

Abstract
Activated immune cell infiltration into organs contributes to the development and maintenance of hypertension. Studies targeting specific immune cell populations or reducing their inflammatory signalling have demonstrated a reduction in BP. Lymphatic vessels play a key role in immune cell trafficking and in resolving inflammation, but little is known about their role in hypertension. Studies from our laboratory and others suggest that inflammation‐associated or induction of lymphangiogenesis is organ protective and anti‐hypertensive. This review provides the basis for hypertension as a disease of chronic inflammation in various tissues and highlights how renal lymphangiogenesis is a novel regulator of kidney health and BP.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- APC
antigen‐presenting cell
- DC
dendritic cell
- DOCA
deoxycorticosterone acetate
- IAL
inflammation‐associated lymphangiogenesis
- LEC
lymphatic endothelial cell
- LHTN
l‐NAME‐induced hypertension
- LT
lymphotoxin
- PVAT
perivascular adipose tissue
- SFO
subfornical organ
- SHR
spontaneously hypertensive rat
- SSHTN
salt‐sensitive hypertension
- TonEBP
tonicity‐responsive enhancer‐binding protein
- UUO
unilateral ureteral obstruction
Introduction
The increasing prevalence of hypertension in children and adults is a significant public health issue. A major concern in hypertensive disease and what makes its investigation necessary is target organ damage. Hypertension actively contributes to the development of heart, kidney, brain, eye and peripheral vascular diseases. Endothelial cell activation by inflammatory factors and attenuated endothelium‐dependent vasodilatation are underlying events in the pathogenesis of hypertension (Sprague and Khalil, 2009). Blood vessels undergo remodelling wherein the lumen diameter decreases and the ratio of medial to intimal thickness increases. Chronic hypertension increases the workload on the heart, which eventually undergoes hypertrophy and fibrosis leading to increased wall stiffness and dysfunction (Drazner, 2011). The systemic elevated BP load eventually reaches the renal microvasculature leading to nephrosclerosis and renal damage. Changes in the microvascular structure of the cerebral circulation are associated with an increased risk of death, ischaemic stroke and decline in cognitive abilities (Gąsecki et al., 2013).
Over the years, a better understanding of the pathophysiology of hypertensive end‐organ damage has been attained, which aids the design of better therapeutics. However, the incidence of hypertension continues to rise globally, indicating the need to revisit current treatment options. Hypertension and cardiovascular diseases have gained appreciation as a low‐grade inflammatory disease, and studies on human hypertension support this association (Solak et al., 2016). Aberrant immune system activation and inflammation is now recognized to have a mechanistic role in the progression of hypertension and should pave the way for designing more effective therapeutics to attenuate end‐organ damage in hypertensive patients (Miguel et al., 2015; Lopez Gelston and Mitchell, 2017).
Immune system activation in hypertension
The contribution of both the innate and adaptive immune systems in the pathogenesis of hypertension has been recognized for the past 50 years. Pioneering studies by White and Grollman showed that immunosuppression blunted hypertension in rats with partial renal infarction (White and Grollman, 1964; Okuda and Grollman, 1967) and that the transfer of lymphocytes from lymph nodes of rats with renal infarction triggered the elevation of BP in normal recipient rats (Okuda and Grollman, 1967). Subsequently, studies by Olsen (1970) reported the presence of inflammatory cells in the vasculature of angiotensin II‐infused rats. In another study, it was demonstrated that thymectomized or athymic nude mice with renal infarction did not maintain hypertension (Svendsen, 1976). Transplant of the thymus from Wistar–Kyoto rats to spontaneously hypertensive rats (SHRs) lowered BP (Bendich et al., 1981), and similar results were obtained by treatment with anti‐thymocyte drugs or the immunosuppressive drug cyclophosphamide (Dzielak, 1991). Studies also demonstrated that transfer of splenocytes from deoxycorticosterone acetate (DOCA)‐salt hypertensive rats to normal recipient rats triggered hypertensive responses in the recipient rats (Olsen, 1980).
Following the evidence of immune system activation in hypertension, immunosuppression became a popular tool of choice to lower BP in experimental models of hypertension. Administration of the immunosuppressive agent mycophenolate mofetil blunts the development of several forms of hypertension including salt‐induced hypertension after angiotensin II infusion (Rodriguez‐Iturbe et al., 2001), SHRs (Rodriguez‐Iturbe et al., 2002), salt‐induced hypertension after NOS inhibition (Quiroz et al., 2001), Dahl salt‐sensitive rats (Mattson et al., 2006) and hypertensive patients (Herrera et al., 2006). All of these studies had demonstrated renal injury associated with the increased BP; the treatments reduced renal inflammation and injury and were accompanied by a lowered BP.
Inflammation in the cardiovascular system and the brain is also an important event in the pathogenesis of hypertension. Immune cells accumulate in the adventitia and perivascular adipose tissue (PVAT) of the larger vessels and of the smaller resistance vessels during hypertension. The PVAT then releases factors that modulate the tone of these vessels and also secretes factors leading to inflammation (Guzik et al., 2007). Several forms of hypertension are associated with increased vascular wall expression of chemokines like CCL2 (also known as MCP‐1) and adhesion molecules including VCAM‐1 and ICAM‐1 (Ebrahimian et al., 2011). Another important target of activated immune cells in hypertension is the heart. Activation of T‐cells found in the heart of angiotensin II‐infused hypertensive mice caused cardiac inflammation, hypertrophy and fibrosis (Kvakan et al., 2009). BP regulation is also governed by central mechanisms, in part, through innervation of the blood vessels and the kidney. Strong evidence for the involvement of the CNS was demonstrated by the study showing that renal denervation abolishes hypertension in humans (Schlaich et al., 2009). Intracerebroventricular administration of the anti‐inflammatory antibiotic minocycline reduces levels of TNF‐α, IL‐1β and IL‐6 in the paraventricular nucleus and reduces angiotensin II‐dependent hypertension (Shi et al., 2010).
