

Abstract
Background and Purpose
Angiotensin II (AngII) and NO regulate the cerebral circulation. AngII AT1 receptors exert ligand‐dependent and ligand‐independent (myogenic tone [MT]) vasoconstriction of cerebral vessels. NO induces post‐translational modifications of proteins such as S‐nitrosation (redox modification of cysteine residues). In cultured cells, S‐nitrosation decreases AngII's affinity for the AT1 receptor. The present work evaluated the functional consequences of S‐nitrosation on both AngII‐dependent and AngII‐independent cerebrovascular responses.
Experimental Approach
S‐Nitrosation was induced in rat isolated middle cerebral arteries by pretreatment with the NO donors, S‐nitrosoglutathione (GSNO) or sodium nitroprusside (SNP). Agonist‐dependent activation of AT1 receptors was evaluated by obtaining concentration–response curves to AngII. Ligand‐independent activation of AT1 receptors was evaluated by calculating MT (active vs. passive diameter) at pressures ranging from 20 to 200 mmHg in the presence or not of a selective AT1 receptor inverse agonist.
Key Results
GSNO or SNP completely abolished the AngII‐dependent AT1 receptor‐mediated vasoconstriction of cerebral arteries. GSNO had no impact on responses to other vasoconstrictors sharing (phenylephrine, U46619) or not (5‐HT) the same signalling pathway. MT was reduced by GSNO, and the addition of losartan did not further decrease MT, suggesting that GSNO blocks AT1 receptor‐dependent MT. Ascorbate (which reduces S‐nitrosated compounds) restored the response to AngII but not the soluble GC inhibitor ODQ, suggesting that these effects are mediated by S‐nitrosation rather than by S‐nitrosylation.
Conclusions and Implications
In rat middle cerebral arteries, GSNO pretreatment specifically affects the AT1 receptor and reduces both AngII‐dependent and AngII‐independent activation, most likely through AT1 receptor S‐nitrosation.
Abbreviations
- AngII
angiotensin II
- AT1 receptor
angiotensin II type 1 receptor
- SNO
S‐nitrosoglutathione
- ID
internal diameter
- L‐NAME
N ω‐nitro‐l‐arginine methyl ester
- MCA
middle cerebral artery
- MT
myogenic tone
- ODQ
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one
- PE
phycoerythrin
- Phe
phenylephrine
- PSS
physiological salt solution
- SNP
sodium nitroprusside
- sGC
soluble GC
What is already known about this subject
Exposure of transfected cells to NO‐donors reduces AT1 receptor affinity for angiotensin II through S‐nitrosation.
What this study adds
S‐nitrosation specifically abolishes ligand‐dependent and ‐independent contractile responses mediated by AT1 in cerebral vessels.
What is the clinical significance
This AT1 regulation is of potential interest in cerebrovascular diseases involving excessive AT1 receptor‐mediated responses.
1. INTRODUCTION
NO is a gaseous radical well known for its vascular dilator effect through the nitrosylation (coordination of NO to the ferrous haem prosthetic group) of soluble GC (sGC) and cGMP production (Rapoport, Draznin, & Murad, 1983). Besides, NO also induces the S‐nitrosation of proteins (Stamler et al., 1992). The S‐nitrosation is a redox‐based modification of proteins resulting from the covalent bond between NO and the sulfhydryl function of cysteine residues. This post‐translational modification of proteins is involved in the regulation of the activity of many proteins (see the SNObase database [Zhang et al., 2012] for an exhaustive review).
In the large field of NO‐donors, S‐nitrosothiols, in which the NO moiety is grafted on a sulfur atom, display low toxicity and no tolerance phenomenon (Munzel, Daiber, & Gori, 2011). In particular, S‐nitrosoglutathione (GSNO), a low MW S‐nitrosothiol formed by nitrosation of reduced GSH, mediates protein S‐nitrosation and plays an important role in vascular signalling (Marozkina & Gaston, 2012). Several studies, including ours, have shown the therapeutic potential of exogenous GSNO in a preclinical model of ischaemic stroke (Khan et al., 2005; Parent et al., 2015). S‐Nitrosation of target proteins seems to contribute to these cerebroprotective effects (Khan & Singh, 2016).
In 2006, Leclerc et al. showed that short exposure to NO donors of cells overexpressing the angiotensin II (AngII) type 1 receptor (AT1 receptor) led to S‐nitrosation of this receptor on cysteine 289, leading to a decrease in its affinity for AngII (Leclerc et al., 2006). The AT1 receptor is involved in cerebral blood flow autoregulation in cerebrovascular structure and functions (Dupuis, Atkinson, Limiñana, & Chillon, 2005). At the cerebrovascular level, two of the main functions of the AT1 receptor are (a) an AngII‐dependent vasoconstriction and (b) an AngII‐independent involvement in myogenic tone (MT, i.e., contractile response of arteries consecutive to an increase in arterial pressure; Mederos y Schnitzler et al., 2008). Both functions are inhibited by AT1 receptor blockers. Following stroke and the consecutive increase in brain levels of AngII (Kagiyama, Kagiyama, & Phillips, 2003; Li et al., 2014), these contractile functions of AT1 receptors might be deleterious if they reduce blood flow and result in enlarged infarct size. Reducing AT1 receptor activity with AT1 receptor blockers has protective effects against stroke in preclinical (Omura‐Matsuoka et al., 2009) and clinical studies (Diener, 2009).
Based on the above, we hypothesized that S‐nitrosation of the AT1 receptor, by reducing its activity, might be an important mechanism that contributes to the cerebroprotective effects of GSNO.
The aim of this study was thus to determine the impact of exogenous GSNO administration on AT1 receptor functions: AngII‐dependent vasoconstriction and AngII‐independent MT. We exposed isolated rat middle cerebral arteries (MCAs) to GSNO (Alencar et al., 2003) and tried to decipher the mechanisms involved in the altered AT1 receptor functions.
First, concentration–response curves to AngII were built using isolated and perfused rat MCAs and compared to other vasoconstrictors. Specificity towards the AT1 receptor was assessed using several vasoconstrictors that do or do not share the AT1 receptor signalling pathway. Phenylephrine (Phe) and the TxA2 agonist U46619 were chosen since both mediate vasoconstriction through receptors activating heterotrimeric Gq/11, like AT1 receptors (Schleifenbaum et al., 2014). 5‐HT served as a positive control as its effects mainly involve the 5‐HT1B receptor (Watts, Morrison, Davis, & Barman, 2012), which is coupled to Gi/Go (Lin, Setya, Johnson‐Farley, & Cowen, 2002).
Second, we evaluated the effect of GSNO on AT1 receptor‐dependent MT in arteries using the AT1 receptor inverse agonist losartan.
Third, as GSNO can release both NO and GSH, we verified that the observed effect was only linked to the exogenous release of NO. Moreover, the role of endogenous NO production was assessed. As NO acts through two distinct mechanisms: nitrosylation and S‐nitrosation, both of these pathways were investigated in order to determine GSNO's mode of action.
Finally, as receptor internalization is important in receptor re‐sensitization and activation of downstream effectors (Hunyady, Catt, Clark, & Gaborik, 2000), we evaluated whether GSNO could modify basal or activated AT1 receptor cell surface localization in a model of heterologous expression in transfected HEK293 cells.
2. METHODS
2.1. Ethics statement and animals
The experiments were conducted on 4‐ to 5‐month‐old (500 ± 50 g) male normotensive outbred Wistar rats (HanRj:WI, Janvier, Le Genest‐Saint‐Isles, France, RGD Cat# 13792727, RRID:RGD_13792727), a commonly used physiological model. Only male rats were used to avoid any gender parameters that could influence AngII response (De Silva, Broughton, Drummond, Sobey, & Miller, 2009). Animals were housed in a standard animal unit at 23°C (three animals per cage, carton tunnels as enrichment), exposed to 12 hr of light (lights on at 06:00 and off at 18:00 hr) and allowed free access to food and fluid. Animals were randomly allocated to the various pretreatments and protocols.
They ate standard rat chow (A04, Safe, Villemoison‐Epinay sur Orge, France) and drank water (Aqua‐clear®, Culligan, Northbrook, USA) ad libitum. All experiments were performed in accordance with the European Community guidelines (2010/63/EU) for the use of experimental animals in the respect of the 3Rs' requirements for Animal Welfare. The project untitled “Nitro‐Vivo” was positively evaluated by the regional ethical committee for animal experiments and approved by the French Ministry of Research (No. APAFIS#1614‐2015090216575422v2). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology.
2.2. Vasoactive experiments
2.2.1. Preparation of MCAs
Rats were anaesthetized with sodium pentobarbitone (100 mg·kg−1, i.p.), injected with heparin (1000 IU·kg−1, i.v.), and then killed by decapitation. The brain was placed in a physiological salt solution (PSS) at 4°C (NaCl 119, NaHCO3 24, KCl 4.7, MgSO4 1.17, KH2PO4 1.18, CaCl2 1.6, and glucose 5.10−3 mol·L−1).
MCAs were chosen as a classical model of isolated cerebral vessels. The first branch‐free segment of the MCA immediately proximal to the circle of Willis was dissected out and mounted on two glass micropipettes (diameter, 125–150 μm) in a small vessel arteriograph (model CH/1/AU/SH, Living Systems Instrumentation, Burlington, VT, USA) and secured with silk thread (diameter, 20 μm, 2–0, 48 mm, 2540, Ethicon, Issy‐les‐Moulineaux, France). The vessel was then pressurized (intraluminal pressure of 80 mmHg) and perfused (intraluminal flow rate of 100 μl·min−1) with PSS at 37°C. Arteries were placed in an organ bath with PSS at 37°C gassed with 5% CO2/95% O2 (extraluminal flow rate of 10 ml·min−1) for measurement of isotonic vasoconstriction and assessment of viability.
Following initial pressurization, arteries were allowed to recover their basal MT for 60 min. Baseline internal diameter (ID) was measured using a video dimension analyser (V94, Living Systems Instrumentation, Burlington, VT, USA); signals were digitized and stored with WinDataq DI 720 data acquisition software (Dataq Instruments, Akron, OH, USA). Viability of the MCA was then assessed with potassium chloride (KCl, 4 x10−2 mol·L−1) added to the extraluminal bath. The contractile response rapidly reached a steady state and was measured after a 5‐min exposure. Responses were expressed as percentage change in ID from baseline ID (arteries showing less than 20% of contraction were excluded). Perfusion with KCl was followed by a 15‐min washout period so that the ID returns to baseline.
