

Abstract
Over the last few years, evidence has accumulated to suggest that hypertension is, at least in part, an immune‐mediated inflammatory disorder. Many links between immunity and hypertension have been established and provide a complex framework of mechanistic interactions contributing to the rise in BP. These include immune‐mediated inflammatory processes affecting regulatory brain nuclei and interactions with other mediators of cardiovascular regulation such as the sympathetic nervous system. Sympathoexcitation differentially regulates T‐cells based upon activation status of the immune cell as well as the resident organ. Exogenous and endogenous triggers activate signalling pathways in innate and adaptive immune cells resulting in pro‐inflammatory cytokine production and activation of T‐lymphocytes in the cardiovascular and renal regions, now considered major factors in the development of essential hypertension. The inflammatory cascade is sustained and exacerbated by the immune flow via the brain–bone marrow–spleen–gastrointestinal axis and thereby further aggravating immune‐mediated pathways resulting in a vicious cycle of established hypertension and target organ damage. This review summarizes the evidence and recent advances in linking immune‐mediated inflammation, sympathetic activation and their bidirectional interactions with the development of hypertension.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- Ang II
angiotensin II
- APCs
antigen‐presenting cells
- CKD
chronic kidney disease
- CVO
circumventricular organ
- SHR
spontaneously hypertensive rat
- TLRs
toll‐like receptors
- TMAO
trimethylamine N‐oxide
Introduction
Hypertension is a major cause of mortality and morbidity worldwide affecting more than one billion adults. It is the most common modifiable risk factor for cardiovascular events and death. With an underlying cause established for only ~10% of cases (i.e. secondary hypertension), the majority of the hypertensive population suffers from essential hypertension, which is frequently characterized by sustained sympathetic activation. More recently, immune mechanisms have been proposed to contribute to the development of hypertension. Immune reactivity in experimental and human hypertension was initially believed to be a consequence, rather than a cause, of BP elevation. It has now become clear that immune‐mediated oxidative stress can initiate renal interstitial inflammation, loss of peritubular capillaries and medullary hypoxia, which in turn causes impaired pressure natriuresis and increased BP (Guyton et al., 1972; Franco et al., 2013). These findings laid the foundations for further investigations into the role of immunity‐induced pathways as a causal mechanism in the development of hypertension. Indeed, Franco et al. (2013) demonstrated a close relationship between renal immune cell infiltration and impaired pressure natriuresis, which was preventable by mycophenolate mofetil‐dependent reduction of immune infiltration in a salt‐sensitive hypertension rat model.
There has since been a shift from the perception of hypertension and hypertension‐mediated end‐organ damage merely being the result of elevated pressure on the vessel wall to a much more complex scenario involving immune mechanisms. Experimentally, immunological approaches for the treatment of BP have yielded promising results and provided evidence for a concerted interplay of innate and adaptive immunity. The objective of this review is to summarize the evidence and recent advances in linking immune‐mediated inflammation, sympathetic activation and their bidirectional interactions, with the development of hypertension.
Immunity and hypertension: compelling evidence for immune‐mediated hypertension
Experimental suppression of immune mechanisms is an elegant approach to explore their role in BP regulation. Indeed, immunosuppression or thymectomy blunted the development and maintenance of hypertension (White and Grollman, 1964; Okuda and Grollman, 1967; Harrison, 2014). BP and urinary excretion of inflammatory cytokines were decreased in hypertensive patients treated with mycophenolate mofetil for rheumatoid arthritis or psoriasis over a 3 month period, despite unchanged antihypertensive therapy or salt intake, but reversed when treatment was discontinued (Herrera et al., 2006). Human immunodeficiency virus‐positive patients with low CD4+ T‐cell counts were less prone to hypertension than those on antiretroviral therapy with normal CD4+ T‐cell counts, who developed hypertension to a similar degree as the normal population (Seaberg et al., 2005). Thymic transplantation from normotensive Wistar Kyoto rats reduced hypertension in spontaneously hypertensive rats (SHRs) (Ba et al., 1982). The requirement of a functional thymus for the maintenance of hypertension and the inability of thymectomized or athymic nude mice with renal infarction to maintain hypertension (Svendsen, 1976; Svendsen, 1977; Bataillard et al., 1986) support a pathogenic role of the immune system in hypertension. Moreover, mice with severe combined immunodeficiency were protected from developing hypertension (Crowley et al., 2010). The passive transfer of lymphocytes from hypertensive rat models to normotensive rats induced hypertension (Okuda and Grollman, 1967). Furthermore, hypertension was suppressed by anti‐thymocyte serum or immunosuppressive cyclophosphamide in SHR (Bendich et al., 1981; Dzielak, 1991), suggesting that inflammation is a cause rather than merely a consequence of hypertension.
In another important line of research, in mice with deletion of the recombinase activating gene 1 (Rag1−/−), which lack T‐lymphocytes and B‐lymphocytes, hypertension failed to develop when they were chronically exposed to angiotensin II (Ang II) (Guzik et al., 2007). Rag1 deletion also attenuated hypertension in Dahl salt‐sensitive rats (Mattson et al., 2013) further supporting a role of immune cells in hypertension development. However, in a more recent publication, Ji et al. (2017) reported that these Rag1−/− mice on a C57BL/6J background lost their resistance to Ang II‐induced hypertension, which was independent of T‐cells. Enhanced sensitivity and up‐regulation of the renal angiotensin AT1 receptor activity was identified as the most likely explanation, and this was attributed to the universal drive for genetic variation in inbred animals. Clearly, this phenotypic change in the Rag1 knockout C57BL/6J mice model has implications for investigators using this strain to study mechanisms of T‐cell modulation of Ang II‐dependent BP control. However, it does not necessarily compromise the findings from studies investigating the role of T‐cell modulation in Ang II‐induced hypertension, prior to this occurrence. Furthermore, there is strong evidence that T‐cells modulate other important physiological processes mediated by Ang II such as endothelial dysfunction and microvascular injury (Mian et al., 2016).
T‐cells as critical immune players in the genesis of hypertension
Sympathoexcitation differentially regulates T‐lymphocytes, and it has been shown that sympathetic stimulation induces noradrenaline‐mediated T‐cell activation and vascular inflammation (Marvar et al., 2010). The activation of T‐lymphocytes involves two steps: (i) T‐cell receptor interaction with antigen presented by the major histocompatibility complex and (ii) co‐stimulation via CD28, bound by the B7 ligands, CD80 and CD86 of the antigen‐presenting cells (APCs) – the absence of which induces T‐cell apoptosis (Frauwirth and Thompson, 2002). Depletion of neutrophils and monocytes with diphtheria toxin in mice eliminates the hypertensive response to Ang II, and hypertension is restored with repletion of monocytes but not granulocytes, highlighting the important role of monocyte antigen presentation to T‐cells (Wenzel et al., 2011; Harrison, 2014). Inhibition of co‐stimulation by CTLA4‐Ig, a pharmacological agent that binds B7 ligands on APCs, attenuated the associated BP rise as well as T‐cell activation, vascular infiltration and T‐cell‐mediated TNF‐α and IFN‐γ production in both Ang II and deoxycorticosterone acetate (DOCA) hypertension models (Vinh et al., 2010). Hypertension as a T‐cell‐driven inflammatory process became evident when passive transfer of T‐cells and not B‐cells restored hypertension following Ang II infusion accompanied by elevated levels of memory and circulating T‐cell markers including CD69+, CCR5 + and CD44high (Guzik et al., 2007). Furthermore, the role of T‐cells in the pathogenesis of hypertension has recently been confirmed in humanized mice, in which the murine immune system is replaced by the human immune system (Itani et al., 2016).
In all models of salt‐sensitive hypertension, the tubulointerstitium of rat kidney is intensively infiltrated with lymphocytes and macrophages, which correlates with the degree of hypertension (Rodríguez‐Iturbe et al., 2004; Heijnen et al., 2014). Lymphocyte infiltration was also seen in perivascular aortic tissue in Ang II‐induced hypertension, DOCA‐salt hypertension and aldosterone‐induced hypertension (Guzik et al., 2007; Kasal et al., 2012). Significant immune infiltration precedes hypertension and intensifies with increasing severity of hypertension, suggestive of a cause and effect relationship (Rodríguez‐Iturbe et al., 2004; Franco et al., 2013). Blocking of immune infiltration using mycophenolate mofetil prevents hypertension in various animal models, such as SHR (Rodríguez‐Iturbe et al., 2001), Dahl salt‐sensitive rats (De Miguel et al., 2011) and salt‐driven hypertension models induced by protein overload glomerular disease (Alvarez et al., 2002), and in the Page kidney model when the renal parenchyma is subjected to external compression (Vanegas et al., 2005).
