and pathological differences among diseases, such as between sepsis and hypertension. In the same trial, it was reported that eritoran clearance is affected by the patient's weight, HDL levels and age, features that are highly important factors over the course of hypertension. Eritoran inhibits MD2‐mediated TLR4 stimulation by binding to a large pocket of this adaptor protein. As previously discussed, this mechanism of action is similar to the one reported to be used by AngII to activate TLR4. To date, the possible benefits of TLR4 blockade by eritoran are still unknown in hypertensive patients. Thus, it is possible to speculate that this drug might have therapeutic applications in the management of hypertension.
One of the most widely used TLR4 antagonists in basic research is TAK‐242, which binds to the TIR domain at Cys747 and, consequently, inhibits TLR4's ability to recruit both adaptor proteins (TIRAP/MAL and TRAM) responsible for mediating TLR4 actions (Matsunaga et al., 2011). Despite its consistent inhibitory effects in animal models, the drug failed to decrease sepsis symptoms when tested in human subjects (NCT00633477). Nevertheless, TAK‐242 may yet have a potential application in hypertension treatment due to the different natures of these pathologies. This drug has been tested in different animal models of hypertension and has produced exciting results. TAK‐242 has been reported to block NF‐κB, reduce oxidative stress, decrease cell migration and proliferation and lower BP (De Batista et al., 2014; Hernanz et al., 2015; Zhang et al., 2015). Overall, the pharmacological benefits of this inhibitor in animal models of hypertension are clear. However, it remains unknown whether the same effects would be observed in hypertensive human subjects.
Another drug used to block TLR4 signalling, NI‐0101, is a monoclonal antibody currently being assessed in a clinical trial for rheumatoid arthritis (NCT01808469). In cultured cells, the drug has been shown to decrease pro‐inflammatory cytokine secretion (Page et al., 2011). It is not yet known whether the drug would be effective when used systemically in animal models of hypertension. Further exploring the available data using TLR4 antibodies in basic research, there are promising outcomes regarding their abilities to ameliorate the inflammatory state and reduce oxidative stress, which, together, blunt the deleterious effects of hypertension in the major organs studied.
Repurposing drugs is a key process in unearthing new options in the treatment of a disease. In this sense, statins, which are primarily prescribed to lower cholesterol, have produced promising results in modulating the TLR4 pathway. In particular, atorvastatin has been shown to decrease TLR4 activation, which abolishes inflammation in atherosclerosis, and fluvastatin has been demonstrated to prevent myocardial ischaemia and reperfusion (Yang et al., 2011; Fang et al., 2014). Based on these data and the findings of other studies, it can be predicted that, because treated animals had decreased TLR4 levels in the atorvastatin and fluvastatin studies, they might also have had decreased oxidative stress. As hypertension and atherosclerosis have overlapping pathways, it would be of interest to explore whether the same outcomes would be observed in hypertensive animal models.
The pharmacological effects of the AT1 receptor blockers, valsartan and candesartan, have also been examined in animal models (Lv et al., 2009; Yang et al., 2009a). These drugs were shown to reduce oxidative stress and decrease cytokine profiles. These studies were conducted before it was shown that AngII could activate TLR4 by interacting with MD2. It is still unclear whether AngII has two independent mechanisms to modulate TLR4, one through MD2 and the other via AT1 receptor crosstalk. Additionally, it has been suggested that, when AT1 receptor blockers are used, AngII levels might accumulate and favour TLR4 activation (Campbell, 1996). However, increased AngII levels also contribute to the formation of angiotensin‐(1–7), another important peptide of the RAS system that opposes the effects of AngII (Santos, 2014).
The drug ST2825, a mutated form of MyD88 that inhibits MyD88 homodimerization, has also been tested. In a model of hypertension, it was observed that ST2825 decreases NADPH oxidase activity, which in turn ameliorates oxidative stress (Hernanz et al., 2015). When used in a model of myocardial infarction, ST2825 blocked NF‐κB activity (Ha et al., 2006). While the effects of ST2825 differed between these studies, both showed treatment outcomes that are favourable in the management of hypertension. When comparing the results of TLR4 blockade with the use of adaptor molecule inhibitors, it must be taken into consideration that TLR4 signal transduction occurs through two arms, one led by MyD88 and the other by TRIF. This means that targeting MyD88 rather than TLR4 itself would only partially inhibit TLR4's signalling, which may prove to be an advantage depending on the clinical application.
We have presented evidence supporting the role of TLR4 in hypertension. TLR4‐mediated hypertension involves many different aspects of cardiovascular, renal and CNSs, much of which require further investigation. Indeed, there are many questions regarding TLR4 and hypertension that remain to be addressed. A paramount question is whether the use of immunological suppressants would produce better results in hypertensive patients when compared with the well‐established pharmacological drugs available in the market. Thus far, it is possible to speculate that such a treatment could be promising in patients who do not respond to standard pharmacotherapy and inevitably succumb to end‐organ damage.