2.2.2. Pretreatment with GSNO
After assessment of viability, MCA were exposed for 30 min to GSNO (2 x10−6 mol·L−1), which increased the ID. GSNO was then removed and the vessels submitted to a 1‐hr washout period (PSS changed every 20 min, adapted from Alencar et al. (2003) that allowed MCA ID to return to baseline (data not shown). According to Alencar et al., S‐nitrosation of cysteine residues is maintained after the washout (Alencar et al., 2003).
2.2.3. AngII‐dependent response and Gq11, Gi/Go pathways
Concentration–response curves to AngII (5 min per concentration, from 10−12 to 10−6 mol·L−1) were compared following PSS or GSNO pretreatment. Phe (10−12 to 10−4 mol·L−1), U46619 (10−12 to 10−5 mol·L−1), and 5‐HT (10−12 to 10−5 mol·L−1) concentration–response curves were performed in the same conditions. Then, for the following series of experiments, in order to shorten the duration of the protocols, MCA were successively exposed to two equiactive concentrations of AngII (10−10 and 10−9 mol·L−1) and 5‐HT (3.10−9 and 10−8 mol·L−1, concentrations determined from the previous concentration–responses curves). In order to rule out any time‐dependent effect, we reversed the order of administration of AngII and 5‐HT (n = 2). Similar results were obtained regardless of the order AngII or 5‐HT perfusion (Figure S1). Given the lack of available selective AT1 agonists, the specific impact of GSNO on AT1 receptor‐mediated vasoactive responses was assessed using AngII in the presence of the AT2 receptor antagonist PD123319 (10−6 mol·L−1; Näveri, Strömberg, & Saavedra, 1994).
2.2.4. AT1 receptor‐dependent MT experiment
MT was evaluated comparing the evolution of ID at pressures ranging from 20 to 200 mmHg under active or passive (20 mmol·L−1 EDTA) conditions in order to build MT pressure curves (Foulquier, Lartaud, & Dupuis, 2014). MCAs were exposed or not to the AT1 inverse agonist losartan (10−5 mol·L−1) to confirm the involvement of AT1 receptors in MT (Schleifenbaum et al., 2014). MCA were pretreated with GSNO before exposure or not to losartan. The AUC (arbitrary unit) of MT as a function of pressure was obtained for each MCA using the following formula: MT = (ID − IDEDTA) × 100/IDEDTA.
2.2.5. NO involvement in AT1 receptor‐attenuated response
In order to rule out any antioxidant effect of GSNO (via GSH release), MCA were exposed to GSH (30 min, 2 x 10−6 mol·L−1) instead of GSNO. To confirm the involvement of NO, MCA were exposed to another NO donor (sodium nitroprusside, SNP, 2.10−6 mol·L−1) in the same conditions. Control MCA followed the same protocol with PSS instead of NO donors. Implication of basal level of NO was assessed using N ω‐nitro‐l‐arginine methyl ester (L‐NAME) an endothelial NOS inhibitor. The concentration of L‐NAME was fixed at 10−4 mol·L−1 (Briones, Alonso, Marin, & Salaices, 1999).
2.2.6. Role of S‐nitrosation
As most effects of NO are mediated by sGC nitrosylation, its implication was assessed using the sGC inhibitor 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ). The concentration of ODQ was fixed at 10−5 mol·L−1 (Yu, Sun, Maier, Harder, & Roman, 2002). A series a MCA underwent DMSO (i.e., ODQ solvent) treatment as control (final concentration 0.001%).
To confirm the involvement of S‐nitrosation in the effect of GSNO, MCA were exposed to sodium ascorbate (20 min, 5.10−6 mol·L−1, based on Figueiredo‐Freitas et al., 2015) immediately after GSNO pretreatment to specifically reduce S–NO link. This was followed by 40 min PSS before exposure to AngII and 5‐HT.
2.3. Stable expression of fluorescently labelled EGFP‐AT 1 receptor in HEK293 cells
HEK293 cells (ATCC Cat# CRL‐1573, RRID:CVCL_0045) were cultured to ~80% confluence in T‐75‐cm2 flasks in MEM with Earle's salt supplemented with 10% fetal calf serum, 2 mM glutamine, and 1% antibiotics (penicillin/streptomycin) and re‐plated twice a week.
The expression vector pIRES (Clontech) encoding for the fluorescent EGFP‐AT1 human receptor was a gift from Prof. Haiech and Prof. Kilhoffer (University of Strasbourg) and PCBIS UMS3286 CNRS‐University of Strasbourg. The vector was constructed using a ligation‐independent cloning strategy. Briefly, the receptor is expressed with, at its N‐terminus, the fluorophore preceded by a signal peptide for translocation of the fusion protein to the membrane of the endoplasmic reticulum (Vollmer, Alix, Chollet, Takeda, & Galzi, 1999). The construct was verified by DNA sequencing before transfection.
For stable transfection of the EGFP‐AT1 receptor, cells were plated 1 day before on 10‐cm dishes and transfected with 18 μg of plasmids using the calcium phosphate precipitation method (described in Vollmer et al., 1999). Selection with antibiotic started 2 days later with 600 μg·ml−1 G418 (Invitrogen). Clones were collected after 2 weeks of selection.
2.4. Internalization experiments
Internalization were carried out according to Gicquiaux et al. (2002). Briefly, cells expressing stably EGFP‐AT1 receptors were suspended in HEPES‐BSA buffer at a concentration of 106 cells·ml−1, and the suspension was separated into tubes of 1 ml. Cells were either untreated (control) or treated with AngII (10−9 M) or GSNO (2.10−6 M) for 0, 5, 15, and 30 min at 37°C. A series of cells were pretreated for 30 min with GSNO at 37°C before activation with AngII (same times and concentrations). At the end of the incubations, cells were fixed with PBS containing 1% paraformaldehyde and 0.1% sodium azide then pelleted by centrifugation (at 4°C and suspended in PBS 0.2% BSA on ice to block nonspecific antibody labelling). Immunolabelling of EGFP‐AT1 receptors expressed at the cell surface was done on intact cells using a monoclonal mouse anti‐GFP (Thermo Fisher Scientific Cat# A‐11120, RRID:AB_221568; 1/100 dilution) for 30 min at room temperature and secondary labelling done using a R‐phycoerythrin‐conjugated AffiniPure F (ab′)2 fragment goat anti‐mouse IgG (Thermo Fisher Scientific Cat# 12‐4010‐82, RRID:AB_11063706; 1/100 dilution). Phycoerythrin (PE) staining was quantified by flow cytometric analysis (20,000 cells per sample) on a FACSCalibur cytometer (BD FACSCalibur Flow Cytometry System, RRID:SCR_000401). Using FACStar data software, mean PE fluorescence intensity was calculated after subtraction of the mean PE fluorescence of nonspecific staining by the two antibodies measured on non‐transfected HEK cells. Results are expressed as percent variation of mean PE fluorescence between activated versus EGFP‐AT1 cells not exposed to agonists (100%) control for unwanted sources of variation such as non‐specific staining of non‐transfected cells or a slight drop in the number of cell surface receptors of the cell population as a function of culture time.
2.5. Biotin switch technique
S‐Nitrosated proteins were purified by the biotin switch technique as previously described with some adaptations (Belcastro et al., 2017; Forrester, Foster, Benhar, & Stamler, 2009).
EGFP‐AT1 receptor expressing HEK293 cells or untransfected HEK293 cells (as negative controls) were cultured for 2 days in T10 plates. Then, cells were lysed in 1 ml of cold 50 mM Tris (pH 6.8) buffer containing 0.15 M NaCl, 1% (v/v) Triton ×100, 0.1% (v/v) SDS, 1 mM EDTA, 0.1 mM neocuproine and protease inhibitor cocktail (EDTA‐free Roche). Briefly, S‐nitrosation was performed at 37°C on a wheel for 1 hr by 1 mM GSNO (or PBS as control). After a 10,000× g centrifugation for 15 min at 4°C, the free thiols in cell lysate supernatants were blocked with 50 mM of N‐ethylmaleimide for 50 min at 40°C. Samples were precipitated by acetone. Pellets were then incubated with 10 mM sodium ascorbate to liberate thiols from the S–NO bonds, and free new thiols were immediately biotinylated. As negative controls, sodium ascorbate was omitted in one sample treated or not with GSNO. Under such conditions, S–NO bonds are not reduced and S‐nitrosated proteins cannot be labelled by the biotin‐HPDP. Excess biotin‐HPDP was removed by acetone precipitation. Biotin‐HPDP‐labelled proteins were purified with High Capacity NeutrAvidin beads overnight on a wheel at 4°C. The following day, NeutrAvidinAgarose beads were washed three times in a high‐salt containing buffer (50 mM Tris pH 7.5, 0.6 M NaCl, 1% [v/v] Triton ×100, 0.1% [v/v] SDS, 1 mM EDTA, 0.1 mM neocuproine). Beads were suspended in a 2× Laemmli buffer (25 mM Tris pH 6, 8, 3 M urea, 1.5% SDS, 3% [v/v] 2‐mercaptoethanol) vortexed for 15 min at room temperature, heated for 20 min at 37°C, centrifuged for 3 min at 10,000× g. Biotin‐HPDP‐labelled proteins eluted from the beads were separated in 10% acrylamide gels then transferred onto PVDF membrane for 2 hr. The detection of the EGFP‐AT1 receptor proteins among the proteins that had been precipitated by streptavidin beads, indicating that the receptor was biotinylated during the biotin switch, was performed by immunoblotting with anti‐GFP‐HRP antibodies (Abcam Cat# ab6663, RRID:AB_305636): incubation at 1/25,000 overnight revelation with Immobilon Western HRP and image acquisition on a LAS4000 camera (GE‐Healthcare). The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.6. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology. Blinding of the operator could not be done as the various pretreatments could be easily distinguishable (colour of the solution for GSNO, impact of the pretreatment on basal MCA diameter such as dilatation induced by GSNO or SNP, or constriction induced by L‐NAME). The values of ID were measured automatically with no intervention by the operator (except to set the start of the recording), ensuring reliable and objective measurements. Statistical analysis was performed using Graphpad Prism version 6.00 (RRID:SCR_002798).