A distinct infiltrating T‐cell phenotype, characterized by amplified production of the chemokine CCL2, promotes leukocyte recruitment and enhances ROS and generation of cytokines, thereby intensifying the local and advanced inflammation causing organ dysfunction and overt hypertension (Wei et al., 2014). Elevated CCL2 levels were found in organs regulating BP (Bush et al., 2000), and T‐cells were identified to be an important source of CCL2 contributing to inflammation associated with hypertension. Leukocytes express the CCL2 receptor, CCR2, which binds CCL2 and several other chemokines such as CCL8, CCL13 and CCL27. CCR2 inhibition reduces vascular macrophage accumulation and reverses the pressor responses in DOCA‐salt‐induced hypertension (Chan et al., 2012). CCR2‐deficient mice exhibit decreased macrophage and monocyte infiltration into the arterial wall during Ang II‐induced hypertension (Bush et al., 2000), suggesting a crucial role of CCR2–CCL2 signalling in immune‐mediated inflammation of hypertension and associated organ dysfunction, which may be initially driven by infiltrating T‐cells leading to a pro‐inflammatory cascade.
B‐cells as additional players in the genesis of hypertension
Whereas much evidence supports an important role for T‐cells, the role of B‐cells has been less well documented. Only quite recently did Chan et al. demonstrate that B‐cells and the associated IgG production contributed to Ang II‐induced hypertension. Interestingly, a clinically available anti‐CD20 antibody approach reduced hypertension, providing an interesting potential therapeutic perspective for hypertension (Chan et al., 2015).
The concerted interplay of adaptive and innate immunity in the development of hypertension
The T‐cell‐dominant adaptive immune response in hypertension, initiated and coordinated by the innate immune components such as phagocytes, dendritic cells, monocytes/macrophages, NK cells and the pattern recognition toll‐like receptors (TLRs), is critical to the induction and sustenance of hypertension. These events are mediated through the downstream immune components such as ROS and reactive nitrogen species (Ryan, 2013). The peripheral blood monocytes of hypertensive patients have elevated mRNA levels of TLR2 and TLR4, which are reduced by intensive antihypertensive treatment, indicative of a relevant role of the innate immune response (Hartupee et al., 2007).
Oxidative stress has been observed in all experimental models of hypertension. ROS act as second messengers in Ang II signalling and i.c.v. injections of adenovirus encoding for SOD‐ablated Ang II‐dependent changes in heart rate and BP (Zimmerman et al., 2002). Furthermore, SOD deletion in the circumventricular organ (CVO) increases baseline BP and augments the Ang II‐dependent hypertensive response and promotes T‐cell and leukocyte infiltration as well as vascular inflammation, partly by modulating sympathetic outflow. (Lob et al., 2010). With respect to ROS, a feedforward loop between NADPH oxidase (NOX) and mitochondria results in the induction and maintenance of diffuse inflammation in organs regulating BP (Datla and Griendling, 2010; Dikalov and Nazarewicz, 2013). This inflammatory milieu is characterized by elevated levels of Ang II, aldosterone, cytokines and altered mechanical forces, stimulated enzyme sources such as the NOXs, uncoupled NOS and the mitochondrial ROS, which contribute to the development of hypertension (Harrison and Gongora, 2009).
Several cytokines are implicated in the pathogenesis of hypertension. Inflammatory markers (Chae et al., 2001; Engstrom et al., 2002) and T‐cell‐specific cytokines such as IL‐4, IL‐6, IL‐7, IFN‐γ‐inducible protein‐10, TNF‐α and IL‐17 (Madhur et al., 2010; Stumpf et al., 2011) are elevated in patients with essential hypertension. IL‐17 infusion caused endothelial dysfunction and hypertension in mice (Nguyen et al., 2013) and also induced oxidative stress in the placenta of pregnant rats, thereby promoting hypertension (Dhillion et al., 2012). The TNF‐α antagonist etanercept effectively prevented hypertension in animal models (Tran et al., 2009; Venegas‐Pont et al., 2010), and IL‐6‐deficient mice appear to be protected in certain hypertension models (Lee et al., 2006; Schrader et al., 2007; Brands et al., 2010). Short‐term administration of pressor doses of Ang II resulted in renal infiltration and activation of immune cells with subsequent development of salt‐sensitive hypertension in male Sprague–Dawley rats (Rodríguez‐Iturbe et al., 2001). The Ang II‐induced vascular dysfunction and arterial hypertension were driven by IFN‐γ‐mediated immune recruitment and activation of NK cells and monocytes (Kossmann et al., 2013). Furthermore, IFN‐γ is up‐regulated in kidneys of hypertensive mice (Crowley et al., 2010), and deletion of IFN‐γ receptor (IFNGR) prevented Ang II‐dependent end‐organ damage (Marko et al., 2012).
The ongoing metabolic and oxidative cellular damage in hypertension can result in the oxidative modification of endogenous molecules, exposure of intramolecular sites by protein cleavage that are otherwise not exposed to the immune system and release reactive intracellular molecules further propagating the damage (Harrison et al., 2011). Highly reactive oxidatively modified isoketals accumulate in the dendritic cells and activate T‐cell‐mediated immuno‐inflammation and promote hypertension in mice (Kirabo et al., 2014). Certain molecules such as oxidized LDL, heat shock proteins (HSP) and platelet glycoproteins have been suggested to elicit immune responses in atherosclerosis (Gotsman et al., 2008). Abnormal HSP responses have been noted in patients with essential hypertension (Kunes et al., 1992; Pockley et al., 2002; Pockley et al., 2003; Li et al., 2009), and increased expression of HSP70 has been observed in circulating lymphocytes of hypertensive subjects (Kunes et al., 1992) that caused clonal expansion of CD4 + T‐cells and induced delayed hypersensitivity in models of salt‐sensitive hypertension (Pons et al., 2013). HSP70 gene polymorphisms, namely, HSPA1A, HSPA1B and HSPA1L, predispose to hypertension in individuals of Uygur ancestry (Li et al., 2009). Furthermore, auto‐antibodies against AT1 receptors and α1‐adrenoceptors have been detected in patients with hypertension (Liao et al., 2002) indicative of a role for protein modification. Agonistic antibodies directed to the second extracellular loop of AT1 receptors, initially described in pre‐eclampsia, have also been detected in patients with essential hypertension (Fu et al., 2000; Wei et al., 2011). Circulating anti‐endothelial IgG and IgM has also been observed in hypertension (Frostegard et al., 1998; Papadopoulos et al., 2006). Taken together, the ‘first hit’ phase is likely to be initiated by cytokine‐dependent oxidative damage and protein modification resulting in the generation of neoantigens, which contribute to the initiation of the inflammatory cascade of hypertension (Figure 1).
Figure 1.
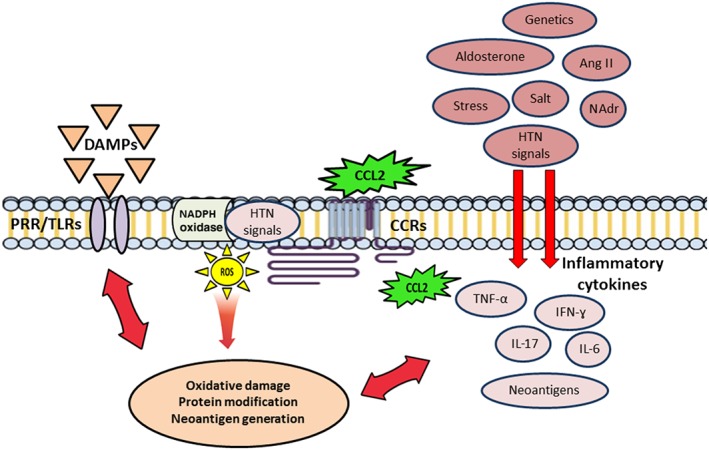
The ‘first hit pro‐inflammatory phase’ of pre‐ hypertension. Hypertensive (HTN) signals, damage‐associated molecular patterns (DAMPs) and cytokine‐dependent ROS signalling causes protein modification generating new neoantigens such as oxidized LDL, modified receptor proteins, HSPs and platelet glycoproteins that elicit an immunological response in the cardiovascular target tissues via the activation of T‐cells. This phase initiates the inflammatory cascade in organs associated with BP regulation. NAdr, noradrenaline.
Interaction of the sympathetic nervous system and immunity in the development of hypertension
Physiological integration and processing of the interaction between the autonomic nervous system and the immune system determines the balance of the sympathetic responses at the peripheral sites including the lymphoid targets. Neural and non‐neural communication pathways keep the central autonomic nervous system informed of the peripheral immune status. In addition, the micro‐environment created by diverse signalling mechanisms mediated through neurotransmitters and neuromodulators including adrenoceptors, immune cells, cytokines and pathogens maintains neural communication. Importantly, immune cells express adrenoceptors and nicotinic receptors (nAChRs), which bind the sympathetic neurotransmitters to initiate immunomodulatory responses. Of interest in this context is the observation that bilateral ablation of renal sympathetic nerves with phenol prevented immune activation and renal inflammation and attenuated Ang II‐induced hypertension in mice (Xiao et al., 2015). Furthermore, catheter‐based renal denervation applied to lower BP by reducing sympathetic nervous system activation has recently been demonstrated to reduce monocyte activation and inflammatory markers in patients with essential hypertension (Zaldivia et al., 2017).
