

Abstract
Arterial hypertension represents a serious public health problem, being a major cause of morbidity and mortality worldwide. The availability of many antihypertensive therapeutic strategies still fails to adequately treat around 20% of hypertensive patients, who are considered resistant to conventional treatment. In the pathogenesis of hypertension, immune system mechanisms are activated and both the innate and adaptive immune responses play a crucial role. However, what, when and how the immune system is triggered during hypertension development is still largely undefined. In this context, this review highlights scientific advances in the manipulation of the immune system in order to attenuate hypertension and end‐organ damage. Here, we discuss the potential use of immunosuppressants and immunomodulators as pharmacological tools to control the activation of the immune system, by non‐specific and specific mechanisms, to treat hypertension and improve end‐organ damage. Nevertheless, more clinical trials should be performed with these drugs to establish their therapeutic efficacy, safety and risk–benefit ratio in hypertensive conditions.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- 2K1C
two kidneys, one clip
- AngII
angiotensin II
- APC
antigen‐presenting cell
- C
complement
- CD40L
ligand of CD40
- CsA
cyclosporine
- CTLA4
cytotoxic T‐lymphocyte‐associated protein 4
- DAMPs
damage‐associated molecular patterns
- DOCA
deoxycorticosterone acetate
- DSS
Dahl salt‐sensitive
- KO
knockout
- mAb
monoclonal antibody
- MAC
membrane attack complex
- MR
mineralocorticoid receptor
- mTOR
mammalian target of rapamycin
- nAb
neutralizing antibody
- NLR
nucleotide‐binding oligomerization domain‐like receptor
- NLRP
nucleotide‐binding oligomerization domain‐like receptor family pyrin domain‐containing protein
- NOD
nucleotide‐binding oligomerization domain
- OKT3
muromonab‐CD3
- PAMPs
pathogen‐associated molecular patterns
- PDTC
pyrrolidine dithiocarbamate
- PRR
pattern recognition receptor
- SHR
spontaneously hypertensive rats
- SLE
systemic lupus erythematosus
- TCR
T‐cell receptor
- TLRs
toll‐like receptors
- Treg
T regulatory cell
- WT
wild‐type
Introduction
For many years, cardiovascular diseases have been the major cause of death and disability worldwide. Raised levels of BP amplify the risk of cerebrovascular and coronary heart disease, making BP control the primary goal in the treatment of any cardiovascular disease. Based on the predictive value of BP level thresholds for adverse cardiac outcomes, some American guidelines have assumed more aggressive values for the definition of hypertension, which now are above 130–139 for systolic or 80–89 mmHg for diastolic pressures (Whelton et al., 2018).
Interventions that effectively lower BP directly affect hypertension‐associated morbidity and mortality. In pharmacological terms, several classes of drug are clinically available, based on the classical view of BP control by the nervous system, vascular and renal mechanisms. Sympatholytic agents, natural alkaloids (reserpine) and semi‐synthetic agents (methyldopa) were the earlier antihypertensive drugs, forerunners of the modern treatments (Sever and Messerli, 2011). Progressively, they were replaced by safer agents in an attempt to avoid their central depressor effects. From the 1950s to 2010, vasodilators, such as hydralazine, calcium channel blockers and renin–angiotensin system (RAS) blockers, emerged from the progress in our understanding of the mechanisms of vascular tone control, aiming to decrease peripheral vascular resistance (Sever and Messerli, 2011). Almost in parallel, thiazides and thiazide‐like diuretics were introduced into the management of hypertension (Moser and Feig, 2009), whose antihypertensive effect results from the diuretic‐induced reduction in systolic volume and an non‐specific reduction in vascular resistance.
Certainly, the modern therapy of hypertension has reduced fatal and non‐fatal outcomes and decreased end‐organ damage (Ettehad et al., 2016). However, the current treatments have some limitations. For instance, ethnicity (Brewster et al., 2016), genetic variability (Chan et al., 2012b; Shahin and Johnson, 2016; Heidari et al., 2017; Ware et al., 2017) and phenotypic differences still determine tolerability and efficacy of antihypertensive drugs. Moreover, about 5 to 20% of hypertensive patients do not achieve BP control, even with combined agents from different pharmacological classes, and are categorized in a resistant or refractory phenotype of the disease (Siddiqui and Calhoun, 2017). Both terms refer to a failure to reduce BP, the former with three or more and the latter with five or more medications. At present, the clinical pharmacological principle of combination therapy is to target different mechanisms of BP control (nervous system, vascular and renal), and the existence of resistant and refractory patients illustrates the need for additional therapies.
With this background, studies on the immune basis of hypertension in animal models have discovered several targets and have encouraged studies in human populations using immunosuppressant agents. However, limited progress has been made in the clinical settings. Even traditional antihypertensive therapies have immunomodulatory effects that play a role in hypertension. This review highlights scientific advances made in the manipulation of the immune system, as a target in the attenuation of hypertension, vascular inflammation and end‐organ damage. Here, we discuss the potential use of immunosuppressants and immunomodulators as pharmacological tools to control the activation of the immune system, by non‐specific and specific mechanisms, to treat hypertension and improve end‐organ damage. We also provide a risk/benefit evaluation for these drugs (Figure 2), based on the preclinical and clinical evidence.
Figure 2.
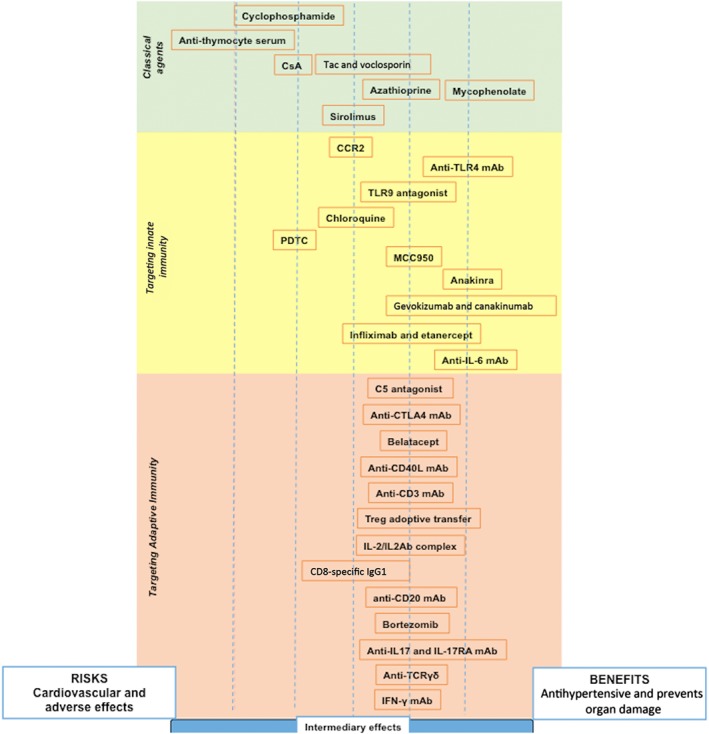
Differential effects of immunosuppressants and immunomodulators on hypertension. Classical immunosuppressant drugs are in the green section, immunomodulators targeting the innate immune response are in the yellow section and immunomodulators targeting the adaptive immune response are in the red section. Drugs shifted to the right have these effects: (i) reduced BP, (ii) decreased organ damage and/or (iii) there is clinical evidence of cardiovascular benefits. Drugs on the left of the figure indicate that one or more of these conditions were met: (i) does not have clinical evidence in hypertension, only experimental; (ii) it works in one hypertensive model and others had no effect and/or (iii) it is toxic to the cardiovascular system. The centre position indicates intermediary effects, with risks and benefits to hypertension. Overall, this figure correlates the risk–benefit ratio to treat hypertension. Anti‐IL‐17AR, IL 17A receptor antibody; BAS, basiliximab; MMF, mycophenolate mofetil; Tac, tacrolimus.
An overview of immune responses in hypertension
The main function of the immune system is defending a host against antigens. For this purpose, the organism relies on a complex interplay between the two main arms of the immune system: the innate immunity, which mediates the early reactions, and the adaptive immunity, which is a late and more specific response (Figure 1). Damage‐associated molecular patterns (DAMPs) and pathogen‐associated molecular patterns (PAMPs) are the antigens that initially activate the immune system. In hypertension, DAMPs are known to be increased and are responsible for the chronic inflammation present in this condition. The origins of DAMPs are uncertain and much discussed. Angiotensin II (AngII), HMGB‐1, HSP60 and HSP70, fibrinogen, uric acid and mitochondrial DNA (see McCarthy et al., 2014) are the main molecules recognized as DAMPs in hypertension, whose levels are chronically elevated. DAMPs and PAMPs are recognized by pattern recognition receptors (PRRs) and start the immune response activation, as described below.
Figure 1.
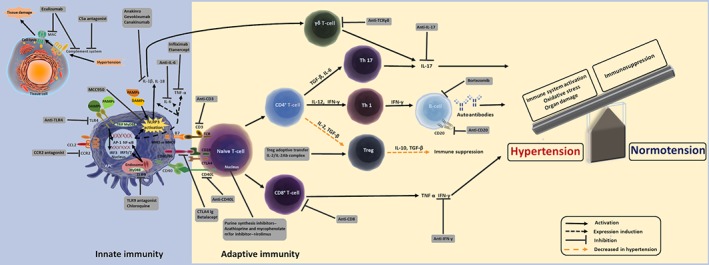
Targets in the immune system for the treatment of hypertension. The unregulated and persistent activation of the immune system, cytokine release and oxidative stress induce cardiovascular organ damage and, consequently, hypertension. Drugs available to modulate the immune response and likely to control BP and improve end‐organ damage are shown in the grey boxes. The dashed orange arrows indicate a diminished response, while black arrows indicate activated mechanisms. More details about the drugs are found in the main text. Based on patient risk/benefit ratio profile of targeted therapies, immune system modulation may provide a new strategy to treat hypertension. AP‐1, activator protein 1; IRF3, IFN regulatory factor 3; MyD88, myeloid differentiation response 88 protein; Tac, tacrolimus; TRIF, toll/IL‐1 receptor homologous region domain‐containing adapter‐inducing IFN‐β.
Innate immune response
The immune system has different cell types to perform its function. Macrophages, dendritic cells (DCs) and neutrophils are the main cell types involved in the innate immune response. DCs are seen as a source of DAMPs in hypertension, as there is an enhancement in NADPH oxidase‐dependent superoxide production in DCs, which leads to the formation of highly reactive γ‐ketoaldehydes, also known as isoketals or isolevuglandins (Kirabo et al., 2014). These compounds rapidly form self‐proteins adducts, which are processed and presented as DAMPs by DCs, promoting renal and vascular dysfunction and hypertension.
Macrophage infiltration into vascular wall, the kidneys and myocardium (Justin Rucker and Crowley, 2017; Hulsmans et al., 2018) has been described in experimental models and patients with hypertension. Also, genetic deficiency of macrophages protected mice from vascular dysfunction and attenuated BP increase induced by AngII (De Ciuceis et al., 2005) and deoxycorticosterone acetate (DOCA)‐salt hypertension (Ko et al., 2007). Macrophages are classified as ‘M1’ and ‘M2’ according to the effects that these two populations have on the differentiation of T‐lymphocytes. Specifically, M1 macrophages are known to activate and guide Th1 lymphocytes, and M2 macrophages are associated with the induction of Th2 lymphocyte responses. Despite some data showing the contribution of macrophages in hypertension, there is a paucity of information about the polarization of M1 and M2 phenotype in this disease. Nevertheless, based on the T‐lymphocyte interaction with macrophage subsets, there is indirect evidence of the role of macrophage polarization in hypertension.
NK cells are a cytotoxic cell type with non‐specific functions and, for this reason, it is considered to be part of the innate response. NK cells produce a variety of inflammatory cytokines and growth factors, such as IFN‐γ, TNF‐α and GM‐CSF. Depletion of NK cells, using an anti‐NK antibody, protects against AngII‐induced vascular dysfunction (Kossmann et al., 2013).
The complement system is an essential component of humoral and natural immunity. Complement activation occurs through classical, alternative and lectin pathways. These events converge in the activation of the complement (C)3 convertase and formation of C5 convertase, which cleaves C5 into the C5a, C5b and other fragments. The complement fragments act as potent pro‐inflammatory anaphylatoxins stimulating immune cell recruitment and activation and, especially, leading to membrane attack complex (MAC) formation. The formation of MAC results in cell lysis and also triggers inflammation (Horiuchi and Tsukamoto, 2016). In hypertensive mice, complement activation was associated with renal damage (Raij et al., 1989; Shagdarsuren et al., 2005) but was not involved in aortic and cardiac remodelling (Coles et al., 2007). Also, MAC deposition in renal biopsies from systemic lupus erythematosus (SLE) patients correlates with higher systolic BP (Wang et al., 2018b). Therefore, complement inhibition can contribute to control of organ damage in hypertension.
