

Abstract
Traditionally, arterial hypertension and subsequent end‐organ damage have been attributed to haemodynamic factors, but increasing evidence indicates that inflammation also contributes to the deleterious consequences of this disease. The immune system has evolved to prevent invasion of foreign microorganisms and to promote tissue healing after injury. However, this beneficial activity comes at a cost of collateral damage when the immune system overreacts to internal injury, such as prehypertension. Over the past few years, important findings have revolutionized hypertension research. Firstly, in 2007, a seminal paper showed that adaptive immunity is involved in the pathogenesis of hypertension. Secondly, salt storage in the skin and its consequences for cardiovascular physiology were discovered. Thirdly, after the discovery that salt promotes the differentiation of CD4+ T cells into TH17 cells, it was demonstrated that salt directly changes several cells of the innate and adaptive immune system and aggravates autoimmune disease but may improve antimicrobial defence. Herein, we will review pathways of activation of immune cells by salt in hypertension as the framework for understanding the multiple roles of salt and immunity in arterial hypertension and autoimmune disease.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- Ang II
angiotensin II
- gp91phox
cytochrome b‐245 heavy chain
- NCC
Na‐Cl cotransporter
- NFAT5
nuclear factor of activated T‐cells 5
- p47phox
neutrophil cytosol factor 1
- RAG‐1
recombination‐activating gene‐1
- SGK1
serum and glucocorticoid‐regulated kinase 1
Introduction
High blood pressure afflicts more than 1 billion people worldwide. Although the borders between normotension and hypertension are arbitrary, the recent Systolic Blood Pressure Intervention Trial illustrated that strict blood pressure control in hypertensive populations can significantly improve cardiovascular outcomes. Thus, in the recent update of the ‘Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults’, the border for stage 1 hypertension was lowered by 10 to 130 mmHg (Whelton et al., 2018). In turn, the number of hypertensive patients increased dramatically, for example, in the United States overnight from 32 to 43% of the American population. Despite this high prevalence and decades of research, the aetiology of most cases of hypertension remains undefined, and blood pressure remains uncontrolled in up to 50% of some hypertensive populations (Montaniel and Harrison, 2016). Meanwhile, it is evident that hypertension and hypertensive end organ damage are mediated through blood pressure‐dependent and ‐independent mechanisms. Over the last few years, three important findings have revolutionized hypertension research. Firstly, in 2007, the seminal paper by the group of David Harrison showed that adaptive immunity makes a key contribution to the pathogenesis of hypertension (Guzik et al., 2007). Secondly, Titze and colleagues demonstrated nonosmotic sodium storage in the skin and its consequences for cardiovascular physiology. Thirdly, it was established that salt directly alters the polarization and activation of cells in the innate and adaptive immune system with consequent aggravation of autoimmune disease but improvements in antimicrobial defence.
The role of immunity in arterial hypertension has been comprehensively reviewed elsewhere (Wenzel et al., 2016; Foss et al., 2017; Wenzel et al., 2017; Norlander et al., 2018; Rucker et al., 2018). Below, we will focus on the cooperative roles of salt, IL‐17 and the inflammatory response in autoimmune disease and hypertension.
Adaptive immunity
In 2007, Guzik and colleagues reported that the increase in blood pressure caused by chronic angiotensin II (Ang II) infusion or DOCA/salt administration was significantly blunted in recombination‐activating gene‐1 (RAG1, also known as SLC50A1)‐deficient mice that lack T and B lymphocytes, both key constituents of the adaptive immune system. In these experiments, adoptive transfer of T cells, but not beta cells, restored the hypertensive response, indicating that T cells play an important role in the generation of arterial hypertension (Guzik et al., 2007). These results have been confirmed in severe combined immunodeficiency mice and in Dahl salt‐sensitive rats in which the RAG1 or the essential T cell receptor component CD3 have been deleted using zinc finger nuclease technology (Crowley et al., 2010; Mattson et al., 2013). Collectively, these reports have further established that lymphocytes promote blood pressure elevation by inducing vascular endothelial dysfunction and renal sodium retention. Recently, it has been reported that since 2015, the Jackson B6.RAG1−/− mouse line lost its resistance to Ang II‐induced hypertension which was attributed to spontaneous mutations (Ji et al., 2017). RAG1−/− mice have an ‘empty niche’ where secondary lymphoid organs and the bone marrow, devoid of lymphocytes, could be populated by other immune cells. In particular, these mice have innate lymphoid cells and an expanded population of natural killer (NK) cells that can assume many of the roles of T cells. The role of NK cells can be studied using double knockout mice that lack RAG1 and IL‐2 receptor γ chain. Experiments are urgently warranted to examine the role of innate lymphoid cells and NK cells in hypertension. In addition, recent data suggest that beta cells/IgGs are also crucial for the development of Ang II‐induced hypertension and vessel remodelling in mice (Chan et al., 2015).
IL‐17
IL‐17 is the defining cytokine of TH17 cells. Several isoforms of IL‐17 exist, with IL‐17A and IL‐17F being the most abundant. The role of IL‐17 in hypertension is controversial. Ang II infusion increases IL‐17A production by T cells and IL‐17 protein in the aortic media and the heart. The initial hypertensive response to Ang II infusion is similar in IL‐17A−/− mice and wild type mice. However, hypertension is not sustained in IL‐17A−/− mice, settling 30 mmHg lower than in wild type controls after 4 weeks of Ang II infusion (Madhur et al., 2010). In addition, mice lacking IL‐17A are also protected against aortic stiffening and cardiac fibrosis after infusion of Ang II (Wu et al., 2014). Amador et al. demonstrated an immediate decrease in blood pressure after the injection of neutralizing anti‐IL‐17 antibodies in rats (Amador et al., 2014). Dermal overexpression of IL‐17A induces systemic endothelial dysfunction, vascular oxidative stress and arterial hypertension (Karbach et al., 2014). In contrast, Marko et al. did not observe any lowering of blood pressure upon anti‐IL‐23 or anti‐IL‐17 antibody treatment (Marko et al., 2012). This latter study is in agreement with our report showing that genetic disruption of the IL‐23/IL‐17A axis does not alter hypertension after Ang II treatment in the presence of excess mineralocorticoids (Krebs et al., 2014). One alternative explanation for this inconsistency in demonstrating the role of IL‐17 in hypertension could be due to a lack of characterization of the gut microbiome in these studies.
