

Abstract
The role of myelomonocytic cells appears to be critical for the initiation, progression and manifestation of arterial hypertension. Monocytes can induce vascular inflammation as well as tissue remodelling and (mal)adaptation by secreting chemokines and cytokines, producing ROS, expressing coagulation factors and transforming into macrophages. A multitude of adhesion molecules promote the infiltration and accumulation of monocytes into the kidney, heart, brain and vasculature in hypertension. All these facets offer the possibility to pharmacologically target monocytes and may represent novel therapeutic ways to treat hypertension, attenuate hypertension‐associated end organ damage or prevent the development or worsening of high blood pressure.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- AngII
angiotensin II
- CCR
CC chemokine receptor
- CVD
cardiovascular disease
- ICAM‐1
intercellular adhesion molecule‐1
- MCP‐1
monocyte chemoattractant protein 1
- NOX
NADPH oxidase
- SHRs
spontaneously hypertensive rats
- TF
tissue factor
- VCAM‐1
vascular cell adhesion molecule 1
Introduction
Arterial hypertension is currently regarded as the leading risk factor for morbidity and mortality worldwide (Lim et al., 2012). In particular, it is the main pathophysiological and epidemiological driver of heart failure, ischaemic stroke and ischaemic heart disease including acute myocardial infarction, the single most frequent cause of death in the world. The immune system is central in the development and perpetuation of high blood pressure and in hypertension‐related end organ damage (Norlander et al., 2018). Interestingly, individuals with a constitutive overexpression of inflammasome‐related genes, in particular IL‐1β, have higher blood pressure, vascular stiffness and experience premature death (Furman et al., 2017). While there is a myriad of interactions of the immune system with all critical players in blood pressure regulation (CNS, kidney, microvasculature, conductance vessels, heart), the role of myelomonocytic cells is inherent in all of them.
Role of monocytes in high blood pressure and hypertensive end organ damage
In the brain, angiotensin II (AngII) signalling mainly through the AngII receptor type 1 (AT1 receptor) increases neuronal activity in brain areas such as the rostral ventrolateral medulla or subfornical organ with efferent projections into the periventricular nucleus of the hypothalamus, to the solitary tract and others. This results in increased systemic sympathetic outflow, thereby inducing, maintaining or amplifying high blood pressure (Ferguson, 2009; de Kloet et al., 2015). The microglia representing the tissue resident myelomonocytic cell type in the brain may play a critical role in blood pressure control. Hypertension induces the activation and proliferation of microglia in the brain, an essential feature of neuroinflammation, with concomitant intracerebral up‐regulation of IL‐1β, IL‐6 and TNF‐α expression as well as ROS formation (Wu et al., 2012). Ablation of CD11b+ cells by a diphtheria toxin‐mediated approach blocked AngII‐driven microgliosis as well as inflammatory cytokine expression and attenuated arterial hypertension (Shen et al., 2015). Neuroinflammation is also a hallmark of Alzheimer's disease and other forms of dementia, significantly associated with hypertension. Intriguingly, it is an early step in hypertension preceding amyloid deposition (Carnevale et al., 2012a), making myeloid cells in the brain central in both the development of hypertension and end organ damage (Carnevale et al., 2012b).
The kidneys are the major site of renin release, blood volume control and electrolyte homeostasis and are central to the development of hypertension (Goldblatt, 1947). They are inflamed in chronic hypertension (Wilson, 1963), leading to hypertensive kidney disease (McMaster et al., 2015). However, renal inflammation may also precede development of hypertension (Rodriguez‐Iturbe et al., 2004), and the infusion of AngII leads to a persistent accumulation of myelomonocyte populations in the renal medulla and cortex (Ozawa et al., 2007). For example, Ly6C+ monocytes and macrophages infiltrating the kidneys promote sodium reabsorption via the NKCC2 co‐transporter in the nephron, which can be alleviated by blockade of IL‐1 receptor 1 (Zhang et al., 2016).