Pattern recognition receptors
The involvement of PRR in hypertension has been extensively investigated and has become determinant (Bomfim et al., 2017). Initially, PAMPs or DAMPs are recognized by immune cells by the PRR, expressed in cellular membranes or inside the cells. The main families of PRR that have been described are toll‐like receptors (TLRs), retinoic acid‐inducible gene I ‐like helicases and nucleotide‐binding oligomerization domain (NOD)‐like receptor (NLR). Despite the fact that other PRRs have been reported, only two types have been investigated in hypertension: TLR and NLR. These receptors recognize PAMPs and DAMPs and initiate the immune response of the host (Liew et al., 2005; Takeda and Akira, 2005). Their activation leads to the production of pro‐inflammatory cytokines, chemokines, tissue‐destructive enzymes and type I IFNs by host cells. In the case of NLR, some subtypes as NLR family pyrin domain‐containing protein (NLRP)1 and NLRP3 induce inflammasome activation, which leads to IL‐1β and IL‐18 maturation.
Several TLR subtypes seem to be involved in hypertension such as TLR2, TLR3, TLR4, TLR7/TLR8 and TLR9. TLR4 was up‐regulated in the cardiovascular system of different hypertensive models, such as in spontaneously hypertensive rats (SHR) (Bomfim et al., 2012), and hypertension induced by l‐NAME (Eissler et al., 2011). Regardless of the hypertensive model used, they all display hypertension‐associated end‐organ damage (Eissler et al., 2011). In general, TLR4 plays a role in vascular dysfunction, cardiac and renal injury and dysregulation of the CNS in hypertension (Biancardi et al., 2017). Treatment with anti‐TLR4 showed good results decreasing BP in different models of hypertension (Bomfim et al., 2012; Hernanz et al., 2015) (Table 1). Indeed, l‐NAME‐induced hypertension was blunted in TLR4 knockout (KO) when compared with wild‐type (WT) mice (Sollinger et al., 2014). However, TLR4‐deficient mice infused with AngII presented marked inhibition of vascular remodelling, although there was no change in BP levels when compared with WT mice infused with AngII (Nakashima et al., 2015). Also, high‐salt‐diet‐induced pre‐hypertension was associated with increased TLR4 and pro‐inflammatory cytokines in the paraventricular nucleus (PVN) of rats. More importantly, PVN infusion of a TLR4 antagonist reduced BP, pro‐inflammatory cytokine expression and oxidative stress (Wang et al., 2018a).
Table 1.
Drugs aimed at the innate and adaptive immune systems as pharmacological targets in hypertension
| Immune target | Drug | Experimental model | Experimental outcome | Clinical perspective or limitation |
|---|---|---|---|---|
| Classical agents | ||||
| All immune cells (alkylating agent) | Cyclophosphamide | SHR and DOCA‐salt (Khraibi et al., 1984) | ↓ BP in SHR but not in DOCA‐salt rats. | Intense cardiotoxic effects (Curigliano et al., 2016). |
| Thymocyte | Anti‐thymocyte serum | SHR (Bendich et al., 1981) | ↓ BP. | Serum sickness‐like reactions are common adverse effects, and cytokine release syndrome appears to be involved (Chen et al., 2015). |
| mTORC1 | Rapamycin (sirolimus) | DSS rats (Kumar et al., 2017) | ↓ BP, ↓ kidney injury and hypertrophy and ↓ kidney T‐lymphocytes (CD3+) and macrophage (ED1+) infiltration. | Associated with high incidence of hypertension (Olyaei et al., 2005). |
| Purine metabolism | Azathioprine | DOCA‐salt pregnant rats (Tinsley et al., 2009) | ↓ BP, ↓ proteinuria, ↓ IL‐2, ↓ IL‐12, ↓ IFN‐γ, ↓ chemokines and ↓ endothelial dysfunction. | Associated with high incidence of hypertension (Olyaei et al., 2005). |
| IMPDH | Mycophenolate | SHR (Rodriguez‐Iturbe et al., 2002), DOCA‐salt pregnant rats (Tinsley et al., 2009), DOCA‐salt rats (Boesen et al., 2010) and SLE mice (Taylor and Ryan, 2017) | ↓ BP, ↓ proteinuria, ↓ IL‐2, ↓ IL‐12, ↓ IFN‐γ, ↓ chemokines, ↓ endothelial dysfunction, ↓ renal cortical T‐lymphocyte and macrophage infiltration, ↓ albuminuria and creatinine clearance, selectively depleted CD45R+ B‐cells and ↓ subsequent autoantibody production. | ↓ hypertension in patients with autoimmune diseases, probably through the reduction of TNF‐α, chemokines and oxidative stress (Herrera et al., 2006). |
| Innate immune response | ||||
| TLR signalling | ||||
| TLR4 | Anti‐TLR4 | SHR (Bomfim et al., 2012) | ↓ BP, ↓ TLR4 expression, ↓ serum IL‐6, ↓ endothelial dysfunction, ↓ Cox‐2 expression and ↓ TxA2 release. | Phase I studies demonstrated a safe profile of anti‐TLR4 (Monnet et al., 2017). |
| SHR (Echem et al., 2015) | BP not altered, ↓ TLR4 expression, ↓ MyD88 expression, ↓ NF‐κB activation, ↓ IL‐1β and TNF‐α cardiac expression, ↓ cardiac hypertrophy, ↓ cardiac fibrosis and ↓ cardiac remodelling. | |||
| AngII‐infused mice (Hernanz et al., 2015) | ↓ BP, ↓ endothelial dysfunction, ↓ vascular remodelling, ↓ collagen deposition, ↓ NOX‐1 mRNA levels, ↓ JNK1/2, ↓ NF‐κB activation, ↓ IL‐1β, ↓ TNF‐α, ↓ CCL2, ↓ NADPH oxidase activity, ↓ oxidative stress and ↑ NO release. | |||
| TLR9 | Chloroquine | SHR (McCarthy et al., 2017) | ↓ BP, ↓ MyD88 and TRAF6, ↓ NF‐κB activation, ↓ LVH, ↓ aortic immune cell recruitment and infiltration, ↓ aortic total leukocytes (CD45+), ↓ serum T‐cells (CD3+) and ↓ CD44+ on T‐cells (CD3+). | Long therapeutic regimes with chloroquine occasionally lead to cardiac conduction disorders and cardiomyopathy (Al‐Bari, 2015). |
| NF‐κB | PDTC | SHR (Rodriguez‐Iturbe et al., 2005) | ↓ BP, ↓ NF‐κB activation, ↓ ICAM‐1, ↓ MCP‐1, ↓ lymphocyte and macrophage renal infiltration and ↓ renal malondialdehyde. | No tolerability test was performed in humans. |
| 2K1C rats (Cau et al., 2015; Cau et al., 2011) | ↓ BP, ↓ endothelial dysfunction, ↓ aortic and cardiac remodelling, ↓ oxidative stress and ↓ MMP‐2 and MMP‐9 levels and activity. | |||
| TNF‐α | Infliximab | SHR (Filho et al., 2013) | ↓ BP and ↓ LVH, ↑ AKT/eNOS phosphorylation and ↓ JNK and ↓ NF‐κB activation. | Infliximab infusion may result in cytokine release syndrome, anaphylactic reactions and fever with degranulation of mast cells and basophils (Lichtenstein et al., 2015). TNF‐α blockade was associated with higher risk of infection diseases, heart failure and elevation of PVR, SBP and DBP (Sinagra et al., 2013; Zhao et al., 2015). |
| TNFR2 | Etanercept | Fructose‐fed hypertensive rats (Tran et al., 2009), | ↓ BP, ↑ vascular function and restored eNOS expression. | |
| SLE mice (Venegas‐Pont et al., 2010) | ↓ BP, ↓ NF‐κB activation, ↓ BP, ↓ macrophage renal infiltration and ↓ albuminuria. | |||
| TNFR2/IL‐1R | TFI | AngII‐infused mice (Wang et al., 2014) | No changes in BP, ↓ cardiac damage and ↓ leukocyte adhesion and migration responses. | |
| IL‐6 | IL‐6 nAb | DSS rats (Hashmat et al., 2016) | ↓ BP, ↓ renal monocyte and macrophage (CD11b/c+) infiltration and ↓ glomerular injury index. | Tocilizumab treatment did not increase cardiovascular risk or events (Kim et al., 2017). |
| NLR signalling | ||||
| NLRP3 | MCC950 | DOCA‐salt mice (Krishnan et al., 2016) | ↓ BP, ↓ renal IL‐6, ↓ IL‐17A, ↓ TNF and ↓ osteopontin. | Canakinumab has just been approved for clinical use and has proved to lower chances of recurrent cardiovascular events (Shah et al., 2018). |
| IL‐1β | Gevokizumab | I.c.v. injection in DSS rats (Qi et al., 2016) | ↓ BP, ↓ HR, ↓ plasma noradrenaline, ↓ oxidative stress and restored cytokine balance. | |
| Anakinra | DOCA‐salt in mice (Ling et al., 2017) | ↓ BP, ↓ renal CCL2, CCL5, ↓ renal collagen deposition but ↑ renal hypertrophy. | ||
| Ang II‐infused mice (Zhang et al., 2016) | ↓ BP and ↓ cardiac hypertrophy. | |||
| Adaptive immune response | ||||
| T‐cell and APC interaction | ||||
| CTLA4 | CTLA4‐Ig | AngII‐infused and DOCA‐salt mice | ↓ BP, ↓ T‐cell activation, ↓ cytokine release and ↓ vascular inflammation. | Risk of infections, gastrointestinal disorders and headache; belatacept was associated with no changes in BP but ↓ aortic augmentation pressure (Seibert et al., 2014). |
| CD40L | Anti‐CD40L (Cornelius et al., 2015) | Pre‐eclampsia in rats | ↓ BP, ↓ placental oxidative stress and ↓ AT1R. | Incidence of thrombotic events (Sidiropoulos and Boumpas, 2004). |
| Ubiquitin–proteasome system | ||||
| Proteasome | Bortezomib | AngII‐infused mice (Li et al., 2013) and SLE mice (Taylor et al., 2018a) | ↓ BP, ↓ vascular remodelling, ↓ ROS generation and ↓ VCAM‐1. | Haematological, gastrointestinal and neurological toxicities (Bross et al., 2004). |
| T‐cells and B‐cells | ||||
| CD3 | Muromonab | SLE in female mice (Mathis et al., 2017) | ↓ BP and spleen weight, ↓ expression of IL‐17RA, but it did not reduce renal injury nor TNF‐α expression. | Risk of cytokine release syndrome, which can result in multiple organ damage (Bugelski and Martin, 2012). |
| Treg | IL‐2/IL‐2Ab complex | AngII‐infused mice (Majeed et al., 2014) | No effect in BP but ↓ vascular stiffness, ↑ Treg in spleen, ↓ Th17 and macrophages in aorta. | No human tests so far. |
| Transverse aortic constriction (Wang et al., 2016) | No effect in BP but ↓ congestive heart failure. | |||
| Treg adoptive transfer | AngII‐infused mice (Barhoumi et al., 2011; Kvakan et al., 2009; Matrougui et al., 2011) | ↓ BP, ↓ cardiac hypertrophy, inflammation and fibrosis, ↓ vascular injury and improved coronary arteriolar dysfunction. | Clinical limitation due to the difficulty in acquiring Treg cells for treatment. | |
| CD8 | Anti‐CD8 | AngII‐infused mice (Ma et al., 2014) | No effect in BP but ↓ cardiac profibrogenic inflammation. | Weak evidence for its use in hypertension, and no clinical tests were performed yet. |
| CD20 | Anti‐CD20 | AngII‐infused mice (Chan et al., 2015) | ↓ BP and ↓ vessel remodelling. | Immunosuppression, infections, renal toxicity and cardiac arrhythmias (Hansel et al., 2010). |
| TCRγδ | anti‐TCRγδ | AngII‐infused mice (Caillon et al., 2017) | ↓ BP, γδ T‐cell depletion and ↓ endothelial dysfunction. | In vitro assays showed that anti‐TCRγδ stimulate human γδ T‐cells from peripheral blood, which brings the question if anti‐TCRγδ would be safe in hypertension (Zhou et al., 2012). |
| IL‐17 | IL‐17A mAb | AngII‐infused mice (Saleh et al., 2016) | ↓ BP, ↓ albuminuria, ↓ renal and aortic infiltration of total leukocytes, ↓ total T‐cells (CD3+), ↓ T helper (CD4+), ↓ T‐cytotoxic (CD8+), ↓ renal TGF‐β, blunted TH17, ↓ ROS, ↓ autoantibodies, ↑ Tregs in spleen and lymph node and ↓ aortic endothelial dysfunction. | In normotensive patients, brodalumab (IL‐17RA mAb) demonstrated cardiovascular safety (Papp et al., 2016). |
| IL‐17 nAb | Calcineurin inhibitor‐induced hypertension (Chiasson et al., 2017) | |||
| IL‐17RC | Pre‐eclampsia in rats (Cornelius et al., 2013) | |||
| IL‐17AR | IL‐17AR mAb | AngII‐infused mice (Saleh et al., 2016) | ↓ BP, ↓ albuminuria, ↓ renal and aortic infiltration of total leukocytes, ↓ total T‐cells (CD3+), ↓ T helper (CD4+), ↓ T‐cytotoxic (CD8+), ↓ renal TGF‐β and ↓ organ damage. | |
| IFN | IFN‐γ nAb (Sun et al., 2017) | Mice overexpressing MR after AngII infusion | ↓ BP. | Increase the patient's risk of infection (Kelchtermans et al., 2008). |
AT1R, angiotensin II AT1 receptor; DBP, diastolic BP; ED1+, macrophage marker; eNOS, endothelial NOS; HR, heart rate; ICAM‐1, intercellular adhesion molecule 1; IL‐17RC, IL‐17 recombinant receptor C; IMPDH, inosine monophosphate dehydrogenase; LVH, left ventricular hypertrophy; mTORC1, mammalian target of rapamycin complex 1; MyD88, myeloid differentiation response 88 protein; NOX‐1, catalytic subunit of NADPH oxidase (also known as gp91phox); PVR, peripheral vascular resistance; SBP, systolic BP; TFI, TNF receptor 2 fragment crystallization IL‐1 receptor antagonist; TNFR2, TNF receptor 2; TRAF6, TNF receptor‐associated factor 6; VCAM‐1, vascular adhesion molecule 1.