TLR4 and hypertension‐associated diseases
Pulmonary hypertension
Pulmonary hypertension (PH) of different aetiologies and prognoses share a central pathogenesis characterized by persistent pulmonary vasoconstriction, vascular remodelling and thrombosis in situ. Pulmonary vascular remodelling, through pulmonary arterial (PA) smooth muscle cell (PASMC) proliferation and fibrosis, plays a critical role in PH development and involves a chronic imbalance in vasoactive substances. Pulmonary arterial hypertension (PAH), in which small pulmonary arteries progressively narrow, leading to increases in pulmonary vascular resistance and pressure, right heart failure and ultimately death, is strongly associated with dysregulated immunity and pulmonary vascular inflammation largely regulated through TLR4 signalling pathways (Gerges and Lang, 2018). In response to local injury (acute lung injury) or stress (hypoxia; cold exposure), TLR4 stimulation causes PA ECs to produce and secrete fractalkine (CX3CL1), a chemokine that attracts immune cells (Amsellem et al., 2017; Florentin and Dutta, 2017). Activation of TLR4 has been demonstrated to regulate MMP‐9 production in lungs after hypoxia exposure, increasing ECM degradation and PASMC migration and proliferation (Young et al., 2010). PA ECs also release TLR4 endogenous agonists, such as HMGB1, which activate platelets and stimulate their aggregation at the injured/stressed PA sites (Bauer et al., 2013; Sun, 2014). The TLR4‐activated platelets produce and secrete vasoactive substances (5‐HT and TxA2), mitogenic and growth factors (PDGF, TGF‐β and VEGF) and pro‐inflammatory cytokines (IL‐1α, IL‐1β and TNF‐α) (Bauer et al., 2013; Sun, 2014). HMGB1 has also been shown to promote both pulmonary vascular remodelling and right ventricular hypertrophy through stimulation of TLR4 (Hilbert et al., 2017). The pathogenesis of PAH has been further linked with Notch, TGF‐β, PI3K/Akt and Hippo signalling, many of which point to a dysregulation in either ligands or substrates or TLR4 signalling (Li et al., 2017). However, it is yet to be determined whether TLR4‐induced inflammation is a primary or secondary mechanism in PAH pathogenesis, what may be the implications of these findings in other forms of PH or how these pathways might be therapeutically targeted.
Pre‐eclampsia
Pre‐eclampsia (PE) is a severe complication of pregnancy characterized by the development of hypertension and proteinuria (Whelton et al., 2018). The maternal immune system, specifically maternal systemic inflammation, is strongly implicated as a key contributor to the pathogenesis of PE (Bounds et al., 2015). The excessive pro‐inflammatory response is suggested to be mediated through TLR recognition of DAMPs and other danger signals from placental dysfunction, metabolic syndrome and/or vascular dysfunction (Yeh et al., 2013; Bounds et al., 2015; Zhao et al., 2017). TLR4, MyD88 and NF‐κB are widely expressed in the maternal–fetal interface, and their expressions are increased in the placenta of women with PE (Zhu et al., 2013; Bounds et al., 2015; Qian et al., 2015; Xue et al., 2015). Similarly, the cytokines TNF‐α, IL‐6 and MCP‐1, are increased in PE patients, both systemically and within placental tissue (Xue et al., 2015). Zhao et al. (2017) found both TLR4 and NF‐κB serum expressions to be elevated in PE patients and demonstrated that either could be used as a serum marker for PE diagnosis. In vitro studies showed that binding of LPS by TLR4 in trophoblasts increased cytokine secretion significantly and resulted in monocyte chemotaxis (Yeh et al., 2013). Furthermore, TNF‐α infusion in pregnant mice was associated with placental TLR4 up‐regulation and resulted in the development of both hypertension and proteinuria (Bomfim et al., 2017). Preliminary investigations into the potential therapeutic application of immune modulation in PE have yielded promising results. Qian et al. (2015) showed vitamin D3 supplementation to decrease peripheral blood monocyte TLR4 expression, serum pro‐inflammatory cytokines and PE incidence in at‐risk pregnant women. Additionally, down‐regulation of the TLR4 signalling pathway with curcumin attenuated both high BP and proteinuria in a rat model of PE (Gong et al., 2016). While still very few, these results highlight the potential therapeutic application of TLR4 modulation in preventing PE. Further investigation is warranted to understand whether the supplements tested would provide similar findings in different models/types of hypertension.