IL‐17 isoforms bind as homo‐ and heterodimers to a receptor complex composed of IL‐17 receptor A and IL‐17 receptor C subunits. Whereas the role of IL‐17A has been intensively examined in hypertension, less is known about the role of other isoforms and the IL‐17 receptors. Genetic knockouts of IL‐17 receptors are available but have not been examined in hypertension. One report found that inhibition of IL‐17 receptor unit A by a neutralizing antibody lowered blood pressure in Ang II‐infused mice. In contrast, in the same report, antibodies to the IL‐17F isoform did not lower blood pressure in Ang II infused mice and lowered marginally albuminuria but not renal induction of TGF‐β (Saleh et al., 2016). This confirmed previous work from the same group showing that genetic IL‐17F deficiency has little or no effect in Ang II‐infused mice (Norlander et al., 2016). It is of interest that using IL‐17F gene‐deficient mice, IL‐17F‐neutralizing antibodies and adoptive transfer experiments into Rag−/− mice demonstrated that CD4+ T cell‐derived IL‐17F drives strongly renal tissue injury in acute crescentic nephritis (Riedel et al., 2016).
The mechanisms through which IL‐17A causes hypertension are receiving considerable scrutiny to identify which target cells and receptors transmit IL‐17A signalling to modulate blood pressure. These studies have revealed that IL‐17A and IFN‐γ released from TH17 or TH1 cells stimulate or up‐regulate transport channels in the tubules of the kidney. This includes the sodium hydrogen exchanger 3 (NHE3) in the proximal tubule, the Na‐K‐2Cl‐cotransporter (NKCC2) in the thick ascending limb, the Na‐Cl cotransporter (NCC) in the distal tubule and the epithelial sodium channel (ENaC) in the collecting duct as reviewed recently by Norlander et al. and shown in Figure 1. This in turn can cause sodium and volume retention (Kamat et al., 2015; Norlander et al., 2016). Within the vasculature, IL‐17A deficiency protects against aortic stiffening in Ang II‐infused mice. Since lowering of blood pressure by hydralazine and hydrochlorothiazide also prevents aortic stiffening, it remains open whether IL‐17A causes aortic stiffening only by raising blood pressure or by direct effects in the vasculature (Wu et al., 2014). However, if the effects of IL‐17A on the vasculature extend to resistance vessels, IL‐17A could also modulate sodium retention in the kidney through alterations in renal haemodynamics.
Figure 1.
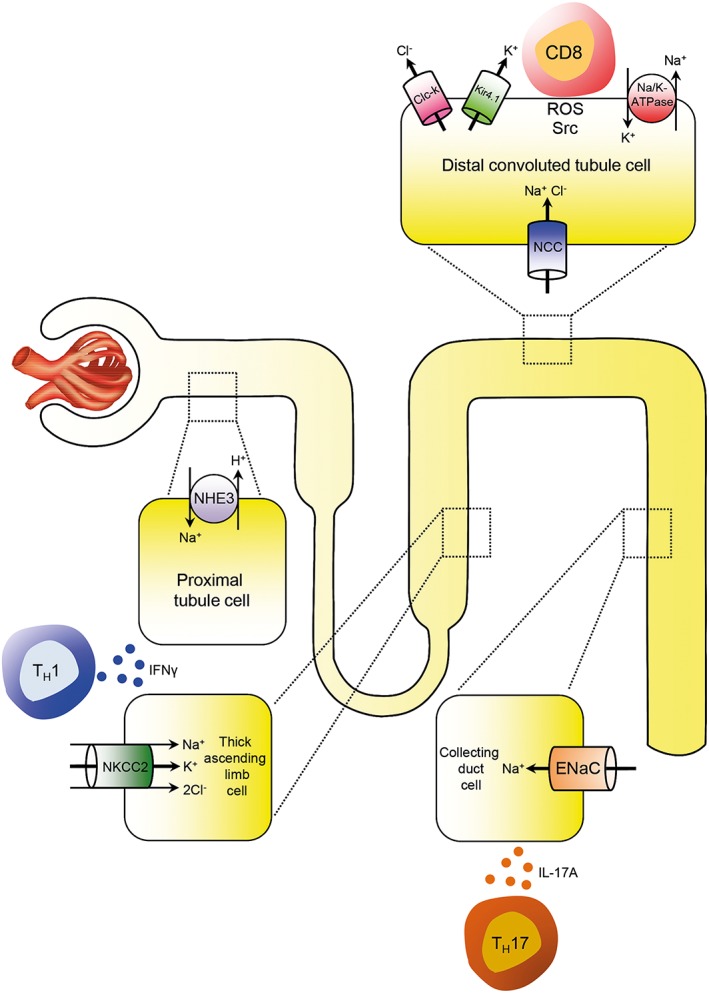
Inflammation and renal sodium transporters. Infiltrating TH1 and TH17 cells modulate sodium transportation throughout the tubular system by secretion of cytokines like IFNγ and IL‐17A. This includes the sodium hydrogen exchanger 3 (NHE3) in the proximal tubular cells, the Na‐K‐2Cl‐cotransporter (NKCC2) in cells of the thick ascending limb, the Na‐Cl cotransporter (NCC) in distal tubular cells and the epithelial sodium (Na) channel (ENaC) in the collecting duct. A direct cell–cell contact between CD8+ T cell and tubular cells as a prerequisite for activation of the transporter has been shown for the NCC in distal tubule cells (modified from Norlander and Madhur, 2017).
Although TH17 cells are considered to be the principal source of IL‐17 other cells like CD8+ cells, γδ T cells, NKT cells and type 3 innate lymphoid cells have been reported to produce IL‐17. γδ T cells are T cells that have a distinctive T‐cell receptor on their surface and play important roles in mucosal defences and autoimmunity. They serve as a ‘first line of defence’ or ‘bridge between innate and adaptive responses’. Recent data suggest genetic knockout of γδ T cells and antibody‐induced γδ T cell depletion can blunt Ang II‐induced increases in blood pressure and endothelial dysfunction suggesting these cells may play a causal role in elevations in blood pressure (Li et al., 2014; Caillon et al., 2017).