The immune system is constituted of both the innate immune system, consisting of macrophages, dendritic cells (DCs), mast cells, granulocytes etc, and the adaptive immune system, consisting of T and B lymphocytes. Components from both classes of the immune system are implicated in hypertension (Norlander et al., 2018). Guzik et al. (2007) conducted a study that dissected the role of T and B lymphocytes in hypertension. They demonstrated that angiotensin II‐induced hypertension was blunted in Rag1−/− mice that are deficient in T and B lymphocytes. Vascular superoxide production and endothelial dysfunction were also blunted in these mice. The adoptive transfer of T but not B lymphocytes restored hypertension, indicating the importance of T‐cells in the initiation of hypertension. T‐cells are also required for the development of DOCA salt‐induced and noradrenaline‐induced hypertension (Marvar et al., 2010). Similarly, Crowley et al. demonstrated that severe combined immune deficiency mice deficient in lymphocytes are protected against hypertension. The lymphocyte‐deficient mice displayed diminished cardiac damage and renal injury in response to angiotensin II infusion. More recently, Mattson et al. (2013) used Dahl salt‐sensitive rats with deleted Rag1 gene to demonstrate that these rats have attenuated BP, kidney damage and albuminuria. Mice lacking the macrophage colony‐stimulating factor, also called osteoporotic mice (Op/Op), have blunted hypertensive responses to chronic angiotensin II infusion. The endothelial dysfunction, vascular remodelling and oxidative stress associated with wild‐type controls were reduced in these Op/Op mice (De Ciuceis et al., 2005). Deletion of monocytes using diphtheria toxin prevented hypertension and reduced the expression of markers of aortic inflammation (Wenzel et al., 2011). Harwani et al. (2012) demonstrated that the cholinergic agonist nicotine promoted the development of pro‐inflammatory responses in splenic macrophages from SHRs and also induced toll‐like receptor‐mediated cytokine release. Most recent studies have also demonstrated the involvement of the gut microbiome in hypertension. Karbach et al. (2016) reported that germ‐free mice had reduced BP and reduced leukocyte infiltration in the kidney and vasculature in response to angiotensin II infusion when compared to normal mice.
Cytokines and antigens in hypertension
Once localized in the target organs, immune cells release inflammatory cytokines such as TNF‐α, IL‐1β, IL‐6, IL‐17 and IFN‐γ. In generalized terms, T helper 1 (Th1) cells secrete pro‐inflammatory cytokines, and Th2 cells secrete anti‐inflammatory cytokines. Th1 cytokines mediate the pathogenic effects leading to end‐organ damage, and evidence exists for elevated levels of these cytokines in hypertensive models (Trott and Harrison, 2014). Direct infusion of IL‐17A, for example, has been reported to induce hypertension and endothelial dysfunction in mice (Nguyen et al., 2013). IL‐17−/− mice exhibited an initial increase in BP in response to angiotensin II, similar to wild‐type mice; however, BP dropped in IL‐17−/− mice after a week. Increases in vascular oxidative stress and endothelial dysfunction were also blunted in IL‐17−/− mice (Madhur et al., 2010). IL‐6 promotes the polarization of T‐cells towards Th17 cells that secrete IL‐17. IL‐6−/− mice also display blunted responses to angiotensin II infusion (Lee et al., 2006). Etanercept, a TNF‐α antagonist, prevents vascular dysfunction and the development of hypertension (Guzik et al., 2007). Following angiotensin II infusion, IFN‐γ is elevated in the kidneys of hypertensive mice, and inhibition of IFN‐γ prevents the end‐organ damage induced by angiotensin II infusion (Garcia et al., 2012). Accordingly, adoptive transfer of anti‐inflammatory regulatory T‐cells did not affect hypertension but attenuated the cardiac hypertrophy, fibrosis and inflammation induced by chronic angiotensin II infusion (Kvakan et al., 2009). IL‐10 is an anti‐inflammatory cytokine that stimulates the differentiation of regulatory T‐cells and is also produced by the same cells; studies have reported that carotid arteries from IL‐10−/− mice have marked endothelial dysfunction in response to angiotensin II. Vascular superoxide production is also increased in IL‐10−/− mice (Kassan et al., 2011).
The complete mechanisms by which inflammatory cytokines mediate end‐organ damage are not well understood. In the kidneys, the accumulation of inflammatory cytokines leads to a loss of peritubular capillaries resulting in medullary hypoxia and increased oxidative stress (Rodriguez‐Iturbe et al., 2013). The infiltrating lymphocytes also express angiotensin II, and increased renal interstitial angiotensin II and oxidative stress are factors well known to impair pressure‐induced natriuresis, thereby affecting renal function. Inflammation and oxidative stress are inextricably linked, and the generation of ROS reduces the bioavailability of NO. Chabrashvili et al. (2002) demonstrated that mRNA levels of the components of NADPH oxidase were increased in the kidneys of SHRs prior to the development of hypertension. The kidney of the SHR has an exaggerated tubuloglomerular feedback response, which may be due to the diminished availability of NO. In the vasculature, inflammatory cytokines released around blood vessels can alter the rates of synthesis and degradation of vasoconstrictors and vasodilators, including NO. TNF‐α inhibits endothelial NOS and reduces the capacity of the endothelium to produce NO leading to an impairment of vasodilator responses. Accordingly, inhibiting TNF‐α restored endothelium‐dependent vasodilatation (Zhang et al., 2006). IL‐17 has also been demonstrated to cause inhibition of endothelial NOS activity, thereby increasing vascular tone and leading to endothelial dysfunction (Nguyen et al., 2013). Vascular collagen deposition and aortic stiffening happen as a consequence of increased oxidative stress (Wu et al., 2014). IFN‐γ has been reported to induce the expression of angiotensinogen in renal proximal tubular cells (Satou et al., 2012), which when converted to angiotensin II promotes sodium reabsorption in the nephron.
What activates the immune system in hypertension and causes the infiltration of immune cells in target organs is a more recent subject of investigation. The concept of self‐antigens promoting hypertension was introduced following the discovery of agonistic antibodies to adrenoceptors and angiotensin receptors. Endogenous antigens like heat shock proteins and γ‐ketoaldehydes (isoketals) generated by the lipid peroxidation of arachidonic acid are increased in the kidneys of hypertensive animals (Kirabo et al., 2014). Isoketals can react with intracellular proteins and form protein adducts, which could then be taken up by DCs and presented to MHC type I receptors, thus activating the immune system. The production of isoketals also promotes the production of cytokines like IL‐1β, IL‐6 and IL‐23 by DCs, which further affect the polarization of T‐cells (Kirabo et al., 2014). Studies have also examined the effects of antigen recognition by specifically blocking the interaction between DC CD80 and CD86 with the T‐cell receptor CD28; this reduced vascular T‐cell accumulation and prevented hypertension (Vinh et al., 2010). Seminal work by Zimmerman et al. (2004) reported that hypertension is caused in part by increased sympathetic outflow following elevated levels of ROS in the subfornical organ (SFO) of the brain in response to angiotensin II infusion. Adenoviral overexpression of cytoplasmic SOD in the SFO reduced ROS and lowered BP as potently as direct i.c.v. infusion of losartan, highlighting the crucial role of CNS inflammation in hypertension. Collectively, mounting evidence has changed the previously existing notion that immune cell infiltration and inflammation are a consequence of hypertension but, instead, suggest that the inflammation in the kidneys and other organs are central to the development of hypertension.