Besides TLR4, other subtypes also contribute to hypertension. Expression of TLR2 was augmented in the kidney of rats infused with AngII (Ahn et al., 2007) and TLR9 activation was associated with vascular dysfunction in SHR (McCarthy et al., 2015). TLR3 and TLR7/TLR8 have not been directly investigated in a hypertensive model, but are important in pre‐eclampsia, which is a pregnancy‐specific hypertensive syndrome characterized by excessive maternal immune system activation, inflammation and endothelial dysfunction. Genetic expression levels of TLR3, TLR7 and TLR8 were significantly increased in women with pre‐eclampsia compared with normotensive pregnant women (Chatterjee et al., 2012). Treatment of human trophoblasts with a TLR3 (polyinosine–polycytidylic acid), TLR7 [imiquimod (R‐837)] or TLR7/TLR8 (CL097) agonists enhanced the expression levels of TLR3/TLR7/TLR8 and caused pregnancy‐dependent hypertension (Chatterjee et al., 2012). TLR9 activation (CpG ODN) during gestation caused maternal hypertension and increased contractile responses in resistance arteries (Goulopoulou et al., 2016).
NLRP3 is the most studied NLR in the context of cardiovascular diseases. NLRP3 gene polymorphisms were associated with higher systolic and diastolic BP in 50‐year‐old patients (Kunnas et al., 2015). The two kidneys, one clip (2K1C) and DOCA‐salt hypertensive mouse models exhibited enhanced renal expression of NLRP3 and IL‐1β. Accordingly, NLRP3 KO mice did not develop hypertension when submitted to 2K1C hypertension and DOCA‐salt treatment (Wang et al., 2012).
Other NLR subtypes have also been investigated in hypertension. Stimulation of NOD1 in mice promoted haemodynamic changes such as decreased BP, increased heart rate and vascular hyporesponsivity to contractile agonists (Cartwright et al., 2007). In humans, the NOD2 gene locus has been associated with hypertension (Gu et al., 2007) and the NLRP6 angiotensin–vasopressin receptor loci may contribute to susceptibility to essential hypertension, because vasopressin binds to NLRP6 with high affinity (Glorioso et al., 2013).
Adaptive immune responses
The contribution of adaptive immune responses, represented by lymphocytes and antibodies, to hypertension has been extensively investigated in the last decade. Hypertension in humans and SHR is associated with enhanced serum levels of IgG, IgA or IgM antibodies (Hilme et al., 1989; Chen and Schachter, 1993).
Many researchers have used animal models with depletion of adaptive immune cells and cytokines to show the association of lymphocytes with hypertension. For example, mice with inactivation of the recombination‐activating gene 1 (Guzik et al., 2007), which lack T‐lymphocytes and B‐lymphocytes; severe combined immunodeficiency mice lacking lymphocyte responses (Crowley et al., 2010); IL‐17 KO mice (Kamat et al., 2015); mice with adoptive transfer of T regulatory cell (Treg) (Barhoumi et al., 2011); and mice genetically deficient in B‐cells, B‐cell‐activating factor receptor KO (Chan et al., 2015), all exhibit attenuated hypertension in response to AngII.
B‐cells play an essential role in adaptive immunity to detect and process antigens with further differentiation in plasma cells with antibody production. AngII infusion was associated with increased splenic B‐cells expressing CD86, splenic plasma cell, circulating IgG and marked IgG accumulation in the aortic adventitia (Chan et al., 2015).
Regarding CD4 T‐cell participation in hypertension, studies demonstrated a Th1 and Th17 lymphocyte polarizing response, which may be characterized by IFN‐γ and IL‐17 release from Th1 and Th17 respectively. Also, hypertension decreased responses of Th2 and Treg lymphocytes.
IFN‐γ KO mice were protected from AngII‐induced vascular and kidney dysfunction (Kossmann et al., 2013; Kamat et al., 2015). Th1 cells are important IFN‐γ producers. Th17 and γδ T‐cells are the primary sources of IL‐17A in AngII‐induced hypertension (Saleh et al., 2016) and play an essential role in the pathogenesis of hypertension in specific cases. IL‐17 KO mice demonstrated preserved endothelial function and attenuation of hypertension after exposure to AngII (Madhur et al., 2010). DOCA‐salt rats exhibited elevated levels of RAR‐related orphan receptor γ T, the transcription factor for Th17 and increased IL‐17 expression in spleen, the heart and the kidney (Amador et al., 2014). Accordingly, hypertension induced by AngII was not sustained in IL‐17 KO mice, which displayed preserved vascular function, decreased superoxide production and reduced aortic T‐cell infiltration (Kamat et al., 2015). Although Th17 cells are a source of IL‐17, Chen et al. (2014) suggested that γδ T‐cells are the most important source of IL‐17 in hypertension, representing a crucial cell type during the development of hypertension.
Regulatory T‐lymphocytes (Treg) are cells that suppress the immune response and maintain self‐tolerance. Many mechanisms have been proposed for the suppressor function of these cells. These mechanisms include the secretion of suppressive cytokines (IL‐10, TGF‐β and IL‐35), direct cytolysis of effector T‐cell, metabolic disruption through tryptophan catabolites, IL‐2 deprivation and direct interference of co‐stimulation via expression of cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) (Davidson and Shevach, 2011). Treg cells have a protective effect in hypertension, mainly in AngII‐induced hypertension and AngII induced Treg apoptosis (Matrougui et al., 2011). Also, adoptive transfer of Treg prevented AngII‐induced hypertension, vascular damage and vascular immune cell infiltration (Barhoumi et al., 2011). Kvakan et al. (2009) also described decreased cardiac hypertrophy and less cardiac fibrosis with an improvement in arrhythmogenic electric remodelling after adoptive transfer of Treg cells into AngII‐infused hypertensive mice.
CD8 T‐cells have an important specific cytotoxic function. CD8 T‐cells also play a role in hypertension. Recently, Youn et al. (2013) demonstrated that pro‐inflammatory immunosenescent CD8 T‐cells were enhanced in humans with hypertension, suggesting a clonal expansion of a unique population of CD8 T‐cells in hypertension. Another study demonstrated that AngII infusion in WT and CD4 KO mice resulted in increased systolic and diastolic BP. However, BP elevation was blunted in CD8 KO mice under AngII infusion. They also induced DOCA‐salt hypertension in WT, CD8 KO and CD4 KO mice and had similar results of AngII infusion in mice. Also, adoptive transfer of CD4+/CD25− cells from AngII‐treated mice to RAG‐1 KO mice had no effect on BP, but the adoptive transfer of CD8+ cells caused BP elevation (Trott et al., 2014).
Antihypertensive treatments and immune system
Several antihypertensive treatments have been proposed over the years, such as lifestyle modifications, pharmacological therapies and even surgical intervention. To date, the association between the activation of the immune system and hypertension has become clearer. Nevertheless, it is still unclear whether immune activation is a cause or a consequence, of increased BP. In fact, there is indirect evidence that high BP per se may affect the immune system. For example, many antihypertensive treatments may act as modulators of the immune system alongside BP reduction, which may be responsible for some of their beneficial effects. However, to our knowledge, no study has systematically addressed the effects of BP per se on the immune system, and studies addressing this issue are warranted. Therefore, evidence showing modulation of the immune system by antihypertensive treatment will be discussed.
Salt restriction
An important non‐pharmacological strategy to control BP is the reduction of salt consumption. Besides its effects on systolic volume enhancement and, consequently, BP increase, high‐sodium diet is intimately linked to the polarization of Th naïve cells to Th17 and elevated IL‐17A production (Kleinewietfeld et al., 2013). In salt‐dependent and salt‐independent models of hypertension, the deletion of a salt‐sensing enzyme in T‐lymphocytes impairs the development of hypertension, Th17 polarization and end‐organ damage (Norlander et al., 2017). In this sense, salt restriction could represent an intervention to control immune response in hypertension. However, salt‐restricted diets in hypertensive patients led to mild BP attenuation with markers of immune system activation, such as enhanced C‐reactive protein concentration and elevated TNF‐α and IL‐6 levels (Nakandakare et al., 2008). These findings suggest that the BP control by salt restriction is, at least in part, dependent on the down‐regulation of the immune system. Thus, it is possible that hypertensive patients can achieve more efficient BP control with pharmacological therapies focusing on the down‐regulation of the immune system in parallel with salt restriction.
Calcium channel blockers
Calcium channel blockers (CCBs), which control BP mainly through reduction of calcium influx in the peripheral vascular bed, were already known to negatively modulate the immune system in different in vitro (Rodler et al., 1995) and in vivo (Katoh et al., 1997) studies. After that, studies focusing on CCB immunomodulation on the treatment of hypertension appeared. Nifedipine reduced the mRNA for CCL2 by NF‐κB inhibition‐dependent mechanisms in rat vascular smooth muscle cells exposed to AngII (Wu et al., 2006). In addition, amlodipine prevented the expression of aortic NADPH oxidase, vascular cell adhesion molecule 1 and CCL2 in l‐NAME‐induced hypertensive rats in a pressure‐independent manner (Toba et al., 2005). Recently, hypertensive left ventricular hypertrophy was shown to be partly dependent on the activation of a member of the TNF superfamily and that amlodipine could reverse this effect in SHR. (Lu et al., 2016). It is likely that the effects of amlodipine and other CCBs on the cardiovascular system are due, not only to their vasodilator properties, but also through down‐regulation of specific components of innate and adaptive immunity. However, more experimental and clinical studies on the effects of CCBs on hypertension are encouraged, in order to elucidate the connections between the long‐term benefits of CCBs and regulation of the immune system.
Renin–angiotensin aldosterone system (RAS) blockade
Blockade of the RAS with angiotensin‐converting enzyme inhibitors (ACEi) or AT1 receptor antagonists is the first‐choice treatment for hypertension in the present day. These drugs also modulate the immune system, which could contribute to antihypertensive effects and organ damage protection. Under physiological conditions, for example, ACE inhibition by enalapril enhanced splenic CD4+CD103+CD25− Treg production and response in Balb/c mice (Albuquerque et al., 2010). In lupus nephritis‐associated hypertension, the attenuation of BP by ACEi involves the reduction of Th2 polarization, cytokine production and renal damage (De Albuquerque et al., 2004). Also, ACE interferes with both major histocompatibility complex (MHC)I and MHCII antigen‐presenting processes and increases innate and adaptive immune responses (Bernstein et al., 2018). Therefore, it is expected that ACEi would promote a negative modulation on presenting processes of hypertensive patients and, consequently, reduce BP and organ damage. A similar effect was observed by Hevia et al. (2018), as the ablation of antigen‐presenting cells prevented hypertension, cardiac hypertrophy and renal inflammation in mice submitted to AngII plus high‐salt diet. However, this hypothesis remains untested, and the relationship between immune response and other modes of RAS blockade also needs further study.
In SHR‐stroke resistant (SHR‐SR), the AT1 receptor antagonist, telmisartan, was tested at low and high doses, and NLRP3 activation was assessed. Telmisartan attenuated hypertension only at high doses while it inhibited NLRP3 inflammasome from the neurovascular unit in a dose‐dependent manner (Liu et al., 2015). These findings suggest that telmisartan inhibition of NLRP3 is BP‐independent, and for this reason, it could be related to vascular function improvement. In hypertensive patients, losartan decreased T‐cell activity (Sonmez et al., 2001), and in combination with atorvastatin and captopril, it down‐regulated serum levels of IL‐6 (Sepehri et al., 2016).