Innate immunity and hypertension
Innate immunity is considered to be the immune system's first line of defence against invading pathogens. The term ‘innate’ refers to the inherent capacity of this arm of immune defence to be rapidly activated by non‐specific external stimuli without the need for directing antigen‐specific activation of lymphocytes from the adaptive immune system. The essential cellular components of innate immunity are granulocytes, macrophages, mast cells and certain subsets of innate lymphoid cells. The key humoral effectors of innate immunity are the defensins and the complement system. Innate immunity in hypertension has been reviewed recently (Wenzel et al., 2016).
Complement
The self‐amplifying cascade of messenger and effector molecules of the complement system serves as a powerful danger‐sensing system that protects the host from a hostile microbial environment, while maintaining proper tissue and organ function through effective clearance of altered or dying cells. It also plays important roles in the regulation of adaptive immunity. Recent experimental data strongly support a role for complement in arterial hypertension and vascular biology (Zhang et al., 2014; Weiss et al., 2016; Chen et al., 2018; Ren et al., 2018). The remarkably similar clinical and histopathological features of so‐called malignant nephrosclerosis and atypical haemolytic uraemic syndrome, which is driven by complement activation, point to convergent complement‐mechanisms underpinning the development of malignant nephrosclerosis (Timmermans et al., 2017). New discoveries in the complement field refine our appreciation of the close interdependency of ‘ancient’ complement and ‘modern’ adaptive immune mechanisms, but also the role of complement and complement receptors in tissue homeostasis. Complement activation can cause autoimmunity, tissue inflammation and injury. Accordingly, complement‐inhibitory drugs are effective treatments for several inflammatory diseases, and intense research is ongoing to pinpoint how manipulating the complement cascade could afford protection from arterial hypertension (Wenzel et al., 2017).
Is the salt controversy caused by the history of salt?
There is no doubt that the current excess in salt ingestion has profound health consequences including hypertension and cardiovascular target organ damage. Several potential pathophysiological mechanisms relating a high salt diet to cardiovascular disease have been characterized and include changes in the renin angiotensin aldosterone system and ROS. Volume expansion is thought to be the major effect through which salt increases blood pressure. The famous Yellow Emperor already wrote in approximately 3000 BC ‘If too much salt is used in food, the pulse hardens, tears make their appearance and the complexion changes’. Guyton later hypothesized that the kidney was crucial in mediating this relationship between salt and hypertension. He argued that through its functions to regulate volume homeostasis and sodium reabsorption, the kidney could preserve normal blood pressure via pressure natriuresis and that persistent hypertension reflected a failure of the kidney to appropriately excrete sodium (Guyton, 1991). There is still vigorous debate about the optimal salt intake and the importance of dietary salt reduction. Such an emotional discussion is rather unusual in scientific questions. Could it be that the emotional energy surrounding the issue of salt accrues from the cultural and historical significance of this substance over the last several thousand years (Ritz, 1996)?
Salt has been regarded in some societies as a symbol of purity, incorruptibility and even immortality. In the past, physicians recognized the potential therapeutic effects of salt, attributable to bacteriostatic properties accruing from its osmolarity and ionic strength. The Latin words for health and healthy, ‘salutem’ and ‘salubris’, are actually derived from ‘sal’ (salt). It is probable that the French, when they greet each other with the word ‘salut’, still realize subconsciously today that this word comes from salt. The word salad also goes back to the salting of green plants and vegetables in Roman cuisine. Salt was also a symbol of virility and potency. This explains, for example, the statement in William Shakespeare's ‘The merry wives of Windsor’: ‘Though we are justices we have some salt of our youth in us.’ In Bavaria, Germany, some salt used to be sprinkled into the bridal bed on the day of a wedding to increase the bride's fertility. Salt, the ‘white gold’ was an object of high politics. The high commercial value of salt is reflected by the fact that enormous efforts were made to transport it to the customer. Ancient Rome had special salt streets (via salaria) which were maintained by salt officials (salarii). The Hanseatic League owes its outstanding trading position in Northern Europe, as well as its wealth, primarily to the fish trade. Fish had to be salted for preservation. Thus, the Hanseatic League had access to herring in the Baltic Sea and the North Sea on the one hand and to high‐quality salt from the Lüneburg saltworks in Northern Germany on the other hand. With the introduction of alternative methods, especially the refrigerator, salt is no longer critical to conserve food. Today, salt is cheap to produce and has completely lost its role as a luxury item and valuable commodity. Nevertheless, by traditionally adopting historical behaviours, individuals in modern times consume far more salt than is physiologically necessary.
New salt concepts
Salt induced changes in the immune system
Salt provokes an increase in blood pressure and damage in target organs at least in part by polarizing adaptive and innate immune cells towards a pro‐inflammatory phenotype. For example, in 2013, Kleinewietfeld et al. and Wu et al. independently found that an elevated sodium chloride concentration (40–80 mM) in an otherwise isotonic culture medium promotes the differentiation of CD4+ T cells into TH17 cells in vitro. The authors demonstrated that a high‐salt diet accelerated neuropathology in experimental autoimmune encephalomyelitis, a mouse model of the autoimmune disease multiple sclerosis (Kleinewietfeld et al., 2013; Wu et al., 2013). The link between sodium chloride and TH17 differentiation was the transcription factor nuclear factor of activated T‐cells 5 (NFAT5) and serum and glucocorticoid‐regulated kinase 1 (SGK1). SGK1 is a kinase important for sensing and responding to changes in extracellular Na+. It was cloned as a gene regulated by the hydration state of a cell (Binger et al., 2015b). SGK1 is a downstream target of NFAT5. Under physiological conditions, SGK1 is expressed at low levels but is significantly up‐regulated during glucocorticoid or mineralocorticoid excess and hypertonicity. In vitro a TH17‐promoting cytokine cocktail together with increased salt concentrations substantially accelerate the induction of TH17 cells (Figure 2). TH1 and TH2 polarization is not affected by Na+‐induced hypertonicity, suggesting a specific effect of salt on TH17 differentiation. Blockade or genetic knockdown of either NFAT5 or SGK1 prevents Na+‐mediated TH17 induction, thus identifying NFAT5 and SGK1 as the intracellular signalling pathways by which excess salt enhances TH17 differentiation. IL‐23 has a critical role in stabilizing and reinforcing the TH17 phenotype by increasing the expression of IL‐23 receptors. SGK1 is also an essential node downstream of IL‐23 signalling by regulating IL‐23 receptor expression. (Kleinewietfeld et al., 2013). It is of interest that IL‐17A‐induced signalling also seems to be SGK1‐dependent. Norlander et al. found that IL‐17A increased the activity of the sodium‐chloride cotransporter in mouse distal convoluted tubule cells – by a SGK1‐dependent pathway. Moreover, knockout of SGK1 in CD4+ T cells results in a blunted hypertensive response to Ang II and DOCA/salt and attenuated renal and vascular inflammation (Norlander et al., 2016). Recent studies have also established a strong correlation between eating a fast food diet and an increased number of TH17 cells in the circulation (Manzel et al., 2014).