The lymphatic system
Lymphatic vessels transport immune cells and soluble antigens out of the peripheral interstitium to the draining lymph nodes, where further acquired immune responses are initiated. Soluble antigens reach the lymph nodes faster than they reach antigen‐presenting cells (APCs) like DCs and are thought to prime the lymph node for the arrival of APCs (Randolph et al., 2017). By also taking up fluid extravasated from the blood vasculature, lymphatic vessels regulate tissue fluid homeostasis (Levick and Michel, 2010; Aspelund et al., 2016). Lymphatic capillaries, also called initial lymphatic vessels, are blind‐ended structures present in nearly all tissues. They consist of a single layer of oak leaf‐shaped lymphatic endothelial cells (LECs) and are equipped with thin fibrillar structures called anchoring filaments that permit expansion of these vessels with increased interstitial fluid pressure. The capillaries lack mural cell coverage, have little basement membrane coverage, and button‐like cell–cell junctions (Trzewik et al., 2001; Baluk et al., 2007). As such, lymphatic capillaries are the preferred route for uptake of fluid and macromolecules in what is generally assumed to be a nonselective process (although mechanisms for selective uptake may exist, Triacca et al., 2017).
Lymphatic capillaries coalesce into larger collecting vessels, which unlike the capillaries have tight zipper‐like junctions with a continuous basement membrane (Wiig and Swartz, 2012). The collecting vessels have intraluminal valves and also possess smooth muscle cell coverage. The inter‐endothelial flaps in the capillaries and the intraluminal valves in the collecting vessels are responsible for the unidirectional transport of lymph. Unlike the extrinsic contraction of veins, lymphatic smooth muscles cells provide an intrinsic pump to propel fluid along against a gradually increasing pressure before returning lymph to the blood circulation (Zawieja, 2009; Wiig and Swartz, 2012). Given these important roles of the lymphatic vessels, defects in their function disrupt fluid and immune homeostasis causing oedema and inflammatory diseases. Indeed, the respiratory and gastrointestinal systems, which are readily exposed to foreign antigens, have a dense network of lymphatic vessels stressing their importance in immune surveillance and defence mechanisms (Aspelund et al., 2016).
Lymphatic vessels are not passive conduits for the transport of antigens and leukocytes, but rather, the process is actively regulated by LECs (Card et al., 2014). LECs express an array of chemokines and most notably, and nearly LEC‐specific, are CCL21 and CCL19. The receptor for these chemokines, CCR7, is expressed on DCs and other leukocytes, and this ligand–receptor interaction aids the recruitment of immune cells into and through the lymphatic vessels to the lymph nodes (Forster et al., 2008; Card et al., 2014). Recent studies have also reported that LECs can themselves participate in the induction of peripheral immune tolerance through antigen archiving and presentation through MHC class I and class II molecules (Cohen et al., 2010; Dubrot et al., 2014; Tamburini et al., 2014). LECs have also recently been demonstrated to have direct interactions with DCs and T‐cells altering their maturation, differentiation and cytokine repertoires (elegantly reviewed in Maisel et al., 2017).
Inflammation‐associated lymphangiogenesis
Inflammation is a complex biological process that occurs as a protective mechanism against harmful agents and tissue remodelling. Inflammation is associated with the migration and activation of leukocytes, increased vascular permeability and an accumulation of excess interstitial fluid. To relieve the tissue of this hostile micro‐environment, excess fluid, cells and antigens must be cleared and hence there is an increased demand for lymphatic drainage. To keep up with this need, lymphangiogenesis occurs at sites of persistent inflammation given that lymphatics are structurally suited to be the physiological route for removal of increased antigens, fluid, cytokines and macromolecules from the site of inflammation. The process has been termed inflammation‐associated lymphangiogenesis (IAL) (Medzhitov, 2010; Lim et al., 2013; Kim et al., 2014). The rate at which these new vessels grow and the nature of these new vessels are highly tissue‐specific and stimulus‐specific. IAL has been observed in inflammatory conditions in several organs including the skin, intestine, heart, kidney and airway tract, and the consequences of IAL vary with the disease state and type of tissue in question (excellently reviewed in Kim et al., 2014; Abouelkheir et al., 2017).
In the search for the mechanisms of such postnatal lymphangiogenesis, several mediators of IAL have been found in different inflammatory diseases (Tan et al., 2014; Maisel et al., 2017); however, the most common of these are the potent lymphangiogenic VEGFs VEGFC and VEGFD. VEGFC and VEGFD signal through the receptor VEGFR‐3. The sources of these mediators may be either leukocytes or stromal cells. Macrophages in particular have been demonstrated to be sources of VEGFC in several cases. In a mouse model of tail lymphoedema, CD68+ macrophages were reported to be sources of VEGFC (Gousopoulos et al., 2017), and depletion of macrophages reduced IAL in a mouse model of acute colitis (Becker et al., 2016). More recently, DCs and neutrophils have also emerged as sources of lymphangiogenic signals (Baluk et al., 2005). Besides leukocytes, epithelial cells (Wuest and Carr, 2010), keratinocytes (Halin et al., 2007) and fibroblastic reticular cells (Chyou et al., 2008) have also been reported to be sources of VEGFC in inflammation. In the kidney, VEGFC staining was observed in tubular epithelial cells during unilateral ureteral obstruction (UUO) (Lee et al., 2013). Other inflammatory stimuli like LPS (Kang et al., 2009), lymphotoxin [α (TNFSF1) and β (TNFSF3) ) and inflammatory cytokines like IL‐17 and IL‐8 (Chauhan et al., 2011; Choi et al., 2013) can also mediate IAL. Some cytokines like IFN‐γ exhibit inhibitory effects on IAL, and TGFβ has been implicated in both stimulation and inhibition of lymphangiogenesis in disease states (Zampell et al., 2012; Kinashi et al., 2013). Inhibition of TGFβ induces lymphangiogenesis in a mouse model of chronic peritonitis (Oka et al., 2008) and also improves lymphatic function in a model of lymphoedema (Avraham et al., 2010), whereas in a model of peritoneal fibrosis, inhibiting the TGFβ receptor TGFBR2 suppressed lymphangiogenesis (Kinashi et al., 2013). Thus, the outcome of IAL is dependent on the balance of pro‐lymphangiogenic and anti‐lymphangiogenic factors and cytokines (Zampell et al., 2012). The new lymphatic vessels that are formed may proliferate from existing vessels (Wirzenius et al., 2007), sprout as new vessels (Kataru et al., 2009; Flister et al., 2010) or form from the transdifferentiation of bone marrow‐derived cells (Maruyama et al., 2005; Kerjaschki et al., 2006). Interestingly, this seems to depend on the type of lymphangiogenic stimulus. Although controversial, evidence exists for the transdifferentiation of macrophages into LECs in LPS‐induced peritonitis (Hall et al., 2012) and corneal inflammation (Maruyama et al., 2005). Moreover, activated murine peritoneal CD11b+ macrophages in vitro were reported to form tube‐like structures and express LEC‐specific markers (Maruyama et al., 2005).
Is inflammation‐associated lymphangiogenesis good or bad?