Aldosterone, through action at the mineralocorticoid receptor (MR), is the main mediator of volume retention after RAS activation. Spironolactone, an MR antagonist, demonstrated a different immunomodulatory pattern from other RAS blockers. In DOCA‐salt hypertension, spironolactone treatment reduced Th17 response and IL‐17 production and enhanced Treg response (Amador et al., 2014). This mechanism appears to be related to spironolactone‐prevented hypertension, as anti‐IL‐17 treatment effectively attenuated hypertension, as did spironolactone (Amador et al., 2014). Spironolactone also demonstrated potent inhibition of leukocyte migration (Hofbauer et al., 2002), which may have contributed to its down‐regulation of the immune system. However, the exact mechanism(s) involved in this down‐regulation of the immune system by MR antagonists is not entirely understood. Although there is only limited knowledge from clinical studies on immunomodulation by MR antagonists, in hypertensive patients, spironolactone efficiently controlled BP, preserved endothelial function and reduced inflammation, as shown by reduced C‐reactive protein (Yamanari et al., 2009).
Diuretics
Besides MR antagonists, thiazide and loop diuretics are also prescribed for BP control, with or without other antihypertensive drugs. Although the effect of diuretics on the immunomodulation in hypertension is controversial, it is possible to find relevant evidence on diuretics as hypertensive treatments and their effects on the immune system.
For instance, thiazide diuretics up‐regulated the immune system of recently diagnosed hypertensive patients, enhancing IgM anti‐ApoB‐D autoantibodies (Fonseca et al., 2015). On the other hand, Toledo et al. (2015) observed that diuretic treatment down‐regulated circulating levels of pro‐inflammatory cytokines IL‐6 and TNF‐α in elderly hypertensive patients (Toledo et al., 2015). Both studies claim that diuretic treatment improved BP and endothelial function in hypertensive patients, but they disagree concerning the immunomodulation triggered by diuretic drugs. It is possible that age differences may have influenced the findings and more studies are required to test the exact mechanism of immunomodulation by diuretics in hypertensive patients. In Dahl salt‐sensitive (DSS) rats, for instance, hydrochlorothiazide and chlorthalidone were administered, and although BP was efficiently controlled, these diuretics did not attenuate oxidative stress and inflammation caused by hypertension (Zhou et al., 2008). Combination of diuretics with other antihypertensive therapies has demonstrated to negatively immunomodulate and, consequently, improve hypertension diuretic treatment in both experimental (Jin et al., 2014) and clinical studies (Agabiti‐Rosei et al., 2014). Therefore, long‐term inflammation caused by diuretics should be considered as a potentially threatening condition, and additional pharmacological therapies to down‐regulate the immune system in parallel to diuretic therapy may contribute to the treatment of hypertension.
Renal sympathetic denervation
Renal sympathetic denervation (RSD), a therapeutic strategy used in patients with resistant hypertension (Coppolino et al., 2017), has also been reported as a modulator of the immune system (Xiao et al., 2015; Zaldivia et al., 2017). In addition to the attenuation of sympathetic activity and BP, bilateral RSD in AngII‐exposed mice reduced albuminuria, dendritic cell activation, renal Th and T‐cytotoxic infiltration, fibrosis, ROS, IL‐1α, IL1‐β and IL‐6 (Xiao et al., 2015). Similar effects on BP and inflammatory status were observed in clinical patients after 3 and 6 months of RSD procedure, in which the RSD significantly attenuated BP, plasma levels of CCL2, monocyte activation, IL‐12, IL‐1β and TNF‐α (Zaldivia et al., 2017). Also, a positive relationship was found between sympathetic activity and monocyte activation before and after RSD (Zaldivia et al., 2017), which suggests that the therapeutic strategy of RSD partly includes the attenuation of monocyte activity and inflammation in hypertension. As a proof of concept, nebivolol, a selective antagonist of β1‐adrenoceptors triggered similar immunomodulation. Nebivolol efficiently reduced innate immune responses in hypertensive patients after treatment, as shown by decreased levels of circulating neutrophils (Hussain et al., 2017), oxidative stress and intercellular adhesion molecule 1 (Serg et al., 2012).
The down‐regulation of the immune system appears as a key factor in the treatment of hypertension. Therefore, there is enough evidence that antihypertensive treatments may benefit hypertensive patients not only through its classical pathways but also through down‐regulation of the immune system. Although different hypertensive therapies result in different pathways of immunomodulation, these pieces of evidence support the idea of immunosuppressant agents as potential therapeutic strategies for the treatment of hypertension. Furthermore, it suggests that immunosuppressant drugs may be beneficial to hypertensive patients in specific cases.
Immunosuppressants as antihypertensives
Classical agents
The contribution of immunological mechanisms to hypertension is now clearly demonstrated. Therefore, treatment with immunosuppressant drugs is the most intuitive attempt to translate basic science to the treatment of hypertensive patients. Immunosuppressant drugs t decrease the activity of the immune system and they were introduced in clinical practice mainly to prevent acute rejection of organ transplants, thus avoiding graft loss (Allison, 2000). They also comprise the treatment of autoimmune diseases, such as rheumatoid arthritis and SLE. From a classical view, these drugs can be grouped on the basis of their mechanisms of action (Allison, 2000): (i) regulators of gene expression (glucocorticoids), (ii) alkylating agents (cyclophosphamide), (iii) kinase and phosphatase inhibitors (cyclosporine A, tacrolimus and rapamycin) and (iv) inhibitors of nucleic acids synthesis (azathioprine, mycophenolate mofetil and leflunomide). Some of these drugs have also been tested in experimental models of hypertension, as summarized in Table 1.
The first pharmacological attempts to treat hypertension with immunosuppressants were performed in the 1980s when a polyclonal anti‐thymocyte globulin and cyclophosphamide were given to SHR and hypertension was reversed (Bendich et al., 1981; Khraibi et al., 1984). Both studies with non‐specific immunosuppressant drugs demonstrated that lymphocyte depletion was the primary mechanism involved in this mitigation of hypertension. Nowadays, these therapies are considered unacceptable, due to the cytokine release syndrome, general toxicity and extensive exposure to infectious diseases.
The drugs in the third class of immunosuppressant include calcineurin inhibitors, cyclosporin A (CsA), tacrolimus and mammalian target of rapamycin (mTOR) inhibitors. CsA and tacrolimus are out of consideration as arterial hypertension and kidney dysfunction are well‐recognized long‐term side effect of these compounds (Hoskova et al., 2017). These effects are not associated with the immunosuppression per se but with activation of the RAS and endothelin systems, increased sympathetic activity and diminished NO bioavailability (Hoorn et al., 2012; Hoskova et al., 2017). Also, a new calcineurin inhibitor, voclosporin, which was developed to achieve higher efficacy and lower toxicity, did not show any significant difference regarding the incidence of hypertension and adverse events after kidney transplantation compared with tacrolimus (Busque et al., 2011).
mTOR inhibitors such as everolimus, temsirolimus and sirolimus appear as less cytotoxic pharmacological therapies to inhibit lymphocyte proliferation in a non‐specific manner (see Figure 1) (Shimobayashi and Hall, 2014). Sirolimus attenuated hypertension and renal infiltration by T‐lymphocytes and macrophages, in DSS rats (Kumar et al., 2017) and decreased cardiac hypertrophy in SHR (Soesanto et al., 2009) and heart transplant recipients (Kushwaha et al., 2008). In contrast, the administration of sirolimus for 7 weeks induced hypertension in normotensive rats in consequence of its side effects such as dyslipidaemia, hyperglycaemia and proteinuria (Reis et al., 2009). Therefore, sirolimus has clear limitations as an alternative treatment for hypertension (Figure 2).
The inhibitors of nucleic acid synthesis, such as azathioprine, leflunomide and mycophenolate mofetil, represent the last class of immunosuppressant, shown in Figures 1 and 2. Azathioprine, a purine analogue that inhibits de novo purine synthesis, attenuated proteinuria and hypertension, as well as reducing chemokines and pro‐inflammatory cytokines in a model of pregnancy‐induced hypertension in rats (Tinsley et al., 2009). Similarly, mycophenolate mofetil, an inosine monophosphate dehydrogenase inhibitor, attenuated hypertension in DOCA‐salt rats (Boesen et al., 2010), SHR (Rodriguez‐Iturbe et al., 2002) and an SLE model (Taylor and Ryan, 2017). These effects were brought about by the improvement of renal function as a result of reduced renal infiltration of T‐lymphocyte and macrophages (Tinsley et al., 2009; Boesen et al., 2010; Taylor and Ryan, 2017). Importantly, the treatment of patients with autoimmune diseases with mycophenolate mofetil was associated with BP reduction and reduced TNF‐α and oxidative stress levels (Herrera et al., 2006). Taken together, all these data demonstrate that, among the classical immunosuppressant drugs, mycophenolate mofetil has the most desirable risk/benefit balance (Figure 2).
As summarised above, the classical immunosuppressant drugs were disappointing in terms of their antihypertensive action. It is possible that more specific drugs, targeting particular molecules involved directly in the organization of immune responses, could overcome these limitations and be considered as treatments for hypertension.
Immune targets for the treatment of hypertension involving the innate immune response
Monocyte/macrophage inhibition
Macrophages and neutrophils compose the most important effector cells of the innate immune response. The chemokine CCL2 is a chemotactic factor for monocytes and a potential intervention point to treat hypertension. The absence of CCL2 reduced monocyte recruitment in the hearts of mice exposed to the hypertrophic stimulus and protected against diastolic dysfunction (Hulsmans et al., 2018). In addition, treatment with the selective CCL2 receptor antagonist, RS‐102895 (Figure 1), attenuated kidney injury in renovascular and AngII‐infused hypertensive mice. However, the effects on BP were disappointing (Elmarakby et al., 2007; Kashyap et al., 2016). In fact, the potential role of macrophages on the pathophysiology of hypertension seems to be more related to vascular, renal and neurovascular damage (Justin Rucker and Crowley, 2017) than BP elevation per se. On the other hand, antagonism of the chemokine receptor, CCR2, with INCB3344 reversed DOCA‐salt‐induced elevations in BP (Chan et al., 2012a), which may be related to the improvement of vascular function. However, neither RS‐102895 nor INCB3344 have been tested in humans (Figure 2).
TLR inhibition
Several attempts have been made to inhibit the receptors involved in innate immunity and their downstream pathways by pharmacological interventions (Figure 1). Treatment of SHR (Bomfim et al., 2012) and AngII‐infused mice (Hernanz et al., 2015) with neutralizing antibodies to TLR4 (Figure 1) ameliorated vascular dysfunction and lowered BP, presumably by decreasing vascular resistance. Also, anti‐TLR4‐Ig reversed cardiac hypertrophy (Echem et al., 2015). In SRH, the earlier the anti‐TLR‐4 treatment starts, the greater is the reduction of BP (Bomfim et al., 2012; Echem et al., 2015). Although we are still far from clinical use of anti‐TLR4‐Ig, the safety profile of this agent shown in a recent phase I study conducted in 73 healthy volunteers (Monnet et al., 2017) should encourage further clinical trials in hypertensive patients (Figure 2). Although there are data to support TLR4 as a target in hypertension, the TLR4 antagonist, eritoran, has not been tested either in experimental models of hypertension or in clinical studies.
In SHR, TLR9 contributes to dysfunctional vascular reactivity and treatment of SHRs with the TLR9 antagonist, ODN2088, lowered BP (McCarthy et al., 2015). Similarly, chloroquine caused a significant decrease in BP by inhibiting TLR9 downstream signalling proteins (McCarthy et al., 2017). Chloroquine, which was developed primarily to treat malaria, has anti‐inflammatory and immunomodulatory properties, in part related to the inhibition of endosomal TLR9 (Figure 1), allowing its application to the treatment of inflammatory disease (Al‐Bari, 2015) and making it an attractive candidate to be tested in hypertension. Nonetheless, the abnormal cardiac conduction disorders and cardiomyopathy related to long‐term treatment with chloroquine (Al‐Bari, 2015) must be disadvantages, particularly in patients with existing cardiovascular disease. As a result, there is limited interest in this drug, in the context of hypertension (Figure 2).
NF‐κB inhibition
Activation of the NF‐κB pathway is a key component of the stimulation of innate immunity by PRR recognition of PAMPs/DAMPs, which in turn results in the expression of pro‐inflammatory mediators including cytokines such as TNF‐α and IL‐1β, chemokines, adhesion molecules and enzymes. Oxidative stress is also a critical trigger to amplify NF‐κB activation, which plays an essential role in the pathogenesis of chronic inflammatory diseases and hypertension. Antioxidants, such as pyrrolidine dithiocarbamate (PDTC), are the most commonly used compounds for the pharmacological inhibition of NF‐κB and this compound is known to exert antihypertensive effects (Rodriguez‐Iturbe et al., 2005). PDTC also protected against hypertension‐induced vascular and cardiac hypertrophy through a mechanism involving down‐regulation of MMP (Cau et al., 2015; Cau et al., 2011). Although it is effective, PDTC safety is still a barrier to clinical studies (Figure 2). Historically, dithiocarbamates have been used as insecticides and fungicides, and to our knowledge, no safety assessments of PDTC in humans have yet been carried out.