Figure 2.
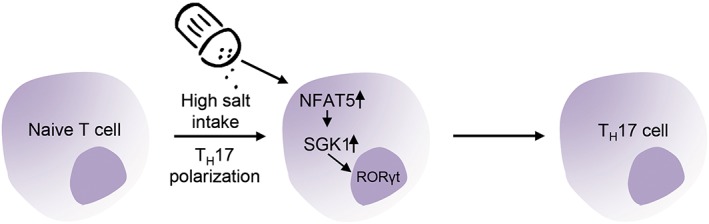
Salt and T cells. In vitro a TH17‐promoting cytokine cocktail together with increased NaCl‐concentrations substantially accelerates the induction of TH17 cells. The enhanced induction of TH17 cells evoked by excess salt is mediated by an up‐regulation of NFAT5 and SGK1 (modified from Binger et al., 2015b).
Dendritic cells play an important role in the genesis of hypertension through their capacity to stimulate T cell activation (Kirabo et al., 2014; Hevia et al., 2018). Recent data show that a high sodium diet can prime dendritic cells to cause hypertension. Sodium enters dendritic cells through the amiloride‐sensitive epithelial sodium channel and the sodium hydrogen exchanger 1. This leads to calcium influx via the sodium calcium exchanger, activation of PKC, phosphorylation of p47phox and association of p47phox with gp91phox. The NADPH oxidase produces superoxide with subsequent formation of immunogenic isolevuglandin‐protein adducts. Dendritic cells activated by excess sodium produce increased IL‐1β and promote T cell production of cytokines IL‐17A and IFN‐γ. When adoptively transferred into naive mice, dendritic cells primed with high salt trigger hypertension in response to a subpressor dose of Ang II (Barbaro et al., 2017).
In a simplified paradigm, macrophages can be dichotomized into pro‐inflammatory M1 and anti‐inflammatory M2 phenotypes. M2 macrophages have been shown to play central roles in mediating TH2 immunity, wound healing and the suppression of effector T cell function. NaCl blunts the activation of M2 macrophages (Binger et al., 2015a). Similarly, bone marrow‐derived dendritic cells developed ex vivo in sodium chloride‐enriched medium acquire a M2‐like signature (Chessa et al., 2016). FOXP3+ regulatory T cells are another population of anti‐inflammatory immune cells central for the maintenance of self‐tolerance and suppression of inflammation. Increasing NaCl, either in vitro or in murine models via diet, markedly impairs regulatory T cell function in a SGK1‐dependent manner (Hernandez et al., 2015). These studies show that excess dietary salt intake might therefore represent an environmental risk factor for the development (or exacerbation) of autoimmune diseases and arterial hypertension by disrupting the balance between the suppressive and inflammatory actions of the immune system. Salt stimulates the induction of pro‐inflammatory cells like TH17 and M1 macrophages and curtails the reparative actions of regulatory T cells and M2 macrophages. Moreover, the inflammasome, which is also critical for the generation of a TH17 response, can induce widespread inflammatory responses after exposure to a high‐salt environment (Ip and Medzhitov, 2015). What are the mechanisms by which salt‐induced changes in immune cells cause hypertension? Previous data suggested that infiltrating lymphocytes secrete cytokines like IL‐17A and IFN‐γ that modulate sodium reabsorption as shown in Figure 1. In addition, an interesting finding has been reported by Liu et al. These authors showed that CD8+ T cells induced an up‐regulation and activation of the NCC in distal convoluted tubules, which leads to salt‐sensitive hypertension. The up‐regulation and activation of NCC occurred via direct cell–cell contact by ROS‐induced Src activation, chloride efflux via regulation of the potassium channel Kir4.1 and the chloride channel ClC‐K on the basolateral cell membrane as shown in Figure 1 (Liu et al., 2017).
Salt and the microbiome
Recent studies have revealed that salt may provoke TH17 immunity in vivo through effects on the gut microbiota. Dietary shifts in sodium intake can have widespread effects on the gut by causing changes in gut architecture and immune profiles. Wilck and colleagues recently showed that a high salt intake affects the gut microbiome in mice by depleting Lactobacillus murinus. The altered microbiome results in an altered metabolome. For example, less indoles are generated from tryptophan in the gut lumen. Indoles inhibit the generation of TH17 cells. Consequently, treatment of mice with Lactobacillus prevented salt‐induced exacerbations of actively‐induced experimental autoimmune encephalomyelitis and salt‐sensitive hypertension by inducing an up‐regulation of TH17 cells. In line with these findings, a moderate high‐salt challenge in a pilot study in humans reduced intestinal survival of Lactobacillus, increased TH17 cells and increased blood pressure (Wilck et al., 2017). The high salt‐induced decrease in Lactobacillus levels was confirmed by Miranda et al. Moreover, this decrease was accompanied by decreased butyrate production and an up‐regulation of pro‐inflammatory genes in the intestine. A high salt diet has been shown to accelerate the induction of chemically‐induced forms of colitis. The pro‐inflammatory effects of dietary salt were not found in germ free mice. This clearly indicates that salt induced effects are mediated by changes in the gut microbiota (Miranda et al., 2018). In addition to changes in gut microbiota, high salt also induces changes in host and bacterial proteins in the gut, which will alter protein digestion (Wang et al., 2017). Increased dietary salt promotes neurovascular and cognitive dysfunctions. Interestingly, Faracao and colleagues found an increased number of IL‐17+ lymphocytes in the lamina propria of the small intestine and IL‐17A in the circulation in response to a high salt diet. These salt‐induced neurovascular changes were ameliorated in IL‐17A−/− and lymphocyte‐deficient RAG1−/− mice suggesting that dietary salt induces the polarization of TH17 cells in the small intestine resulting in cardiovascular changes (Faraco et al., 2018).