Vasoactive responses of the MCA are expressed as percentage changes in ID from the value measured during the washout preceding administration of the drug. This normalization enables control for unwanted sources of variation such as anatomical, differences in MCA diameter. For each MCA experiment, the initial sample size was n = 8 to take into account experimental losses based on the predetermined viability criteria described above and a final sample size of at least n = 5 was obtained. The only exception is the group receiving DMSO, for which the sample size was reduced to n = 3 (given the low variability in this control group and to avoid unnecessary animal deaths) and was thus excluded from the statistical analyses. For MT determination, the sample size is greater due to a higher variability and the subsequent inclusion of partial preliminary data for the control group (n = 18). Concentration–response curves to vasoactive agents were built for each MCA and half maximal effective concentration (EC50) was calculated using Hill's logistic equation:
where E min and E max are minimal and maximal response reached in each concentration–response curve. The value of the Hill slope for all experiments was 1.0 ± 0.1. Then −logEC50 values (pD2 values) were calculated.
For functional studies after pretreatments (PSS, GSH, SNP GSNO ± ascorbate, L‐NAME, or ODQ), differences in the responses to AngII ± PD123319, 5‐HT, PHE, or U46619 (mean ± SEM) were determined by two‐way ANOVAs (drug concentration, pretreatment) followed by a Bonferroni post‐test (P < 0.05). For AT1 receptor‐dependent MT, IDs (active or passive) of MT or AUC were compared using one‐way ANOVA (treatment) followed by a Bonferroni post‐test (P < 0.05).
For internalization studies, differences in the responses to GSNO, AngII ± GSNO were determined by two‐way ANOVAs (time, treatment) followed by a Bonferroni post‐test (P < 0.05).
2.7. Materials
All reagents were purchased from Sigma Chemical Company (St Louis, USA), U46619 from Enzo life Sciences (Farmingdale, USA), ODQ from Alfa Aesar (Haverhill, USA), sodium pentobarbital from Sanofi‐Aventis (Libourne, France), High Capacity NeutrAvidin beads and biotin‐HPDP from Fisher Scientific (Illkirch, France), anti‐GFP‐HRP antibodies from Abcam (Paris, France), Immobilon Western HRP from Promega (Charbonnières‐les‐Bains, France), primary monoclonal anti‐GFP and secondary goat anti‐mouse PE antibodies from Thermofisher (Waltham, USA). GSNO was prepared extemporaneously as previously described (Parent et al., 2013). All manipulations and assays involving GSH, ascorbate, and GSNO were performed under conditions of subdued light, in order to minimize light‐induced degradation.
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Christopoulos et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. NO acts specifically on AT 1 receptor signalling rather than on other G q11 or G i/G o signalling pathways
The data obtained on AngII‐mediated vasoconstriction are in accordance with the literature (De Silva et al., 2009; Vincent et al., 2005). GSNO pretreatment specifically reduced the AngII‐mediated vasoconstriction of the MCA (Figure 1). This was not linked to an increase in EC50 as GSNO completely abolished the response to AngII even at high concentrations. GSNO did not change the responses to other vasoconstrictors sharing the same Gq11 pathway (U46619, Phe) or other pathways (5‐HT; Figure 1). Moreover, the abolition of AngII vasoconstriction obtained in the presence of the AT2‐antagonist PD123319 confirmed a decrease of AT1 receptor‐mediated vasoconstriction rather than an increase of AT2‐mediated vasodilation (Figure 2).
Figure 1.
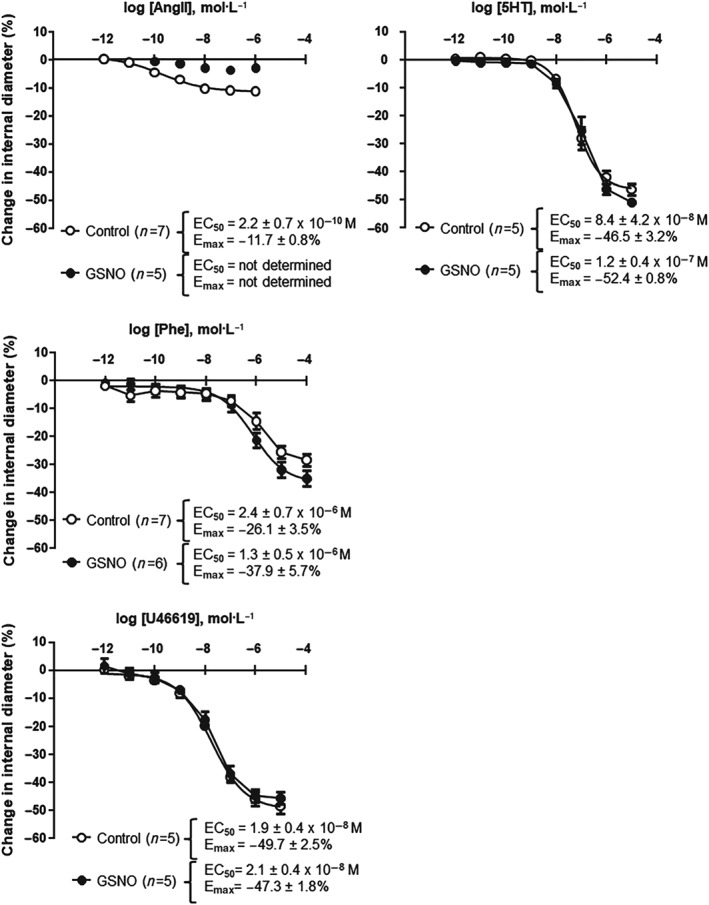
S‐nitrosoglutathione (GSNO) effect lies specifically on AT1 receptor signalling rather than on other Gq11 or Gi/Go signalling pathways. Contractile responses of isolated and perfused rat middle cerebral arteries to different vasoconstrictor agonists were measured. Concentration–response curves, EC50, and maximal effect (E max) values in response to (a) angiotensin II (AngII), α1‐adrenoceptor agonist phenylephrine (Phe), TxA2 agonist (U46619), and (b) 5‐HT)were obtained after exposure (30 min) to either physiological salt solution (control) or GSNO (2 x 10−6 mol·L−1), followed by a 1‐hr washout. Fitting to Hill's logistic equation was used. Values for EC50 (mol·L−1) and E max (%) are expressed as means ± SEM
Figure 2.
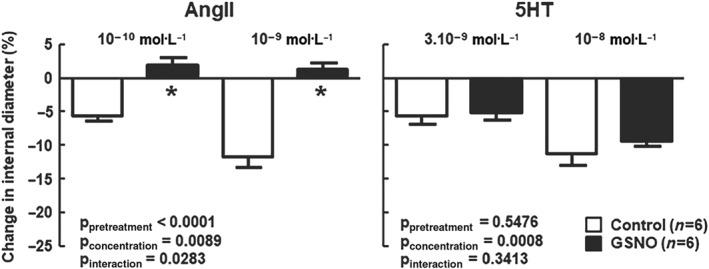
S‐nitrosoglutathione (GSNO) specifically abolishes AT1 receptor‐induced vasoconstriction. Specific AT1 receptor‐induced vasoconstriction was assessed in response to angiotensin II (AngII) in the presence of PD123319 (a selective AT2 receptor antagonist, 10−6 mol·L−1) and compared to 5‐HT after exposure (30 min) to either physiological salt solution (control) or GSNO (2 x 10−6 mol·L−1), followed by a 1‐hr washout. Changes in internal diameter (%) are expressed as means ± SEM. Two‐way ANOVAs (pretreatment, concentration) were used to calculate P values; *P < 0.05 versus control (Bonferroni post‐test)
3.2. GSNO abolishes AT1 receptor‐mediated agonist‐independent MT
As previously observed by others (Schleifenbaum et al., 2014), losartan, an inverse agonist of the AT1 receptor, shifted the MT‐pressure curve towards lower values (Figure 3a) and significantly reduced the AUC of the MT–pressure relationships versus controls (Figure 3b). This confirms the implication of AT1 receptor on MT. GSNO significantly reduced MT to a lower extent than that obtained with losartan alone (Figure 3a,b). The reduction of the AUC induced by GSNO + losartan was equivalent to that obtained by GSNO alone (Figure 3a,b) suggesting that GSNO decreased AT1 receptor‐dependent MT.
Figure 3.
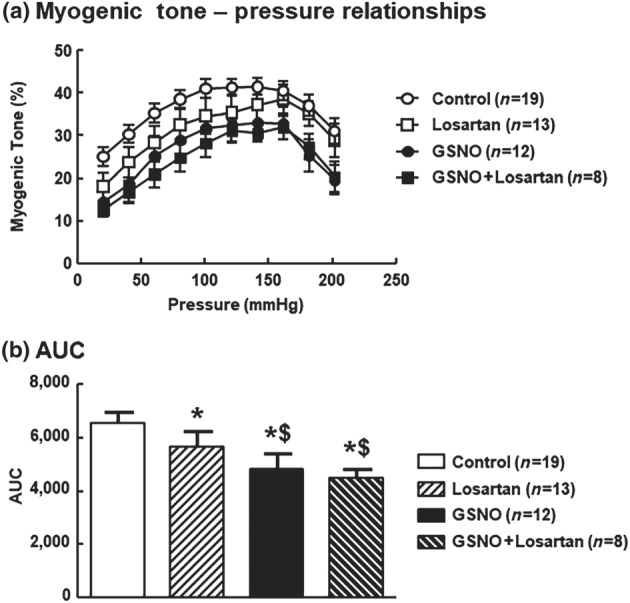
S‐nitrosoglutathione (GSNO) decreases AT1 receptor‐dependant myogenic tone. Impact of an exposure to GSNO (2 x 10−6 mol·L−1, 30 min followed by a 1‐hr washout), losartan (10−5 mol·L−1), or the combination GSNO + losartan on (a) myogenic tone and (b) AUC of myogenic tone of middle cerebral arteries isolated from Wistar rats. Values are expressed as means ± SEM. One‐way ANOVA (treatment) was used to calculate P values; *P < 0.05 versus control; $ P < 0.05 versus losartan; *P < 0.05 versus control (Bonferroni post‐test)
3.3. GSNO abolishes AT1 receptor functions through NO release
We ruled out any antioxidant properties of GSNO (via GSH release) as exposure to GSH did not change vasoconstriction induced by AngII (Figure 4a). Involvement of NO was confirmed as SNP (another NO donor) induced similar results to those produced by GSNO (Figure 4b). Involvement of endogenous NO was excluded as treatment with L‐NAME (endothelial NOS inhibitor) did not restore vasoconstriction to AngII following GSNO pretreatment (Figure 4c).