NLR inhibition
Hypertension also triggers NLR signalling and activation of NLR induces a complex intracellular signalling structure, the inflammasome, that results in the production of highly pro‐inflammatory cytokines: IL‐1β and IL‐18. Treatment of DOCA‐salt hypertensive mice with MCC950, an inhibitor of NLRP3 inflammasome oligomerization (Figure 1), significantly reduced systolic BP and renal pro‐inflammatory cytokines (Krishnan et al., 2016). However, aged mice with AngII‐induced hypertension exhibited enhanced expression of NLRP3 inflammasome components in the kidney but had no hypotensive response to treatment with MCC950 (Dinh et al., 2017). Nevertheless, this is a very novel NLRP3 inflammasome inhibitor and a promising compound. The most commonly used drugs for blocking the inflammasome target one of its products, IL‐1β through neutralizing antibodies (nAbs) or the recombinant IL‐1β soluble receptor (Figure 1). The relevance of IL‐1β to hypertensive heart disease has been supported by studies showing that anti‐IL‐1β‐Ig blocks AngII‐induced cardiac hypertrophy with no effects on BP (Wang et al., 2014). Human nAbs for IL‐1β, such as gevokizumab and canakinumab, have been approved for clinical use. Importantly, canakinumab led to significantly lower recurrent cardiovascular events (Shah et al., 2018). Moreover, the injection of gevokizumab in the paraventricular nucleus of DSS rats led to lower BP, heart rate, plasma levels of noradrenaline and oxidative stress and restored cytokine balance (Qi et al., 2016). Additionally, anakinra, which is a recombinant form of the human IL‐1β receptor antagonist (IL‐1Ra), improved renal function and ameliorated hypertension in AngII‐infused (Zhang et al., 2016) and DOCA‐salt hypertensive mice (Ling et al., 2017). Anakinra has been an approved drug since 2001 to treat the signs of rheumatoid arthritis, and recent preclinical findings have confirmed its efficacy in cardiovascular disease (Abbate et al., 2013; Van Tassell et al., 2017), making the possible clinical applications of anakinra in hypertension, closer than the other drugs mentioned (Figure 2).
TNF‐α and IL‐6 inhibition
TNF‐α and IL‐6 are essential effectors of innate immunity, but they are also involved in the adaptive response. Genetic KO animals for both cytokines have already been tested, and both resulted in reduced inflammation, organ damage and BP in AngII‐induced hypertension (Sriramula et al., 2008; Brands et al., 2010). Infliximab, a monoclonal antibody (mAb) against TNF‐α, and etanercept, a soluble recombinant TNF receptor 2 fusion protein, prevented hypertension in experimental models (Filho et al., 2013; Tran et al., 2009), as summarized in Table 1. The inhibition of IL‐6 with a nAb (Table 1) also attenuated hypertension and decreased glomerular inflammation (Hashmat et al., 2016). Regarding efficacy, both TNF‐α blockers and IL‐6 inhibitor normalized BP in experimental studies by controlling important pathways in the pathophysiology of hypertension. However, clinical studies also have demonstrated that infliximab infusions might result in the cytokine release syndrome, anaphylactic reactions and fever with degranulation of mast cells and basophils (Lichtenstein et al., 2015), whereas the mAb anti‐IL6 receptor, tocilizumab, did not change cardiovascular risk or events in rheumatoid patients (Kim et al., 2017). Taken together, these results compromise the risk/benefit balance for both of these immunosuppressant drugs (Figure 2).
Complement system inhibition
Eculizumab, an anti‐C5 mAb (Figure 1), has been used for the treatment of paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome (Horiuchi and Tsukamoto, 2016). In a case report, treatment with eculizumab for atypical haemolytic uraemic syndrome attenuated severe hypertension and led to the cessation of dialysis in an infant (Ohta et al., 2015). However, the effects of eculizumab in hypertension, associated or not with autoimmune diseases, still need further assessment (Figure 2). The high cost of eculizumab is one of its disadvantages, as well as increased susceptibility to severe infection from encapsulated bacteria (Wiseman, 2016).
Targeting adaptive immune response
Since the development of highly specific drugs targeting molecules involved in the adaptive immune response, several options to interfere with this type of response in the immune component of hypertension are possible (Table 1), as discussed below.
Agents targeting co‐stimulatory molecules
The expression of CD80/CD86 on antigen‐presenting cell (APC) is required to interact with CD28 on the T‐cell, reinforcing T‐cell activation. Once an effective immune response is established, T‐cells express cytotoxic T‐lymphocyte‐associated protein 4 (CTLA4) on their surface, and a competitive binding occurs to CD80/CD86 to down‐regulate T‐cell activation. In AngII‐induced and DOCA‐salt‐induced hypertension, the blockade of B7‐dependent co‐stimulation with CTLA4‐Ig decreased BP, T‐cell cytokine production, accumulation and, consequently, vascular inflammation (Vinh et al., 2010). Abatacept is a CTLA4 human IgG (Figure 1) used to mimic this effect on T‐cells and has been approved for the treatment of rheumatoid arthritis. The most common adverse effects in humans include headache, nasopharyngitis, upper respiratory tract infection, gastrointestinal infections and, especially, a higher risk of infections (Bugelski and Martin, 2012). Belatacept is a drug more selective as a blocker of T‐cell co‐stimulation. A clinical study, comparing belatacept with cyclosporine, demonstrated amelioration of central aorta haemodynamics, regardless of the effects on arterial stiffness (Seibert et al., 2014). The absence of major clinical trials limits the use of lymphocyte co‐stimulation inhibitors in the treatment of hypertension (Figure 2).
The CD40 on APC interacts with the ligand of CD40 (CD40L) on activated T‐cells and leads to up‐regulation of CD80/CD86 on APC (Figure 1), amplifying T‐cell co‐stimulatory signalling (Wiseman, 2016). An anti‐CD40L in pre‐eclamptic Sprague–Dawley rats reduced hypertension, placental oxidative stress and AngII type 1 receptors (Cornelius et al., 2015). No tests have been performed in other models of hypertension. However, the clinical use of anti‐CD40L treatment for autoimmune disorders was discontinued because of the incidence of thrombotic events and myocardial infarction (Sidiropoulos and Boumpas, 2004).
Anti‐T‐cell receptor drugs
Muromonab‐CD3 (OKT3) is an anti‐CD3 murine mAb (Figure 1), which recognizes the ε chain of the CD3 on T‐cells. The T‐cells are unable to respond to an antigen or target cells and are opsonized afterwards. OKT3 has been used for the prevention of allograft rejection in renal, heart and hepatic transplantation (Wiseman, 2016). Mathis et al. (2017) demonstrated that anti‐CD3 therapy resulted in attenuation of the development of hypertension in female mice with SLE. The main limitation of OKT3 is the development of the cytokine release syndrome, a condition with symptoms similar to those of severe infection, such as fever and hypotension (Bugelski and Martin, 2012).
The anti‐CD25 antibodies, such as basiliximab and daclizumab, are another example of T‐cell blockers (Van Gelder et al., 2004), based on the fundamental role played by IL‐2 in lymphocyte proliferation. These drugs bind to the CD25 chain of the IL‐2 receptor, preventing IL‐2 interaction with its receptor on the surface of T‐lymphocytes (Van Gelder et al., 2004). Both are approved drugs for immunosuppressant proposes, but there is little information about their cardiovascular effects. The favourable safety profile of anti‐CD25 antibodies enhances the interest of its use for trials in hypertensive patients (Figure 2).
Treg increase
Treg cell therapy has emerged as a strategy to induce tolerance in organ transplantation and autoimmune disease. In hypertension, adoptive transfer of Treg prevents AngII‐induced hypertension and vascular injury (Barhoumi et al., 2011) and improves coronary artery endothelial dysfunction (Matrougui et al., 2011). However, the use of Treg adoptive transfer as a therapy in humans may be limited due to the low cell numbers present in donor lymphoid organs, requiring a large number of donors or in vitro expansion.
In another approach, the IL‐2/IL‐2 antibody complex binds to the small population of CD4+ T‐lymphocytes expressing CD25− and induces in vivo expansion of CD4+CD25+ forkhead box P3+ Treg cells (Webster et al., 2009), as illustrated in Figure 1. Accordingly, IL‐2/IL‐2 antibody complex induced Treg expansion in AngII‐infused mice and decreased vascular stiffness (Majeed et al., 2014) and attenuated the progression of congestive heart in mice after transverse aortic constriction (Wang et al., 2016). Despite this effect, it does not seem to be an appropriate option to control BP, as the IL‐2/IL‐2Ab complex did not lower the high BP of AngII‐infused mice or that of mice with transverse aortic constriction. However, IL‐2/IL‐2 antibody complex is still an experimental approach.
CD8 T‐cell inhibition
Although recent papers have shown the role for CD8 T‐cells in hypertension, few studies have assessed the effects of blocking CD8 T‐cell activation in this condition. Ma et al. (2014) used WT mice infused with AngII treated with the CD8‐specific IgG1 antibody (Figure 1), which caused an efficient depletion of CD8+ T‐cells. They found that treatment reduced cardiac profibrogenic inflammation, but they did not measure BP in the treated animals. This study used only CD8 KO mice infused with AngII. According to these findings, CD8 cells have an important role in hypertension. However, any clinical applications will depend on a better knowledge of how to control this cell type.
B‐cell targeting
Mature B‐cells have a CD20 antigen expressed on its surface. Hence, anti‐CD20 mAbs are useful for the treatment of lymphoproliferative diseases, such as follicular non‐Hodgkin's lymphoma and autoimmune haematological disorders (Hansel et al., 2010). The first generation of anti‐CD20 mAbs includes rituximab, while the second humanized generation has lower immunogenicity and comprises ofatumumab, veltuzumab and ocrelizumab (Wiseman, 2016). Little data of their efficacy against hypertension is available. However, an anti‐CD20 (analogous to the clinically used drug) reduced BP in AngII hypertensive mice (Chan et al., 2015). In terms of adverse effects, anti‐CD20 Abs are associated with immunosuppression, infections, renal toxicity, cardiac arrhythmias and several other effects (Wiseman, 2016).
Targeting B‐cell differentiation is another mechanism of B‐cell blockade, which is primarily used in autoimmune diseases and is accomplished by drugs like belimumab and atacicept (Wiseman, 2016). The principle is to impair the binding of B‐cell‐activating factors to their receptors, thus inhibiting B‐cell maturation (Wiseman, 2016). However, the role of these immunomodulators in the context of hypertension, associated or not with SLE, still needs further investigation, as their use has been limited in autoimmune diseases.
The ubiquitin–proteasome system controls protein turnover, and its inhibition induces apoptosis, particularly in effector B‐cells (Wiseman, 2016). The proteasome inhibitors PSI and MG132 demonstrated antihypertensive effects and suppression of hypertension‐induced cardiac fibrosis, respectively (Takaoka et al., 1998; Meiners et al., 2004), but, in both cases, their effects on B‐cell activity were not addressed. Bortezomib (Figure 1), a proteasome inhibitor approved for the treatment of multiple myeloma, attenuated BP increase and aortic vascular remodelling in AngII‐infused mice (Li et al., 2013). Also, recent data on a rat model of SLE demonstrated significant reduction on BP and improvement of renal function by bortezomib, and these effects were associated with the bortezomib‐induced decrease of plasma immunoglobulin levels and B‐cell and T‐cell infiltration in the kidneys (Taylor et al., 2018a). Although bortezomib emerges as a modulator of the adaptive immune responses driven by B‐cells and T‐cells in hypertension, common adverse effects in patients with myeloma include dose‐limiting toxicity leading to sensory neuropathy, asthenic conditions, gastrointestinal disorders and no dose‐limiting responses such as thrombocytopenia and anaemia (Bross et al., 2004). For all the reasons discussed above, anti‐B‐cell drugs have a risk/benefit balance, limiting their use in hypertension (Figure 2).
IL‐17 inhibition
The response of Th17 and γδ T‐cells in producing IL‐17 has gained importance in hypertension. For example, both T‐cell receptor (TCR) δ gene deletion and treatment with anti‐TCRγδ (Figure 1) blunted endothelial dysfunction and hypertension in AngII‐infused mice (Caillon et al., 2017). It has been suggested that IL‐17 plays a fundamental role in these effects. In addition, direct inhibition of IL‐17 with Ab therapy (IL‐17 nAb; IL‐17A mAb) or the soluble specific receptor for IL‐17, attenuated hypertension in different experimental models (Cornelius et al., 2013; Saleh et al., 2016; Chiasson et al., 2017). The mechanisms involved in hypertension control induced by direct inhibitors of IL‐17 seem to include the enhancement of Treg cells in spleen and lymph node, reduction of renal and vascular infiltration of total T‐cells (CD3+), Th17 and T‐cytotoxic (CD8+). Also, IL‐17 inhibition provides a decrease in TGF‐β, ROS, albuminuria, organ damage and vascular dysfunction (Cornelius et al., 2013; Saleh et al., 2016; Chiasson et al., 2017). Similarly, inhibition, using IL‐17A receptor mAb, attenuated hypertension, albuminuria, renal and aortic infiltration of total leukocytes, total T‐cell (CD3+), T‐cell (CD4+), T‐cytotoxic (CD8+), renal TGF‐β and organ damage (Saleh et al., 2016). In normotensive patients, IL‐17RA mAb therapy showed an acceptable cardiovascular safety (Papp et al., 2016). However, IL‐17 seems to play a protective role in renal disease (Mohamed et al., 2016), and its inhibition could accelerate renal injury in specific cases of hypertension (Krebs et al., 2014). Therefore, although IL‐17 is not the only cytokine involved on the pathophysiology of hypertension, it has the potential to be one of the targets for the treatment of hypertension, by immunosuppressant drugs in the future.