Salt and skin
The extracellular fluid volume in skin and muscle together constitute 60% of the body's extracellular fluid volume. Large amounts of sodium are stored outside of the vasculature in the interstitium. As the skin is a large interstitial reservoir for sodium storage, it may play a previously overlooked role in blood pressure homeostasis, salt sensitivity and infection. In rodents fed a high salt diet, sodium accumulates in the skin, creating a local micro‐environment that is hypertonic relative to plasma. Interestingly, much of this sodium appears to be osmotically inactive and bound to negatively charged glycosaminoglycans in the skin interstitium. In response to osmotic stress macrophage‐derived vascular endothelial growth factor‐C (VEGF‐C) may protect against salt sensitivity by stimulating the angiogenesis of dermal lymphatic vessels to mobilize nonvascular sodium stores back into the circulation for possible excretion by the kidney (Machnik et al., 2009). Moreover, NFAT5 deletion and VEGF receptor blockade induce salt‐sensitive hypertension in mice by impeding lymphatic clearance, substantiating the important role of skin lymphatic electrolyte homeostasis mediated by these macrophage‐derived proteins in blood pressure regulation. Corroborating these findings in humans, 23Na MRI shows that the accumulation of dermal Na+ increases with age and patients with refractory hypertension have an augmented tissue Na+ content compared with normotensive controls (Kopp et al., 2013). Moreover, Schneider et al. demonstrated that although skin sodium content correlated with systolic blood pressure, skin sodium was an even stronger predictor of left ventricular mass in patients with chronic kidney disease (Schneider et al., 2017). Collectively, these studies constitute a paradigm shift in our understanding of salt and water homeostasis by attributing regulatory functions, previously credited only to the kidney, to larger, more ubiquitous organs including the skin and muscle.
Salt and infection
The classically recognized role of the immune system is to protect the body from viral, bacterial, fungal and parasitic infections. Immune cells regulate a hypertonic micro‐environment in the skin; however, the biological advantage of increased skin sodium concentrations was until recently unknown. Jantsch and colleagues recently demonstrated a previously neglected consequence of sodium storage in the skin. They observed an exaggerated accumulation of sodium at the site of bacterial skin infections in humans and in mice. Moreover, they found that increasing sodium content in the skin by a high‐salt diet boosted the activation of macrophages in a NFAT5‐dependent manner and promoted cutaneous antimicrobial defence as illustrated in Figure 3A (Jantsch et al., 2015). Another salt‐induced improvement in antimicrobial defence was recently identified in the kidney. In the renal interstitium, a sodium gradient guides the migration of innate immune cells in the kidney during infections. Lower urinary tract infections are among the most common human bacterial infections, but extension to the kidneys is rare. This protection of the upper urinary tract has been attributed to mechanical forces, such as urine flow, that prevent the ascent of bladder microbes. Hypertonicity in the renal medulla is required for the kidney to function as a urine‐concentrating organ. However, hypertonicity also induces epithelial cells, in a NFAT5‐dependent manner, to produce chemokines that localize monocyte‐derived mononuclear phagocytes to the medulla. In addition, the hypertonic environment in the renal medulla has been proposed to increase the intrinsic bactericidal and neutrophil chemotactic activities of mononuclear phagocytes to generate a zone of defence, as shown in Figure 3B (Berry et al., 2017). Altogether, there is now substantial evidence that local Na+ can act as a danger signal enhancing pro‐inflammatory cell function and dampening anti‐inflammatory immune responses. Thereby, hypertonic micro‐environments serve as a protective fortress against microbial invaders. Hence, an underlying function of Na+ metabolism in the dermal and renal epithelial surfaces may be to strengthen their barrier functions (Schatz et al., 2017). However, the mechanisms that drive local salt storage will require further investigation. In the absence of microbial invaders, salt storage occurs with dietary excess in animals and age in humans. In this context, Na+ ingestion could lead to the unintended consequence of inappropriate pro‐inflammatory immune cell activation, which is supported by the finding that salt exacerbates autoimmune encephalitis and hypertension. Blockade of inflammation‐driven salt accumulation might possibly be used to diminish inflammatory responses and to treat hypertension and autoimmune diseases, but with a possible increase in the risk of mucosal infections (Schatz et al., 2017).
Figure 3.
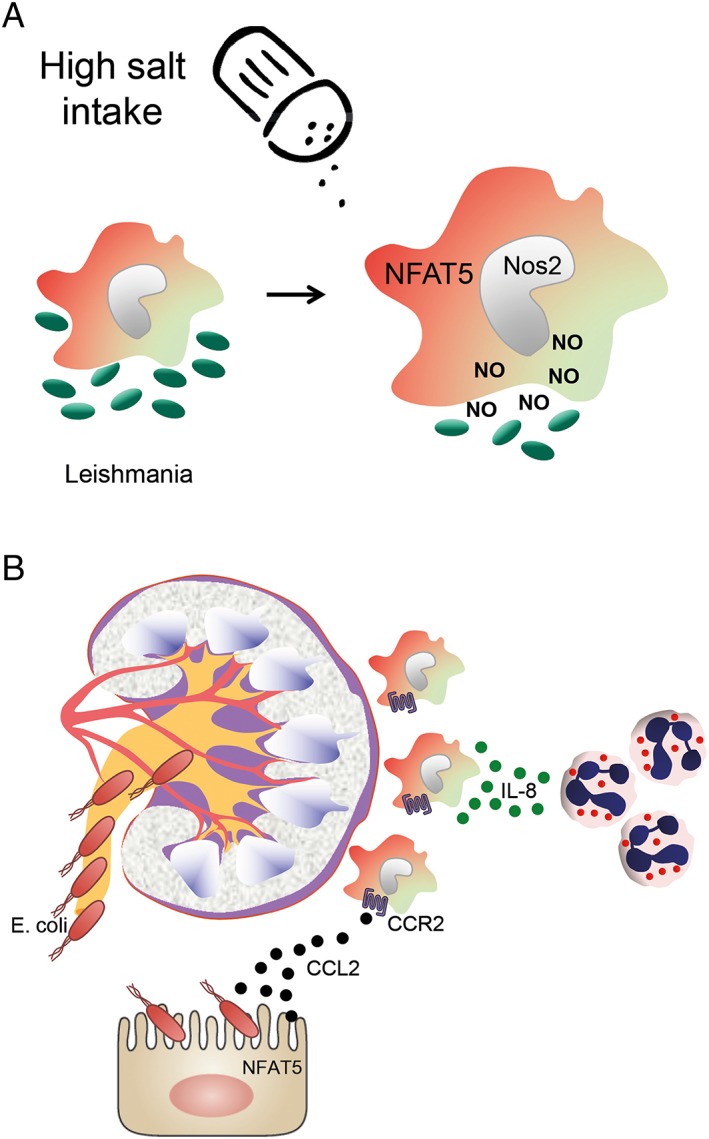
Salt and infection. (A) Increasing the sodium content in the skin by a high‐salt diet boosts the activation of macrophages. Inducible NOS (Nos2) is up‐regulated in a NFAT5‐dependent manner and promotes improved cutaneous antimicrobial defence against leishmaniosis. (B) In the kidney, regional hypertonicity during low fluid intake in the medulla instructs epithelial cells in a NFAT5‐dependent manner to produce chemokines like CCL2 that localize monocyte‐derived mononuclear phagocytes, by binding to the chemokine receptor CCR2, to the medulla. This hypertonic environment also increases the intrinsic bactericidal and neutrophil chemotactic activities of these cells to generate a zone of defence against ascending urinary infection with Escherichia coli.