For more than 45 years, monocytes have been known to infiltrate the vasculature in arterial hypertension and promote proliferation of connective tissue (Olsen, 1971). Remodelling of the (micro)vasculature is the key step in the increase in systemic vascular resistance, a hallmark feature of arterial hypertension (Folkow et al., 1973). The vascular accumulation of monocytes has been widely accepted as a key primary step in atherogenesis, occurring early on in the pathogenesis of the disease (Cybulsky and Gimbrone Jr, 1991). Research has always been centred on monocytes, meaning bone marrow derived circulating cells that represent putative future macrophages infiltrating various organs and tissues. This view traditionally omits the resident macrophages. The role of resident macrophages and that of infiltrating myelomonocytic cells in hypertension is unclear. However, it is well known that yolk sac macrophages express more MerTK genes (encoding for the proto‐oncogene tyrosine‐protein kinase MER) than CC chemokine receptor 2 (CCR2) genes and are early organ resident macrophages, in contrast to bone marrow derived monocytes, that are myelomonocytic cells (Schulz et al., 2012). In principle, yolk sac‐derived macrophages, fetal liver monocytes and bone marrow derived monocytes have the same capacity to populate empty niches and develop into phenotypically ‘identical’ tissue macrophages (van de Laar et al., 2016) In the same vein, some new evidence suggests that cardiac and arterial resident macrophages, which express MerTK, play a protective role by removing apoptotic cells and suppressing vascular inflammation, whereas infiltrating CD115+Ly6hi monocytes play a pathophysiological role in the heart and blood vessels, for instance in the setting of atherosclerosis (Cai et al., 2017; DeBerge et al., 2017). These data point towards the long‐established dichotomy into pro‐inflammatory (Ly6Chi) and reparative (Ly6Clo) monocytes and classically activated ‘M1’ macrophages versus reparative ‘M2’ macrophages (Geissmann et al., 2003; Moore and Tabas, 2011). While the protective role of M2‐like macrophages has been shown in experimental models of atherosclerosis, their role in modulating vascular injury or blood pressure in arterial hypertension remains less clear. A recent review focused on the role of macrophage polarization in hypertension, trying to shed some light on the obscure mechanisms and incomplete concepts (Harwani, 2018). For instance, the group of Grant Drummond has demonstrated that continued exposure to AngII infusion in vivo induces an increase in the population of CD206+F4/80+ macrophages in the vasculature, paralleled by increased aortic expression of arginase‐1 and Fc receptor‐like S scavenger receptor mRNA, compatible with an expansion of the M2 like macrophage population (Moore et al., 2015). In that paper, the exact role of these cells remained unclear; nevertheless, antagonization of CCR2 (see below) reduced both the abundance of this population and reduced blood pressure. To the contrary, Ndisang and Mishra (2013) have demonstrated that pharmacological intervention to increase the M2 macrophage population decreased blood pressure in spontaneously hypertensive rats (SHRs).
In contrast to the yet to be deciphered phenomenon of macrophage polarization in hypertension, it is well known that more advanced atherosclerotic lesions contain a functionally active renin‐angiotensin‐system, facilitating continuous recruitment of monocytes to the plaque and possibly contributing to plaque instability and rupture (Schieffer et al., 2000). Depletion of monocytes or deficiency in the macrophage colony stimulating factor prevents AngII‐induced hypertension and vascular remodelling. The depletion studies also highlighted that the presence of CD11b+F4/80+ macrophages is required for hypertension and that it is linked to the presence of lysozyme positive myloid (precursor) cells (De Ciuceis et al., 2005; Wenzel et al., 2011). As early as 1980, it was shown that experimental hypertension can be transferred from hypertensive animals to normotensive recipients by transfer of splenic mononuclear cells (Olsen, 1980).
Recent work suggests that vascular infiltrating inflammatory Ly6Chi monocytes may trans‐differentiate into CD206+Arg‐1+F4/80+ macrophages or CD11c+ antigen presenting cells in the vasculature or kidneys in response to AngII (Moore et al., 2015; Hevia et al., 2018). This may be the link to communicate with T‐cells and other components of the adaptive immune system that have been shown to play key roles in hypertension (Norlander et al., 2018). Importantly, these tissue remodelling and homeostatic properties of myelomonocytic cells require crosstalk with other cellular components of the vascular wall, such as fibroblasts or vascular smooth muscle cells (Tieu et al., 2009; Sumida et al., 2015).