IFN‐γ inhibition
Although there is little evidence relating to the pharmacological inhibition of IFN in hypertension, it is important to mention that anti‐IFN therapy might play an important role in combination with other drugs. IFN is a cytokine involved in Th1 cell differentiation. Although IFN‐γ receptor KO mice did not display an antihypertensive effect in the model of AngII‐induced hypertension, they did show reduced cardiac macrophages and T‐cell infiltration, fibrosis and hypertrophy, demonstrating cardioprotective effects (Marko et al., 2012). Moreover, inhibition of IFN‐γ (Figure 1) was able to partly prevent the elevation of BP in mice overexpressing the MR after AngII infusion. These results suggest that IFN‐γ participates in the pathophysiology of AngII‐induced hypertension, but it is not the sole factor responsible for BP elevation (Sun et al., 2017). Considering these data together, patients may benefit from the combination of IFN‐γ inhibition with other drugs, as a therapeutic strategy to treat hypertension. Nevertheless, studies are warranted to test this hypothesis.
Conclusion
The recognition that hypertensive disease is accompanied by DAMPs and formation and release of neoantigens, which trigger a sophisticated immune response, has opened a new clinical approach to the treatment of hypertension. The most recent efforts made to determine the involvement of innate and adaptive immunity in hypertension were conducted in KO animals. From a pharmacological standpoint, new strategies to treat hypertension are evolving with the explosion of knowledge about the role of the activation of the immune system, in this condition. As an example, immunosuppressant drugs have been tested in animal models of hypertension, and some clinical information is now available about their cardiovascular safety. Also, the present review has outlined advantages and disadvantages of therapy with classical immunosuppressant drugs and newer targeted therapies that block specific steps of the activation of the immune system, some of which are still in the development phase. Current findings in the literature show a more favourable risk/benefit ratio profile of targeted therapies over classical immunosuppressant drugs, which may decrease BP and protect hypertensive patients from end‐organ damage. In order to implement this challenging clinical translation, we would first propose that more clinical trials evaluating hypertension as an outcome are needed. Secondly, a careful risk–benefit evaluation should be performed before therapy, considering human data about drug safety and efficacy to reduce BP or other cardiovascular damage (Figure 2). The translation of this knowledge, from experimental science to clinical practice, starts with the right choice of the immunomodulator based on the patient's markers of inflammation and with appropriate selection of the patients. Also, there must be caution in interpreting the published results, as the majority of studies correlating activation of the immune system with the development and maintenance of hypertension were carried out in AngII‐infused animals. To our knowledge, no studies have addressed or compared the relevance of all these findings across different hypertensive models. It is possible that contrasting results from different hypertensive conditions may reflect particular inflammatory pathways that are not relevant in all other models. Thus, studies comparing the activation profile of the immune system across different hypertensive models and also with human hypertensive patients may provide insights into the mechanisms triggered in each model. Also, it may allow the development of therapies more likely to translate into human studies.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d, 2017e).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the National Council for Scientific and Technological Development (CNPq), the Minas Gerais Research Foundation (FAPEMIG) and the São Paulo Research Foundation (FAPESP, 2016/11988‐5).
Bomfim G. F., Cau S. B. A., Bruno A. S., Fedoce A. G., and Carneiro F. S. (2019) Hypertension: a new treatment for an old disease? Targeting the immune system, British Journal of Pharmacology, 176, 2028–2048, doi: 10.1111/bph.14436.
References
- Abbate A, Van Tassell BW, Biondi‐Zoccai G, Kontos MC, Grizzard JD, Spillman DW et al (2013). Effects of interleukin‐1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University – Anakinra Remodeling Trial (2) (VCU‐ART2) pilot study]. Am J Cardiol 111: 1394–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agabiti‐Rosei E, Manolis A, Zava D, Omboni S, Group ZS (2014). Zofenopril plus hydrochlorothiazide and irbesartan plus hydrochlorothiazide in previously treated and uncontrolled diabetic and non‐diabetic essential hypertensive patients. Adv Ther 31: 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KO, Lim SW, Li C, Yang HJ, Ghee JY, Kim JY et al (2007). Influence of angiotensin II on expression of toll‐like receptor 2 and maturation of dendritic cells in chronic cyclosporine nephropathy. Transplantation 83: 938–947. [DOI] [PubMed] [Google Scholar]
- Al‐Bari MA (2015). Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother 70: 1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque D, Nihei J, Cardillo F, Singh R (2010). The ACE inhibitors enalapril and captopril modulate cytokine responses in Balb/c and C57Bl/6 normal mice and increase CD4+CD103+CD25negative splenic T cell numbers. Cell Immunol 260: 92–97. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison AC (2000). Immunosuppressive drugs: the first 50 years and a glance forward. Immunopharmacology 47: 63–83. [DOI] [PubMed] [Google Scholar]
- Amador CA, Barrientos V, Pena J, Herrada AA, Gonzalez M, Valdes S et al (2014). Spironolactone decreases DOCA‐salt‐induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension 63: 797–803. [DOI] [PubMed] [Google Scholar]
- Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF et al (2011). T regulatory lymphocytes prevent angiotensin II‐induced hypertension and vascular injury. Hypertension 57: 469–476. [DOI] [PubMed] [Google Scholar]
- Bendich A, Belisle EH, Strausser HR (1981). Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 99: 600–607. [DOI] [PubMed] [Google Scholar]
- Bernstein KE, Khan Z, Giani JF, Cao DY, Bernstein EA, Shen XZ (2018). Angiotensin‐converting enzyme in innate and adaptive immunity. Nat Rev Nephrol 14: 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancardi VC, Bomfim GF, Reis WL, Al‐Gassimi S, Nunes KP (2017). The interplay between angiotensin II, TLR4 and hypertension. Pharmacol Res 120: 88–96. [DOI] [PubMed] [Google Scholar]
- Boesen EI, Williams DL, Pollock JS, Pollock DM (2010). Immunosuppression with mycophenolate mofetil attenuates the development of hypertension and albuminuria in deoxycorticosterone acetate‐salt hypertensive rats. Clin Exp Pharmacol Physiol 37: 1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC et al (2012). Toll‐like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci 122: 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Rodrigues FL, Carneiro FS (2017). Are the innate and adaptive immune systems setting hypertension on fire? Pharmacol Res 117: 377–393. [DOI] [PubMed] [Google Scholar]
- Brands MW, Banes‐Berceli AK, Inscho EW, Al‐Azawi H, Allen AJ, Labazi H (2010). Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster LM, van Montfrans GA, Oehlers GP, Seedat YK (2016). Systematic review: antihypertensive drug therapy in patients of African and South Asian ethnicity. Intern Emerg Med 11: 355–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME et al (2004). Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 10: 3954–3964. [DOI] [PubMed] [Google Scholar]
- Bugelski PJ, Martin PL (2012). Concordance of preclinical and clinical pharmacology and toxicology of therapeutic monoclonal antibodies and fusion proteins: cell surface targets. Br J Pharmacol 166: 823–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busque S, Cantarovich M, Mulgaonkar S, Gaston R, Gaber AO, Mayo PR, Ling S, Huizinga RB, Meier‐Kriesche HU, for the PROMISE Investigators (2011). The PROMISE study: a phase 2b multicenter study of voclosporin (ISA247) versus tacrolimus in de novo kidney transplantation. Am J Transplant 11: 2675–2684. [DOI] [PubMed] [Google Scholar]
- Caillon A, Mian MOR, Fraulob‐Aquino JC, Huo KG, Barhoumi T, Ouerd S et al (2017). γδ T cells mediate angiotensin II‐induced hypertension and vascular injury. Circulation 135: 2155–2162. [DOI] [PubMed] [Google Scholar]
- Cartwright N, Murch O, McMaster SK, Paul‐Clark MJ, van Heel DA, Ryffel B et al (2007). Selective NOD1 agonists cause shock and organ injury/dysfunction in vivo . Am J Respir Crit Care Med 175: 595–603. [DOI] [PubMed] [Google Scholar]
- Cau SB, Guimaraes DA, Rizzi E, Ceron CS, Gerlach RF, Tanus‐Santos JE (2015). The nuclear factor κB inhibitor pyrrolidine dithiocarbamate prevents cardiac remodelling and matrix metalloproteinase‐2 up‐regulation in renovascular hypertension. Basic Clin Pharmacol Toxicol 117: 234–241. [DOI] [PubMed] [Google Scholar]
- Cau SB, Guimaraes DA, Rizzi E, Ceron CS, Souza LL, Tirapelli CR et al (2011). Pyrrolidine dithiocarbamate down‐regulates vascular matrix metalloproteinases and ameliorates vascular dysfunction and remodelling in renovascular hypertension. Br J Pharmacol 164: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A et al (2012a). Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt‐treated mice. Hypertension 60: 1207–1212. [DOI] [PubMed] [Google Scholar]
- Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H et al (2015). Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- Chan SW, Hu M, Tomlinson B (2012b). The pharmacogenetics of β‐adrenergic receptor antagonists in the treatment of hypertension and heart failure. Expert Opin Drug Metab Toxicol 8: 767–790. [DOI] [PubMed] [Google Scholar]
- Chatterjee P, Weaver LE, Doersch KM, Kopriva SE, Chiasson VL, Allen SJ et al (2012). Placental Toll‐like receptor 3 and Toll‐like receptor 7/8 activation contributes to preeclampsia in humans and mice. PLoS One 7: e41884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Schachter D (1993). Elevation of plasma immunoglobulin A in the spontaneously hypertensive rat. Hypertension 21: 731–738. [DOI] [PubMed] [Google Scholar]
- Chen CT, Li Y, Zhang J, Wang Y, Ling HW, Chen KM et al (2014). Association between ambulatory systolic blood pressure during the day and asymptomatic intracranial arterial stenosis. Hypertension 63: 61–67. [DOI] [PubMed] [Google Scholar]
- Chen GD, Lai XQ, Ko DS, Qiu J, Wang CX, Han M et al (2015). Comparison of efficacy and safety between rabbit anti‐thymocyte globulin and anti‐T lymphocyte globulin in kidney transplantation from donation after cardiac death: a retrospective cohort study. Nephrol Ther 20: 539–543. [DOI] [PubMed] [Google Scholar]
- Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM (2017). Regulatory T‐cell augmentation or interleukin‐17 inhibition prevents calcineurin inhibitor‐induced hypertension in mice. Hypertension 70: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles B, Lewis R, Anning PB, Morton J, Baalasubramanian S, Morgan BP et al (2007). CD59 or C3 are not requred for angiotensin II‐dependent hypertension or hypertrophy in mice. Immunology 121: 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppolino G, Pisano A, Rivoli L, Bolignano D (2017). Renal denervation for resistant hypertension. Cochrane Database Syst Rev 2: CD011499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius DC, Castillo J, Porter J, Amaral LM, Campbell N, Paige A et al (2015). Blockade of CD40 ligand for intercellular communication reduces hypertension, placental oxidative stress, and AT1‐AA in response to adoptive transfer of CD4+ T lymphocytes from RUPP rats. Am J Physiol Regul Integr Comp Physiol 309: R1243–R1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J et al (2013). Administration of interleukin‐17 soluble receptor C suppresses TH17 cells, oxidative stress, and hypertension in response to placental ischemia during pregnancy. Hypertension 62: 1068–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curigliano G, Cardinale D, Dent S, Criscitiello C, Aseyev O, Lenihan D et al (2016). Cardiotoxicity of anticancer treatments: epidemiology, detection, and management. CA Cancer J Clin 66: 309–325. [DOI] [PubMed] [Google Scholar]
- Davidson TS, Shevach EM (2011). Polyclonal Treg cells modulate T effector cell trafficking. Eur J Immunol 41: 2862–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Albuquerque DA, Saxena V, Adams DE, Boivin GP, Brunner HI, Witte DP et al (2004). An ACE inhibitor reduces Th2 cytokines and TGF‐β1 and TGF‐β2 isoforms in murine lupus nephritis. Kidney Int 65: 846–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL (2005). Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II‐infused macrophage colony‐stimulating factor‐deficient mice: evidence for a role in inflammation in angiotensin‐induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113. [DOI] [PubMed] [Google Scholar]