Whether IAL serves to resolve or exacerbate inflammation remains a matter of debate. In the skin, IAL has been demonstrated to have functional implications in both acute and chronic inflammation. In acute models of skin inflammation, blocking IAL by the inhibition of VEGFR‐3 signalling increased inflammation (Kajiya and Detmar, 2006), and overexpression of VEGFC induced lymphangiogenesis and limited inflammation in chronic inflammatory conditions (Huggenberger et al., 2011). It has also been demonstrated that in contact hypersensitivity, lymphatic vessels are necessary for the regulation of long‐term immune responses and fluid balance. Upon initial contact, K14‐VEGFR3‐Ig mice, which lack dermal lymphatic vessels, not only develop increased oedema but also fail to tolerize to hypersensitization upon subsequent challenges (Thomas et al., 2012). In the gut, alterations to the lymphatics result in changes not only to immune function but also to lipid transport. Deficiencies in the structure or function of lacteals affect lipid absorption and lead to the leakage of lymph. A proliferation of lymphatic vessels is associated with chronic inflammatory bowel diseases like Crohn's disease, ileitis and colitis, and IAL is unlikely to improve lymphatic drainage as oedema, and lack of DC migration to the draining lymph nodes is evident in such cases (Acedo et al., 2011; Abouelkheir et al., 2017). In airway inflammation, infection leads to robust lymphangiogenesis, and blocking IAL during infection resulted in increased mucosal oedema (Aurora et al., 2005; Baluk et al., 2005). Conversely, stimulating IAL also increased oedema in severe pulmonary lymphangiectasia (Yao et al., 2014). In rat cardiac allografts, newly formed lymphatic vessels participate in the trafficking of immune cells, and presumably donor antigens, to the secondary lymphoid organs; inhibiting VEGFR‐3 using adenoviral VEGFR3‐Ig was reported to increase cardiac allograft survival. Interestingly, this was not linked to reduced lymphangiogenesis but rather to reduced CCL21 production and entry of CD8+ T‐cells into the allograft (Edwards et al., 2018). Thus, the functional implications of IAL are highly disease‐ and tissue‐specific.
Besides the macroscopic expansion, inflammation also affects lymphatic vessels at the cellular level. Under steady state, the expression of adhesion molecules like ICAM‐1, VCAM‐1 and L1CAM by LECs is quite low, whereas their expression is dramatically up‐regulated by increased flow and inflammatory signalling presumably to facilitate the migration of DCs (Johnson et al., 2006; Maddaluno et al., 2009; Miteva et al., 2010; Vigl et al., 2011). Exposure of LECs to TNF‐α in vitro up‐regulates the expression of CCL21, and the up‐regulation of CCL21 has also been demonstrated in vivo under inflammatory conditions (Johnson and Jackson, 2010; Miteva et al., 2010; Vigl et al., 2011). In addition to CCL21, CXCL12 and CX3CL1 are other chemokines implicated in DC migration that are also up‐regulated during inflammation (Johnson et al., 2006; Pegu et al., 2008). In response to inflammation, LECs also increase the expression of molecules that help remove inflammatory chemokines. For example, the chemokine‐scavenging chemokine receptor ACKR2 (also known as D6) is up‐regulated in inflammatory conditions, and in the absence of ACKR2, myelomonocytic cells accumulated around lymphatics vessels and impeded lymph flow (Lee et al., 2011; McKimmie et al., 2013). Inflammation also modulates lymphatic pumping activity. Systemic or i.d. injection of inflammatory cytokines IL‐6, TNF‐α and IL‐1β decreased lymphatic pumping frequency and lymph flow velocity in vivo (Aldrich and Sevick‐Muraca, 2013). However, substance P, a neuropeptide secreted by inflammatory cells, increases the pumping frequency of rat mesenteric vessels (Davis et al., 2008). How inflammation affects lymphatics and vice versa has been and continues to be investigated in great detail.
Lymphatics and hypertension
A previously unknown role for lymphatic vessels and lymphangiogenesis was described by Machnik et al. (2009) in the context of interstitial sodium homeostasis. This study demonstrated that the skin interstitium could act as a reservoir for sodium in order to buffer the effects of sodium overload on BP. In response to a high‐salt challenge, the investigators observed that dermal lymphangiogenesis is associated with sodium accumulation in the skin interstitium. The resulting hypertonicity from sodium accumulation activates the transcription factor, tonicity‐responsive enhancer‐binding protein (TonEBP) in infiltrating macrophages. TonEBP binds to the gene encoding VEGFC and causes macrophages to secrete VEGFC, thereby leading to a robust expansion of the dermal lymphatic network. The lymphatic expansion was dependent on TonEBP secretion from the macrophages; macrophage depletion prevented dermal lymphatic hyperplasia and worsened sodium‐dependent hypertension. Blockade of VEGFC signalling had similar effects in these mice (Wiig and Swartz, 2012). This study not only revealed a novel role for dermal lymphatic vessels but also demonstrated the extrarenal regulation of sodium homeostasis and BP control by the skin interstitium.
In another study, TonEBP‐mediated cardiac lymphangiogenesis and macrophage infiltration was observed in the left ventricles of SHRs that were fed a high‐salt diet (Yang et al., 2014; Yang et al., 2017). Retrovirus‐induced overexpression of VEGFC led to enhanced lymphangiogenesis, reduced myocardial fibrosis and macrophage infiltration, decreased BP and preserved myocardial function. The opposite effects were observed after blocking VEGFC, where diminished lymphangiogenesis was accompanied by increased macrophage infiltration and pronounced left ventricular remodelling (Yang et al., 2014). In another study, it was demonstrated that cardiac lymphangiogenesis also occurs in response to myocardial infarction; however, myocardial infarction induces dysfunction of the epicardial pre‐collector and collecting lymphatic vessels leading to myocardial oedema (Henri et al., 2016). Increasing cardiac lymphangiogenesis through the administration of VEGFC attenuates myocardial oedema, inflammation and fibrosis and improves cardiac function following infarction (Klotz et al., 2015; Henri et al., 2016).
Renal lymphatics and hypertension
In the kidney, lymphangiogenesis has been reported in association with inflammation and infiltrating immune cells in chronic inflammatory kidney diseases such as diabetic nephropathy, IgA nephropathy, tubulointerstitial nephritis and glomerulonephritis with the degree of fibrosis directly correlating to the extent of lymphatic expansion (Yazdani et al., 2014). In acute kidney injury, renal lymphangiogenesis was associated with tubulointerstitial injury, and suppression of renal lymphangiogenesis reduced fibrosis in a mouse model of UUO (Lee et al., 2013). Tubular epithelial cells along with macrophages were demonstrated to be sources of VEGFC in UUO. Hasegawa et al. (2017) were able to reduce inflammation and subsequent fibrosis by the administration of VEGFC for 2 weeks during UUO. In another study, it was reported that lymphangiogenesis might occur in parallel with the onset of proteinuria, yet prior to the development of fibrosis, with activated proximal tubular epithelial cells acting as sources of VEGFC (Yazdani et al., 2012). Renal transplants are also associated with the formation of new lymphatic vessels; however, the consequences of these vessels are a matter of debate. The newly formed vessels could be beneficial by draining out fluid and for immune clearance immediately after operation. In fact, low lymphatic vessel density in the first post‐procedural biopsy is associated with acute rejection. The persistence of these vessels on the other hand may eventually lead to transplant rejection as these vessels help transport APCs to the lymph nodes and initiate immune responses (Kerjaschki, 2004).