Essential hypertension is a clinically heterogeneous entity, and current evidence supports the notion that immunomodulation is specifically relevant in regard to three major factors contributing to the BP rise including (i) increased sympathetic tone, (ii) enhanced sodium and fluid overload and (iii) activation of the renin–angiotensin–aldosterone (RAS) cascade. In cases involving high sympathetic activation, a low‐grade systemic inflammatory environment is established by the amplified release of inflammatory cells into the blood circulation (Heidt et al., 2014). The cells of the innate immune system, monocytes in particular, are implicated in the pathogenesis of hypertension. Acute and long‐term BP elevation is linked to pre‐activated monocytes in animals and patients with hypertension (Dörffel et al., 1994; Wenzel et al., 2011). In fact, genetic depletion of LysM+ monocytes resulted in blunted Ang II‐induced hypertension in mice, whereas adoptive transfer of pro‐inflammatory monocytes restored the BP elevation, supporting the notion of an important role of monocytes in hypertension development (Wenzel et al., 2011). Stimulation of α1‐adrenoceptors has a pro‐inflammatory effect on immune system responses and positively regulates the expression of inflammatory cytokines in the innate immune cells during stress or injury‐dependent release of catecholamines (Grisanti et al., 2011). In addition, the release of pro‐inflammatory cytokines is dependent on activation of adrenoceptors in a number of chronic inflammatory disease states (Maestroni, 2000; Heijnen et al., 2002; Perez et al., 2009). For example, in chronic polyarticular juvenile rheumatoid arthritis, IL‐6 release from mononuclear cells in peripheral circulation is dependent on expression of α1‐adrenoceptors (Heijnen et al., 1996), while activation of these receptors was important for enhanced inflammatory responses in an established model of multiple sclerosis (Brosnan et al., 1985).
Sympathetic activation of the bone marrow (BM) and the spleen and the associated release of haematopoietic cells into the systemic circulation has been identified to play an important role in the pathogenesis of hypertension (Heidt et al., 2014). Sympathetic nerves densely innervate the BM and upon activation can release progenitor cells seeding to the spleen with a subsequent increase in monocyte production (Felten et al., 1984; Felten et al., 1985; Dutta et al., 2012). Enhanced T‐cell infiltration and increased sympathetic ischarge have also been identified in the carotid body of hypertensive patients (McBryde et al., 2013). Exaggerated RAS activation (Rudemiller and Crowley, 2016), with T‐cells and monocytes/macrophages accumulation in the kidney and vasculature, mediates tissue injury and deleterious remodelling in hypertension (Madhur et al., 2010). Activation of the sympathetic nervous system differentially regulates the immune system based upon activation status of the immune cell as well as the resident organ (Case and Zimmerman, 2016). Release of noradrenaline from the sympathetic nerve terminal potentiates the effects of the activated T‐lymphocytes in these cardiovascular‐related organs. In noradrenaline‐infused mice, increased T‐lymphocyte numbers and activation have been observed in the aorta (Marvar et al., 2010), whereas T‐lymphocytes from the spleen in this model demonstrated decreased proliferation that was accompanied by a reduction in IFN‐γ and TNF‐α (Case and Zimmerman, 2015). While stimulation of Th1 cells generated from naïve CD4+ lymphocytes by noradrenaline enhanced IFN‐γ production by more than twofold (Swanson et al., 2001), noradrenaline also caused mitochondrial superoxide‐dependent modulation of the canonical activation of splenic T‐lymphocytes in a model of sympathetically‐driven hypertension (Case and Zimmerman, 2015; Case et al., 2016). Short‐term noradrenaline administration augmented the number of CD8+ circulating T‐lymphocytes, but long‐term administration resulted in decreased T‐lymphocyte numbers (Maisel and Michel, 1989). Taken together, the available evidence suggests that noradrenaline can elicit differential immunomodulatory effects on different subclasses and stages of differentiated T‐lymphocytes. Naïve unchallenged T‐lymphocytes are suppressed by noradrenaline stimulation, whereas the functionality of pre‐existing activated T‐lymphocytes is exacerbated under different conditions such as specific systemic infections, stress or targeted T‐lymphocyte differentiation (Alaniz et al., 1999).
On the contrary, alterations in sympathetic nervous system activity may be involved in stroke‐induced immunosuppression. In experimental stroke, both activation of the hypothalamic–pituitary axis and the sympathetic nervous system and β‐adrenoceptor blockade have been shown to substantially affect stroke‐induced immunomodulation (Prass et al., 2003). It has been suggested that brain injury‐induced generation of inflammatory cytokines such as TNF‐α, IL‐1β and IL‐6 interferes with the autonomic‐immune circuits to result in immune dysfunction (Dirnagl et al., 2007). Direct measurement of sympathetic activation in humans following a stroke using gold standard techniques such as microneurography or noradrenaline spillover (Lambert et al., 2008; Schlaich et al., 2010) has not been applied but are likely to shed some light into the relevance of these findings in a clinical setting. Overall, these findings are indicative of relevant crosstalk between the sympathetic and the immune system to sustain the immune‐mediated inflammation in hypertension (Figure 2).
Figure 2.
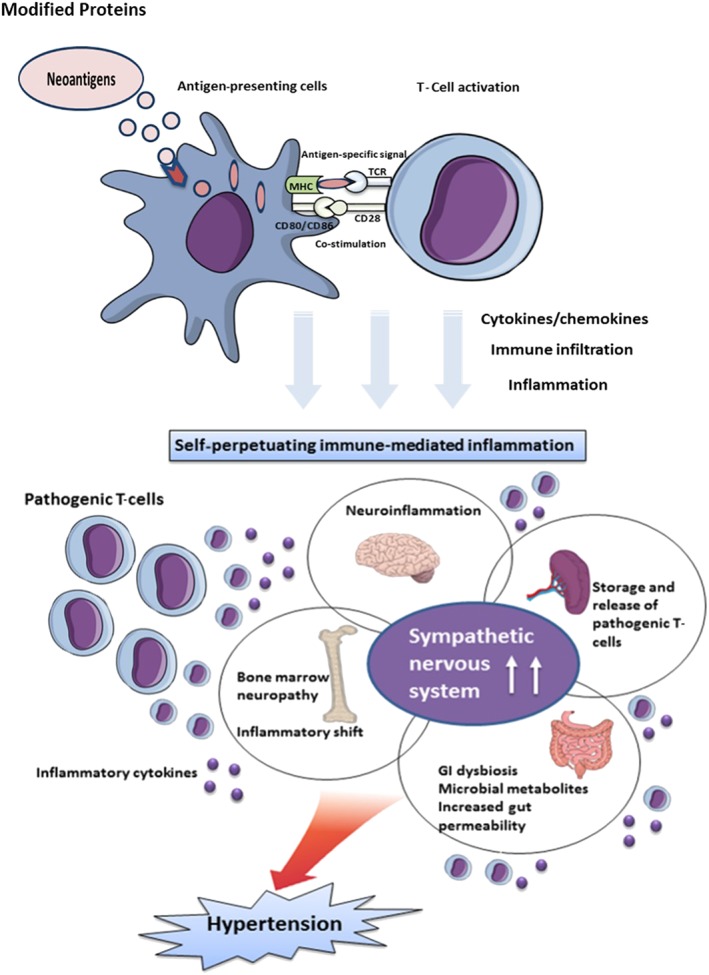
The sympathetic–immune crosstalk results in established hypertension. The brain–BM–spleen–gastrointestinal (GI) axis perpetuates and further exaggerates the immune cascade by causing immune infiltration and immune‐mediated inflammatory damage in the organs regulating BP, ultimately resulting in established hypertension. MHC, major histocompatibility complex; TCR, T‐cell receptor.
The immunomodulatory brain–bone marrow–spleen–gut axis: neuroinflammation is a characteristic feature of hypertension
The sympathetic nervous system is an integrative interface between the brain and the immune system and modulates both haemodynamic and immune responses (Elenkov et al., 2000). Immune infiltration and enhanced inflammation of neural regulatory centres have been suggested to be key players in the interaction between the immune and sympathetic nervous system as demonstrated in the nucleus tractus solitarii of the SHR (Gouraud et al., 2011). Similarly, in Sprague–Dawley rats, chronic Ang II infusion has been demonstrated to activate microglia and increase pro‐inflammatory cytokines in the paraventricular nucleus causing neurogenic hypertension. This effect was attenuated by minocycline, an antibiotic that passes the blood–brain barrier and has been shown to act as an inhibitor of microglia (Shi et al., 2010). Furthermore, i.c.v. infusion of Ang II increased sympathetic discharge of the splenic nerve and increased the expression of IL‐1, IL‐2, IL‐6, IL‐16 and TGF‐β1 in splenocytes, an effect that can be abolished by splenic sympathectomy, thereby linking the CNS to peripheral immune reactivation (Ganta et al., 2005). Anteroventral third ventricle lesions impair the activation of lymphocytes and their homing to the arterial walls and thereby ameliorate Ang II‐induced hypertension (Marvar et al., 2010). Indeed, lesions in this region appear to prevent any form of experimental hypertension (Brody et al., 1978). Chronic i.c.v. infusion of neuropeptides such as substance P and Ang II enhances T‐cell populations in experimental rodents (Fannon and Phillips, 1991).