Obesity‐associated hypertension
Development of obesity‐associated hypertension has been linked with metabolic dysregulation, autonomic dysregulation and vascular dysfunction (endothelial dysfunction and arterial stiffening) (Kang, 2013; Matsuda and Shimomura, 2013; Li et al., 2016b; Reho and Rahmouni, 2017). The chronic low‐grade inflammatory state associated with obesity is characterized by elevations in cytokines, ROS production and secretion and immune cell recruitment (Kang, 2013; Matsuda and Shimomura, 2013; Schneider et al., 2015; Catrysse and van Loo, 2017). Hypertrophy of adipose tissue induces the production of inflammatory mediators (TNF‐α, IL‐6, MCP‐1 and iNOS) from adipocytes, which leads to recruitment of immune cells, especially macrophages and T lymphocytes, to the adipose mass (Kang, 2013; Matsuda and Shimomura, 2013; Schneider et al., 2015; Catrysse and van Loo, 2017). Both adipocytes and recruited cytokine‐producing immune cells continue production of pro‐inflammatory cytokines and chemokines, maintaining local and, eventually, systemic inflammation (Kang, 2013; Matsuda and Shimomura, 2013; Schneider et al., 2015; Catrysse and van Loo, 2017). Ahmad et al. (2012) found a significant elevation in TLR2 and TLR4 expression in adipocytes and peripheral blood mononuclear cells from obese subjects in correlation with increased cytokine levels, supporting the notion that these TLRs may mediate crosstalk between metabolism and the immune system. Obesity is also linked to elevations in TLR4 ligands: Western diets provide pro‐inflammatory free fatty acids and alter the composition of the gut microbiota, increasing intestinal permeability such that LPS can translocate from the gut (Schneider et al., 2015). Once in the bloodstream, these TLR4 ligands can contribute to vascular‐mediated alterations (Schneider et al., 2015). Studies demonstrating elevated oxidative stress and NADPH oxidase subunit expression in the hypothalamus of obese rats, concurrent with antioxidant‐induced reduction of arterial BP and sympathetic nerve activity in obese mice, suggest an important role for sympathetic excitation in the pathogenesis of obesity‐associated hypertension (Matsuda and Shimomura, 2013). Together, oxidative stress and inflammatory signalling are likely to underlie the vascular dysfunction and development of obesity‐associated hypertension. Given that obesity‐associated hypertension is also associated with low‐grade chronic inflammation, the extent to which TLR4 is involved in the genesis and/or maintenance of this disease likely merits further exploration.
Final remarks
As highlighted in this review, the chronic low‐grade inflammation observed in hypertension is due, in large part, to activation of TLR4 and its downstream signalling pathways. Elevations in TLR4 expression have been observed in hypertensive animal models across renal, neural, vascular and myocardial tissues. When combined with the extensive evidence of increased innate pro‐inflammatory mediator profiles, it can be postulated that inhibition of TLR4 signal transduction would be globally beneficial in preventing the lifelong complications associated with this disease. Furthermore, hypertensive models have proven the ability of TLR4 inhibition to ameliorate the deleterious effects of sustained hypertension. In some of the hypertensive models tested, inhibition of TLR4 was also able to normalize elevated BP. Thus, designing antagonists of TLR4 and its downstream signalling components is a compelling strategy to pharmacologically target the dysregulated TLR4 and, feasibly, the progression of hypertension.
There is, however, much to be said for the remaining gaps in our knowledge regarding TLR4 signalling in terms of their application to therapeutic development. In this light, it must be taken into consideration that TLR4, as a facet of the innate immune system, is ultimately involved in both initial and chronic innate immune activation states. The promotion of inflammation is, first and foremost, a beneficial process, protecting against invading pathogens and responding to stress or injury. It is when this initial protective process becomes excessive or chronic that innate immune activation and, specifically, TLR4 signalling, can be calamitous. With overactivation, the disrupted immune homeostasis becomes pathogenic, manifesting as chronic inflammation, such as that observed in hypertension.
In Figure 2, we present several therapeutic targets known to modulate TLR4 signal transduction, as well as potential targets for future pharmaceutical development. As illustrated, activation of TLR4 triggers two signalling cascades, which ultimately lead to the production of pro‐inflammatory cytokines. Based on studies to date, it can be argued that NF‐κB is implicated as the principal downstream component, orchestrating aberrant innate immune system activity in hypertension through TLR4. With similar recent findings of NF‐κB dysregulation in other inflammation‐associated diseases, pharmaceutical constraint of this transcription factor undoubtedly demands further exploration, particularly as a point of target given the multitude of crosstalk within the TLR4 signalling web.
Multidisciplinary investigations suggest a plethora of additional crosstalk mechanisms within and between the two TLR4 pathways, and it is evident that TLR4 signal transduction is yet more intricate than we understand. While not within the scope of this review, it is of note that all TLRs have been shown to induce NF‐κB activity upon stimulation, despite their extensive diversity (Singh et al., 2014). Thus, high specificity in an NF‐κB governing agent is essential to combatting potential activation of this pro‐inflammatory transcription factor either by other TLR pathways or by as of yet undetermined TLR4 mechanisms. On the one hand, our lack of knowledge regarding the extent of TLR4's pathways in hypertension may result in unforeseen complications with putative therapeutics. On the other hand, the potential of supplementary connections could allow for fewer pharmaceutical risks – by targeting the component essential to disease propagation rather than the entire cascade, it is possible that the beneficial effects of TLR4 stimulation will be retained.
Lastly, it is of critical importance that the potential risks of targeting TLR4 in hypertension be considered. As evidenced by its vast array of ligands, the TLR4 pathway plays a substantial role in shaping the immune response, and the consequences of inhibiting this arm of the immune system are still unclear. Essential hypertension is a chronic condition that often requires lifelong treatment and, as such, the potential benefits of targeting TLR4 must be weighed against the risks and disadvantages of immunosuppressant treatment.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2017), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the American Heart Association (grant number 14SDG20400015 to V.C.B. and 12SDG12080023 to K.P.N.).
Nunes K. P., de Oliveira A. A., Mowry F. E., and Biancardi V. C. (2019) Targeting toll‐like receptor 4 signalling pathways: can therapeutics pay the toll for hypertension?, British Journal of Pharmacology, 176, 1864–1879, doi: 10.1111/bph.14438.
Contributor Information
Kenia Pedrosa Nunes, Email: knunes@fit.edu.