Cardiac remodelling and fibrosis are the histomorphological basis of hypertensive heart disease and are also mainly driven by myeloid cells. AngII directly and indirectly increases proliferation of cardiac myocytes and non‐myocytes (McEwan et al., 1998), among them are fibroblasts and myelomonocytic cells (Muller et al., 2000b). Attenuating a pro‐inflammatory milieu by enhancing T regulatory cell responses or by IFN‐γ receptor knockout blunts cardiac infiltration by CD11b+F4/80+ myelomonocytes and reduces cardiac remodelling (Kvakan et al., 2009; Marko et al., 2012).
All these lines of evidence support a strong role for bone marrow derived monocytes in hypertension. Obviously, the role of the myeloid compartment in arterial hypertension is intertwined with other immune cells, such as T‐cells, but also beta cells and NK‐cells (Norlander et al., 2018). In particular, the specific role of T‐cells in arterial hypertension has been covered in a number of reviews. Here, we want to focus on the myelomonocytic cells (in particular monocytes) as possible immune targets in arterial hypertension. Obviously, one has to bear in mind the species differences between mice and man with regard to monocytes and their subsets, which may limit the applicability of many scientific findings to human medicine. However, the growing availability of biodata banks with genetic and tanscriptomic information obtained from monocytes from large population‐based cohorts bears the possibility to enable translation in the field (Wenzel et al., 2015; Zeller et al., 2017).
Monocyte chemotaxis in arterial hypertension
In 1997, Capers et al. showed that monocyte chemoattractant protein 1 (MCP‐1; also known as CCL2) mRNA expression is increased in the aorta of AngII as well as noradrenaline‐infused rats that could be reversed by co‐administration of a diuretic. Supporting experiments in rat isolated aortic smooth muscle cells showed that application of mechanical strain also increased MCP‐1 expression, underlining a fundamental role of this chemokine for chemotaxis in high blood pressure (Capers et al., 1997a). Ishibashi and co‐workers showed the critical role of the MCP‐1 receptor CCR2 on monocytes in arterial hypertension. Blocking the receptor by an antagonizing antibody prevented vascular inflammation and remodelling (Ishibashi et al., 2004). Blocking the MCP‐1/CCR‐2 axis and other CC chemokine pathways (CCR1, CCR5) prevented AngII‐induced monocyte adhesion to the microvasculature (Mateo et al., 2006). Likewise, antagonizing CXC chemokine receptors like CXCR2 also reduces MCP‐1, RANTES (ligand to CCR1 and 5) and MIP1α (CCL3) levels, thereby attenuating AngII‐induced monocyte adhesion in vitro (human umbilical arterial cells) and in vivo (splanchnic circulation) (Abu Nabah et al., 2007).
Monocyte derived cytokine production in arterial hypertension
IL‐6 is a pro‐inflammatory cytokine that can be produced by myelomonocytic cells. It is one of the few cytokines for which an independent association with arterial hypertension has been demonstrated. In epidemiological studies, it was identified to be an independent risk factor for hypertension in apparently healthy individuals (Bautista et al., 2005). IL‐6 may also serve as a biomarker for elevated blood pressure, since it was shown to be significantly elevated in patients with hypertension compared to normotensive controls (Chamarthi et al., 2011). In experimental research, knockout of IL‐6 protected mice from developing high blood pressure in response to AngII and high salt. This was paralleled by partial protection from kidney injury, as assessed by albuminuria (Lee et al., 2006). Pharmacological inhibition of IL‐6 by a neutralizing antibody was able to lower blood pressure and preserve kidney function in salt‐sensitive Dahl rats. Interestingly, this nephroprotection was associated with reduced numbers of monocytes infiltrating the kidney and a decreased abundance of renal macrophages (Hashmat et al., 2016). This suggests that intervening in the IL‐6 signalling pathways may be an elegant way to attenuate end organ damage in hypertension by blocking activation of myelomonocytic cells.
IL‐12 is secreted by activated monocytes and macrophages, typically when exposed to IFN‐γ. In addition, it can also be active as a membrane bound form (Fan et al., 1996). In the setting of AngII‐induced hypertension, it was shown that IFN‐γ derived from NK1.1+ NK cells stimulates monocytes to form IL‐12, which in turn augments the formation of IFN‐γ by NK cells (Kossmann et al., 2013), a classical function of this cytokine (D'Andrea et al., 1992). This mutual activation depends on the transcription factor T‐box expressed in T‐cells (T‐bet), which is critical not only for IFN‐γ transcription in lymphoid cells like T‐cells and NK cells but also for IL‐12 formation by myelomonocytes (Soderquest et al., 2011). The abundance of T‐bet is highest in lymphoid cells such as T‐cells and NK cells. Based on the data of Kossmann et al., targeting of T‐bet specifically in monocytes to block IL‐12 synthesis or in T‐cells and NK cells to block IFN‐γ formation could offer a possible approach to attenuate vascular inflammation in hypertension.