Does the skin affect kidney function?
The observations of salt storage in the skin to buffer free extracellular Na+ suggest that electrolyte homeostasis in the body also relies on extrarenal regulatory mechanisms (Wiig et al., 2018). A low‐salt diet results in a change in skin glycosaminoglycan composition with increased hyaluronan and reduced sulphated proteoglycans, thus lowering the charge density and water‐free Na+ binding to the extracellular matrix. Inversely, a high‐salt diet yields an increase in sulphated glycosaminoglycans and an enhanced capacity for Na+ binding. Interstitial electrolyte balance is not achieved by renal blood purification alone, but instead relies on additional extrarenal regulatory mechanisms within the skin interstitium (gel of glycosaminoglycans). Macrophages act as local osmosensors that regulate local interstitial electrolyte composition via a NFAT5‐ and VEGF‐C‐dependent mechanism, enhancing electrolyte clearance via VEGF‐C and its receptor VEGFR‐3‐mediated modulation of the lymph capillary network in the skin. The Titze group identified an osmolyte gradient from epidermis to dermis in skin that is accentuated during salt accumulation via an urea‐dependent mechanism. This augmented gradient in the face of salt accumulation supports the notion of a countercurrent exchange of osmolytes in the skin (Wiig et al., 2018). The role of urea in this context will be of particular interest to explore in the light of the new observations that the body generates urea to conserve water and excrete salt (Kitada et al., 2017). The observed urea gradient in the skin may contribute to this process. In conclusion, electrolyte homeostasis in the body is not achieved by renal excretion alone, but also involves extrarenal regulatory mechanisms.
Evolutionary aspects
Four questions arise. (i) Why should evolution favour inflammation as a regulator of blood pressure? (ii) Why does salt provoke inflammation? (iii) What is the advantage of storing sodium in the skin? (iv) Why does hypertonicity support antimicrobial defence? Blood pressure control and host defence are essential mechanisms of homeostasis. Infection can cause hypotension via fluid loss during fever, tachypnea and diarrhoea. Septicaemia induces inflammation‐related vascular fluid losses together with vasodilatation, which could culminate in circulatory collapse. Thus, the risk of hypotension related to inflammation might have favoured selection of mechanisms that link sodium accumulation and inflammation to blood pressure increases and resistance to further microbial incursions for short‐term survival benefits. Such an evolutionary force may explain why important antimicrobial effectors can exert direct hypertensive effects by promoting vasoconstriction or sodium retention (Wenzel et al., 2016). In addition, sodium reservoirs may have evolved to facilitate survival during long fasts and in salt‐poor environments just as a camel stores water in its humps. Evolutionary theory suggests that nature ‘preserves’ its most vital components by multi‐tasking them, applying an ‘Ocham's razor’ to heredity for efficient energy expenditure. This may apply to Na+ accumulation in tissue. Important translational questions arise: does exposure to high salt exacerbate the induction of autoimmunity in a genetically‐susceptible individual, and if so, can we therapeutically manipulate these dietary salt effects in patients with autoimmune disease? Conversely, is there any benefit in using anti‐inflammatory or immune‐suppressive agents for the treatment of salt‐sensitive hypertension (Oh et al., 2016)?
Summary and conclusion
An amalgamation of all of the available evidence has led us to propose the following chain of events as a mechanism for hypertension and hypertensive end organ damage (Figure 4) (Wenzel et al., 2016). The skin has the capacity to buffer a certain amount of salt by monocyte‐derived VEGF‐C‐dependent lymphangiogenesis. Small increases in pro‐hypertensive factors like Ang II and aldosterone in combination with high salt intake activate the innate and the adaptive immune system. Na+‐induced hypertonicity activates innate monocytes/macrophages, dendritic cells and adaptive immune cells. T and B cells and also γδ T cells and NK cells are activated and migrate into heart, aorta and kidney. These cells cause local inflammation, sodium reabsorption in the kidney and stiffening in the aorta and resistance vessels. In addition, a high salt intake down‐regulates Lactobacillus murinus in the gut resulting in an altered metabolome and increased generation of TH17 cells that may migrate to the kidney and influence salt retention. Given the importance of sympathetic activation in hypertension and the importance of Na+ exchange in neuronal firing, it seems plausible that interstitial Na+ may also impact immunity‐dependent effects of the CNS on blood pressure.
Figure 4.