Vinicia Campana Biancardi, Email: vbiancardi@auburn.edu.
References
- Abais‐Battad JM, Dasinger JH, Fehrenbach DJ, Mattson DL (2017). Novel adaptive and innate immunity targets in hypertension. Pharmacol Res 120: 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad R, Al‐Mass A, Atizado V, Al‐Hubail A, Al‐Ghimlas F, Al‐Arouj M et al (2012). Elevated expression of the toll like receptors 2 and 4 in obese individuals: its significance for obesity‐induced inflammation. J Inflamm (Lond) 9: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K (2004). Toll‐like receptor signalling. Nat Rev Immunol 4: 499–511. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsellem V, Abid S, Poupel L, Parpaleix A, Rodero M, Gary‐Bobo G et al (2017). Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 chemokine systems in hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol 56: 597–608. [DOI] [PubMed] [Google Scholar]
- Ando H, Zhou J, Macova M, Imboden H, Saavedra JM (2004). Angiotensin II AT1 receptor blockade reverses pathological hypertrophy and inflammation in brain microvessels of spontaneously hypertensive rats. Stroke 35: 1726–1731. [DOI] [PubMed] [Google Scholar]
- Appel SH, Beers DR, Henkel JS (2010). T cell‐microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol 31: 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo M, Wilcox CS (2014). Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal 20: 74–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer EM, Shapiro R, Billiar TR, Bauer PM (2013). High mobility group box 1 inhibits human pulmonary artery endothelial cell migration via a toll‐like receptor 4‐ and interferon response factor 3‐dependent mechanism(s). J Biol Chem 288: 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benicky J, Sanchez‐Lemus E, Honda M, Pang T, Orecna M, Wang J et al (2011). Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology 36: 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benicky J, Sanchez‐Lemus E, Pavel J, Saavedra JM (2009). Anti‐inflammatory effects of angiotensin receptor blockers in the brain and the periphery. Cell Mol Neurobiol 29: 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancardi VC, Bomfim GF, Reis WL, Al‐Gassimi S, Nunes KP (2017). The interplay between angiotensin II, TLR4 and hypertension. Pharmacol Res 120: 88–96. [DOI] [PubMed] [Google Scholar]
- Biancardi VC, Stranahan AM, Krause EG, de Kloet AD, Stern JE (2016). Cross talk between AT1 receptors and toll‐like receptor 4 in microglia contributes to angiotensin II‐derived ROS production in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol 310: H404–H415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC et al (2012). Toll‐like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci (Lond) 122: 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Echem C, Martins CB, Costa TJ, Sartoretto SM, Dos Santos RA et al (2015). Toll‐like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sci 122: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Rodrigues FL, Carneiro FS (2017). Are the innate and adaptive immune systems setting hypertension on fire? Pharmacol Res 117: 377–393. [DOI] [PubMed] [Google Scholar]
- Bounds KR, Newell‐Rogers MK, Mitchell BM (2015). Four pathways involving innate immunity in the pathogenesis of preeclampsia. Front Cardiovasc Med 2: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR (2006). Toll‐like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF‐κB dependent inflammatory response. Cardiovasc Res 72: 384–393. [DOI] [PubMed] [Google Scholar]
- Campbell D (1996). Endogenous angiotensin II levels and the mechanism of action of angiotensin‐converting enzyme inhibitors and angiotensin receptor type 1 antagonists. Clin Exp Pharmacol Physiol Suppl 3: S125–S131. [PubMed] [Google Scholar]
- Catrysse L, van Loo G (2017). Inflammation and the metabolic syndrome: the tissue‐specific functions of NF‐κB. Trends Cell Biol 27: 417–429. [DOI] [PubMed] [Google Scholar]
- Chen P‐G, Guan Y‐J, Zha G‐M, Jiao X‐Q, Zhu H‐S, Zhang C‐Y et al (2017). Swine IRF3/IRF7 attenuates inflammatory responses through TLR4 signaling pathway. Oncotarget 8: 61958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP et al (2006). Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci 103: 17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A et al (2014). Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II‐induced hypertension. Cardiovasc Res 103: 17–27. [DOI] [PubMed] [Google Scholar]
- Dange RB, Agarwal D, Teruyama R, Francis J (2015). Toll‐like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation 12: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Batista PR, Palacios R, Martín A, Hernanz R, Médici CT, Silva MA et al (2014). Toll‐like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One 9: e104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doddapattar P, Gandhi C, Prakash P, Dhanesha N, Grumbach IM, Dailey ME et al (2015). Fibronectin splicing variants containing extra domain A promote atherosclerosis in mice through toll‐like receptor 4. Arteriosclerosis, thrombosis, and vascular biology: ATVBAHA 115.306474. [DOI] [PMC free article] [PubMed]
- Dominiczak AF, Kuo D (2018). Hypertension: update 2018. Hypertension 71: 3–4. [DOI] [PubMed] [Google Scholar]