Figure 4.
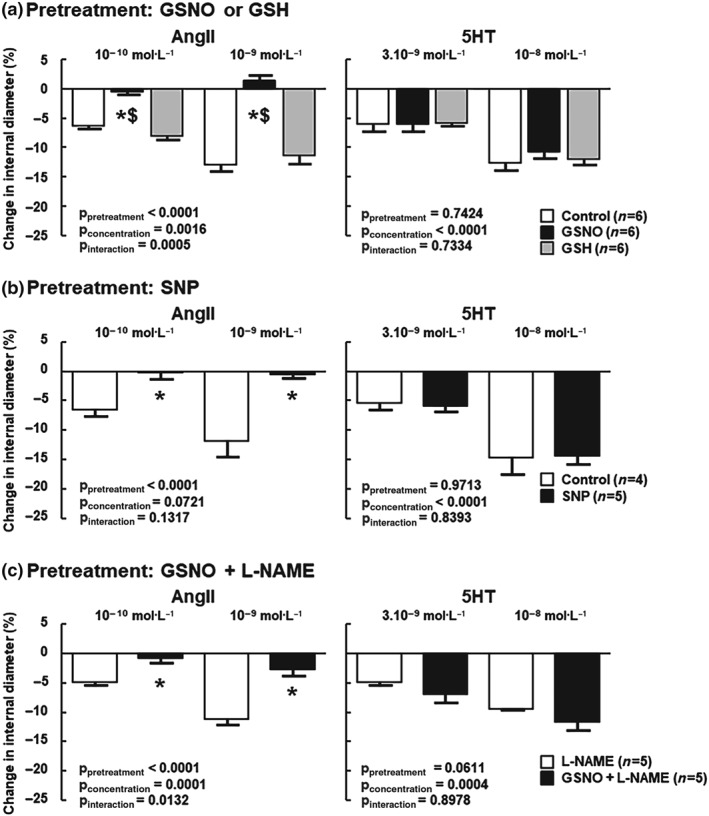
Exogenous NO induced by either S‐nitrosoglutathione (GSNO) or sodium nitroprusside (SNP) is responsible for the loss of AngII response rather than GSH or endogenous NO. In each panel, 5‐HT served as a positive control. (a) Middle cerebral arteries (MCA) were pretreated (30 min) with physiological salt solution (control), GSNO (2.10−6 mol·L−1), or GSH (2.10−6 mol·L−1), followed by a 1‐hr washout. (b) MCA were pretreated (30 min) with physiological salt solution (control) or SNP (2 x 10−6 mol·L−1), followed by a 1‐hr washout. (c) MCA were pretreated (30 min) with physiological salt solution (control) or GSNO, followed by a 1‐hr washout in the presence of L‐NAME ( 10−4 mol·L−1) throughout the experiment. Changes in internal diameter (%) are expressed as means ± SEM. Two‐way ANOVAs (pretreatment, concentration) were used to calculate P values; *P < 0.05 versus unexposed; $ P < 0.05 versus GSH (Bonferroni post‐test)
3.4. NO effects are mediated by a S‐nitrosation mechanism
As most effects of NO are mediated by sGC nitrosylation, its implication was assessed using ODQ, a sGC inhibitor. ODQ did not prevent the loss of AngII vasoconstriction following GSNO pretreatment thus excluding involvement of sGC nitrosylation (Figure 5a). ODQ specifically decreased the response to 5‐HT. However, this response was restored at higher concentration (10−6 mol·L−1; Figure S2). No effect of DMSO (i.e., ODQ solvent) was observed on either AngII or 5‐HT responses.
Figure 5.
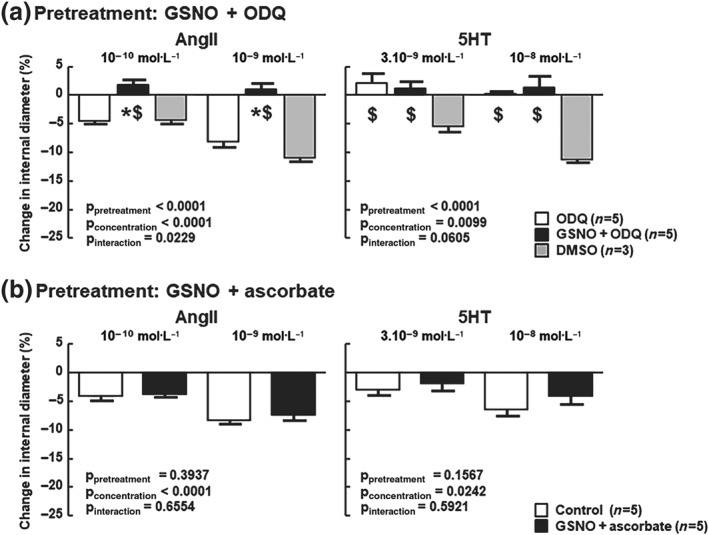
The effect of GSNO is independent of sGC activity as it is mediated by S‐nitrosation. (a) MCA were pretreated (30 min) with physiological salt solution or with S‐nitrosoglutathione (GSNO, 2 x 10−6 mol·L−1) followed by a 1‐hr washout, then ODQ (10−5 mol·L−1) was added throughout the experiment. DMSO (i.e., ODQ solvent was used as control). (b) After pretreatment (30 min) with physiological salt solution (control) or with GSNO, MCA were exposed to sodium ascorbate (5 x 10−6 mol·L−1, 20 min) to specifically reduce the S–NO bonds, followed by a 40‐min washout. Control MCA underwent the same ascorbate washout as ascorbate alters the vasoactive responses to both AngII (Leclerc et al., 2008) and 5‐HT (Muakkassah‐Kelly, Andresen, Shih, & Hochstein, 1982). Changes in internal diameter (%) are expressed as means ± SEM. Two‐way ANOVAs (pretreatment, concentration) were used to calculate P values; *P < 0.05 versus unexposed; $ P < 0.05 versus DMSO (Bonferroni post‐test)
The response of MCA to AngII was restored when ascorbate (used to reduce S–NO bond) was added during the washout after GSNO exposure (Figure 5b) indicating that S‐nitrosation is involved in the decrease of the AngII response following GSNO exposure. No change was observed for 5‐HT.
3.5. AT 1 receptor cell surface localization is not modified by NO
Due to the lack of specific antibodies directed against AT1 receptor (Benicky, Hafko, Sanchez‐Lemus, Aguilera, & Saavedra, 2012; Bouressam, Lartaud, Dupuis, & Lecat, 2018) precluding experiments on isolated MCA, the impact of GSNO on cell surface localization of AT1 receptor was assessed in a heterologous expression system: EGFP‐tagged AT1 receptors expressed in HEK293 cells. Using fluorescence cell sorting analysis, the cell population was controlled to have a homogenous expression of the receptors as detected by a single Gaussian peak of GFP fluorescence as compared with non‐transfected HEK cells (Figure 6a green line vs. black line, respectively). When exposed to GSNO alone (2.10−6 mol·L−1) at 37°C, no variation of cell surface receptors was observed as detected by monoclonal anti‐GFP antibody labelling followed by secondary anti‐mouse antibody coupled to PE (Figure 6b, orange line and Figure 6c). The treatment of EGFP‐AT1 receptor transfected cells with 10−9 mol·L−1 of AngII induced a rapid decrease in the amount of receptors detected at the cell surface of intact cells (measurement of PE fluorescence intensities of the cell population, Figure 6b) while the total amount of receptors remained constant (measurement of GFP fluorescence intensities of the cell population, Figure 6a). The decrease of cell surface receptors upon activation is due to internalization of the receptor. Activated receptors are internalized with a maximal amplitude of 55–60% that is reached almost after 5 min of activation (Figure 6b, blue line and Figure 6c). When cells were treated with GSNO followed by incubation with AngII, the same decrease of PE fluorescence was observed as compared with AngII alone (Figure 6b red line vs. blue line, respectively) indicating that GSNO did not alter AT1 receptor internalization amplitudes (55 vs. 53%, ns; Figure 6b,c). Similar results were obtained with cells exposed to AngII for 5, 15, or 30 min indicating that AT1 receptor internalization kinetics are unaltered as well by GSNO pretreatment (Figure 6c).
Figure 6.
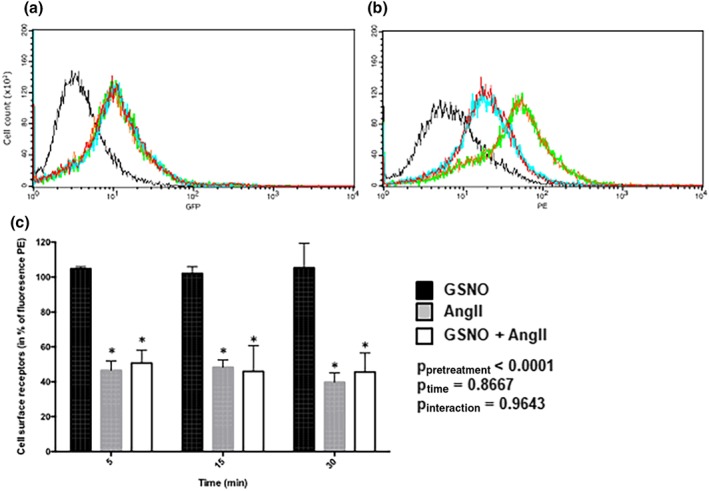
GSNO does not modify AT1 receptor‐internalization kinetics or amplitudes. Cell surface EGFP‐AT1 receptor receptors were evaluated by FACS using anti‐GFP antibodies and secondary phycoerythrin‐labelled antibodies (PE). (a) Cellular GFP or (b) cellular PE fluorescence intensities of non‐transfected HEK cells (black line) and EGFP‐AT1 receptor cells (green line) or EGFP‐AT1 receptor cells activated with either AngII (30 min, 10−9 M—blue line), GSNO (30 min, 2 x 10−6 M—orange line), or GSNO (30 min, 2 x 10−6 M) together with AngII (30 min,10−9 M—red line). (c) Variation of EGFP‐AT1 receptor cell surface receptors normalized versus non activated EGFP‐AT1 receptor cells. Variations in cell surface receptors are expressed as means ± SEM of the mean fluorescence of PE. Two‐way ANOVA (time, treatment) were used to calculate P values; *P < 0.05 versus GSNO (Bonferroni post‐test). n = 2 separate determinations, four points per condition
3.6. GSNO induces S‐nitrosation of AT1 receptor
To directly test if GSNO is able to S‐nitrosate the AT1 receptor, we used the biotin switch technique on EGFP‐tagged AT1 receptor expressing HEK293 cells. This three‐step procedure relies on the ability of ascorbate to selectively reduce S‐nitrosothiol to a free sulfhydryl group, which is subsequently conjugated with a biotin adduct. The biotinylated proteins can then be enriched with avidin‐affinity chromatography and analysed by conventional western blotting with antibodies against the target protein, here using antibodies targeting EGFP because of the lack of specific anti‐AT1 receptor antibodies (Benicky et al., 2012; Bouressam et al., 2018). After the biotin switch, EGFP‐AT1 receptor was specifically detected on western blot among the biotinylated proteins precipitated on streptavidin beads (Figure 7, Lane 7). When lysates were not treated with GSNO (Figure 7, Lanes 2 and 3) or not incubated with ascorbate after GSNO treatment (thus preventing SNO bonds to be labelled by biotin, Figure 7, Lane 5), there was no EGFP‐AT1 receptor detected on the western blot. These results indicate that EGFP‐AT1 receptor is S‐nitrosated by GSNO.