- Dinh QN, Drummond GR, Kemp‐Harper BK, Diep H, De Silva TM, Kim HA et al (2017). Pressor response to angiotensin II is enhanced in aged mice and associated with inflammation, vasoconstriction and oxidative stress. Aging 9: 1595–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echem C, Bomfim GF, Ceravolo GS, Oliveira MA, Santos‐Eichler RA, Bechara LR et al (2015). Anti‐toll like receptor 4 (TLR4) therapy diminishes cardiac remodeling regardless of changes in blood pressure in spontaneously hypertensive rats (SHR). Int J Cardiol 187: 243–245. [DOI] [PubMed] [Google Scholar]
- Eissler R, Schmaderer C, Rusai K, Kuhne L, Sollinger D, Lahmer T et al (2011). Hypertension augments cardiac toll‐like receptor 4 expression and activity. Hypertens Res 34: 551–558. [DOI] [PubMed] [Google Scholar]
- Elmarakby AA, Quigley JE, Olearczyk JJ, Sridhar A, Cook AK, Inscho EW et al (2007). Chemokine receptor 2b inhibition provides renal protection in angiotensin II–salt hypertension. Hypertension 50: 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettehad D, Emdin CA, Kiran A, Anderson SG, Callender T, Emberson J et al (2016). Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta‐analysis. Lancet 387: 957–967. [DOI] [PubMed] [Google Scholar]
- Filho AG, Kinote A, Pereira DJ, Renno A, dos Santos RC, Ferreira‐Melo SE et al (2013). Infliximab prevents increased systolic blood pressure and upregulates the AKT/eNOS pathway in the aorta of spontaneously hypertensive rats. Eur J Pharmacol 700: 201–209. [DOI] [PubMed] [Google Scholar]
- Fonseca HA, Fonseca FA, Lins LC, Monteiro AM, Bianco HT, Brandao SA et al (2015). Antihypertensive therapy increases natural immunity response in hypertensive patients. Life Sci 143: 124–130. [DOI] [PubMed] [Google Scholar]
- Glorioso N, Herrera VL, Didishvili T, Ortu MF, Zaninello R, Fresu G et al (2013). Sex‐specific effects of NLRP6/AVR and ADM loci on susceptibility to essential hypertension in a Sardinian population. PLoS One 8: e77562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulopoulou S, Wenceslau CF, McCarthy CG, Matsumoto T, Webb RC (2016). Exposure to stimulatory CpG oligonucleotides during gestation induces maternal hypertension and excess vasoconstriction in pregnant rats. Am J Physiol Heart Circ Physiol 310: H1015–H1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu CC, Hunt SC, Kardia S, Turner ST, Chakravarti A, Schork N et al (2007). An investigation of genome‐wide associations of hypertension with microsatellite markers in the family blood pressure program (FBPP). Hum Genet 121: 577–590. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ (2010). The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 9: 325–338. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmat S, Rudemiller N, Lund H, Abais‐Battad JM, Van Why S, Mattson DL (2016). Interleukin‐6 inhibition attenuates hypertension and associated renal damage in Dahl salt‐sensitive rats. Am J Physiol Renal Physiol 311: F555–F561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari F, Vasudevan R, Mohd Ali SZ, Ismail P, Arkani M (2017). RAS genetic variants in interaction with ACE inhibitors drugs influences essential hypertension control. Arch Med Res 48: 88–95. [DOI] [PubMed] [Google Scholar]
- Hernanz R, Martinez‐Revelles S, Palacios R, Martin A, Cachofeiro V, Aguado A et al (2015). Toll‐like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II‐induced hypertension. Br J Pharmacol 172: 3159–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera J, Ferrebuz A, MacGregor EG, Rodriguez‐Iturbe B (2006). Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17: S218–S225. [DOI] [PubMed] [Google Scholar]
- Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R et al (2018). Myeloid CD11c+ antigen‐presenting cells ablation prevents hypertension in response to angiotensin II plus high‐salt diet. Hypertension 71: 709–718. [DOI] [PubMed] [Google Scholar]
- Hilme E, Herlitz H, Soderstrom T, Hansson L (1989). Increased secretion of immunoglobulins in malignant hypertension. J Hypertens 7: 91–95. [PubMed] [Google Scholar]
- Hofbauer R, Frass M, Pasching E, Gmeiner B, Kaye AD, Kapiotis S (2002). Furosemide and spironolactone reduce transmigration of leukocytes through endothelial cell monolayers. J Toxicol Environ Health A 65: 685–693. [DOI] [PubMed] [Google Scholar]
- Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH (2012). Pathogenesis of calcineurin inhibitor‐induced hypertension. J Nephrol 25: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Tsukamoto H (2016). Complement‐targeted therapy: development of C5‐ and C5a‐targeted inhibition. Inflamm Regen 36: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskova L, Malek I, Kopkan L, Kautzner J (2017). Pathophysiological mechanisms of calcineurin inhibitor‐induced nephrotoxicity and arterial hypertension. Physiol Res 66: 167–180. [DOI] [PubMed] [Google Scholar]
- Hulsmans M, Sager HB, Roh JD, Valero‐Munoz M, Houstis NE, Iwamoto Y et al (2018). Cardiac macrophages promote diastolic dysfunction. J Exp Med 215: 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Saeed M, Babar MZM, Atif MA, Akhtar L (2017). Nebivolol attenuates neutrophil lymphocyte ratio: a marker of subclinical inflammation in hypertensive patients. Int J Hypertens 2017: 7643628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, O'Boyle S, Kleven DT, Pollock JS, Pollock DM, White JJ (2014). Antihypertensive and anti‐inflammatory actions of combined azilsartan and chlorthalidone in Dahl salt‐sensitive rats on a high‐fat, high‐salt diet. Clin Exp Pharmacol Physiol 41: 579–588. [DOI] [PubMed] [Google Scholar]
- Justin Rucker A, Crowley SD (2017). The role of macrophages in hypertension and its complications. Pflugers Archiv 469: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS et al (2015). Renal transporter activation during angiotensin‐II hypertension is blunted in interferon‐γ−/− and interleukin‐17A−/− mice. Hypertension 65: 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap S, Warner GM, Hartono SP, Boyilla R, Knudsen BE, Zubair AS et al (2016). Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. Am J Physiol Renal Physiol 310: F372–F384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh N, Hirano S, Kishimoto S, Yasuno H (1997). Calcium channel blockers suppress the contact hypersensitivity reaction (CHR) by inhibiting antigen transport and presentation by epidermal Langerhans cells in mice. Clin Exp Immunol 108: 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelchtermans H, Billiau A, Matthys P (2008). How interferon‐γ keeps autoimmune diseases in check. Trends Immunol 29: 479–486. [DOI] [PubMed] [Google Scholar]
- Khraibi AA, Norman RA Jr, Dzielak DJ (1984). Chronic immunosuppression attenuates hypertension in Okamoto spontaneously hypertensive rats. Am J Physiol 247: H722–H726. [DOI] [PubMed] [Google Scholar]
- Kim SC, Solomon DH, Rogers JR, Gale S, Klearman M, Sarsour K et al (2017). Cardiovascular safety of tocilizumab versus tumor necrosis factor inhibitors in patients with rheumatoid arthritis: a multi‐database cohort study. Arthritis Rheum 69: 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J et al (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA et al (2013). Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM et al (2007). Resistance artery remodeling in deoxycorticosterone acetate‐salt hypertension is dependent on vascular inflammation: evidence from m‐CSF‐deficient mice. Am J Physiol Heart Circ Physiol 292: H1789–H1795. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M et al (2013). Angiotensin II‐induced vascular dysfunction depends on interferon‐γ‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 33: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer‐Schwesinger C et al (2014). Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate + angiotensin II‐induced hypertension. Hypertension 63: 565–571. [DOI] [PubMed] [Google Scholar]
- Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D et al (2016). Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt‐induced hypertension in mice. Br J Pharmacol 173: 752–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Wollner C, Kurth T, Bukowy JD, Cowley AW Jr (2017). Inhibition of mammalian target of rapamycin complex 1 attenuates salt‐induced hypertension and kidney injury in Dahl salt‐sensitive rats. Hypertension 70: 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunnas T, Maatta K, Nikkari ST (2015). NLR family pyrin domain containing 3 (NLRP3) inflammasome gene polymorphism rs7512998 (C>T) predicts aging‐related increase of blood pressure, the TAMRISK study. Immun Ageing 12: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha SS, Raichlin E, Sheinin Y, Kremers WK, Chandrasekaran K, Brunn GJ et al (2008). Sirolimus affects cardiomyocytes to reduce left ventricular mass in heart transplant recipients. Eur Heart J 29: 2742–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al (2009). Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 119: 2904–2912. [DOI] [PubMed] [Google Scholar]
- Li S, Wang X, Li Y, Kost CK Jr, Martin DS (2013). Bortezomib, a proteasome inhibitor, attenuates angiotensin II‐induced hypertension and aortic remodeling in rats. PLoS One 8: e78564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein L, Ron Y, Kivity S, Ben‐Horin S, Israeli E, Fraser GM et al (2015). Infliximab‐related infusion reactions: systematic review. J Crohns Colitis 9: 806–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Xu D, Brint EK, O'Neill LA (2005). Negative regulation of toll‐like receptor‐mediated immune responses. Nat Rev Immunol 5: 446–458. [DOI] [PubMed] [Google Scholar]
- Ling YH, Krishnan SM, Chan CT, Diep H, Ferens D, Chin‐Dusting J et al (2017). Anakinra reduces blood pressure and renal fibrosis in one kidney/DOCA/salt‐induced hypertension. Pharmacol Res 116: 77–86. [DOI] [PubMed] [Google Scholar]
- Liu W, Yamashita T, Kurata T, Kono S, Hishikawa N, Deguchi K et al (2015). Protective effect of telmisartan on neurovascular unit and inflammasome in stroke‐resistant spontaneously hypertensive rats. Neurol Res 37: 491–501. [DOI] [PubMed] [Google Scholar]
- Lu J, Liu F, Liu D, Du H, Hao J, Yang X et al (2016). Amlodipine and atorvastatin improved hypertensive cardiac hypertrophy through regulation of receptor activator of nuclear factor κB ligand/receptor activator of nuclear factor κB/osteoprotegerin system in spontaneous hypertension rats. Exp Biol Med 241: 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma F, Feng J, Zhang C, Li Y, Qi G, Li H et al (2014). The requirement of CD8+ T cells to initiate and augment acute cardiac inflammatory response to high blood pressure. J Immunol 192: 3365–3373. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeed B, Tawinwung S, Eberson LS, Secomb TW, Larmonier N, Larson DF (2014). Interleukin‐2/anti‐interleukin‐2 immune complex expands regulatory T cells and reduces angiotensin II‐induced aortic stiffening. Int J Hypertens 2014: 126365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al (2012). Interferon‐γ signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Mathis KW, Taylor EB, Ryan MJ (2017). Anti‐CD3 antibody therapy attenuates the progression of hypertension in female mice with systemic lupus erythematosus. Pharmacol Res 120: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez‐Villalobos RA et al (2011). Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol 178: 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC (2014). Toll‐like receptors and damage‐associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol 306: H184–H196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Wenceslau CF, Goulopoulou S, Baban B, Matsumoto T, Webb RC (2017). Chloroquine suppresses the development of hypertension in spontaneously hypertensive rats. Am J Hypertens 30: 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC et al (2015). Circulating mitochondrial DNA and toll‐like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res 107: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiners S, Hocher B, Weller A, Laule M, Stangl V, Guenther C et al (2004). Downregulation of matrix metalloproteinases and collagens and suppression of cardiac fibrosis by inhibition of the proteasome. Hypertension 44: 471–477. [DOI] [PubMed] [Google Scholar]
- Mohamed R, Jayakumar C, Chen F, Fulton D, Stepp D, Gansevoort RT et al (2016). Low‐dose IL‐17 therapy prevents and reverses diabetic nephropathy, metabolic syndrome, and associated organ fibrosis. J Am Soc Nephrol 27: 745–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf K, Reijers J et al (2017). Evidence of NI‐0101 pharmacological activity, an anti‐TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin Pharmacol Ther 101: 200–208. [DOI] [PubMed] [Google Scholar]
- Moser M, Feig PU (2009). Fifty years of thiazide diuretic therapy for hypertension. Arch Intern Med 169: 1851–1856. [DOI] [PubMed] [Google Scholar]