- Drazner MH (2011). The progression of hypertensive heart disease. Circulation 123: 327–334. [DOI] [PubMed] [Google Scholar]
- Eißler R, Schmaderer C, Rusai K, Kühne L, Sollinger D, Lahmer T et al (2011). Hypertension augments cardiac toll‐like receptor 4 expression and activity. Hypertens Res 34: 551–558. [DOI] [PubMed] [Google Scholar]
- Fang D, Yang S, Quan W, Jia H, Quan Z, Qu Z (2014). Atorvastatin suppresses toll‐like receptor 4 expression and NF‐κB activation in rabbit atherosclerotic plaques. Eur Rev Med Pharmacol Sci 18: 242–246. [PubMed] [Google Scholar]
- Florentin J, Dutta P (2017). Origin and production of inflammatory perivascular macrophages in pulmonary hypertension. Cytokine 100: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freel EM, Connell JM (2004). Mechanisms of hypertension: the expanding role of aldosterone. J Am Soc Nephrol 15: 1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerges C, Lang I (2018). Targeting inflammation and immunity in pulmonary arterial hypertension: any easier after the CANTOS proof‐of‐concept that anti‐inflammation cuts cardiovascular events? Pulm Circ 8: 2045893218754855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Liu M, Hong G, Li Y, Xue P, Zheng M et al (2016). Curcumin improves LPS‐induced preeclampsia‐like phenotype in rat by inhibiting the TLR4 signaling pathway. Placenta 41: 45–52. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font‐Nieves M, Marquez‐Kisinousky L, Santalucia T, Planas AM (2011). Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88‐dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 59: 242–255. [DOI] [PubMed] [Google Scholar]
- Goulopoulou S, McCarthy CG, Webb RC (2016). Toll‐like receptors in the vascular system: sensing the dangers within. Pharmacol Rev 68: 142–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J et al (2006). Blockade of MyD88 attenuates cardiac hypertrophy and decreases cardiac myocyte apoptosis in pressure overload‐induced cardiac hypertrophy in vivo. Am J Phys Heart Circ Phys 290: H985–H994. [DOI] [PubMed] [Google Scholar]
- Ha T, Li Y, Hua F, Ma J, Gao X, Kelley J et al (2005). Reduced cardiac hypertrophy in toll‐like receptor 4‐deficient mice following pressure overload. Cardiovasc Res 68: 224–234. [DOI] [PubMed] [Google Scholar]
- Han J, Zou C, Mei L, Zhang Y, Qian Y, You S et al (2017). MD2 mediates angiotensin II‐induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF‐κB signaling pathway. Basic Res Cardiol 112: 9. [DOI] [PubMed] [Google Scholar]
- Hanke ML, Kielian T (2011). Toll‐like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 121: 367–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2017). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR et al (2011). Inflammation, immunity, and hypertension. Hypertension 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Vinh A, Lob H, Madhur MS (2010). Role of the adaptive immune system in hypertension. Curr Opin Pharmacol 10: 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernanz R, Martinez‐Revelles S, Palacios R, Martin A, Cachofeiro V, Aguado A et al (2015). Toll‐like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II‐induced hypertension. Br J Pharmacol 172: 3159–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbert T, Dornbusch K, Baumgarten G, Hoeft A, Frede S, Klaschik S (2017). Pulmonary vascular inflammation: effect of TLR signalling on angiopoietin/TIE regulation. Clin Exp Pharmacol Physiol 44: 123–131. [DOI] [PubMed] [Google Scholar]
- Idris‐Khodja N, Mian MO, Paradis P, Schiffrin EL (2014). Dual opposing roles of adaptive immunity in hypertension. Eur Heart J 35: 1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israël A (2010). The IKK complex, a central regulator of NF‐κB activation. Cold Spring Harb Perspect Biol 2: a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Liu J, Wang Z, Liu N (2009). Angiotensin II induces inflammatory response partly via toll‐like receptor 4‐dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem 23: 265–276. [DOI] [PubMed] [Google Scholar]
- Kang YS (2013). Obesity associated hypertension: new insights into mechanism. Electrolyte Blood Press 11: 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe T, Sato S, Jung A, Yamamoto M, Matsui K, Kato H et al (2007). Essential role of IRAK‐4 protein and its kinase activity in toll‐like receptor‐mediated immune responses but not in TCR signaling. J Exp Med 204: 1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S (2010). The role of pattern‐recognition receptors in innate immunity: update on toll‐like receptors. Nat Immunol 11: 373–384. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2011). Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650. [DOI] [PubMed] [Google Scholar]
- Kim S‐C, Stice JP, Chen L, Jung JS, Gupta S, Wang Y et al (2009). Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res 105: 1186–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S (2011). Pathogen recognition by the innate immune system. Int Rev Immunol 30: 16–34. [DOI] [PubMed] [Google Scholar]
- Küper C, Beck F‐X, Neuhofer W (2012). Toll‐like receptor 4 activates NF‐κB and MAP kinase pathways to regulate expression of proinflammatory COX‐2 in renal medullary collecting duct cells. Am J Physiol‐Renal Physiol 302: F38–F46. [DOI] [PubMed] [Google Scholar]