Figure 7.
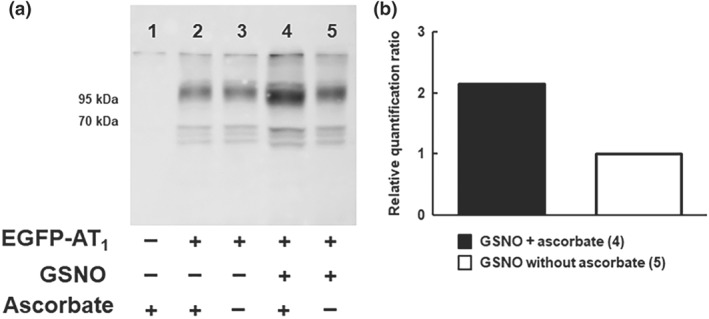
GSNO induces S‐nitrosation of the AT1 receptor. (a) Representative Western blot with anti‐GFP‐HRP antibodies. Cell lysates exposed to S‐nitrosoglutathione (GSNO, 1 mM, 60 min, 37°C) underwent purification of S‐nitrosated proteins with the biotin switch method. Lane 1 is a negative control conducted with untransfected HEK cells. Lanes 2 and 3 are negative controls without GSNO exposure. In Lane 5, the removal of ascorbate prevents SNO bonds being labelled by biotin and thus ensures the specificity of the biotin switch towards S‐nitrosated proteins. (b) Relative quantification of S‐nitrosated EGFP‐AT1 receptor (Lane 4 vs. Lane 5)
4. DISCUSSION
In the present work, we demonstrate that GSNO pretreatment of cerebral arteries specifically abolishes both ligand‐dependent (AngII) and ligand‐independent (MT) AT1 receptor‐mediated functions. Reduced AT1 receptor signalling after GSNO pretreatment may depend on S‐nitrosation, as such a pretreatment of vessels has been reported to induce S‐nitrosation of vascular proteins (Alencar et al., 2003).
We decipher the mechanisms involved using pharmacological tools. First, we argue that the GSNO effect is linked to NO (as SNP, another NO donor, produces similar results) rather than to its antioxidant properties. Indeed, ROS production is an important part of AngII signalling in cerebral arteries contributing to vasoconstriction (De Silva & Faraci, 2013). The effect of GSNO on AngII response could thus be related to a reduced ROS availability after AngII stimulation. The antioxidant properties of GSNO can be related to the release of NO and/or GSH. NO has been shown to reduce ROS production, mainly by inducing the S‐nitrosation of Nox5 (Qian et al., 2012). As ROS production induced by AngII is rather related to activation of Nox1, Nox2, and Nox4 (De Silva & Faraci, 2013), the impact of NO on AngII‐induced ROS production remains unlikely. The lack of effect of GSH pretreatment also rules out any reduction in ROS availability as the main mechanism responsible for the reduced AngII response. Then, we ruled out any involvement of endogenous NO production using the eNOS inhibitor L‐NAME. The abolition of AngII‐induced vasoconstriction of MCA was obtained using 2.10−6 mol·L−1 of GSNO, a concentration very close to its vasodilating EC50 (2.8 ± 0.4 10−6 mol·L−1, Figure S3). Vasodilation involves the activation of the sGC/GMPc pathway through sGC nitrosylation. In our experiments, the 1‐hr washout enables diameters to return to baseline, suggesting that nitrosylation‐dependent activation of sGC disappears and could therefore not account for the reduced AngII constrictive response (Figure S4). This was further confirmed by the use of the sGC inhibitor, ODQ, which does not prevent the abolition of AngII responses, thus suggesting the involvement of other mechanisms.
As Alencar et al. previously showed, using a similar protocol (Alencar et al., 2003), that S‐nitrosation of proteins remains despite a 1‐hr washout, we tested if S‐nitrosation could be involved in the reduced AngII response. Sodium ascorbate treatment, known to specifically reduce the S–NO bond (Forrester et al., 2009; Holmes & Williams, 2000), prevents the abolition of AngII vasoconstriction, thus suggesting that S‐nitrosation contributes to this effect.
The abolition of AngII response following GSNO pretreatment is consecutive to a decreased AT1‐constriction rather than to an increased AT2‐related dilatation, as shown in the presence of AT2 antagonist. This reduced vasoconstriction is not consecutive to a GSNO‐induced alteration of the contractile apparatus, as 5‐HT‐induced vasoconstriction remained unaffected. While the 5‐HT1B receptor involved in cerebrovascular constriction (Watts et al., 2012) is coupled to a Gi/Go G‐protein (Lin et al., 2002), the AT1 receptor, as well as the TxA2 and the α1‐adrenergic receptors are coupled to the Gq11 G‐protein (Schleifenbaum et al., 2014). As GSNO specifically abolished AngII, but not U46619 nor Phe‐induced vasoconstriction, this demonstrates that GSNO produces an alteration upstream of the G‐protein Gq11 and specifically on the AT1 receptor signalling pathway.
Three hypotheses can explain our results. Decrease of AngII affinity for AT1 receptor (Leclerc et al., 2006), uncoupling of G‐protein, and/or changes in AT1 receptor membrane expression. We ruled out the first hypothesis as AngII‐independent response, MT, was also reduced by GSNO pretreatment.
Several studies have previously demonstrated that S‐nitrosation can lead to uncoupling of G‐proteins from their receptors, thus decreasing their activity (Hess, Lin, Freeman, & Norden, 1994; Miyamoto, Laufs, Pardo, & Liao, 1997). Activity of GPCRs could be either up‐regulated (ACh receptor M2/M4) or down‐regulated (purine receptor P2Y12, lysophosphatidic acid receptor LPA1, or cannabinoid receptor CB1) by NO donors such as GSNO or CysNO (Kokkola, Savinainen, Monkkonen, Retamal, & Laitinen, 2005).
Several of our results point towards a probable S‐nitrosation of the AT1 receptor itself that would affect its conformational dynamics inhibiting its stabilization in the active state.
First, a decrease in AngII affinity for AT1 receptor was observed on transfected HEK cells overexpressing AT1 receptor in relation to a probable S‐nitrosation on its Cys289 (Leclerc et al., 2006). Inhibition of isomerization into an active form with a better affinity for agonist could also lead to a decrease in apparent affinity of the receptor, a phenomenon that has been previously described for a mutant of the tachykinin NK2 receptor (Lecat, Bucher, Mely, & Galzi, 2002). This would explain why, in our study, both ligand‐dependent and independent functions are impaired.
Effectively, we report that GSNO also abolished the AngII‐independent response of the AT1 receptor, that is, its involvement in the setting of the cerebrovascular MT. It has been proposed that the molecular mechanism of AT1 receptor activation upon mechanical stress is mediated by a change in the conformation of the receptor in which the transmembrane domain 7 of the receptor shifts into the agonist‐binding pocket and triggers activation independently of agonist (Yasuda et al., 2008). This is why losartan, an inverse agonist that stabilizes the receptor in its inactive form, was shown to block the AT1 receptor's involvement in the setting of the cerebrovascular MT. We observed that pretreatment with GSNO induced a stronger decrease in MT than treatment with losartan alone. When losartan was added to GSNO pretreatment, no further decrease was observed. This cannot be explained by a complete blockade of MT by GSNO (MT remaining relatively high—29% at 80 mmHg—after GSNO pretreatment) and thus suggests that AT1 receptor‐mediated MT was already abolished by GSNO. Thus, these results point towards the hypothesis that GSNO alters the conformational dynamics of the AT1 receptor itself. The stronger decrease in MT compared to losartan alone is most likely related to the impact of GSNO on other mechanosensors, such as other GPCRs (Kauffenstein, Laher, Matrougui, Guerineau, & Henrion, 2012).
The third hypothesis to explain our results relies on changes in the amount of AT1 receptors at the cell surface due to variation in expression, exocytosis, or internalization upon GSNO treatment. It seems unlikely that 30 min exposure to GSNO might affect AT1 receptor transcription or gene expression as these require a much longer treatment (18–24 hr; that could not be detected within the time schedule of our experiments) or higher concentrations (100‐fold more; Cahill, Redmond, Foster, & Sitzmann, 1995; Ichiki et al., 1998). However, general actors of receptor endocytosis, such as β‐arrestins, clathrin, and/or dynamin (Gaborik et al., 2001), were shown to be regulated by S‐nitrosation (Daaka, 2012).
Because there are no antibodies specifically targeting AT1 receptor on the market (Bouressam et al., 2018), we could not test in MCA the two remaining hypotheses, that is, S‐nitrosation of the receptor itself that alters its isomerization properties or the impact of GSNO on the amount of receptor at the cell surface. To directly prove that the AT1 receptor can be S‐nitrosated by GSNO, we had to rely on a heterologous expression system. We chose HEK293 cells, as Leclerc et al. did (2006), and used the biotin switch technique coupled to western blot to purify and identify S‐nitrosated proetins (Belcastro et al., 2017). We were able to detect S‐nitrosated EGFP‐tagged AT1 receptors after GSNO treatment of cell lysates. More sensitive methods, such as the DAN‐Hg2+ method, enable the quantification of intracellular S‐nitrosothiols directly on cells but do not allow the identification of a specific target protein. It would be interesting in the future to test S‐nitrosation of the tagged‐receptor expressed in a cellular context closer to its physiological environment.