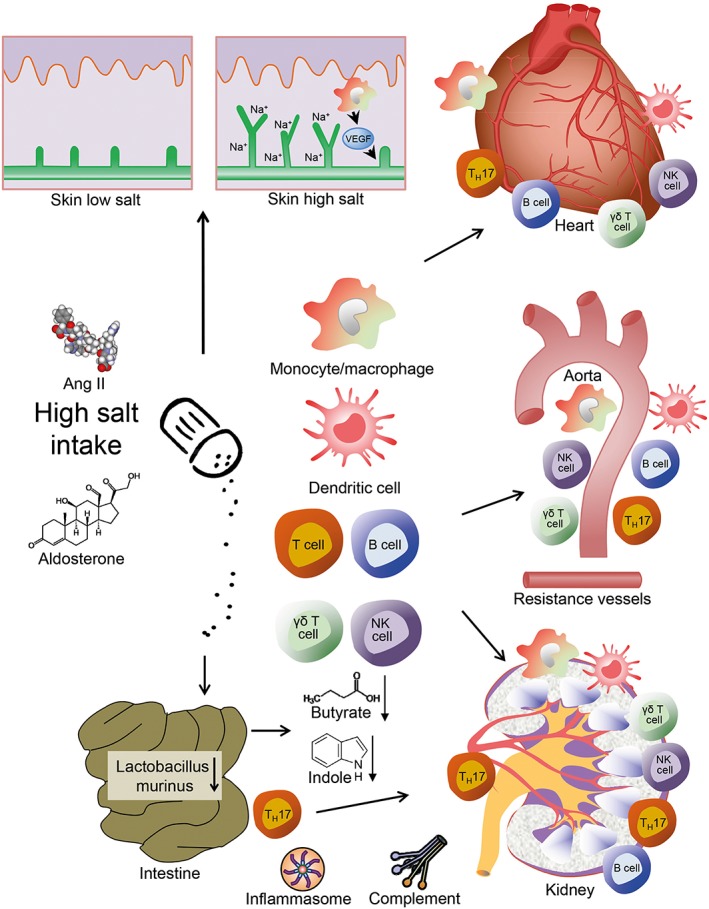
Salt and hypertension. The skin has the capacity to buffer a certain amount of salt by monocyte‐derived VEGF‐C‐dependent lymphangiogenesis. Failure to store salt, or saturation, results in Na+ overload. The high salt intake down‐regulates Lactobacillus murinus and alters the metabolome in the gut resulting in an increased generation of TH17 cells that migrate to the kidney and in the cardiovascular system causing salt retention and dysfunction. In the heart and aorta, they cause inflammation, stiffening and hypertrophy. Na+ hypertonicity generates a pro‐inflammatory environment by influencing cells of the innate and adaptive immune system. In combination with activation of the renin angiotensin aldosterone system, these changes result in arterial hypertension. Moreover, inflammasome and complement activation may cause direct damage or subtly affect the adaptive immunity (modified from Norlander et al., 2017; Rucker and Crowley, 2017; Wenzel et al., 2016).
The major reason to treat hypertension is to limit end organ damage. While blood pressure lowering is clearly important to reach this goal, the research community must also develop strategies to prevent the local inflammation that accompanies hypertensive end organ damage. Since the immune system has pro‐ as well as anti‐inflammatory functions, unspecific inhibition of the immune system may result in unwanted effects on arterial hypertension. In contrast, specific targeting of defined pathways may significantly improve the protection of the heart, brain, kidney and vasculature from hypertensive injury.
Accordingly, the goal of this review was not only to highlight some of the ‘hot areas’ of discovery and surprise in salt, inflammation and cardiovascular research but also specifically raise further awareness of the complex connections between salt, innate and adaptive immunity and arterial hypertension.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017 a,b,c,d,e,f,g).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The work of U. Wenzel, H. Ehmke and C. Kurts is supported by German Research Foundation Grants We 1688/17‐1 (to U. Wenzel) and Deutsche Forschungsgemeinschaft SFB 1192. We thank Steven Crowley for critical reading of the manuscript.
Wenzel U. O., Bode M., Kurts C., and Ehmke H. (2019) Salt, inflammation, IL‐17 and hypertension, British Journal of Pharmacology, 176, 1853–1863, doi: 10.1111/bph.14359.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Other ion channels. Br J Pharmacol 174: S195–S207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017f). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017g). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amador CA, Barrientos V, Pena J, Herrada AA, Gonzalez M et al (2014). Spironolactone decreases DOCA‐salt‐induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 63: 797–803. [DOI] [PubMed] [Google Scholar]
- Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L et al (2017). Dendritic cell amiloride‐sensitive channels mediate sodium‐induced inflammation and hypertension. Cell Rep 21: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MR, Mathews RJ, Ferdinand JR, Jing C, Loudon KW et al (2017). Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell 170 (860–874): e819. [DOI] [PubMed] [Google Scholar]
- Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W et al (2015a). High salt reduces the activation of IL‐4‐ and IL‐13‐stimulated macrophages. J Clin Invest 125: 4223–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binger KJ, Linker RA, Muller DN, Kleinewietfeld M (2015b). Sodium chloride, SGK1, and Th17 activation. Pflugers Arch 467: 543–550. [DOI] [PubMed] [Google Scholar]
- Caillon A, Mian MOR, Fraulob‐Aquino JC, Huo KG, Barhoumi T, Ouerd S et al (2017). Gammadelta T cells mediate angiotensin II‐induced hypertension and vascular injury. Circulation 135: 2155–2162. [DOI] [PubMed] [Google Scholar]
- Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H et al (2015). Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB et al (2018). Deficiency of complement C3a and C5a receptors prevents angiotensin II‐induced hypertension via regulatory T cells. Circ Res 122: 970–983. [DOI] [PubMed] [Google Scholar]
- Chessa F, Mathow D, Wang S, Hielscher T, Atzberger A, Porubsky S et al (2016). The renal microenvironment modifies dendritic cell phenotype. Kidney Int 89: 82–94. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Brea D, Garcia‐Bonilla L, Wang G, Racchumi G, Chang H et al (2018). Dietary salt promotes neurovascular and cognitive dysfunction through a gut‐initiated TH17 response. Nat Neurosci 21: 240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss JD, Kirabo A, Harrison DG (2017). Do high‐salt microenvironments drive hypertensive inflammation? Am J Physiol Regul Integr Comp Physiol 312: R1–R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton AC (1991). Blood pressure control‐special role of the kidneys and body fluids. Science 252: 1813–1816. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N et al (2015). Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest 125: 4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R et al (2018). Myeloid CD11c(+) antigen‐presenting cells ablation prevents hypertension in response to angiotensin II plus high‐salt diet. Hypertension 71: 709–718. [DOI] [PubMed] [Google Scholar]
- Ip WK, Medzhitov R (2015). Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun 6: 6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantsch J, Schatz V, Friedrich D, Schroder A, Kopp C et al (2015). Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage‐driven host defense. Cell Metab 21: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K (2017). Loss of resistance to angiotensin II‐induced hypertension in the Jackson laboratory recombination‐activating gene null mouse on the C57BL/6J background. Hypertension 69: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS et al (2015). Renal transporter activation during angiotensin‐II hypertension is blunted in interferon‐gamma−/− and interleukin‐17A−/− mice. Hypertension 65: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J et al (2014). Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis‐like skin disease. Arterioscler Thromb Vasc Biol 34: 2658–2668. [DOI] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL et al (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T et al (2017). High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest 127: 1944–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA et al (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp C, Linz P, Dahlmann A, Hammon M, Jantsch J, Muller DN et al (2013). 