Monocyte vascular rolling, adhesion, infiltration and accumulation in arterial hypertension
AngII induced the adhesion and binding of monocytes, but not neutrophils, to human and rabbit aortic endothelial cells. This was completely dependent on AT1 receptor‐mediated signal transduction. Interestingly, in this cell culture system, adhesion was independent of the up‐regulation of endothelial vascular cell adhesion molecule 1 (VCAM‐1) or intercellular adhesion molecule‐1 (ICAM‐1) (Kim et al., 1996). Later, Pueyo and co‐workers convincingly showed that AngII does indeed induce VCAM‐1 mRNA expression in endothelial cells in an AT1 receptor‐dependent manner, which was NF‐κB‐ and ROS‐dependent (Pueyo et al., 2000). In vivo models of arterial hypertension later provided strong evidence that adhesion molecules are necessary for attracting monocytes in arterial hypertension. For example, Tummala et al. showed that AngII induced the expression of VCAM‐1 not only in rat cultured aortic smooth muscle cells but also in the vasculature of AngII‐infused rats. This expression was AT1 receptor‐dependent, and proteasome inhibitors were able to block the NF‐κB related transactivation of VCAM‐1 expression (Tummala et al., 1999).
ICAM‐1 is induced by AngII in vivo, at least in part in an NADPH oxidase dependent manner (Liu et al., 2003). In coronary vessels, however, AngII had only small effects on the expression of VCAM‐1 and ICAM‐1; here, it preferentially increased the expression of the adhesion molecule E selectin (Grafe et al., 1997). In the rat mesentery, which represents a classical microvasculature, P‐selectin was induced by AngII both via the AT1 as well as the AT2 receptor (Piqueras et al., 2000). Another important ligand on monocytes promoting endothelial adhesion is Mac‐1 (also known as integrin αMβ2 or CD11b/CD18). It can bind to ICAM‐1 on endothelial cells (Diamond et al., 1990) but also to fibrinogen and junctional adhesion molecule‐3. In addition, it engages with the glycoprotein Ib α on platelets to form monocyte‐platelet‐conjugates (Simon et al., 2000) and is an essential feature in vascular injury (Wang et al., 2005). In stroke‐prone SHRs, administration of an AT1 receptor blocker reduced leukocyte expression of Mac‐1, implying an AngII‐dependent induction of this integrin in arterial hypertension (Takemori et al., 2000).
By using intravital epiflourescence video microscopy, it was revealed that leukocyte rolling and adhesion to the endothelium of both low flow regions of the circulation like arterioles and mesenterics (Piqueras et al., 2000) as well as high flow arterial beds (carotid artery) is a characteristic event in vascular inflammation in AngII‐induced arterial hypertension. By using transgenic mice with conditional expression of yellow fluorescent protein under the transcriptional control of the gene lysozyme M, which is specific for neutrophils, monocytes and macrophages (Gordon et al., 1974; Clausen et al., 1999), Lagrange and co‐workers showed that almost all of these rolling and adhering leukocytes are of myeloid origin (Lagrange et al., 2018). Importantly, this vascular inflammation can be modified by anti‐inflammatory interventions or modulations of anti‐inflammatory systems. A constitutive lack of the anti‐inflammatory and antioxidative enzyme haem oxygenase 1 (HO‐1) not only increased the susceptibility of mice to develop endothelial dysfunction and high blood pressure in response to AngII but also increased leukocyte rolling (Wenzel et al., 2015). This was linked to an augmented vascular accumulation of Cd11b+Ly6Chi monocytes and Cd11b+F4/80+ macrophages. Inversely, germ‐free mice with an attenuated capacity to mount a classical type 1 immune response show significantly less monocyte rolling and adhesion to the endothelium in response to AngII than conventionally reared controls (Karbach et al., 2016). Consequently, these mice do not develop arterial hypertension, endothelial dysfunction and vascular accumulation of Cd11b+Ly6Chi and Cd11b+F4/80+ cells. When VCAM‐1 and its counterpart receptor on leukocytes, the very late antigen 4, are experimentally blocked by antagonizing antibodies in AngII‐infused mice, monocyte rolling and adhesion to the endothelium are effectively blocked in vivo. The same was true for blocking Mac‐1, suggesting that the ICAM‐1/Mac‐1 axis may also be critical for monocyte adhesion in hypertension (Kossmann et al., 2017).