The CVO lacks a blood–brain barrier making it susceptible to the influence of circulating hormones and cytokines. The CVO has abundant AT1 receptors and NOX in the subfornical organ that produces ROS to enhance sympathetic outflow. SOD knockdown induces oxidative stress in the subfornical organ of the hypothalamus and increases vascular infiltration of activated lymphocytes, which is associated with worsening hypertension (Lob et al., 2010). Administration of a dominant negative Ras‐related C3 botulinum toxin substrate 1 (Rac1) has been shown to prevent NOX activation, highlighting the requirement of Rac1‐dependent Ang II effects in the brain (Zimmerman et al., 2004). Similarly, Ang II receptor knockout in the subfornical organ of the brain ameliorates the BP in DOCA‐salt hypertension (Hilzendeger et al., 2013). Suppressing the inflammation of the subfornical organ with minocycline or induction of IL‐10 overexpression ameliorates hypertension (Lob et al., 2013). Intracerebroventricular infusion of etanercept inhibits brain TNF‐α and protects against Ang II‐induced hypertension in rats by restoring the anti‐inflammatory balance and alleviating oxidative stress in the paraventricular nucleus (Venegas‐Pont et al., 2010; Sriramula et al., 2013). The elevation of circulating inflammatory markers characteristic of hypertension (Chae et al., 2001; Engstrom et al., 2002) may well be relevant for inflammatory processes in the CNS, thereby sustaining and further exaggerating the overall systemic inflammation (Wu et al., 2012).
The reciprocal impact: bone marrow‐derived inflammatory cells in hypertension
The bone‐marrow (BM) is extensively innervated by the sympathetic nervous system (Katayama et al., 2006), which regulates immunomodulation (Scheiermann et al., 2013), stem cell niche homeostasis and haematopoiesis (Hanoun et al., 2015). BM from hypertensive subjects is characterized by altered adrenergic signalling with exaggerated sympathetic tone, elevated levels of noradrenaline, loss of circadian rhythmicity and impaired mobilization of haematopoietic stem and progenitor cell populations (Afan et al., 1997; Katayama et al., 2006). The sympathetic overdrive of hypertension shifts the BM equilibrium towards the generation of myeloid progenitor cells, which induce peripheral inflammation and migrate to the brain to differentiate into brain macrophage/microglia to induce and sustain neuroinflammation (Santisteban et al., 2016). Cytokines, chemokines and ROS generated as a consequence of inflammation further accentuate the sympathetic overdrive centrally as well as in the peripheral organs, thereby further perpetuating hypertension. Impaired autonomic input to the BM has been associated with altered inflammatory responses in SHR (Zubcevic et al., 2014). The extravasation of inflammatory progenitors from the BM into the cardioregulatory centres of the brain results in microglial/macrophage activation to cause and sustain hypertension (Santisteban et al., 2015). The inflammatory progenitors from the BM migrate to the brain parenchyma in a CCL2 and CCR2‐dependent manner to exacerbate neuroinflammation (Ataka et al., 2013; Lampron et al., 2013) and infiltrate critical peripheral organs such as the heart and vasculature, carotid body, spleen, kidneys and gut to induce systemic inflammation (Rodríguez‐Iturbe et al., 2004; Gonzalez et al., 2015). It is pertinent to note that the CCL2–CCR2 chemokine system is a crucial player in the migration of T‐lymphocytes, monocytes and NK cells in rheumatoid arthritis and Type 2 diabetes (Madrigal and Caso, 2014). Moreover, Ang II directly stimulates CCL2 production in vascular smooth muscle cells (Chen et al., 1998), monocytes (Tsou et al., 2007) and hypothalamic neurons (Santisteban et al., 2013) via an AT1 receptor‐mediated pathway (Koh et al., 2004; Dai et al., 2007; Marketou et al., 2011). However, the resting microglia of the adult brain express AT1 receptors only when primed by pro‐hypertensive signals (Miyoshi et al., 2008).
The spleen: a neuroimmune hub differentially involved in cardiovascular and metabolic diseases
The spleen contains half of the body's monocyte population and mapping of the genetic, molecular and neurophysiological basis of hypertension‐induced inflammatory reflexes converge in the spleen (Rosas‐Ballina et al., 2008; Rosas‐Ballina et al., 2011). The spleen acts as a reservoir of pathogenic T‐cells, and activation of adrenergic splenic neurons regulates a T‐cell subset in the white pulp that eventually migrates to organs regulating BP. Moreover, the pro‐hypertension signals such as Ang II regulate haematopoietic stem cell proliferation (HSPCs) in the BM and enhance CCR2+ HSPCs, myeloid progenitors and inflammatory monocytes in the spleen, thereby contributing to hypertension (Kim et al., 2016). Hypertensive challenges activate splenic sympathetic discharge to prime the immune response, specifically the vagus–splenic drive mediated by nAChRs (particularly the α7nAChR), which links the brain and spleen (Rosas‐Ballina et al., 2008). The deficiency of α7nAChRs in apolipoprotein E knockout mice or splenic parasympathectomy increases the inflammatory profile characterized by elevated leukocyte counts and increased pro‐inflammatory cytokine expression within the spleen (Kooijman et al., 2015). The neural circuits that modulate the lymphocytes reveal that the action potential generated in the vagus nerve is transmitted to the celiac ganglion (the origin of the splenic nerve) to inhibit macrophage production of TNF‐α and other cytokines (Andersson and Tracey, 2012). Indeed, TNF‐α production is not inhibited by vagus nerve stimulation in splenectomized animals during endotoxemia (Huston et al., 2006), and i.v. administration of LPS increased efferent activity in the splenic and splanchnic sympathetic nerves in rats to induce a strong systemic inflammatory response (Martelli et al., 2014; Martelli et al., 2016), indicating an essential role for the spleen in mediating inflammatory signalling.
Ang II infusion increases the expression of tyrosine hydroxylase , the rate‐limiting enzyme in noradrenaline biosynthesis in adrenergic neurons and noradrenaline levels in the spleen. Splenectomy or splenic nerve ablation prevents the recruitment, activation and egression of pathogenic T‐cells from the spleen and consequent target organ infiltration (Carnevale et al., 2016). Splenic placental growth factor (PlGF)–sirtuin 1 (SIRT1) signalling mediated hypertension development in mice. Furthermore, splenic reimplantation from wild‐type donors restored the hypertensive response to Ang II, whereas mice re‐implanted with spleen from PlGF‐deficient spleen donors continued to be protected against the hypertensive effects of Ang II (Carnevale et al., 2014). Chronic administration of the selective SIRT1 inhibitor, Ex‐527 (selisistat), and genetic silencing of SIRT1 in PlGF‐deficient splenocytes restored the hypertensive response to Ang II (Carnevale et al., 2014). Cumulatively, these data suggest that the spleen is a bidirectional communication hub between the nervous system and immune system, and neural signals culminate in increased PlGF production and increased noradrenaline release resulting in the release of T‐cells and its downstream effectors, thereby contributing to hypertension and tissue damage. Therapeutic strategies targeting the sympathetic innervation of the spleen may be of interest.
The gastrointestinal connection
Recent findings suggest that gut‐related mechanisms may also be of relevance to the development of hypertension. Indeed, hypertension in rodents is accompanied by gut dysbiosis (Yang et al., 2015; Durgan et al., 2016; Santisteban et al., 2017), and a high‐fibre diet induced changes in the gut microbiota and prevented the development of hypertension and heart failure in hypertensive mice (Marques et al., 2017). Furthermore, faecal microbial transplantation from hypertensive patients to germ‐free mice resulted in increased BP, suggesting that alterations in gut microbiota may precede the onset of hypertension (Li et al., 2017). Significant changes in the gut microbiome characterized by decreased butyrate and acetate‐producing bacterial populations and altered Firmicutes/Bacteroidetes ratio have been linked with high BP. Treatment with minocycline not only reduces the neuroinflammation (Shi et al., 2010) but also rectifies the gut dysbiosis in both animal and human hypertension and expanded both acetate‐producing and butyrate‐producing bacteria and lowered BP (Yang et al., 2015) indicative of its potential in the treatment of hypertension. Reductions in the Lactobacillaceae and Prevotellaceae populations were identified in patients with chronic kidney disease (CKD), an important and common consequence of hypertension (Vaziri et al., 2013). Interestingly, patients on maintenance haemodialysis have about 100‐fold higher enterobacteria and enterococci levels than controls (Hida et al., 1996).