23Na magnetic resonance imaging‐determined tissue sodium in healthy subjects and hypertensive patients. Hypertension 61: 635–640. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer‐Schwesinger C et al (2014). Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii‐induced hypertension. Hypertension 63: 565–571. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J et al (2014). GammadeltaT cell‐derived interleukin‐17A via an interleukin‐1beta‐dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension 64: 305–314. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B et al (2017). CD8(+) T cells stimulate Na‐Cl co‐transporter NCC in distal convoluted tubules leading to salt‐sensitive hypertension. Nat Commun 8: 14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K et al (2009). Macrophages regulate salt‐dependent volume and blood pressure by a vascular endothelial growth factor‐C‐dependent buffering mechanism. Nat Med 15: 545–552. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M (2014). Role of “Western diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep 14: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al (2012). Interferon‐gamma signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H (2013). Genetic mutation of recombination activating gene 1 in Dahl salt‐sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda PM, De Palma G, Serkis V, Lu J, Louis‐Auguste MP et al (2018). High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome 6: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaniel KR, Harrison DG (2016). Is hypertension a bone marrow disease? Circulation 134: 1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS (2017). Inflammatory cytokines regulate renal sodium transporters: how, where, and why? Am J Physiol Renal Physiol 313: F141–F144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018). The immunology of hypertension. J Exp Med 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L et al (2016). Interleukin‐17A regulates renal sodium transporters and renal injury in angiotensin II‐induced hypertension. Hypertension 68: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L et al (2017). A salt‐sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end‐organ damage. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh YS, Appel LJ, Galis ZS, Hafler DA, He J, Hernandez AL et al (2016). National heart, lung, and blood institute working group report on salt in human health and sickness: building on the current scientific evidence. Hypertension 68: 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Liu Y, Wang X, Piao C, Ma Y et al (2018). The complement C3a‐C3aR axis promotes development of thoracic aortic dissection via regulation of MMP2 expression. J Immunol 200: 1829–1838. [DOI] [PubMed] [Google Scholar]
- Riedel JH, Paust HJ, Krohn S, Turner JE, Kluger MA, Steinmetz OM et al (2016). IL‐17F promotes tissue injury in autoimmune kidney diseases. J Am Soc Nephrol 27: 3666–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz E (1996). The history of salt‐aspects of interest to the nephrologist. Nephrol Dial Transplant 11: 969–975. [DOI] [PubMed] [Google Scholar]
- Rucker AJ, Crowley SD (2017). The role of macrophages in hypertension and its complications. Pflugers Arch 469: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucker AJ, Rudemiller NP, Crowley SD (2018). Salt, hypertension, and immunity. Annu Rev Physiol 80: 283–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, Norlander AE, Madhur MS (2016). Inhibition of interleukin 17‐A but not interleukin‐17F signaling lowers blood pressure and reduces end‐organ inflammation in angiotensin II‐induced hypertension. JACC Basic Transl Sci 1: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz V, Neubert P, Schroder A, Binger K, Gebhard M et al (2017). Elementary immunology: Na(+) as a regulator of immunity. Pediatr Nephrol 32: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MP, Raff U, Kopp C, Scheppach JB, Toncar S, Wanner C et al (2017). Skin sodium concentration correlates with left ventricular hypertrophy in CKD. J Am Soc Nephrol 28: 1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans S, Abdul‐Hamid MA, Vanderlocht J, Damoiseaux J, Reutelingsperger CP et al (2017). Patients with hypertension‐associated thrombotic microangiopathy may present with complement abnormalities. Kidney Int 91: 1420–1425. [DOI] [PubMed] [Google Scholar]
- Wang C, Huang Z, Yu K, Ding R, Ye K, Dai C et al (2017). High‐salt diet has a certain impact on protein digestion and gut microbiota: a sequencing and proteome combined study. Front Microbiol 8: 1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Rosendahl A, Czesla D, Meyer‐Schwesinger C, Stahl RA et al (2016). The complement receptor C5aR1 contributes to renal damage but protects the heart in angiotensin II‐induced hypertension. Am J Physiol Renal Physiol 310: F1356–F1365. [DOI] [PubMed] [Google Scholar]
- Wenzel UO, Bode M, Kohl J, Ehmke H (2017). A pathogenic role of complement in arterial hypertension and hypertensive end organ damage. Am J Physiol Heart Circ Physiol 312: H349–H354. [DOI] [PubMed] [Google Scholar]
- Wenzel UO, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H (2016). Immune mechanisms in arterial hypertension. J Am Soc Nephrol 27: 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ et al (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension 71: e13–e115. [DOI] [PubMed] [Google Scholar]
- Wiig H, Luft FC, Titze JM (2018). The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol (Oxf) 222. [DOI] [PubMed] [Google Scholar]
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H et al (2017). Salt‐responsive gut commensal modulates TH17 axis and disease. Nature 551: 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y et al (2013). Induction of pathogenic TH17 cells by inducible salt‐sensing kinase SGK1. Nature 496: 513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L et al (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen‐activated protein kinase. Circ Res 114: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li Y, Wang C, Wu Y, Cui W, Miwa T et al (2014). Complement 5a receptor mediates angiotensin II‐induced cardiac inflammation and remodeling. Arterioscler Thromb Vasc Biol 34: 1240–1248. [DOI] [PubMed] [Google Scholar]