- Lehnardt S (2010). Innate immunity and neuroinflammation in the CNS: the role of microglia in toll‐like receptor‐mediated neuronal injury. Glia 58: 253–263. [DOI] [PubMed] [Google Scholar]
- Leong X‐F, Ng C‐Y, Jaarin K (2015). Animal models in cardiovascular research: hypertension and atherosclerosis. Biomed Res Int 2015: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leow‐Dyke S, Allen C, Denes A, Nilsson O, Maysami S, Bowie AG et al (2012). Neuronal toll‐like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J Neuroinflammation 9: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerman LO, Chade AR, Sica V, Napoli C (2005). Animal models of hypertension: an overview. Transl Res 146: 160–173. [DOI] [PubMed] [Google Scholar]
- Li F, Shi W, Wan Y, Wang Q, Feng W, Yan X et al (2017). Prediction of target genes for miR‐140‐5p in pulmonary arterial hypertension using bioinformatics methods. FEBS Open Bio 7: 1880–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H‐B, Li X, Huo C‐J, Su Q, Guo J, Yuan Z‐Y et al (2016a). TLR4/MyD88/NF‐κB signaling and PPAR‐γ within the paraventricular nucleus are involved in the effects of telmisartan in hypertension. Toxicol Appl Pharmacol 305: 93–102. [DOI] [PubMed] [Google Scholar]
- Li S, Strelow A, Fontana EJ, Wesche H (2002). IRAK‐4: a novel member of the IRAK family with the properties of an IRAK‐kinase. Proc Natl Acad Sci 99: 5567–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Wang Y, Liu C, Hu Y, Wu M, Li J et al (2009). MyD88‐dependent nuclear factor‐κB activation is involved in fibrinogen‐induced hypertrophic response of cardiomyocytes. J Hypertens 27: 1084–1093. [DOI] [PubMed] [Google Scholar]
- Li Y, Diao J, Wu WH, Qi CM, Li XH, Su ZP et al (2016b). Patients with metabolic syndrome have higher single tlr4 positive rate of blood mononuclear cells compared with simple hypertension patients. Int J Clin Exp Med 9: 12145–12149. [Google Scholar]
- Liu M, Gu M, Xu D, Lv Q, Zhang W, Wu Y (2010a). Protective effects of toll‐like receptor 4 inhibitor eritoran on renal ischemia–reperfusion injury. 42:1539–1544. [DOI] [PubMed] [Google Scholar]
- Liu N, Liu J, Ji Y, Lu P (2010b). Toll‐like receptor 4 signaling mediates inflammatory activation induced by C‐reactive protein in vascular smooth muscle cells. Cell Physiol Biochem 25: 467–476. [DOI] [PubMed] [Google Scholar]
- Liu N, Liu J, Ji Y, Lu P, Wang C, Guo F (2011). C‐reactive protein induces TNF‐α secretion by p38 MAPK–TLR4 signal pathway in rat vascular smooth muscle cells. Inflammation 34: 283–290. [DOI] [PubMed] [Google Scholar]
- Liu‐Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R (2005). Innate immunity conferred by toll‐like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal‐induced inflammation. Arthritis Rheum 52: 2936–2946. [DOI] [PubMed] [Google Scholar]
- Lopes Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R et al (2016). Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta 1862: 461–471. [DOI] [PubMed] [Google Scholar]
- Lopez Gelston CA, Mitchell BM (2017). Recent advances in immunity and hypertension. Am J Hypertens 30: 643–652. [DOI] [PubMed] [Google Scholar]
- Lv J, Jia R, Yang D, Zhu J, Ding G (2009). Candesartan attenuates angiotensin II‐induced mesangial cell apoptosis via TLR4/MyD88 pathway. Biochem Biophys Res Commun 380: 81–86. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Shimomura I (2013). Increased oxidative stress in obesity: implications for metabolic syndrome, diabetes, hypertension, dyslipidemia, atherosclerosis, and cancer. Obes Res Clin Pract 7: e330–e341. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Umemoto S, Yoshimura K, Itoh S, Murata T, Fukai T et al (2015). Angiotensin II activates MCP‐1 and induces cardiac hypertrophy and dysfunction via toll‐like receptor 4. J Atheroscler Thromb 22: 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga N, Tsuchimori N, Matsumoto T, Ii M (2011). TAK‐242 (resatorvid), a small‐molecule inhibitor of toll‐like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79: 34–41. [DOI] [PubMed] [Google Scholar]
- Matzinger P (2002). The danger model: a renewed sense of self. Science 296: 301–305. [DOI] [PubMed] [Google Scholar]
- McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC (2014). Toll‐like receptors and damage‐associated molecular patterns: novel links between inflammation and hypertension. Am J Phys Heart Circ Phys 306: H184–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGettrick AF, O'Neill LA (2010). Localisation and trafficking of toll‐like receptors: an important mode of regulation. Curr Opin Immunol 22: 20–27. [DOI] [PubMed] [Google Scholar]
- Miller YI, Chang M‐K, Binder CJ, Shaw PX, Witztum JL (2003). Oxidized low density lipoprotein and innate immune receptors. Curr Opin Lipidol 14: 437–445. [DOI] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20: 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ, Liu Z‐g (2011). Crosstalk of reactive oxygen species and NF‐κB signaling. Cell Res 21: 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK (2012). MAP kinase pathways. Cold Spring Harb Perspect Biol 4: a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadruz W (2015). Myocardial remodeling in hypertension. J Hum Hypertens 29: 1–6. [DOI] [PubMed] [Google Scholar]