Sympathetic overdrive is associated with chronic gut inflammation, which in turn enhances gut permeability, dysbiosis and altered microbial metabolites in the systemic circulation. The HSPCs from the BM also migrate to the gut causing local inflammation and immune responses (Santisteban et al., 2016). Intrinsic gut mechanisms regulate the activation of T‐helper 17 cells, which was elevated in hypertension‐inducing marked gut microbial dysbiosis and an increase in circulating inflammatory cells (Kim et al., 2015). The changes in gut microbiota in hypertension were associated with changes in gut inflammatory status (Santisteban et al., 2017). Germ‐free mice were protected from Ang II‐induced arterial hypertension, vascular dysfunction and hypertension‐induced end‐organ damage. This protection was mediated by inhibition of recruitment and activation of the inflammatory myelomonocytic cells in the vasculature and altered cytokine signalling (Karbach et al., 2016). Short‐chain fatty acids (SCFAs) are metabolites of gut microbiota primarily composed of acetate, propionate and butyrate and have been implicated in host–microbiota immune‐inflammatory responses (Xiong et al., 2004; Samuel et al., 2008; Maslowski et al., 2009).
SCFAs directly activate the sympathetic nervous system and are not detectable in the plasma of germ‐free mice (Perry et al., 2016), indicating that all circulating SCFAs are microbial in origin, and any changes in SCFAs indicate an involvement of the gut microbiota. Recent studies analysing different SCFA receptor‐null mice such as those lacking the olfactory receptor 78 (Olfr78) or GPR41 (FFA3 receptor), expressed in renal vasculature, suggest a role of SCFAs in BP regulation (Pluznick, 2017). Olfr78‐null mice have lowered plasma renin levels and lowered baseline BP consistent with its localization in the site of renin storage and secretion (renal afferent arteriole) and in vascular smooth muscle cells of large vessels. GPR41‐null animals display altered vascular tone, functionally stiffer vessels and isolated systolic hypertension (Natarajan et al., 2016). Surprisingly, transplantation of faecal contents from salt‐resistant (R) to salt‐sensitive (S) Dahl rats resulted in consistent and significant elevation in BP during the remaining lifespan of the recipient rat and also shortened lifespan. The genome of R rats was resistant to BP alterations when microbiota was transplanted from the S to the R type. These effects may be mediated by SCFAs as both acetate and heptanoate levels were higher in the R to S transfer group (Mell et al., 2015). Population‐based studies have found that interventions that alter production of SCFAs correlate with changes in BP (Khalesi et al., 2014) indicative of the potential roles of SCFAs in influencing phenotypic changes to regulate BP.
Yet another microbial metabolite that has drawn significant attention is trimethylamine derived from dietary phosphatidylcholine or l‐carnitine, metabolized in the liver following absorption to form trimethylamine N‐oxide (TMAO) and excreted by the kidneys (Wang et al., 2011; Tang et al., 2013). Accumulation of circulating TMAO results in adverse effects such as macrophage/foam cell activation, platelet hyperresponsiveness and vascular and endothelial cell dysfunction and leads to atherosclerosis, thrombosis and cardiorenal impairment (Wang et al., 2011) as well as prolonging the hypertensive effect of Ang II (Ufnal et al., 2014). Also, TMAO concentrations in plasma correlated positively with cardiovascular disease in humans (Koeth et al., 2013; Wang et al., 2015). Moreover, plasma TMAO levels (r = 0.43) were inversely related to vascular endothelial function as assessed by brachial artery flow‐mediated dilation, whereas they were positively correlated with systolic arterial levels (r = 0.47; R. A. Gioscia‐Ryan and D. R. Seals, unpubl. data, Raizada et al., 2017). TMAO levels were high in CKD patients and directly contributed to progressive renal fibrosis and dysfunction in animal models (Tang et al., 2015). Taken together, these studies indicate that (i) the gut microbiome may have a different interaction with the immune system in the prehypertensive and hypertensive states compared with the normotensive state and (ii) dysbiosis of the gut microbiota increases sympathetic activity, and the consequent immune responses are likely to contribute to the chronic systemic inflammation associated with hypertension.
Effect of sympathoinhibitory therapy on immune‐mediated inflammation in hypertension
In addition to its well‐described effects on cardiovascular control, data from laboratory and clinical studies provide substantial evidence for the role of sympathetic nervous system‐mediated immune mechanisms in the pathogenesis of hypertension. Inhibition of sympathetic overdrive in hypertension can be achieved by a number of strategies including weight reduction and exercise, pharmacotherapy and device‐based therapeutic approaches. Disorders such as obesity and the metabolic syndrome are associated with increased sympathetic nerve activity and circulating markers of oxidative stress and inflammation, which can be markedly improved with exercise and weight loss (Festa et al., 2000; Schlaich et al., 2015). Exercise training down‐regulates TLR signalling, attenuates oxidative stress and sympathetic activation and prevents the increase in weight, arterial pressure and white adipose tissue accumulation in rats fed with a high‐fat diet (Li et al., 2015). Pharmacological sympathetic inhibition can be achieved by inhibiting sympathetic outflow at the level of the rostral ventral medulla by means of imidazoline I1 receptor agonists such as moxonidine, which has been demonstrated to exert anti‐inflammatory properties in postmenopausal hypertensive women (Pöyhönen‐Alho et al., 2008) in addition to its beneficial effects on BP and metabolic parameters (Chazova et al., 2006). Another relevant relationship in the current context is the sympathetic nervous system‐induced increase in expression of the sodium/glucose co‐transporter‐2 (SGLT2). Interestingly, SGLT2 inhibition with empagliflozin reduced glucotoxicity‐induced inflammation and fibrosis in human proximal tubular cells (Panchapakesan et al., 2013; Matthews et al., 2017). Interventional approaches to target sympathetic overactivity include device‐based therapeutic strategies such as renal denervation and carotid body ablation. Both carotid body deafferentation and renal denervation reduced T‐cell infiltration in brain tissue and aorta of SHRs (McBryde et al., 2013). The beneficial effects of catheter‐based renal sympathetic denervation applied in patients with resistant hypertension appear at least in part to be mediated by immunomodulation via inhibition of monocyte activation and inflammation (Zaldivia et al., 2017).
Overall, these findings suggest that targeting sympathetic overactivity as a therapeutic principle affects the immune system on various levels, which may at least in part account for the beneficial cardiovascular and metabolic effects observed with these approaches.
Conclusion
The sympathetic nervous system plays a vital role in both activating and limiting immune‐inflammatory responses. This, in turn, encourages thinking that fine tuning of the immune‐inflammatory mechanisms responsible for enhanced sympathetic activity may be useful in the management of hypertension. However, to develop successful therapeutic interventions, an even better understanding and more research focussing on this link is warranted. Another important issue is to determine how modulation of sympathetic nerve activity could be exploited therapeutically to beneficially influence immune‐inflammatory responses. Finally, understanding the immune flow via the brain–BM–spleen–gut axis that perpetuates the immune cascade including immune infiltration and immune‐mediated inflammatory damage in the organs regulating BP will be important. New technologies for manipulating the sympathetic nervous system are already being investigated in clinical trials. Investigating the full consequences of the new modalities used in the treatment of resistant hypertension, such as renal denervation and carotid body ablation, on the immune system is likely to yield relevant insights and is currently being explored.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d, 2017e).
Conflict of interest
The authors declare no conflicts of interest.
Carnagarin R., Matthews V., Zaldivia M. T. K., Peter K., and Schlaich M. P. (2019) The bidirectional interaction between the sympathetic nervous system and immune mechanisms in the pathogenesis of hypertension, British Journal of Pharmacology, 176, 1839–1852, doi: 10.1111/bph.14481.