Our results using EGFP‐AT1 receptor transfected HEK293 cells show that AngII induces AT1 receptor internalization whereas GSNO alone has no effect. Pretreatment of the cells with GSNO did not affect the amplitude or kinetics of activated AT1 receptor internalization. Whether GSNO pretreatment could affect proteins such as ATRAP (Tanaka et al., 2005) or ARAP1 (Guo, Chenier, Tardif, Orlov, & Inagami, 2003), which respectively promote internalization or membrane expression of AT1 receptors, is thus unlikely.
Interestingly, the fact that GSNO alters AT1 receptor‐induced vasoconstriction (i.e., inhibits the Gq signalling pathway) without affecting AT1 receptor internalization (i.e., the β‐arrestin pathway) could also be explained by an alteration in the receptor conformational dynamics. Molecular dynamics simulation of the basal and the active states of AT1 receptor suggests that activation of the Gq11 pathway requires a specific conformational transition (i.e., a movement of the 7th transmembrane domain) to reach an active state, which is stabilized by the agonist. However, surprisingly, the activation of the β‐arrestin pathway, leading to internalization, requires the receptor to be at a basal state (Cabana et al., 2015). This new concept that internalization and activation of AT1 receptors are independently related to different conformational states has been confirmed by the recently developed biased agonist, TRV027, which activates the β‐arrestin pathway while inhibiting the G‐protein pathway (Boerrigter et al., 2011; Boerrigter, Soergel, Violin, Lark, & Burnett, 2012). Further studies are required to demonstrate whether S‐nitrosation of the AT1 receptor, which involves Cys289 (Leclerc et al., 2006) located within the 7th transmembrane domain, impairs these conformational dynamics.
Whether this blockade of the AngII‐induced vasoconstriction occurs in physiological conditions and/or in vivo remains to be established. For example, one could speculate that increases in endogenous NO synthesis following ACh stimulation could reach the AT1 receptor and thus decrease its activity. However, to the best of our knowledge, a reduction in AngII‐induced vasoconstriction following ACh stimulation (a commonly used procedure in pharmacological studies to assess the viability of the endothelium) has never been reported. However, this lack of effect of ACh on AT1 receptor function could be related to a compartmentalization of ACh‐induced NO production targeting directly sGC and vascular smooth muscle cells. Indeed, several reports highlight the pivotal role of subcellular localization of eNOS (Biwer et al., 2016) and of transporters such as Hbα (Straub et al., 2012) or connexin‐formed channels (Figueroa, Lillo, Gaete, Riquelme, & Saez, 2013) for NO to exert its dilating action. This well‐regulated NO compartmentation could preclude ACh‐induced NO synthesis to diffuse and reach proteins such as the AT1 receptor.
In conclusion, we demonstrate a new and unexplored regulation pathway of the cerebrovascular renin angiotensin system: At the cerebrovascular level, GSNO pretreatment induces a decrease in AT1 agonist‐dependent or agonist‐independent vasoconstriction which may depend on AT1 receptor S‐nitrosation. These results were obtained on the MCA, the artery most frequently involved in ischaemic stroke (Hedna et al., 2013), and thus suggest that the redox regulation via S‐nitrosation of the renin angiotensin system components might become an important therapeutic target.
Our preliminary data also indicate that the decrease in AngII‐mediated vasoconstriction produced by GSNO is equivalent in arteries from spontaneously hypertensive rats (SHR) and in those from their normotensive controls (Figure S5). Therefore, besides its neuroprotective properties (Khan et al., 2005), GSNO, if administered after stroke, a situation in which AT1 receptor‐dependent vasoconstriction is exacerbated (Stenman & Edvinsson, 2004) and NO availability is reduced (Chrissobolis, Miller, Drummond, Kemp‐Harper, & Sobey, 2011), could be beneficial by reducing AT1 receptor signalling. We recently demonstrated that the microparticles loaded with GSNO protected the brain from the consequences of ischaemia (Parent et al., 2015). Further experiments are needed to link this cerebroprotection to the S‐nitrosation in AT1 receptor signalling pathway.
4.1. Innovation
AngII is a key regulator of the cerebral circulation. The fine tuning of its AT1 receptor (constrictor) is of critical importance in many pathophysiological processes, such as stroke. The present study is the first to identify S‐nitrosation (the covalent bond between NO and cysteine residues) as a potential mechanism involved in the control of the AT1 receptor‐dependent cerebrovascular functions. GSNO exposure leads to reduced AngII vasoconstriction and MT and might thus represent a new therapeutic approach in cerebrovascular diseases, in which excessive vasoconstriction may lead to deleterious consequences.
AUTHOR CONTRIBUTIONS
M.L.B., C.P.S., I.L., and F.D. conceived the ex vivo experiment and interpreted the results. M.L.B. and S.L. conceived the in vitro experiment. M.L.B. carried out all the experiment. S.L. and A.R. respectively conducted internalization and myogenic tone experiment. C.G. helped for the biotin switch experiments and analysis. All authors equally contributed to the final version of the manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1
Ex vivo exposure to GSNO decreases AngII‐mediated vasoconstriction independently of the order of administration of the drugs. Vasoconstriction (% change in internal diameter) in response to angiotensin II (AngII, 10‐10 mol. L‐1, open bars) and serotonin (5HT, 3.10‐9 mol. L‐1, full bars) in middle cerebral arteries of Wistar rats that were unexposed (panel A, n=7) or exposed (panel B, n=9; panel C, n=2) to Snitrosoglutathione (GSNO, 2.10‐6 mol. L‐1, 30 min followed by 1 hour washout). In contrast with panels A and B, in panel C 5HT was applied before AngII. Results are shown as means ± sem.
Figure S2. Effect of ODQ and DMSO on serotonin vasoconstriction. MCA were pretreated or not with GSNO and 1H‐[1,2,4]Oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ, 10‐5 mol/L) was added throughout the experiments. Dimethyl sulfoxide (DMSO, i.e. ODQ solvent, was used as control). Changes in internal diameter (%) are expressed as means ± SEM. Two‐way ANOVA (pretreatment, concentration) were used to calculate p values; *: p<0.05 vs unexposed; $: p<0.05 vs DMSO (Bonferroni post‐test).
Figure S3. Concentration response curves to S‐nitrosoglutathione. Concentration‐response curves, half maximal effective concentration (EC50) and maximal effect (Emax) in response to S‐nitrosoglutathione (GSNO) of middle cerebral arteries isolated from Wistar rats. Fitting to Hill's logistic equation was used. Values for EC50 (mol/L) and Emax (%) are expressed as means ± SEM. n = 6 middle cerebral arteries.
Figure S4. Impact of S‐nitrosoglutathione (GSNO) pretreatment on internal diameter. Internal diameters of rat middle cerebral arteries were measured before (baseline, open bars), after 30 min of exposure (full bars) to GSNO (2.10‐7 mol/L, 2.10‐6 mol/L or 2.10‐5 mol/L) or physiological salt solution (PSS for controls) and following the on‐hour wash‐out (grey bars). *: p<0.05 vs baseline; $: p<0.05 vs GSNO (paired Student's t‐test). Values are expressed as means ± SEM.
Figure S5. Ex vivo exposure to GSNO decreases Angiotensin II‐mediated vasoconstriction in middle cerebral arteries from Spontaneously Hypertensive Rats. Vasoconstriction of middle cerebral arteries from Wistar Kyoto (WKY) rats or Spontaneous Hypertensive rats (SHR) in response to angiotensin II (AngII, panel A) and serotonin (5HT, panel B). Vessels were unexposed (open bar) or exposed (full bars) to S‐nitrosoglutathione (GSN0, 2.10‐6 mol/L), 30 min followed by a 1‐hour wash out. Changes in internal diameter (%) are expressed as means ± SEM. n=4‐6 middle cerebral arteries per condition.
ACKNOWLEDGEMENTS
The work was funded by the French Ministry of Education, Research and Technology (Paris, France, EA3452). The authors thank José Metzger for help with the vasoactivity experiments, Isabelle Fries for help with preliminary internalization experiments, Jeremy Garwood for English editing, and Prof. Jacques Haiech, Prof. Marie‐Claude Kilhoffer (University of Strasbourg), and PCBIS UMS3286 CNRS‐University of Strasbourg for the vector encoding fluorescent AT1 receptors. The authors acknowledge support of CITHEFOR by the “Impact Biomolecules” project of the “Lorraine Université d'Excellence” (Investissements d'avenir—ANR).