- Nair AR, Ebenezer PJ, Saini Y, Francis J (2015). Angiotensin II‐induced hypertensive renal inflammation is mediated through HMGB1–TLR4 signaling in rat tubulo‐epithelial cells. Exp Cell Res 335: 238–247. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Umemoto S, Yoshimura K, Matsuda S, Itoh S, Murata T et al (2015). TLR4 is a critical regulator of angiotensin II‐induced vascular remodeling: the roles of extracellular SOD and NADPH oxidase. Hypertens Res 38: 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018). The immunology of hypertension. J Exp Med 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosalski R, McGinnigle E, Siedlinski M, Guzik TJ (2017). Novel immune mechanisms in hypertension and cardiovascular risk. Curr Cardiovasc Risk Rep 11: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes KP, Bomfim GF, Toque HA, Szasz T, Webb RC (2017). Toll‐like receptor 4 (TLR4) impairs nitric oxide contributing to angiotensin II‐induced cavernosal dysfunction. Life Sci 191: 219–226. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Ghosh S (2009). The NF‐κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1: a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Aoki K (1963). Development of a strain of spontaneously hypertensive rats. Jpn Circ J 27: 282–293. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173: 3916–3924. [DOI] [PubMed] [Google Scholar]
- Page G, Buatois T, Daubeuf V, Chatel B, Cons L, Lippens L (2011). Ni‐0101, a therapeutic Tlr4 monoclonal antibody for rheumatoid arthritis. Arthritis Rheum 63: 977. [Google Scholar]
- Park BS, Song DH, Kim HM, Choi B‐S, Lee H, Lee J‐O (2009). The structural basis of lipopolysaccharide recognition by the TLR4–MD‐2 complex. Nature 458: 1191–1195. [DOI] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim J‐Y, Strassheim D, Ishizaka A et al (2004). Involvement of toll‐like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279: 7370–7377. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C (2010). Microglia in neurodegenerative disease. Nat Rev Neurol 6: 193–201. [DOI] [PubMed] [Google Scholar]
- Phillips M, Kagiyama S (2002). Angiotensin II as a pro‐inflammatory mediator. Current opinion in investigational drugs (London, England: 2000) 3: 569–577. [PubMed]
- Pockley AG, Wu R, Lemne C, Kiessling R, de Faire U, Frostegård J (2000). Circulating heat shock protein 60 is associated with early cardiovascular disease. Hypertension 36: 303–307. [DOI] [PubMed] [Google Scholar]
- Pushpakumar S, Ren L, Kundu S, Gamon A, Tyagi SC, Sen U (2017). Toll‐like receptor 4 deficiency reduces oxidative stress and macrophage mediated inflammation in hypertensive kidney. Sci Rep 7: 6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Wang H, Wu F, Li M, Chen W, Lv L (2015). Vitamin D3 alters toll‐like receptor 4 signaling in monocytes of pregnant women at risk for preeclampsia. Int J Clin Exp Med 8: 18041–18049. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ramani V, Madhusoodhanan R, Kosanke S, Awasthi S (2013). A TLR4‐interacting SPA4 peptide inhibits LPS‐induced lung inflammation. Innate Immun 19: 596–610. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Brown MA (2012). Innate immunity in the central nervous system. J Clin Invest 122: 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reho JJ, Rahmouni K (2017). Oxidative and inflammatory signals in obesity‐associated vascular abnormalities. Clin Sci (Lond) 131: 1689–1700. [DOI] [PubMed] [Google Scholar]
- Ruoff G (1989). ACE inhibitors and diuretics: the benefits of combined therapy for hypertension. Postgrad Med 85: 127–139. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG et al (2009). A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation‐induced death. Cell 137: 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchorawala H, Keaney JF (1997). Physiology of vascular homeostasis In: Textbook of Coronary Thrombosis and Thrombolysis. Springer, pp. 139–153. [Google Scholar]
- Santos RA (2014). Angiotensin‐(1–7). Hypertension 63: 1138–1147. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL (2008). Oxidative stress, nitric oxide synthase, and superoxide dismutase: a matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension 51: 31–32. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL (2012). Vascular remodeling in hypertension: mechanisms and treatment. Hypertension 59: 367–374. [DOI] [PubMed] [Google Scholar]
- Schneider S, Hoppmann P, Koch W, Kemmner S, Schmaderer C, Renders L et al (2015). Obesity‐associated hypertension is ameliorated in patients with TLR4 single nucleotide polymorphism (SNP) rs4986790. J Inflamm (Lond) 12: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q et al (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MV, Chapleau MW, Harwani SC, Abboud FM (2014). The immune system and hypertension. Immunol Res 59: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley ST, King JA, Hancock WW (2001). Fibrinogen stimulates macrophage chemokine secretion through toll‐like receptor 4. J Immunol 167: 2887–2894. [DOI] [PubMed] [Google Scholar]
- Souza AC, Tsuji T, Baranova IN, Bocharov AV, Wilkins KJ, Street JM et al (2015). TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Phys Rep 3: e12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z (2014). Platelet TLR4: a critical link in pulmonary arterial hypertension. Circ Res 114: 1551–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C et al (2002). Severe impairment of interleukin‐1 and toll‐like receptor signalling in mice lacking IRAK‐4. Nature 416: 750–754. [DOI] [PubMed] [Google Scholar]