The importance of renal lymphatics in fluid balance was demonstrated as early as the 1960s. Renal lymphatic ligation was reported to result in renal oedema, increased urine volume and, surprisingly, increased BP in several studies (Barer and Ward‐Mcquaid, 1957; Lilienfeld et al., 1967). Zhang et al. (2008) later demonstrated that dual renal lymphatic ligation was as detrimental to kidney function as a nephrectomy. With renal immune cell infiltration and accumulation a part of the pathogenesis of hypertension, IAL in the kidney should be evident and necessary to remediate inflammation. Our laboratory investigated this relationship and reported that the kidneys of SHRs demonstrate increased renal lymphatic vessel density in association with inflammation and infiltration of CD68+ macrophages (Kneedler et al., 2017). Interestingly, a strain of SHRs (SHR‐B2) that were hypertensive but resistant to renal injury displayed lower lymphatic vessel density than their age‐matched controls. Fischer 344 rats are a strain of rats that are normotensive but display age‐associated renal injury. Kidneys from these rats at 20 or 24 months of age demonstrated increased renal lymphatic vessel density accompanying age‐associated renal injury compared with 4‐month‐old controls (Kneedler et al., 2017). Thus, the renal inflammation evident in hypertensive and aged kidneys is associated with an increase in renal lymphatic vessel density.
Following this genetic model of hypertension, our lab also tested if this increase in renal lymphatic density occurs in two other models of hypertension, salt‐sensitive (SSHTN) and NOS inhibition‐induced hypertension, induced by the administration of L‐NAME (LHTN). In both hypertensive models, we observed a significant increase in renal lymphatic vessel density (Figure 1) (Lopez Gelston et al., 2018). In SSHTN, the increase in lymphatic density was associated with renal infiltration of macrophages and Th1 cells, whereas in LHTN, this was associated with an increase in renal macrophages and DCs. Furthermore, we hypothesized that while minimal renal lymphatic expansion may be indicative of inflammation, it is insufficient to clear infiltrating immune cells from the cortical interstitium and that further augmenting renal lymphangiogenesis should therefore be beneficial. Enhancing renal lymphatic expansion prior to the onset of hypertension using an inducible genetic model of kidney‐specific lymphangiogenesis (Lammoglia et al., 2016) completely prevented the development of hypertension associated with high‐salt diet and NOS inhibition (Lopez Gelston et al., 2018). Importantly, in both of these models, augmenting renal lymphatic vessel density reduced the immune cell populations previously found to be increased, suggesting increased trafficking from hypertensive kidneys. This study emphasizes the role of renal lymphatics in BP regulation and provides a potential new therapeutic or diagnostic target for the treatment of hypertension.
Figure 1.
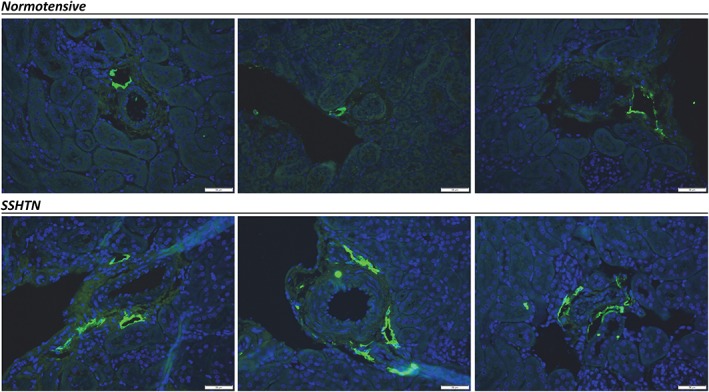
Increased renal lymphatic density in hypertension. In the normotensive kidney cortex, lymphatic vessels are limited to the interlobular arteries with 1–2 identifiable lymphatic lumen (green, Lyve‐1) in each vascular bundle. During SSHTN, the endogenous lymphatic network expands, with increased numbers of lymphatic lumen. Blue, DAPI. Bars = 50 μm.
Conclusions
In conclusion, studies to date demonstrate that lymphatics and expanded lymphatic density are important not only in combating peripheral inflammation but are also key players in BP regulation (Figure 2). Targeting immune cell exfiltration from various tissues through the lymphatics may represent a unique method to reduce diseases of chronic inflammation and ameliorate hypertension.
Figure 2.
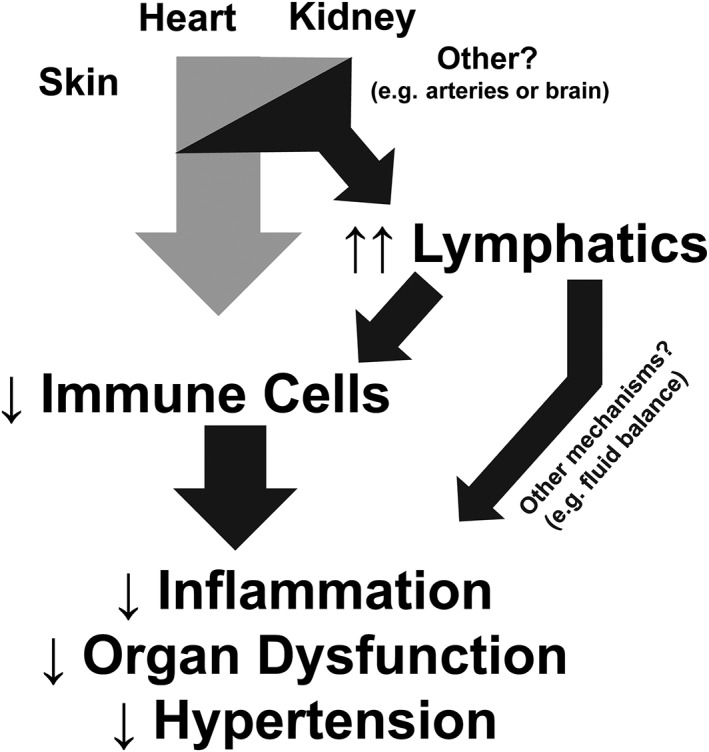
Mechanism by which increased lymphatics decrease inflammation, organ dysfunction and BP.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are supported by the Texas A&M University Health Science Center College of Medicine and the Texas A&M University Health Science Center Department of Medical Physiology. J.M.R. is supported by the American Heart Association (17GRNT33671220) and Lipedema Foundation.
Balasubbramanian D., Lopez Gelston C. A., Rutkowski J. M., and Mitchell B. M. (2019) Immune cell trafficking, lymphatics and hypertension, British Journal of Pharmacology, 176, 1978–1988, 10.1111/bph.14370.