References
- Afan AM, Broome CS, Nicholls SE, Whetton AD, Miyan JA (1997). Bone marrow innervation regulates cellular retention in the murine haemopoietic system. Br J Haematol 98: 569–577. [DOI] [PubMed] [Google Scholar]
- Alaniz RC, Thomas SA, Perez‐Melgosa M, Mueller K, Farr AG, Palmiter RD et al (1999). Dopamine β‐hydroxylase deficiency impairs cellular immunity. Proc Natl Acad Sci U S A 96: 2274–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017e). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez V, Quiroz Y, Nava M, Pons H, Rodríguez‐Iturbe B (2002). Overload proteinuria is followed by salt‐sensitive hypertension caused by renal infiltration of immune cells. Am J Physiol Renal Physiol 283: F1132–F1141. [DOI] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ (2012). Neural reflexes in inflammation and immunity. J Exp Med 209: 1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataka K, Asakawa A, Nagaishi K, Kaimoto K, Sawada A, Hayakawa Y et al (2013). Bone marrow‐derived microglia infiltrate into the paraventricular nucleus of chronic psychological stress‐loaded mice. PLoS One 8: e81744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ba D, Takeichi N, Kodama T, Kobayashi H (1982). Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol 128: 1211–1216. [PubMed] [Google Scholar]
- Bataillard A, Freiche JC, Vincent M, Touraine JL, Sassard J (1986). Effects of neonatal thymectomy on blood pressure and immunological characteristics of genetically hypertensive rats of the Lyon strain. J Hypertens 4: 5445–5447. [Google Scholar]
- Bendich A, Belisle EH, Strausser HR (1981). Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 99: 600–607. [DOI] [PubMed] [Google Scholar]
- Brands MW, Banes‐Berceli AK, Inscho EW, Al‐Azawi H, Allen AJ, Labazi H (2010). Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and Janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody M, Fink G, Buggy J, Haywood J, Gordon F et al (1978). Critical role of the AV3V region in development and maintenance of experimental hypertension In: Schmitt H, Meyers P. (eds). Perspectives in Nephrology and Hypertension. Wiley and Flammarion: New York, NY, pp. 76–84. [Google Scholar]
- Brosnan CF, Goldmuntz EA, Cammer W, Factor SM, Bloom BR, Norton WT (1985). Prazosin, an α1‐adrenergic receptor antagonist, suppresses experimental autoimmune encephalomyelitis in the Lewis rat. Proc Natl Acad Sci U S A 82: 5915–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush E, Maeda N, Kuziel WA, Dawson TC, Wilcox JN, DeLeon H et al (2000). CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II‐induced hypertension. Hypertension 36: 360–363. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Pallante F, Fardella V, Fardella S, Iacobucci R, Federici M et al (2014). The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 41: 737–752. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Perrotta M, Pallante F, Fardella V, Iacobucci R, Fardella S et al (2016). A cholinergic‐sympathetic pathway primes immunity in hypertension and mediates brain‐to‐spleen communication. Nat Commun 7: 13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Roessner CT, Tian J, Zimmerman MC (2016). Mitochondrial superoxide signaling contributes to norepinephrine‐mediated T‐lymphocyte cytokine profiles. PLoS One 11: e0164609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Zimmerman MC (2015). Redox‐regulated suppression of splenic T‐lymphocyte activation in a model of sympathoexcitation. Hypertension 65: 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Zimmerman MC (2016). Sympathetic‐mediated activation versus suppression of the immune system: consequences for hypertension. J Physiol 594: 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae CU, Lee RT, Rifai N, Ridker PM (2001). Blood pressure and inflammation in apparently healthy men. Hypertension 38: 399–403. [DOI] [PubMed] [Google Scholar]
- Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A et al (2012). Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt‐treated mice. Hypertension 60: 1207–1212. [DOI] [PubMed] [Google Scholar]
- Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H et al (2015). Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- Chazova I, Almazov VA, Shlyakhto E (2006). Moxonidine improves glycaemic control in mildly hypertensive, overweight patients: a comparison with metformin. Diabetes Obes Metab 8: 456–465. [DOI] [PubMed] [Google Scholar]
- Chen XL, Tummala PE, Olbrych MT, Alexander RW, Medford RM (1998). Angiotensin II induces monocyte chemoattractant protein‐1 gene expression in rat vascular smooth muscle cells. Circ Res 83: 952–959. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P (2010). Lymphocyte responses exacerbate angiotensin II‐dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Xu M, Yao M, Sun B (2007). Angiotensin AT1 receptor antagonists exert anti‐inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol 152: 1042–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datla SR, Griendling KK (2010). Reactive oxygen species, NADPH oxidases and hypertension. Hypertension 56: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Miguel C, Guo C, Lund H, Feng D, Mattson DL (2011). Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300: F734–F742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J et al (2012). IL‐17‐mediated oxidative stress is an important stimulator of AT1‐AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol 303: R353–R358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov SI, Nazarewicz RR (2013). Angiotensin II‐induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal 19: 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T et al (2007). Stroke‐induced immunodepression: experimental evidence and clinical relevance. Stroke 38: 770–773. [DOI] [PubMed] [Google Scholar]
- Dörffel Y, Lätsch C, Stuhlmüller B, Schreiber S, Scholze S et al (1994). Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension 34: 113–117. [DOI] [PubMed] [Google Scholar]
- Durgan DJ, Ganesh BP, Cope JL, Ajami NJ, Phillips SC, Petrosino JF et al (2016). Role of the gut microbiome in obstructive sleep apnea‐induced hypertension. Hypertension 67: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R et al (2012). Myocardial infarction accelerates atherosclerosis. Nature 487: 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzielak DJ (1991). Immune mechanisms in experimental and essential hypertension. Am J Physiol 260: R459–R467. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RI, Chrousos GP, Vizi S (2000). The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52: 595–638. [PubMed] [Google Scholar]
- Engstrom G, Janzon L, Berglund G, Lind P, Stavenow L et al (2002). Blood pressure increase and incidence of hypertension in relation to inflammation‐sensitive plasma proteins. Arterioscler Thromb Vasc Biol 22: 2054–2058. [DOI] [PubMed] [Google Scholar]
- Fannon LD, Phillips MI (1991). Chronic ICV infusion of neuropeptides alters lymphocyte populations in experimental rodents. Regul Pept 34: 189–195. [DOI] [PubMed] [Google Scholar]
- Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S (1985). Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol 135: 755s–765s. [PubMed] [Google Scholar]
- Felten DL, Livnat S, Felten SY, Carlson SL, Bellinger DL, Yeh P (1984). Sympathetic innervation of lymph nodes in mice. Brain Res Bull 13: 693–699. [DOI] [PubMed] [Google Scholar]
- Festa A, D'Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM (2000). Chronic sublinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 102: 42–47. [DOI] [PubMed] [Google Scholar]
- Franco M, Tapia E, Bautista R, Pacheco U, Santamaria J, Quiroz Y et al (2013). Impaired pressure natriuresis resulting in salt‐sensitive hypertension is caused by tubulointerstitial immune cell infiltration in the kidney. Am J Physiol Renal Physiol 304: F982–F990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Thompson CB (2002). Activation and inhibition of lymphocytes by costimulation. J Clin Invest 109: 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frostegard J, Wu R, Gillis‐Haegerstrand C, Lemne C, de Faire U (1998). Antibodies to endothelial cells in borderline hypertension. Circulation 98: 1092–1098. [DOI] [PubMed] [Google Scholar]
- Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P et al (2000). Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens 18: 945–953. [DOI] [PubMed] [Google Scholar]
- Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L et al (2005). Central angiotensin II‐enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol 289: H1683–H1691. [DOI] [PubMed] [Google Scholar]
- Gonzalez GE, Rhaleb NE, D'Ambrosio MA, Nakagawa P, Liu Y et al (2015). Deletion of interleukin‐6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II‐high salt‐induced hypertension. J Hypertens 33: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotsman I, Sharpe AH, Lichtman AH (2008). T‐cell costimulation and coinhibition in atherosclerosis. Circ Res 103: 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouraud SS, Waki H, Bhuiyan ME, Takagishi M, Cui H et al (2011). Down‐regulation of chemokine Ccl5 gene expression in the NTS of SHR may be pro‐hypertensive. J Hypertens 29: 732–740. [DOI] [PubMed] [Google Scholar]
- Grisanti LA, Woster AP, Dahlman J, Sauter ER, Combs CK, Porter JE (2011). α1‐Adrenergic receptors positively regulate toll‐like receptor cytokine production from human monocytes and macrophages. J Pharmacol Exp Ther 338: 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton AC, Coleman TG, Granger HJ (1972). Circulation: overall regulation. Annu Rev Physiol 34: 13–46. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A et al (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoun M, Maryanovich M, Arnal‐Estapé A, Frenette PS (2015). Neural regulation of hematopoiesis, inflammation, and cancer. Neuron 86: 360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DG (2014). The immune system in hypertension. Trans Am Clin Climatol Assoc 125: 130–140. [PMC free article] [PubMed] [Google Scholar]
- Harrison DG, Gongora MC (2009). Oxidative stress and hypertension. Med Clin North Am 93: 621–635. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR et al (2011). Inflammation, immunity, and hypertension. Hypertension 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartupee J, Liu C, Novotny M, Li X, Hamilton T (2007). IL‐17 enhances chemokine gene expression through mRNA stabilization. J Immunol 179: 4135–4141. [DOI] [PubMed] [Google Scholar]
- Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A et al (2014). Chronic variable stress activates hematopoietic stem cells. Nat Med 20: 754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijnen BF, Van Essen H, Schalkwijk CG, Janssen BJ, Struijker‐Boudier HA (2014). Renal inflammatory markers during the onset of hypertension in spontaneously hypertensive rats. Hypertens Res 37: 100–109. [DOI] [PubMed] [Google Scholar]
- Heijnen CJ, Rouppe van der Voort C, van de Pol M, Kavelaars A (2002). Cytokines regulate α1‐adrenergic receptor mRNA expression in human monocytic cells and endothelial cells. J Neuroimmunol 125: 66–72. [DOI] [PubMed] [Google Scholar]
- Heijnen CJ, Rouppe van der Voort C, Wulffraat N, van der Net J, Kuis W, Kavelaars A (1996). Functional α1‐adrenergic receptors on leukocytes of patients with polyarticular juvenile rheumatoid arthritis. J Neuroimmunol 71: 223–226. [DOI] [PubMed] [Google Scholar]
- Herrera J, Ferrebuz A, García‐Macgregor E, Rodríguez‐Iturbe B (2006). Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17: S218–S225. [DOI] [PubMed] [Google Scholar]
- Hida M, Aiba Y, Sawamura S, Suzuki N, Satoh T, Koga Y (1996). Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin®, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 74: 349–355. [DOI] [PubMed] [Google Scholar]
- Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL et al (2013). CHBPR: Angiotensin AT1a receptors in the subfornical organ are required for DOCA‐Salt hypertension. Hypertension 61: 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston JM, Ochani M, Rosas‐Ballina M, Liao H, Ochani K, Pavlov VA et al (2006). Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 203: 1623–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP et al (2016). Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 68: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K (2017). Loss of resistance to angiotensin II‐induced hypertension in the Jackson Laboratory recombination‐activating gene null mouse on the C57BL/6J background. Hypertension 69: 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbach SH, Schonfelder T, Brandao I, Wilms E, Hormann N et al (2016). Gut microbiota promote angiotensin II‐induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 5: e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A et al (2012). T regulatory lymphocytes prevent aldosterone‐induced vascular injury. Hypertension 59: 324–330. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA et al (2006). Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 124: 407–421. [DOI] [PubMed] [Google Scholar]
- Khalesi S, Sun J, Buys N, Jayasinghe R (2014). Effect of probiotics on blood pressure: a systematic review and meta‐analysis of randomized, controlled trials. Hypertension 64: 897–903. [DOI] [PubMed] [Google Scholar]
- Kim S, Rodriguez V, Santisteban M, Yang T, Qi Y, Raizada M et al (2015). Hypertensive patients exhibit gut microbial dysbiosis and an increase in Th17 cells. J Hypertens 33: e77–e78. [Google Scholar]
- Kim S, Zingler M, Harrison JK, Scott EW, Cogle CR, Luo D et al (2016). Angiotensin II regulation of proliferation, differentiation, and engraftment of hematopoietic stem cells. Hypertension 67: 574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL et al (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT et al (2013). Intestinal microbiota metabolism of l‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19: 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KK, Quon MJ, Han SH, Chung WJ, Ahn JY, Seo YH et al (2004). Additive beneficial effects of losartan combined with simvastatin in the treatment of hypercholesterolemic, hypertensive patients. Circulation 110: 3687–3692. [DOI] [PubMed] [Google Scholar]
- Kooijman S, Meurs I, Van Beek L, Khedoe PP, Giezekamp A et al (2015). Splenic autonomic denervation increases inflammatory status but does not aggravate atherosclerotic lesion development. Am J Physiol Heart Circ Physiol 309: H646–H654. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M et al (2013). Angiotensin II‐induced vascular dysfunction depends on interferon‐γ‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 33: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Kunes J, Poirier M, Tremblay J, Hamet P (1992). Expression of HSP70 gene in lymphocytes from normotensive and hypertensive humans. Acta Physiol Scand 146: 307–311. [DOI] [PubMed] [Google Scholar]
- Lambert EA, Eikelis N, Esler M, Dawood T, Schlaich MP, Bayles R et al (2008). Altered sympathetic nervous reactivity and norepinephrine transporter expression in patients with the postural tachycardia syndrome. Circ Arrhythm Electrophysiol 1: 103–109. [DOI] [PubMed] [Google Scholar]
- Lampron A, Pimentel‐Coelho PM, Rivest S (2013). Migration of bone marrow‐derived cells into the central nervous system in models of neurodegeneration. J Comp Neurol 521: 3863–3876. [DOI] [PubMed] [Google Scholar]
- Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS et al (2006). Angiotensin II hypertension is attenuated in interleukin‐6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940. [DOI] [PubMed] [Google Scholar]
- Li G, Liu JY, Zhang HX, Li Q, Zhang SW (2015). Exercise training attenuates sympathetic activation and oxidative stress in diet‐induced obesity. Physiol Res 64: 355–367. [DOI] [PubMed] [Google Scholar]
- Li J, Zhao F, Wang Y, Chen J, Tao J et al (2017). Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 5: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX, Tang BP, Sun HP, Feng M, Cheng ZH, Niu WQ (2009). Interacting contribution of the five polymorphisms in three genes of Hsp70 family to essential hypertension in Uygur ethnicity. Cell Stress Chaperones 14: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX (2002). Autoantibodies against AT1‐receptor and α1‐adrenergic receptor in patients with hypertension. Hypertens Res 25: 641–646. [DOI] [PubMed] [Google Scholar]
- Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA et al (2010). Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 55: 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG (2013). Role of the NADPH oxidases in the subfornical organ in I‐induced hypertension. Hypertension 61: 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y et al (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrigal JL, Caso JR (2014). The chemokine (C–C motif) ligand 2 in neuroinflammation and neurodegeneration. Adv Exp Med Biol 824: 209–219. [DOI] [PubMed] [Google Scholar]
- Maestroni GJ (2000). Dendritic cell migration controlled by α1b‐adrenergic receptors. J Immunol 165: 6743–6747. [DOI] [PubMed] [Google Scholar]
- Maisel AS, Michel MC (1989). β‐Adrenergic receptors in congestive heart failure: present knowledge and future directions. Cardiology 76: 338–346. [DOI] [PubMed] [Google Scholar]
- Matthews VB, Elliot RH, Rudnicka C, Hricova J, Herat L, Schlaich MP (2017). Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J Hypertens 35: 2059–2068. [DOI] [PubMed] [Google Scholar]
- Marketou ME, Kontaraki JE, Tsakountakis NA, Zacharis EA, Kochiadakis GE, Arfanakis DA et al (2011). Differential effect of telmisartan and amlodipine on monocyte chemoattractant protein‐1 and peroxisome proliferator‐activated receptor‐γ gene expression in peripheral monocytes in patients with essential hypertension. Am J Cardiol 107: 59–63. [DOI] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al (2012). Interferon‐γ signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Marques FZ, Nelson E, Chu PY, Horlock D, Fiedler A, Ziemann M et al (2017). High‐fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 135: 964–977. [DOI] [PubMed] [Google Scholar]
- Martelli D, Farmer DG, Yao ST (2016). The splanchnic anti‐inflammatory pathway: could it be the efferent arm of the inflammatory reflex? Exp Physiol 101: 1245–1252. [DOI] [PubMed] [Google Scholar]
- Martelli D, Yao ST, McKinley MJ, McAllen RJ (2014). Reflex control of inflammation by sympathetic nerves, not the vagus. J Physiol 592: 1677–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA et al (2010). Central and peripheral mechanisms of T‐lymphocyte activation and vascular inflammation produced by angiotensin II‐induced hypertension. Circ Res 107: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H (2013). Genetic mutation of recombination activating gene 1 in Dahl salt‐sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBryde FD, Abdala AP, Hendy EB, Pijacka W, Marvar P et al (2013). The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat Commun 4: 2395. [DOI] [PubMed] [Google Scholar]
- Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y et al (2015). Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics 47: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL (2016). Deficiency of T‐regulatory cells exaggerates angiotensin II‐induced microvascular injury by enhancing immune responses. J Hypertens 34: 97–108. [DOI] [PubMed] [Google Scholar]
- Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T (2008). Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide‐induced stimulation of rat microglial cells by suppressing nuclear factor κB and activator protein‐1 activation. Eur J Neurosci 27: 343–351. [DOI] [PubMed] [Google Scholar]
- Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE et al (2016). Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein‐coupled receptor 41. Physiol Genomics 48: 826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM (2013). Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Grollman A (1967). Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med 25: 257–264. [PubMed] [Google Scholar]
- Papadopoulos DP, Makris TK, Krespi P, Papazachou U, Stavroulakis G, Hatzizacharias A et al (2006). Antiendothelial cell antibody levels in healthy normotensives with high normal blood pressure. Clin Exp Hypertens 28: 663–667. [DOI] [PubMed] [Google Scholar]