- Szasz T, Ogbi S, Webb RC (2016). Activation of toll‐like receptor 4 (TLR4) by high mobility group box 1 (HMGB1) leads to oxidative stress and inflammation in human aortic endothelial cell. FASEB J 30: 1282.1283–1282.1283. [Google Scholar]
- Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL (2004). Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem 279: 17079–17084. [DOI] [PubMed] [Google Scholar]
- Tian J, Guo X, Liu X‐M, Liu L, Weng Q‐F, Dong S‐J et al (2013). Extracellular HSP60 induces inflammation through activating and up‐regulating TLRs in cardiomyocytes. Cardiovasc Res 98: 391–401. [DOI] [PubMed] [Google Scholar]
- Vaure C, Liu Y (2014). A comparative review of toll‐like receptor 4 expression and functionality in different animal species. Front Immunol 5: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Yu XJ, Li XG, Pang DZ, Su Q, Saahene RO et al (2018). Blockade of TLR4 within the paraventricular nucleus attenuates blood pressure by regulating ROS and inflammatory cytokines in prehypertensive rats. Am J Hypertens; 10.1093/ajh/hpy074 [DOI] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C et al (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 71: e127–e248. [DOI] [PubMed] [Google Scholar]
- Wolf G, Bohlender J, Bondeva T, Roger T, Thaiss F, Wenzel UO (2006). Angiotensin II upregulates toll‐like receptor 4 on mesangial cells. J Am Soc Nephrol 17: 1585–1593. [DOI] [PubMed] [Google Scholar]
- Xu Z, Li W, Han J, Zou C, Huang W, Yu W et al (2017). Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein‐2 (MD2). Sci Rep 7: 44911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue PP, Zheng MM, Gong P, Lin CM, Zhou JJ, Li YJ et al (2015). Single administration of ultra‐low‐dose lipopolysaccharide in rat early pregnancy induces TLR4 activation in the placenta contributing to preeclampsia. PLoS One 10: e0124001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T et al (2002). Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 420: 324–329. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T et al (2003). TRAM is specifically involved in the toll‐like receptor 4‐mediated MyD88‐independent signaling pathway. Nat Immunol 4: 1144–1150. [DOI] [PubMed] [Google Scholar]
- Yang J, Jiang H, Yang J, Ding J‐W, Chen L‐H, Li S et al (2009a). Valsartan preconditioning protects against myocardial ischemia–reperfusion injury through TLR4/NF‐κB signaling pathway. Mol Cell Biochem 330: 39–46. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang X‐D, Yang J, Ding J‐W, Liu Z‐Q, Li S‐G et al (2011). The cardioprotective effect of fluvastatin on ischemic injury via down‐regulation of toll‐like receptor 4. Mol Biol Rep 38: 3037–3044. [DOI] [PubMed] [Google Scholar]
- Yang Z‐C, Wang K‐S, Wu Y, Zou X‐Q, Xiang Y‐Y, Chen X‐P et al (2009b). Asymmetric dimethylarginine impairs glucose utilization via ROS/TLR4 pathway in adipocytes: an effect prevented by vitamin E. Cell Physiol Biochem 24: 115–124. [DOI] [PubMed] [Google Scholar]
- Yeh CC, Chao KC, Huang SJ (2013). Innate immunity, decidual cells, and preeclampsia. Reprod Sci 20: 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim HE, Yoo KH (2008). Renin–angiotensin system – considerations for hypertension and kidney. Electrolyte Blood Pressure 6: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Davisson RL (2015). Angiotensin‐II, the brain, and hypertension: an update. Hypertension 66: 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KC, Hussein SM, Dadiz R, deMello D, Devia C, Hehre D et al (2010). Toll‐like receptor 4‐deficient mice are resistant to chronic hypoxia‐induced pulmonary hypertension. Exp Lung Res 36: 111–119. [DOI] [PubMed] [Google Scholar]
- Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB (2008). TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 19: 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Peng W, Ao X, Dai H, Yuan L, Huang X et al (2015). TAK‐242, a toll‐like receptor 4 antagonist, protects against aldosterone‐induced cardiac and renal injury. PloS one 10: e0142456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao LT, Wang H, Zhang WM, Tian XH, Sun Q (2017). Serum NF‐κBp65, TLR4 as biomarker for diagnosis of preeclampsia. Open Medicine 12: 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang C, Wei X, Li P, Cui Y, Qin Y et al (2015). Heat shock protein 60 stimulates the migration of vascular smooth muscle cells via toll‐like receptor 4 and ERK MAPK activation. Sci Rep 5: 15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Pavel J, Macova M, Yu ZX, Imboden H, Ge L et al (2006). AT1 receptor blockade regulates the local angiotensin II system in cerebral microvessels from spontaneously hypertensive rats. Stroke 37: 1271–1276. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wu M, Wu CY, Xia GQ (2013). Role of progesterone in TLR4–MyD88‐dependent signaling pathway in pre‐eclampsia. J Huazhong Univ Sci Technolog Med Sci 33: 730–734. [DOI] [PubMed] [Google Scholar]
- Zubcevic J, Waki H, Raizada MK, Paton JF (2011). Autonomic–immune–vascular interaction: an emerging concept for neurogenic hypertension. Hypertension 57: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]