References
- Abouelkheir GR, Upchurch BD, Rutkowski JM (2017). Lymphangiogenesis: fuel, smoke, or extinguisher of inflammation's fire? Exp Biol Med 242: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acedo SC, Gotardo EM, Lacerda JM, de Oliveira CC, de Oliveira Carvalho P, Gambero A (2011). Perinodal adipose tissue and mesenteric lymph node activation during reactivated TNBS‐colitis in rats. Dig Dis Sci 56: 2545–2552. [DOI] [PubMed] [Google Scholar]
- Aldrich MB, Sevick‐Muraca EM (2013). Cytokines are systemic effectors of lymphatic function in acute inflammation. Cytokine 64: 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K (2016). Lymphatic system in cardiovascular medicine. Circ Res 118: 515–530. [DOI] [PubMed] [Google Scholar]
- Aurora AB, Baluk P, Zhang D, Sidhu SS, Dolganov GM, Basbaum C et al (2005). Immune complex‐dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol 175: 6319–6326. [DOI] [PubMed] [Google Scholar]
- Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, Rockson SG et al (2010). Blockade of transforming growth factor‐β1 accelerates lymphatic regeneration during wound repair. Am J Pathol 177: 3202–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S et al (2007). Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 204: 2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ et al (2005). Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest 115: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barer GR, Ward‐Mcquaid JN (1957). Demonstration of renal lymphatics in vivo by intravenous injection of dye: the effect of lymphatic ligature on the blood‐pressure. BJU Int 29: 171–174. [DOI] [PubMed] [Google Scholar]
- Becker F, Kurmaeva E, Gavins FNE, Stevenson EV, Navratil AR, Jin L et al (2016). A critical role for monocytes/macrophages during intestinal inflammation‐associated lymphangiogenesis. Inflamm Bowel Dis 22: 1326–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich A, Belisle EH, Strausser HR (1981). Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 99: 600–607. [DOI] [PubMed] [Google Scholar]
- Card CM, Yu SS, Swartz MA (2014). Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest 124: 943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T et al (2002). Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39: 269–274. [DOI] [PubMed] [Google Scholar]
- Chauhan SK, Jin Y, Goyal S, Lee HS, Fuchsluger TA, Lee HK et al (2011). A novel pro‐lymphangiogenic function for Th17/IL‐17. Blood 118: 4630–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, Lee YS, Chung HK, Choi D, Ecoiffier T, Lee HN et al (2013). Interleukin‐8 reduces post‐surgical lymphedema formation by promoting lymphatic vessel regeneration. Angiogenesis 16: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyou S, Ekland EH, Carpenter AC, Tzeng TC, Tian S, Michaud M et al (2008). Fibroblast‐type reticular stromal cells regulate the lymph node vasculature. J Immunol 181: 3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JN, Guidi CJ, Tewalt EF, Qiao H, Rouhani SJ, Ruddell A et al (2010). Lymph node‐resident lymphatic endothelial cells mediate peripheral tolerance via Aire‐independent direct antigen presentation. J Exp Med 207: 681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Lane MM, Davis AM, Durtschi D, Zawieja DC, Muthuchamy M, Gashev AA et al (2008). Modulation of lymphatic muscle contractility by the neuropeptide substance P. Am J Physiol Heart Circ Physiol 295: H587–H597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL (2005). Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II‐infused macrophage colony‐stimulating factor‐deficient mice: evidence for a role in inflammation in angiotensin‐induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113. [DOI] [PubMed] [Google Scholar]
- Drazner MH (2011). The progression of hypertensive heart disease. Circulation 123: 327–334. [DOI] [PubMed] [Google Scholar]
- Dubrot J, Duraes FV, Potin L, Capotosti F, Brighouse D, Suter T et al (2014). Lymph node stromal cells acquire peptide–MHCII complexes from dendritic cells and induce antigen‐specific CD4+ T cell tolerance. J Exp Med 211: 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzielak DJ (1991). Immune mechanisms in experimental and essential hypertension. Am J Physiol 260: R459–R467. [DOI] [PubMed] [Google Scholar]
- Ebrahimian T, Li MW, Lemarie CA, Simeone SM, Pagano PJ, Gaestel M et al (2011). Mitogen‐activated protein kinase‐activated protein kinase 2 in angiotensin II‐induced inflammation and hypertension: regulation of oxidative stress. Hypertension 57: 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LA, Nowocin AK, Jafari NV, Meader L, Brown K, Sarde A et al (2018). Chronic rejection of cardiac allografts is associated with increased lymphatic flow and cellular trafficking. Circulation 137: 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flister MJ, Wilber A, Hall KL, Iwata C, Miyazono K, Nisato RE et al (2010). Inflammation induces lymphangiogenesis through up‐regulation of VEGFR‐3 mediated by NF‐κB and Prox1. Blood 115: 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R, Davalos‐Misslitz AC, Rot A (2008). CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol 8: 362–371. [DOI] [PubMed] [Google Scholar]
- Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N et al (2012). Interferon‐γ ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol 303: H587–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gąsecki D, Kwarciany M, Nyka W, Narkiewicz K (2013). Hypertension, brain damage and cognitive decline. Curr Hypertens Rep 15: 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gousopoulos E, Proulx ST, Bachmann SB, Dieterich LC, Scholl J, Karaman S et al (2017). An important role of VEGF‐C in promoting lymphedema development. J Invest Dermatol 137: 1995–2004. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halin C, Tobler NE, Vigl B, Brown LF, Detmar M (2007). VEGF‐A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood 110: 3158–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KL, Volk‐Draper LD, Flister MJ, Ran S (2012). New model of macrophage acquisition of the lymphatic endothelial phenotype. PLoS One 7: e31794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM (2012). Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 111: 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S, Nakano T, Torisu K, Tsuchimoto A, Eriguchi M, Haruyama N et al (2017). Vascular endothelial growth factor‐C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab Invest 97: 1439–1452. [DOI] [PubMed] [Google Scholar]
- Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards‐Levy F et al (2016). Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation 133: 1484–1497 discussion 1497. [DOI] [PubMed] [Google Scholar]
- Herrera J, Ferrebuz A, MacGregor EG, Rodriguez‐Iturbe B (2006). Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol: JASN 17: S218–S225. [DOI] [PubMed] [Google Scholar]
- Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M et al (2011). An important role of lymphatic vessel activation in limiting acute inflammation. Blood 117: 4667–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. (2006). An inflammation‐induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med 203: 2763–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Jackson DG (2010). Inflammation‐induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin‐mediated dendritic cell transmigration. Int Immunol 22: 839–849. [DOI] [PubMed] [Google Scholar]