- Panchapakesan U, Pegg K, Gross S, Komala MG, Mudaliar H, Forbes J et al (2013). Effects of SGLT2 inhibition in human kidney proximal tubular cells – renoprotection in diabetic nephropathy? PLoS ONE 8: e54442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DM, Papay RS, Shi T (2009). α1‐Adrenergic receptor stimulates interleukin‐6 expression and secretion through both mRNA stability and transcriptional regulation: involvement of p38 mitogen‐activated protein kinase and nuclear factor‐κB. Mol Pharmacol 76: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL et al (2016). Acetate mediates a microbiome–brain–β‐cell axis to promote metabolic syndrome. Nature 534: 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluznick JL (2017). Microbial short‐chain fatty acids and blood pressure regulation. Curr Hypertens Rep 19: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pockley AG, De Faire U, Kiessling R, Lemne C, Thulin T, Frostegard J (2002). Circulating heat shock protein and heat shock protein antibody levels in established hypertension. J Hypertens 20: 1815–1829. [DOI] [PubMed] [Google Scholar]
- Pockley AG, Georgiades A, Thulin T, De Faire U, Frostegård J (2003). Serum heat shock protein 70 levels predict the development of atherosclerosis in subjects with established hypertension. Hypertension 42: 235–238. [DOI] [PubMed] [Google Scholar]
- Pons H, Ferrebuz A, Quiroz Y, Romero‐Vasquez F, Parra G, Johnson RJ et al (2013). Immune reactivity to heat shock protein 70 expressed in the kidney is cause of salt‐sensitive hypertension. Am J Physiol Renal Physiol 304: F289–F299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöyhönen‐Alho MK, Manhem K, Katzman P, Kibarskis A, Antikainen RL, Erkkola RU et al (2008). Central sympatholytic therapy has anti‐inflammatory properties in hypertensive postmenopausal women. J Hypertens 26: 2445–2449. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Hoflich C, Braun J, Halle E et al (2003). Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1‐like immunostimulation. J Exp Med 198: 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raizada MK, Joe B, Bryan NS, Chang EB, Dewhirst FE et al (2017). Report of the national heart, lung, and blood institute working group on the role of microbiota in blood pressure regulation: Current status and future directions. Hypertension 70: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M et al (2001). Mycophenolate mofetil prevents salt‐sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND (2004). Evolution of renal interstitial inflammation and NF‐κB activation in spontaneously hypertensive rats. Am J Nephrol 24: 587–594. [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Iturbe B, Vaziri ND, Herrera‐Acosta J, Johnson RJ (2004). Oxidative stress, renal infiltration of immune cells, and salt‐sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol 286: F606–F616. [DOI] [PubMed] [Google Scholar]
- Rosas‐Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM et al (2008). Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A 105: 11008–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas‐Ballina M, Olofsson PS, Ochani M, Valdes‐Ferrer SI, Levine YA, Reardon C et al (2011). Acetylcholine‐synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudemiller NP, Crowley SD (2016). Interactions between the immune and renin–angiotensin systems in hypertension. Hypertension 68: 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MJ (2013). Immune mechanisms in hypertension In: Granger ND, Granger J. (eds). Colloquium Series on Integrated System Physiology. Morgan & Claypool Life Sciences: San Rafael, CA, pp. 33–44. [Google Scholar]
- Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK et al (2008). Effects of the gut microbiota on host adiposity are modulated by the short‐chain fatty‐acid binding G protein‐coupled receptor, Gpr41. Proc Natl Acad Sci U S A 105: 16767–16772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban MM, Ahmari N, Carvajal JM, Zingler MB, Qi Y, Kim S et al (2015). Involvement of bone marrow cells and neuroinflammation in hypertension. Circ Res 117: 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban MM, Kim S, Pepine CJ, Raizada MK (2016). Brain–gut–bone marrow axis: implications for hypertension and related therapeutics. Circ Res 118: 1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V et al (2017). Hypertension‐linked pathophysiological alterations in the gut. Circ Res 120: 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santisteban MM, Zubcevic J, Baekey DM, Raizada MK (2013). Dysfunctional brain–bone marrow communication: a paradigm shift in the pathophysiology of hypertension. Curr Hypertens Rep 15: 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiermann C, Kunisaki Y, Frenette PS (2013). Circadian control of the immune system. Nat Rev Immunol 13: 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaich MP, Krum H, Sobotka PA (2010). Renal sympathetic nerve ablation: the new frontier in the treatment of hypertension. Curr Hypertens Rep 12: 39–46. [DOI] [PubMed] [Google Scholar]
- Schlaich M, Straznicky N, Lambert E, Lambert G (2015). Metabolic syndrome: a sympathetic disease? Lancet Diabetes Endocrinol 3: 148–157. [DOI] [PubMed] [Google Scholar]
- Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP (2007). IL‐6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol 27: 2576–2581. [DOI] [PubMed] [Google Scholar]
- Seaberg EC, Muñoz A, Lu M, Detels R, Margolick JB, Riddler SA et al (2005). Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS 19: 953–960. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q et al (2010). Microglial cytokines in neurogenic hypertension. Hypertension 56: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP, Francis J (2013). Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II‐induced hypertension. PLoS One 8: e63847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf C, Auer C, Yilmaz A, Lewczuk P, Klinghammer L, Schneider M et al (2011). Serum levels of the Th1 chemoattractant interferon‐γ‐inducible protein (IP) 10 are elevated in patients with essential hypertension. Hypertens Res 34: 484–488. 10.1038/hr.2010.258. [DOI] [PubMed] [Google Scholar]
- Svendsen UG (1976). Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand 84: 523–528. [DOI] [PubMed] [Google Scholar]
- Svendsen UG (1977). The importance of thymus in the pathogenesis of the chronic phase of hypertension in mice following partial infarction of the kidney. Acta Pathol Microbiol Scand A 85: 539–547. [DOI] [PubMed] [Google Scholar]
- Swanson MA, Lee WT, Sanders VM (2001). IFN‐γ production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol 166: 232–240. [DOI] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA et al (2015). Gut microbiota‐dependent trimethylamine N‐oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res 116: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB et al (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 368: 1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran LT, MacLeod KM, McNeill JH (2009). Chronic etanercept treatment prevents the development of hypertension in fructose‐fed rats. Mol Cell Biochem 330: 219–228. [DOI] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP et al (2007). Critical roles for CCR2 and MCP3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117: 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufnal M, Jazwiec R, Dadlez M, Drapala A, Sikora M, Skrzypecki J (2014). Trimethylamine‐N‐oxide: a carnitine‐derived metabolite that prolongs the hypertensive effect of angiotensin II in rats. Can J Cardiol 30: 1700–1705. [DOI] [PubMed] [Google Scholar]
- Vanegas V, Ferrebuz A, Quiroz Y, Rodríguez‐Iturbe B (2005). Hypertension in Page (cellophane wrapped) kidney is due to interstitial nephritis. Kidney Int 68: 1161–1170. [DOI] [PubMed] [Google Scholar]
- Vaziri ND, Wong J, Pahl M, Piceno YM, Yuan J, DeSantis TZ et al (2013). Chronic kidney disease alters intestinal microbial flora. Kidney Int 83: 308–315. [DOI] [PubMed] [Google Scholar]
- Venegas‐Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C et al (2010). Tumor necrosis factor‐α antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ et al (2010). Inhibition and genetic ablation of the B7/CD28 T‐cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B et al (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E et al (2015). Non‐lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163: 1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Jia XJ, Yu SQ, Gu Y, Wang L, Guo XM et al (2011). Candesartan versus imidapril in hypertension: a randomised study to assess effects of anti‐AT1 receptor autoantibodies. Heart 97: 479–484. [DOI] [PubMed] [Google Scholar]
- Wei Z, Spizzo I, Diep H, Drummond GR, Widdop RE, Vinh A (2014). Differential phenotypes of tissue‐infiltrating T cells during angiotensin II‐induced hypertension in mice. PLOS ONE 9: e114895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- White FN, Grollman A (1964). Autoimmune factors associated with infarction of the kidney. Nephron 1: 93–102. [DOI] [PubMed] [Google Scholar]
- Wu KLH, Chan SHH, Chan JYH (2012). Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation 9: 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L et al (2015). Renal denervation prevents immune cell activation and renal inflammation in angiotensin II‐induced hypertension. Circ Res 117: 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Miyamoto N, Shibata K, Valasek MA, Motoike T, Kedzierski RM et al (2004). Short‐chain fatty acids stimulate leptin production in adipocytes through the G protein‐coupled receptor GPR41. Proc Natl Acad Sci U S A 101: 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM et al (2015). Gut dysbiosis is linked to hypertension. Hypertension 65: 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaldivia MT, Rivera J, Hering D, Marusic P, Sata Y et al (2017). Renal denervation reduces monocyte activation and monocyte‐platelet aggregate formation: an anti‐inflammatory effect relevant for cardiovascular risk. Hypertension 69: 323–331. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR et al (2002). Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res 91: 1038–1045. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL (2004). Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95: 210–216. [DOI] [PubMed] [Google Scholar]
- Zubcevic J, Jun JY, Kim S, Perez PD, Afzal A, Shan Z et al (2014). Altered inflammatory response is associated with an impaired autonomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension 63: 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]