- Nakandakare ER, Charf AM, Santos FC, Nunes VS, Ortega K, Lottenberg AM et al (2008). Dietary salt restriction increases plasma lipoprotein and inflammatory marker concentrations in hypertensive patients. Atherosclerosis 200: 410–416. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Umemoto S, Yoshimura K, Matsuda S, Itoh S, Murata T et al (2015). TLR4 is a critical regulator of angiotensin II‐induced vascular remodeling: the roles of extracellular SOD and NADPH oxidase. Hypertension Research: Official Journal of the Japanese Society of Hypertension 38: 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L et al (2017). A salt‐sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end‐organ damage. JCI insight 2 10.1172/jci.insight.92801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Urayama K, Tada Y, Furue T, Imai S, Matsubara K et al (2015). Eculizumab in the treatment of atypical hemolytic uremic syndrome in an infant leads to cessation of peritoneal dialysis and improvement of severe hypertension. Pediatr Nephrol 30: 603–608. [DOI] [PubMed] [Google Scholar]
- Olyaei AJ, Demattos AM, Bennett WM (2005). Cardiovascular complications of immunosuppressive agents in renal transplant recipients. Expert Opin Drug Saf 4: 29–44. [DOI] [PubMed] [Google Scholar]
- Papp KA, Reich K, Paul C, Blauvelt A, Baran W, Bolduc C et al (2016). A prospective phase III, randomized, double‐blind, placebo‐controlled study of brodalumab in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol 175: 273–286. [DOI] [PubMed] [Google Scholar]
- Qi J, Zhao XF, Yu XJ, Yi QY, Shi XL, Tan H et al (2016). Targeting interleukin‐1β to suppress sympathoexcitation in hypothalamic paraventricular nucleus in Dahl salt‐sensitive hypertensive rats. Cardiovasc Toxicol 16: 298–306. [DOI] [PubMed] [Google Scholar]
- Raij L, Dalmasso AP, Staley NA, Fish AJ (1989). Renal injury in DOCA‐salt hypertensive C5‐sufficient and C5‐deficient mice. Kidney Int 36: 582–592. [DOI] [PubMed] [Google Scholar]
- Reis F, Parada B, Teixeira de Lemos E, Garrido P, Dias A, Piloto N et al (2009). Hypertension induced by immunosuppressive drugs: a comparative analysis between sirolimus and cyclosporine. Transplant Proc 41: 868–873. [DOI] [PubMed] [Google Scholar]
- Rodler S, Roth M, Nauck M, Tamm M, Block LH (1995). Ca2+‐channel blockers modulate the expression of interleukin‐6 and interleukin‐8 genes in human vascular smooth muscle cells. J Mol Cell Cardiol 27: 2295–2302. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND (2005). Early and sustained inhibition of nuclear factor‐κB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther 315: 51–57. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera‐Acosta J et al (2002). Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Renal Physiol 282: F191–F201. [DOI] [PubMed] [Google Scholar]
- Saleh MA, Norlander AE, Madhur MS (2016). Inhibition of interleukin 17‐A but not interleukin‐17F signaling lowers blood pressure and reduces end‐organ inflammation in angiotensin II‐induced hypertension. JACC Basic Transl Sci 1: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert FS, Steltzer J, Melilli E, Grannas G, Pagonas N, Bauer F et al (2014). Differential impact of belatacept and cyclosporine A on central aortic blood pressure and arterial stiffness after renal transplantation. Clin Transplant 28: 1004–1009. [DOI] [PubMed] [Google Scholar]
- Sepehri Z, Masoumi M, Ebrahimi N, Kiani Z, Nasiri AA, Kohan F et al (2016). Atorvastatin, losartan and captopril lead to upregulation of TGF‐β, and downregulation of IL‐6 in coronary artery disease and hypertension. PLoS One 11: e0168312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serg M, Kampus P, Kals J, Zagura M, Zilmer M, Zilmer K et al (2012). Nebivolol and metoprolol: long‐term effects on inflammation and oxidative stress in essential hypertension. Scand J Clin Lab Invest 72: 427–432. [DOI] [PubMed] [Google Scholar]
- Sever PS, Messerli FH (2011). Hypertension management 2011: optimal combination therapy. Eur Heart J 32: 2499–2506. [DOI] [PubMed] [Google Scholar]
- Shagdarsuren E, Wellner M, Braesen JH, Park JK, Fiebeler A, Henke N et al (2005). Complement activation in angiotensin II‐induced organ damage. Circ Res 97: 716–724. [DOI] [PubMed] [Google Scholar]
- Shah SR, Abbasi Z, Fatima M, Ochani RK, Shahnawaz W, Asim Khan M et al (2018). Canakinumab and cardiovascular outcomes: results of the CANTOS trial. J Community Hosp Intern Med Perspect 8: 21–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin MH, Johnson JA (2016). Mechanisms and pharmacogenetic signals underlying thiazide diuretics blood pressure response. Curr Opin Pharmacol 27: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN (2014). Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 15: 155–162. [DOI] [PubMed] [Google Scholar]
- Siddiqui M, Calhoun DA (2017). Refractory versus resistant hypertension. Curr Opin Nephrol Hypertens 26: 14–19. [DOI] [PubMed] [Google Scholar]
- Sidiropoulos PI, Boumpas DT (2004). Lessons learned from anti‐CD40L treatment in systemic lupus erythematosus patients. Lupus 13: 391–397. [DOI] [PubMed] [Google Scholar]
- Sinagra E, Perricone G, Romano C, Cottone M (2013). Heart failure and anti tumor necrosis factor‐α in systemic chronic inflammatory diseases. Eur J Intern Med 24: 385–392. [DOI] [PubMed] [Google Scholar]
- Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S et al (2009). Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension 54: 1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollinger D, Eissler R, Lorenz S, Strand S, Chmielewski S, Aoqui C et al (2014). Damage‐associated molecular pattern activated toll‐like receptor 4 signalling modulates blood pressure in l‐NAME‐induced hypertension. Cardiovasc Res 101: 464–472. [DOI] [PubMed] [Google Scholar]
- Sonmez A, Kisa U, Uckaya G, Eyileten T, Comert B, Koc B et al (2001). Effects of losartan treatment on T‐cell activities and plasma leptin concentrations in primary hypertension. J Renin Angiotensin Aldosterone Syst 2: 112–116. [DOI] [PubMed] [Google Scholar]
- Sriramula S, Haque M, Majid DS, Francis J (2008). Involvement of tumor necrosis factor‐α in angiotensin II‐mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ et al (2017). T‐cell mineralocorticoid receptor controls blood pressure by regulating interferon‐γ. Circ Res 120: 1584–1597. [DOI] [PubMed] [Google Scholar]
- Takaoka M, Okamoto H, Ito M, Nishioka M, Kita S, Matsumura Y (1998). Antihypertensive effect of a proteasome inhibitor in DOCA‐salt hypertensive rats. Life Sci 63: PL65–PL70. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S (2005). Toll‐like receptors in innate immunity. Int Immunol 17: 1–14. [DOI] [PubMed] [Google Scholar]
- Taylor EB, Barati MT, Powell DW, Turbeville HR, Ryan MJ (2018a). Plasma cell depletion attenuates hypertension in an experimental model of autoimmune disease. Hypertension 71: 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, Ryan MJ (2017). Immunosuppression with mycophenolate mofetil attenuates hypertension in an experimental model of autoimmune disease. J Am Heart Assoc 6: e005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinsley JH, Chiasson VL, South S, Mahajan A, Mitchell BM (2009). Immunosuppression improves blood pressure and endothelial function in a rat model of pregnancy‐induced hypertension. Am J Hypertens 22: 1107–1114. [DOI] [PubMed] [Google Scholar]
- Toba H, Nakagawa Y, Miki S, Shimizu T, Yoshimura A, Inoue R et al (2005). Calcium channel blockades exhibit anti‐inflammatory and antioxidative effects by augmentation of endothelial nitric oxide synthase and the inhibition of angiotensin converting enzyme in the NG‐nitro‐l‐arginine methyl ester‐induced hypertensive rat aorta: vasoprotective effects beyond the blood pressure‐lowering effects of amlodipine and manidipine. Hypertens Res 28: 689–700. [DOI] [PubMed] [Google Scholar]
- Toledo JO, Moraes CF, Souza VC, Tonet‐Furioso AC, Afonso LC, Cordova C et al (2015). Tailored antihypertensive drug therapy prescribed to older women attenuates circulating levels of interleukin‐6 and tumor necrosis factor‐α. Clin Interv Aging 10: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran LT, MacLeod KM, McNeill JH (2009). Chronic etanercept treatment prevents the development of hypertension in fructose‐fed rats. Mol Cell Biochem 330: 219–228. [DOI] [PubMed] [Google Scholar]
- Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE et al (2014). Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 64: 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gelder T, Warle M, Ter Meulen RG (2004). Anti‐interleukin‐2 receptor antibodies in transplantation: what is the basis for choice? Drugs 64: 1737–1741. [DOI] [PubMed] [Google Scholar]
- Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C et al (2017). Interleukin‐1 blockade in recently decompensated systolic heart failure: results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 10 10.1161/CIRCHEARTFAILURE.117.004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas‐Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC et al (2010). Tumor necrosis factor‐α antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ et al (2010). Inhibition and genetic ablation of the B7/CD28 T‐cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hou L, Kwak D, Fassett J, Xu X, Chen A et al (2016). Increasing regulatory T cells with interleukin‐2 and interleukin‐2 antibody complexes attenuates lung inflammation and heart failure progression. Hypertension 68: 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Yu XJ, Li XG, Pang DZ, Su Q, Saahene RO et al (2018a). Blockade of TLR4 within the paraventricular nucleus attenuates blood pressure by regulating ROS and inflammatory cytokines in prehypertensive rats. Am J Hypertens. 10.1093/ajh/hpy074. [DOI] [PubMed] [Google Scholar]
- Wang Q, So A, Nussberger J, Tschopp J, Burnier M (2012). Impact of NLRP3 inflammasome on the development of hypertension and renal and cardiac hypertrophy in 2K1C and DOCA/salt mice. Kidney Res Clin Pract 31: A83. [Google Scholar]
- Wang S, Wu M, Chiriboga L, Zeck B, Belmont HM (2018b). Membrane attack complex (mac) deposition in lupus nephritis is associated with hypertension and poor clinical response to treatment. Semin Arthritis Rheum. 10.1016/j.semarthrit.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li Y, Wu Y, Jia L, Wang J, Xie B et al (2014). 5TNF‐α and IL‐1β neutralization ameliorates angiotensin II‐induced cardiac damage in male mice. Endocrinology 155: 2677–2687. [DOI] [PubMed] [Google Scholar]
- Ware JS, Wain LV, Channavajjhala SK, Jackson VE, Edwards E, Lu R et al (2017). Phenotypic and pharmacogenetic evaluation of patients with thiazide‐induced hyponatremia. J Clin Invest 127: 3367–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD et al (2009). In vivo expansion of T reg cells with IL‐2‐mAb complexes: induction of resistance to EAE and long‐term acceptance of islet allografts without immunosuppression. J Exp Med 206: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C et al (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71: 1269–1324. [DOI] [PubMed] [Google Scholar]
- Wiseman AC (2016). Immunosuppressive medications. Clin J Am Soc Nephrol 11: 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Iwai M, Li Z, Li JM, Mogi M, Horiuchi M (2006). Nifedipine inhibited angiotensin II‐induced monocyte chemoattractant protein 1 expression: involvement of inhibitor of nuclear factor κB kinase and nuclear factor κB‐inducing kinase. J Hypertens 24: 123–130. [DOI] [PubMed] [Google Scholar]
- Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F et al (2015). Renal denervation prevents immune cell activation and renal inflammation in angiotensin II‐induced hypertension. Circ Res 117: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanari H, Nakamura K, Miura D, Yamanari S, Ohe T (2009). Spironolactone and chlorthalidone in uncontrolled elderly hypertensive patients treated with calcium antagonists and angiotensin II receptor‐blocker: effects on endothelial function, inflammation, and oxidative stress. Clin Exp Hypertens 31: 585–594. [DOI] [PubMed] [Google Scholar]
- Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY et al (2013). Immunosenescent CD8+ T cells and C‐X‐C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62: 126–133. [DOI] [PubMed] [Google Scholar]
- Zaldivia MT, Rivera J, Hering D, Marusic P, Sata Y, Lim B et al (2017). Renal denervation reduces monocyte activation and monocyte‐platelet aggregate formation: an anti‐inflammatory effect relevant for cardiovascular risk. Hypertension 69: 323–331. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA et al (2016). Interleukin‐1 receptor activation potentiates salt reabsorption in angiotensin II‐induced hypertension via the NKCC2 co‐transporter in the nephron. Cell Metab 23: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Hong D, Zhang Y, Sang Y, Yang Z, Zhang X (2015). Association between anti‐TNF therapy for rheumatoid arthritis and hypertension: a meta‐analysis of randomized controlled trials. Medicine 94: e731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kang N, Cui L, Ba D, He W (2012). Anti‐γδ TCR antibody‐expanded γδ T cells: a better choice for the adoptive immunotherapy of lymphoid malignancies. Cell Mol Immunol 9: 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou MS, Schulman IH, Jaimes EA, Raij L (2008). Thiazide diuretics, endothelial function, and vascular oxidative stress. J Hypertens 26: 494–500. [DOI] [PubMed] [Google Scholar]