- Kajiya K, Detmar M (2006). An important role of lymphatic vessels in the control of UVB‐induced edema formation and inflammation. J Invest Dermatol 126: 920–922. [DOI] [PubMed] [Google Scholar]
- Kang S, Lee SP, Kim KE, Kim HZ, Memet S, Koh GY (2009). Toll‐like receptor 4 in lymphatic endothelial cells contributes to LPS‐induced lymphangiogenesis by chemotactic recruitment of macrophages. Blood 113: 2605–2613. [DOI] [PubMed] [Google Scholar]
- Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S et al (2016). Gut microbiota promote angiotensin II‐induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 5: e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassan M, Galan M, Partyka M, Trebak M, Matrougui K (2011). Interleukin‐10 released by CD4+ CD25+ natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol 31: 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE et al (2009). Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood 113: 5650–5659. [DOI] [PubMed] [Google Scholar]
- Kerjaschki D (2004). Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol 15: 603–612. [DOI] [PubMed] [Google Scholar]
- Kerjaschki D, Huttary N, Raab I, Regele H, Bojarski‐Nagy K, Bartel G et al (2006). Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat Med 12: 230–234. [DOI] [PubMed] [Google Scholar]
- Kim H, Kataru RP, Koh GY (2014). Inflammation‐associated lymphangiogenesis: a double‐edged sword? J Clin Invest 124: 936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi H, Ito Y, Mizuno M, Suzuki Y, Terabayashi T, Nagura F et al (2013). TGF‐β1 promotes lymphangiogenesis during peritoneal fibrosis. J Am Soc Nephrol 24: 1627–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J et al (2014). DC isoketal‐modified proteins activate T cells and promote. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz L, Norman S, Vieira JM, Masters M, Rohling M, Dube KN et al (2015). Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature 522: 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneedler SC, Phillips LE, Hudson KR, Beckman KM, Lopez Gelston CA, Rutkowski JM et al (2017). Renal inflammation and injury are associated with lymphangiogenesis in hypertension. Am J Physiol Renal Physiol 312: F861–F869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al (2009). Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 119: 2904–2912. [DOI] [PubMed] [Google Scholar]
- Lammoglia GM, Van Zandt CE, Galvan DX, Orozco JL, Dellinger MT, Rutkowski JM (2016). Hyperplasia, de novo lymphangiogenesis, and lymphatic regression in mice with tissue‐specific, inducible overexpression of murine VEGF‐D. Am J Physiol Heart Circ Physiol 311: H384–H394. [DOI] [PubMed] [Google Scholar]
- Lee AS, Lee JE, Jung YJ, Kim DH, Kang KP, Lee S et al (2013). Vascular endothelial growth factor‐C and ‐D are involved in lymphangiogenesis in mouse unilateral ureteral obstruction. Kidney Int 83: 50–62. [DOI] [PubMed] [Google Scholar]
- Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS et al (2006). Angiotensin II hypertension is attenuated in interleukin‐6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940. [DOI] [PubMed] [Google Scholar]
- Lee KM, McKimmie CS, Gilchrist DS, Pallas KJ, Nibbs RJ, Garside P et al (2011). D6 facilitates cellular migration and fluid flow to lymph nodes by suppressing lymphatic congestion. Blood 118: 6220–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levick JR, Michel CC (2010). Microvascular fluid exchange and the revised Starling principle. Cardiovasc Res 87: 198–210. [DOI] [PubMed] [Google Scholar]
- Lilienfeld RM, Friedenberg RM, Herman JR (1967). The effect of renal lymphatic ligation on kidney and blood pressure. Radiology 88: 1105–1109. [DOI] [PubMed] [Google Scholar]
- Lim HY, Thiam CH, Yeo KP, Bisoendial R, Hii CS, McGrath KC et al (2013). Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR‐BI‐mediated transport of HDL. Cell Metab 17: 671–684. [DOI] [PubMed] [Google Scholar]
- Lopez Gelston CA, Balasubbramanian D, Abouelkheir GR, Lopez AH, Hudson KR, Johnson ER et al (2018). Enhancing renal lymphatic expansion prevents hypertension in mice. Circ Res 122: 1094–1101. [DOI] [PubMed] [Google Scholar]
- Lopez Gelston CA, Mitchell BM (2017). Recent advances in immunity and hypertension. Am J Hypertens 30: 643–652. [DOI] [PubMed] [Google Scholar]
- Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K et al (2009). Macrophages regulate salt‐dependent volume and blood pressure by a vascular endothelial growth factor‐C‐dependent buffering mechanism. Nat Med 15: 545–552. [DOI] [PubMed] [Google Scholar]
- Maddaluno L, Verbrugge SE, Martinoli C, Matteoli G, Chiavelli A, Zeng Y et al (2009). The adhesion molecule L1 regulates transendothelial migration and trafficking of dendritic cells. J Exp Med 206: 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisel K, Sasso MS, Potin L, Swartz MA (2017). Exploiting lymphatic vessels for immunomodulation: rationale, opportunities, and challenges. Adv Drug Deliv Rev 114: 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K, Ii M, Cursiefen C, Jackson DG, Keino H, Tomita M et al (2005). Inflammation‐induced lymphangiogenesis in the cornea arises from CD11b‐positive macrophages. J Clin Invest 115: 2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C et al (2010). Central and peripheral mechanisms of T‐lymphocyte activation and vascular inflammation produced by angiotensin II‐induced hypertension. Circ Res 107: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson DL, James L, Berdan EA, Meister CJ (2006). Immune suppression attenuates hypertension and renal disease in the Dahl salt‐sensitive rat. Hypertension 48: 149–156. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H (2013). Genetic mutation of recombination activating gene 1 in Dahl salt‐sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKimmie CS, Singh MD, Hewit K, Lopez‐Franco O, Le Brocq M, Rose‐John S et al (2013). An analysis of the function and expression of D6 on lymphatic endothelial cells. [DOI] [PubMed]
- Medzhitov R (2010). Inflammation 2010: new adventures of an old flame. Cell 140: 771–776. [DOI] [PubMed] [Google Scholar]
- Miguel CD, Rudemiller NP, Abais JM, Mattson DL (2015). Inflammation and hypertension: new understandings and potential therapeutic targets. Curr Hypertens Rep 17: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miteva DO, Rutkowski JM, Dixon JB, Kilarski W, Shields JD, Swartz MA (2010). Transmural flow modulates cell and fluid transport functions of lymphatic endothelium. Circ Res 106: 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM (2013). Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018). The immunology of hypertension. J Exp Med 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Iwata C, Suzuki HI, Kiyono K, Morishita Y, Watabe T et al (2008). Inhibition of endogenous TGF‐β signaling enhances lymphangiogenesis. Blood 111: 4571–4579. [DOI] [PubMed] [Google Scholar]
- Okuda T, Grollman A (1967). Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med 25: 257–264. [PubMed] [Google Scholar]