Bouressam M‐L, Lecat S, Raoul A, et al. S‐nitrosoglutathione inhibits cerebrovascular angiotensin II‐dependent and ‐independent AT1 receptor responses: A possible role of S‐nitrosation. Br J Pharmacol. 2019;176:2049–2062. 10.1111/bph.14644
REFERENCES
- Alencar, J. L. , Lobysheva, I. , Chalupsky, K. , Geffard, M. , Nepveu, F. , Stoclet, J. C. , & Muller, B. (2003). S‐Nitrosating nitric oxide donors induce long‐lasting inhibition of contraction in isolated arteries. The Journal of Pharmacology and Experimental Therapeutics, 307, 152–159. 10.1124/jpet.103.052605 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The concise guide to pharmacology 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174(Suppl 1), S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). THE CONCISE GUIDE TO PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcastro, E. , Wu, W. , Fries‐Raeth, I. , Corti, A. , Pompella, A. , Leroy, P. , … Gaucher, C. (2017). Oxidative stress enhances and modulates protein S‐nitrosation in smooth muscle cells exposed to S‐nitrosoglutathione. Nitric Oxide, 69, 10–21. 10.1016/j.niox.2017.07.004 [DOI] [PubMed] [Google Scholar]
- Benicky, J. , Hafko, R. , Sanchez‐Lemus, E. , Aguilera, G. , & Saavedra, J. M. (2012). Six commercially available angiotensin II AT1 receptor antibodies are non‐specific. Cellular and Molecular Neurobiology, 32, 1353–1365. 10.1007/s10571-012-9862-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biwer, L. A. , Taddeo, E. P. , Kenwood, B. M. , Hoehn, K. L. , Straub, A. C. , & Isakson, B. E. (2016). Two functionally distinct pools of eNOS in endothelium are facilitated by myoendothelial junction lipid composition. Biochimica et Biophysica Acta, 1861, 671–679. 10.1016/j.bbalip.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerrigter, G. , Lark, M. W. , Whalen, E. J. , Soergel, D. G. , Violin, J. D. , & Burnett, J. C. Jr. (2011). Cardiorenal actions of TRV120027, a novel β‐arrestin‐biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: A novel therapeutic strategy for acute heart failure. Circulation. Heart Failure, 4, 770–778. 10.1161/CIRCHEARTFAILURE.111.962571 [DOI] [PubMed] [Google Scholar]
- Boerrigter, G. , Soergel, D. G. , Violin, J. D. , Lark, M. W. , & Burnett, J. C. Jr. (2012). TRV120027, a novel β‐arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circulation. Heart Failure, 5, 627–634. 10.1161/CIRCHEARTFAILURE.112.969220 [DOI] [PubMed] [Google Scholar]
- Bouressam, M. L. , Lartaud, I. , Dupuis, F. , & Lecat, S. (2018). No answer to the lack of specificity: Mouse monoclonal antibody targeting the angiotensin II type 1 receptor AT1 fails to recognize its target. Naunyn‐Schmiedeberg's Archives of Pharmacology, 391, 883–889. 10.1007/s00210-018-1522-4 [DOI] [PubMed] [Google Scholar]
- Briones, A. M. , Alonso, M. J. , Marin, J. , & Salaices, M. (1999). Role of iNOS in the vasodilator responses induced by l‐arginine in the middle cerebral artery from normotensive and hypertensive rats. British Journal of Pharmacology, 126, 111–120. 10.1038/sj.bjp.0702281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabana, J. , Holleran, B. , Leduc, R. , Escher, E. , Guillemette, G. , & Lavigne, P. (2015). Identification of distinct conformations of the angiotensin‐II type 1 receptor associated with the Gq/11 protein pathway and the β‐arrestin pathway using molecular dynamics simulations. The Journal of Biological Chemistry, 290, 15835–15854. 10.1074/jbc.M114.627356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill, P. A. , Redmond, E. M. , Foster, C. , & Sitzmann, J. V. (1995). Nitric oxide regulates angiotensin II receptors in vascular smooth muscle cells. European Journal of Pharmacology, 288, 219–229. 10.1016/0922-4106(95)90197-3 [DOI] [PubMed] [Google Scholar]
- Chrissobolis, S. , Miller, A. A. , Drummond, G. R. , Kemp‐Harper, B. K. , & Sobey, C. G. (2011). Oxidative stress and endothelial dysfunction in cerebrovascular disease. Frontiers in Bioscience, 16, 1733–1745. 10.2741/3816 [DOI] [PubMed] [Google Scholar]
- Daaka, Y. (2012). S‐Nitrosylation‐regulated GPCR signaling. Biochimica et Biophysica Acta, 1820, 743–751. 10.1016/j.bbagen.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva, T. M. , Broughton, B. R. , Drummond, G. R. , Sobey, C. G. , & Miller, A. A. (2009). Gender influences cerebral vascular responses to angiotensin II through Nox2‐derived reactive oxygen species. Stroke, 40, 1091–1097. 10.1161/STROKEAHA.108.531707 [DOI] [PubMed] [Google Scholar]
- De Silva, T. M. , & Faraci, F. M. (2013). Effects of angiotensin II on the cerebral circulation: Role of oxidative stress. Frontiers in Physiology, 3, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener, H. C. (2009). Preventing stroke: The PRoFESS, ONTARGET, and TRANSCEND trial programs. Journal of Hypertension. Supplement, 27, S31–S36. 10.1097/01.hjh.0000357906.60778.7f [DOI] [PubMed] [Google Scholar]
- Dupuis, F. , Atkinson, J. , Limiñana, P. , & Chillon, J. M. (2005). Comparative effects of the angiotensin II receptor blocker, telmisartan, and the angiotensin‐converting enzyme inhibitor, ramipril, on cerebrovascular structure in spontaneously hypertensive rats. Journal of Hypertension, 23, 1061–1066. 10.1097/01.hjh.0000166848.95592.a5 [DOI] [PubMed] [Google Scholar]
- Figueiredo‐Freitas, C. , Dulce, R. A. , Foster, M. W. , Liang, J. , Yamashita, A. M. , Lima‐Rosa, F. L. , … Pinto, J. R. (2015). S‐Nitrosylation of sarcomeric proteins depresses myofilament Ca2+ sensitivity in intact cardiomyocytes. Antioxidants & Redox Signaling, 23, 1017–1034. 10.1089/ars.2015.6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa, X. F. , Lillo, M. A. , Gaete, P. S. , Riquelme, M. A. , & Saez, J. C. (2013). Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin‐based channels. Neuropharmacology, 75, 471–478. 10.1016/j.neuropharm.2013.02.022 [DOI] [PubMed] [Google Scholar]
- Forrester, M. T. , Foster, M. W. , Benhar, M. , & Stamler, J. S. (2009). Detection of protein S‐nitrosylation with the biotin‐switch technique. Free Radical Biology & Medicine, 46, 119–126. 10.1016/j.freeradbiomed.2008.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulquier, S. , Lartaud, I. , & Dupuis, F. (2014). Impact of short‐term treatment with telmisartan on cerebral arterial remodeling in SHR. PLoS ONE, 9, e110766 10.1371/journal.pone.0110766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaborik, Z. , Szaszak, M. , Szidonya, L. , Balla, B. , Paku, S. , Catt, K. J. , … Hunyady, L. (2001). β‐arrestin‐ and dynamin‐dependent endocytosis of the AT1 angiotensin receptor. Molecular Pharmacology, 59, 239–247. 10.1124/mol.59.2.239 [DOI] [PubMed] [Google Scholar]
- Gicquiaux, H. , Lecat, S. , Gaire, M. , Dieterlen, A. , Mely, Y. , Takeda, K. , … Galzi, J. L. (2002). Rapid internalization and recycling of the human neuropeptide Y Y1 receptor. The Journal of Biological Chemistry, 277, 6645–6655. 10.1074/jbc.M107224200 [DOI] [PubMed] [Google Scholar]
- Guo, D. F. , Chenier, I. , Tardif, V. , Orlov, S. N. , & Inagami, T. (2003). Type 1 angiotensin II receptor‐associated protein ARAP1 binds and recycles the receptor to the plasma membrane. Biochemical and Biophysical Research Communications, 310, 1254–1265. 10.1016/j.bbrc.2003.09.154 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedna, V. S. , Bodhit, A. N. , Ansari, S. , Falchook, A. D. , Stead, L. , Heilman, K. M. , & Waters, M. F. (2013). Hemispheric differences in ischemic stroke: Is left‐hemisphere stroke more common? J Clin Neurol, 9, 97–102. 10.3988/jcn.2013.9.2.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess, D. T. , Lin, L. H. , Freeman, J. A. , & Norden, J. J. (1994). Modification of cysteine residues within Go and other neuronal proteins by exposure to nitric oxide. Neuropharmacology, 33, 1283–1292. 10.1016/0028-3908(94)90028-0 [DOI] [PubMed] [Google Scholar]
- Holmes, A. J. , & Williams, D. L. H. (2000). Reaction of ascorbic acid with S‐nitrosothiols: Clear evidence for two distinct reaction pathways. Journal of the Chemical Society, Perkin Transactions 2, 1639–1644. 10.1039/b004028m [DOI] [Google Scholar]
- Hunyady, L. , Catt, K. J. , Clark, A. J. , & Gaborik, Z. (2000). Mechanisms and functions of AT1 angiotensin receptor internalization. Regulatory Peptides, 91, 29–44. 10.1016/S0167-0115(00)00137-3 [DOI] [PubMed] [Google Scholar]
- Ichiki, T. , Usui, M. , Kato, M. , Funakoshi, Y. , Ito, K. , Egashira, K. , & Takeshita, A. (1998). Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension, 31, 342–348. 10.1161/01.HYP.31.1.342 [DOI] [PubMed] [Google Scholar]
- Kagiyama, T. , Kagiyama, S. , & Phillips, M. I. (2003). Expression of angiotensin type 1 and 2 receptors in brain after transient middle cerebral artery occlusion in rats. Regulatory Peptides, 110, 241–247. 10.1016/S0167-0115(02)00223-9 [DOI] [PubMed] [Google Scholar]
- Kauffenstein, G. , Laher, I. , Matrougui, K. , Guerineau, N. C. , & Henrion, D. (2012). Emerging role of G protein‐coupled receptors in microvascular myogenic tone. Cardiovascular Research, 95, 223–232. 10.1093/cvr/cvs152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M. , Sekhon, B. , Giri, S. , Jatana, M. , Gilg, A. G. , Ayasolla, K. , … Singh, I. (2005). S‐Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. Journal of Cerebral Blood Flow and Metabolism, 25, 177–192. 10.1038/sj.jcbfm.9600012 [DOI] [PubMed] [Google Scholar]
- Khan, M. , & Singh, I. (2016). Inhibition of the AMPK/nNOS pathway for neuroprotection in stroke. Neural Regeneration Research, 11, 398–399. 10.4103/1673-5374.179039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola, T. , Savinainen, J. R. , Monkkonen, K. S. , Retamal, M. D. , & Laitinen, J. T. (2005). S‐Nitrosothiols modulate G protein‐coupled receptor signaling in a reversible and highly receptor‐specific manner. BMC Cell Biology, 6, 21 10.1186/1471-2121-6-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecat, S. , Bucher, B. , Mely, Y. , & Galzi, J. L. (2002). Mutations in the extracellular amino‐terminal domain of the NK2 neurokinin receptor abolish cAMP signaling but preserve intracellular calcium responses. The Journal of Biological Chemistry, 277, 42034–42048. 10.1074/jbc.M203606200 [DOI] [PubMed] [Google Scholar]