- Olsen F (1970). Type and course of the inflammatory cellular reaction in acute angiotensin‐hypertensive vascular disease in rats. Acta Pathol Microbiol Scand A 78: 143–150. [DOI] [PubMed] [Google Scholar]
- Olsen F (1980). Transfer of arterial hypertension by splenic cells from DOCA‐salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C 88: 1–5. [DOI] [PubMed] [Google Scholar]
- Pegu A, Qin S, Fallert Junecko BA, Nisato RE, Pepper MS, Reinhart TA (2008). Human lymphatic endothelial cells express multiple functional TLRs. J Immunol 180: 3399–405. [DOI] [PubMed] [Google Scholar]
- Quiroz Y, Pons H, Gordon KL, Rincon J, Chavez M, Parra G et al (2001). Mycophenolate mofetil prevents salt‐sensitive hypertension resulting from nitric oxide synthesis inhibition. Am J Physiol Renal Physiol 281: F38–F47. [DOI] [PubMed] [Google Scholar]
- Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP (2017). The lymphatic system: integral roles in immunity. Annu Rev Immunol 35: 31–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Franco M, Johnson RJ (2013). Impaired pressure natriuresis is associated with interstitial inflammation in salt‐sensitive hypertension. Curr Opin Nephrol Hypertens 22: 37–44. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M et al (2001). Mycophenolate mofetil prevents salt‐sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera‐Acosta J et al (2002). Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 282: F191–F201. [DOI] [PubMed] [Google Scholar]
- Satou R, Miyata K, Gonzalez‐Villalobos RA, Ingelfinger JR, Navar LG, Kobori H (2012). Interferon‐γ biphasically regulates angiotensinogen expression via a JAK‐STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J 26: 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaich MP, Sobotka PA, Krum H, Whitbourn R, Walton A, Esler MD (2009). Renal denervation as a therapeutic approach for hypertension: novel implications for an old concept. Hypertension 54: 1195–1201. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q et al (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solak Y, Afsar B, Vaziri ND, Aslan G, Yalcin CE, Covic A et al (2016). Hypertension as an autoimmune and inflammatory disease. Hypertens Res 39: 567–573. [DOI] [PubMed] [Google Scholar]
- Sprague AH, Khalil RA (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol 78: 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen UG (1976). The role of thymus for the development and prognosis of hypertension and hypertensive vascular disease in mice following renal infarction. Acta Pathol Microbiol Scand A 84: 235–243. [DOI] [PubMed] [Google Scholar]
- Tamburini BA, Burchill MA, Kedl RM (2014). Antigen capture and archiving by lymphatic endothelial cells following vaccination or viral infection. Nat Commun 5: 3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KW, Chong SZ, Angeli V (2014). Inflammatory lymphangiogenesis: cellular mediators and functional implications. Angiogenesis 17: 373–381. [DOI] [PubMed] [Google Scholar]
- Thomas SN, Rutkowski JM, Pasquier M, Kuan EL, Alitalo K, Randolph GJ et al (2012). Impaired humoral immunity and tolerance in K14‐VEGFR‐3‐Ig mice that lack dermal lymphatic drainage. J Immunol 189: 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triacca V, Guc E, Kilarski WW, Pisano M, Swartz MA (2017). Transcellular pathways in lymphatic endothelial cells regulate changes in solute transport by fluid stress. Circ Res 120: 1440–1452. [DOI] [PubMed] [Google Scholar]
- Trott DW, Harrison DG (2014). The immune system in hypertension. Adv Physiol Educ 38: 20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzewik J, Mallipattu SK, Artmann GM, Delano FA, Schmid‐Schonbein GW (2001). Evidence for a second valve system in lymphatics: endothelial microvalves. FASEB J 15: 1711–1717. [DOI] [PubMed] [Google Scholar]
- Vigl B, Aebischer D, Nitschke M, Iolyeva M, Rothlin T, Antsiferova O et al (2011). Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus‐dependent manner. Blood 118: 205–215. [DOI] [PubMed] [Google Scholar]
- Vinh A, Chen W, Blinder Y, Weiss D, Robert Taylor W, Goronzy J et al (2010). Inhibition and genetic ablation of the B7/CD28 T cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- White FN, Grollman A (1964). Autoimmune factors associated with infarction of the kidney. Nephron 1: 93–102. [DOI] [PubMed] [Google Scholar]
- Wiig H, Swartz MA (2012). Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev 92: 1005–1060. [DOI] [PubMed] [Google Scholar]
- Wirzenius M, Tammela T, Uutela M, He Y, Odorisio T, Zambruno G et al (2007). Distinct vascular endothelial growth factor signals for lymphatic vessel enlargement and sprouting. J Exp Med 204: 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L et al (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen‐activated protein kinase. Circ Res 114: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuest TR, Carr DJ (2010). VEGF‐A expression by HSV‐1‐infected cells drives corneal lymphangiogenesis. J Exp Med 207: 101–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang GH, Zhou X, Ji WJ, Liu JX, Sun J, Dong Y et al (2017). VEGF‐C‐mediated cardiac lymphangiogenesis in high salt intake accelerated progression of left ventricular remodeling in spontaneously hypertensive rats. Clin Exp Hypertens 39: 740–747. [DOI] [PubMed] [Google Scholar]
- Yang GH, Zhou X, Ji WJ, Zeng S, Dong Y, Tian L et al (2014). Overexpression of VEGF‐C attenuates chronic high salt intake‐induced left ventricular maladaptive remodeling in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 306: H598–H609. [DOI] [PubMed] [Google Scholar]
- Yao LC, Testini C, Tvorogov D, Anisimov A, Vargas SO, Baluk P et al (2014). Pulmonary lymphangiectasia resulting from vascular endothelial growth factor‐C overexpression during a critical period. Circ Res 114: 806–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani S, Navis G, Hillebrands JL, van Goor H, van den Born J (2014). Lymphangiogenesis in renal diseases: passive bystander or active participant? Expert Rev Mol Med e15: 16. [DOI] [PubMed] [Google Scholar]
- Yazdani S, Poosti F, Kramer AB, Mirkovic K, Kwakernaak AJ, Hovingh M et al (2012). Proteinuria triggers renal lymphangiogenesis prior to the development of interstitial fibrosis. PLoS One 7: e50209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampell JC, Avraham T, Yoder N, Fort N, Yan A, Weitman ES et al (2012). Lymphatic function is regulated by a coordinated expression of lymphangiogenic and anti‐lymphangiogenic cytokines. Am J Physiol Cell Physiol 302: C392–C404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawieja DC (2009). Contractile physiology of lymphatics. Lymphat Res Biol 7: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Xu X, Potter BJ, Wang W, Kuo L, Michael L et al (2006). TNF‐α contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol 26: 475–480. [DOI] [PubMed] [Google Scholar]
- Zhang T, Guan G, Liu G, Sun J, Chen B, Li X et al (2008). Disturbance of lymph circulation develops renal fibrosis in rats with or without contralateral nephrectomy. Nephrol Ther 13: 128–138. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL (2004). Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95: 210–216. [DOI] [PubMed] [Google Scholar]