- Leclerc, P. C. , Lanctot, P. M. , Auger‐Messier, M. , Escher, E. , Leduc, R. , & Guillemette, G. (2006). S‐Nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. British Journal of Pharmacology, 148, 306–313. 10.1038/sj.bjp.0706725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc, P. C. , Proulx, C. D. , Arguin, G. , Belanger, S. , Gobeil, F. Jr. , Escher, E. , … Guillemette, G. (2008). Ascorbic acid decreases the binding affinity of the AT1 receptor for angiotensin II. American Journal of Hypertension, 21, 67–71. 10.1038/ajh.2007.1 [DOI] [PubMed] [Google Scholar]
- Li, J. , He, J. , Du, Y. , Cui, J. , Ma, Y. , & Zhang, X. (2014). Electroacupuncture improves cerebral blood flow and attenuates moderate ischemic injury via angiotensin II its receptors‐mediated mechanism in rats. BMC Complementary and Alternative Medicine, 14, 441 10.1186/1472-6882-14-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S. L. , Setya, S. , Johnson‐Farley, N. N. , & Cowen, D. S. (2002). Differential coupling of 5‐HT1 receptors to G proteins of the Gi family. British Journal of Pharmacology, 136, 1072–1078. 10.1038/sj.bjp.0704809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marozkina, N. V. , & Gaston, B. (2012). S‐Nitrosylation signaling regulates cellular protein interactions. Biochimica et Biophysica Acta, 1820, 722–729. 10.1016/j.bbagen.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederos y Schnitzler, M. , Storch, U. , Meibers, S. , Nurwakagari, P. , Breit, A. , Essin, K. , … Gudermann, T. (2008). Gq‐coupled receptors as mechanosensors mediating myogenic vasoconstriction. The EMBO Journal, 27, 3092–3103. 10.1038/emboj.2008.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto, A. , Laufs, U. , Pardo, C. , & Liao, J. K. (1997). Modulation of bradykinin receptor ligand binding affinity and its coupled G‐proteins by nitric oxide. The Journal of Biological Chemistry, 272, 19601–19608. 10.1074/jbc.272.31.19601 [DOI] [PubMed] [Google Scholar]
- Muakkassah‐Kelly, S. F. , Andresen, J. W. , Shih, J. C. , & Hochstein, P. (1982). Decreased [3H]serotonin and [3H]spiperone binding consequent to lipid peroxidation in rat cortical membranes. Biochemical and Biophysical Research Communications, 104, 1003–1010. 10.1016/0006-291X(82)91349-3 [DOI] [PubMed] [Google Scholar]
- Munzel, T. , Daiber, A. , & Gori, T. (2011). Nitrate therapy: New aspects concerning molecular action and tolerance. Circulation, 123, 2132–2144. 10.1161/CIRCULATIONAHA.110.981407 [DOI] [PubMed] [Google Scholar]
- Näveri, L. , Strömberg, C. , & Saavedra, J. M. (1994). Angiotensin II AT1 receptor mediated contraction of the perfused rat cerebral artery. Neuroreport, 5, 2278–2280. 10.1097/00001756-199411000-00018 [DOI] [PubMed] [Google Scholar]
- Omura‐Matsuoka, E. , Yagita, Y. , Sasaki, T. , Terasaki, Y. , Oyama, N. , Sugiyama, Y. , … Kitagawa, K. (2009). Postischemic administration of angiotensin II type 1 receptor blocker reduces cerebral infarction size in hypertensive rats. Hypertension Research, 32, 548–553. 10.1038/hr.2009.69 [DOI] [PubMed] [Google Scholar]
- Parent, M. , Boudier, A. , Perrin, J. , Vigneron, C. , Maincent, P. , Violle, N. , … Dupuis, F. (2015). In situ microparticles loaded with S‐nitrosoglutathione protect from stroke. PLoS ONE, 10, e0144659 10.1371/journal.pone.0144659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent, M. , Dahboul, F. , Schneider, R. , Clarot, I. , Maincent, P. , Leroy, P. , & Boudier, A. (2013). A complete physicochemical identity card of S‐nitrosoglutathione. Current Pharmaceutical Analysis, 9, 31–42. [Google Scholar]
- Qian, J. , Chen, F. , Kovalenkov, Y. , Pandey, D. , Moseley, M. A. , Foster, M. W. , … Fulton, D. J. R. (2012). Nitric oxide reduces NADPH oxidase 5 (Nox5) activity by reversible S‐nitrosylation. Free Radical Biology & Medicine, 52, 1806–1819. 10.1016/j.freeradbiomed.2012.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, R. M. , Draznin, M. B. , & Murad, F. (1983). Endothelium‐dependent relaxation in rat aorta may be mediated through cyclic GMP‐dependent protein phosphorylation. Nature, 306, 174–176. 10.1038/306174a0 [DOI] [PubMed] [Google Scholar]
- Schleifenbaum, J. , Kassmann, M. , Szijarto, I. A. , Hercule, H. C. , Tano, J. Y. , Weinert, S. , … Gollasch, M. (2014). Stretch‐activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circulation Research, 115, 263–272. 10.1161/CIRCRESAHA.115.302882 [DOI] [PubMed] [Google Scholar]
- Stamler, J. S. , Simon, D. I. , Osborne, J. A. , Mullins, M. E. , Jaraki, O. , Michel, T. , … Loscalzo, J. (1992). S‐Nitrosylation of proteins with nitric oxide: Synthesis and characterization of biologically active compounds. Proceedings of the National Academy of Sciences of the United States of America, 89, 444–448. 10.1073/pnas.89.1.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman, E. , & Edvinsson, L. (2004). Cerebral ischemia enhances vascular angiotensin AT1 receptor‐mediated contraction in rats. Stroke, 35, 970–974. 10.1161/01.STR.0000121642.53822.58 [DOI] [PubMed] [Google Scholar]
- Straub, A. C. , Lohman, A. W. , Billaud, M. , Johnstone, S. R. , Dwyer, S. T. , Lee, M. Y. , … Isakson, B. E. (2012). Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature, 491, 473–477. 10.1038/nature11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y. , Tamura, K. , Koide, Y. , Sakai, M. , Tsurumi, Y. , Noda, Y. , … Umemura, S. (2005). The novel angiotensin II type 1 receptor (AT1R)‐associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Letters, 579, 1579–1586. 10.1016/j.febslet.2005.01.068 [DOI] [PubMed] [Google Scholar]
- Vincent, J. M. , Kwan, Y. W. , Chan, S. L. , Perrin‐Sarrado, C. , Atkinson, J. , & Chillon, J. M. (2005). Constrictor and dilator effects of angiotensin II on cerebral arterioles. Stroke, 36, 2691–2695. 10.1161/01.STR.0000190002.79052.bf [DOI] [PubMed] [Google Scholar]
- Vollmer, J. Y. , Alix, P. , Chollet, A. , Takeda, K. , & Galzi, J. L. (1999). Subcellular compartmentalization of activation and desensitization of responses mediated by NK2 neurokinin receptors. The Journal of Biological Chemistry, 274, 37915–37922. 10.1074/jbc.274.53.37915 [DOI] [PubMed] [Google Scholar]
- Watts, S. W. , Morrison, S. F. , Davis, R. P. , & Barman, S. M. (2012). Serotonin and blood pressure regulation. Pharmacological Reviews, 64, 359–388. 10.1124/pr.111.004697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda, N. , Miura, S. , Akazawa, H. , Tanaka, T. , Qin, Y. , Kiya, Y. , … Komuro, I. (2008). Conformational switch of angiotensin II type 1 receptor underlying mechanical stress‐induced activation. EMBO Reports, 9, 179–186. 10.1038/sj.embor.7401157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Sun, C. W. , Maier, K. G. , Harder, D. R. , & Roman, R. J. (2002). Mechanism of cGMP contribution to the vasodilator response to NO in rat middle cerebral arteries. American Journal of Physiology. Heart and Circulatory Physiology, 282, H1724–H1731. 10.1152/ajpheart.00699.2001 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Huang, B. , Zhang, L. , Zhang, Y. , Zhao, Y. , Guo, X. , … Chen, C. (2012). SNObase, a database for S‐nitrosation modification. Protein & Cell, 3, 929–933. 10.1007/s13238-012-2094-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Ex vivo exposure to GSNO decreases AngII‐mediated vasoconstriction independently of the order of administration of the drugs. Vasoconstriction (% change in internal diameter) in response to angiotensin II (AngII, 10‐10 mol. L‐1, open bars) and serotonin (5HT, 3.10‐9 mol. L‐1, full bars) in middle cerebral arteries of Wistar rats that were unexposed (panel A, n=7) or exposed (panel B, n=9; panel C, n=2) to Snitrosoglutathione (GSNO, 2.10‐6 mol. L‐1, 30 min followed by 1 hour washout). In contrast with panels A and B, in panel C 5HT was applied before AngII. Results are shown as means ± sem.
Figure S2. Effect of ODQ and DMSO on serotonin vasoconstriction. MCA were pretreated or not with GSNO and 1H‐[1,2,4]Oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ, 10‐5 mol/L) was added throughout the experiments. Dimethyl sulfoxide (DMSO, i.e. ODQ solvent, was used as control). Changes in internal diameter (%) are expressed as means ± SEM. Two‐way ANOVA (pretreatment, concentration) were used to calculate p values; *: p<0.05 vs unexposed; $: p<0.05 vs DMSO (Bonferroni post‐test).
Figure S3. Concentration response curves to S‐nitrosoglutathione. Concentration‐response curves, half maximal effective concentration (EC50) and maximal effect (Emax) in response to S‐nitrosoglutathione (GSNO) of middle cerebral arteries isolated from Wistar rats. Fitting to Hill's logistic equation was used. Values for EC50 (mol/L) and Emax (%) are expressed as means ± SEM. n = 6 middle cerebral arteries.
Figure S4. Impact of S‐nitrosoglutathione (GSNO) pretreatment on internal diameter. Internal diameters of rat middle cerebral arteries were measured before (baseline, open bars), after 30 min of exposure (full bars) to GSNO (2.10‐7 mol/L, 2.10‐6 mol/L or 2.10‐5 mol/L) or physiological salt solution (PSS for controls) and following the on‐hour wash‐out (grey bars). *: p<0.05 vs baseline; $: p<0.05 vs GSNO (paired Student's t‐test). Values are expressed as means ± SEM.
Figure S5. Ex vivo exposure to GSNO decreases Angiotensin II‐mediated vasoconstriction in middle cerebral arteries from Spontaneously Hypertensive Rats. Vasoconstriction of middle cerebral arteries from Wistar Kyoto (WKY) rats or Spontaneous Hypertensive rats (SHR) in response to angiotensin II (AngII, panel A) and serotonin (5HT, panel B). Vessels were unexposed (open bar) or exposed (full bars) to S‐nitrosoglutathione (GSN0, 2.10‐6 mol/L), 30 min followed by a 1‐hour wash out. Changes in internal diameter (%) are expressed as means ± SEM. n=4‐6 middle cerebral arteries per condition.