In order to allow infiltration of leukocytes into the vascular wall or the perivascular space, the biochemical activity of proteases is needed. These enzymes cleave components of the endothelial barrier, the basal membrane and the extracellular matrix, such as elastin and collagen. In atherosclerotic lesions, enigmatic for vascular remodelling, high expression and activity of elastases, collagenases and gelatinases have been detected. Among the proteolytic enzymes, MMPs, serine proteases and cytein proteases (such as lysosomal cathepsin S and K) have received the highest attention (Liu et al., 2004). The role of proteases as prerequisite to allow leukocyte infiltration in arterial hypertension is less well understood. Our data on platelet‐localized vascular thrombin amplification in arterial hypertension in the absence of overt clotting suggest an involvement of protease‐activated receptor signalling in vascular inflammation induced by AngII (Kossmann et al., 2017). In a mouse model of acute aortic dissection induced by AngII, MMP‐9 was significantly up‐regulated in the diseased aorta. Pharmacological inhibition of MMP‐9 as well as genetic deficiency of MMP‐9 largely protected from acute aortic dissection. Both dissection and MMP‐9 activity were reduced by depleting Gr‐1+ cells (comprising both neutrophils and Ly6Chi monocytes) with a monoclonal antibody (Kurihara et al., 2012). Similarly, monocytes of thrombospondin‐1‐deficient mice had reduced migratory capacity and endothelial adhesion in response to AngII, and thrombospondin‐1−/− mice were largely protected from AngII‐induced aortic aneurysm, in part explained by the lack of thrombospondin‐1‐mediated proteolytic damage in the vessel wall (Liu et al., 2015).
Taken together, pharmacological interventions to block rolling, adhesion and vascular infiltration of monocytes could be a promising way to limit immune‐mediated vascular in jury in hypertension (Figure 1).
Figure 1.
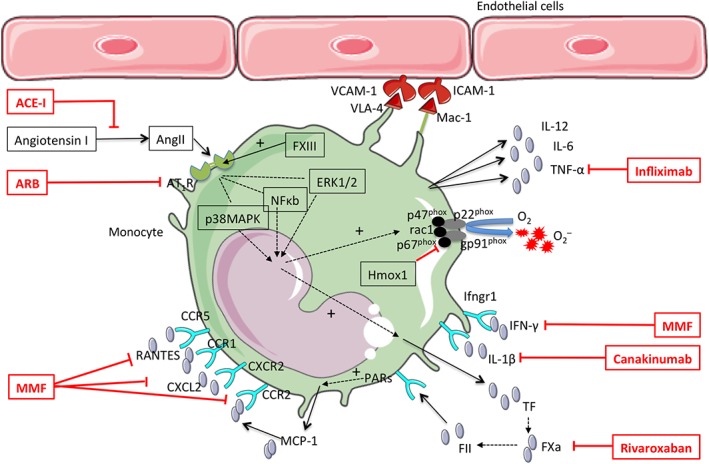
Potential immunotargets on monocytes in arterial hypertension. This figure illustrates the molecular and cellular targets that might be feasible for pharmacological approaches in the treatment of hypertension. The framed drug names indicate pharmaceuticals that have already been shown to lower blood pressure or related disease in humans. MMF, mycophenolate mofetil; O2‐, superoxide radical; PARs, protease activated receptors; VLA‐4, very late antigen 4. All other abbreviations are explained in the text.
Coagulation factors, monocytes and hypertension
While the role of coagulation factors in the propagation of vascular inflammation has been appreciated for many years, its role in hypertension has only recently gained more attention. Tissue factor (TF) kicks off the coagulation cascade in the classical scheme of extrinsic coagulation (‘tissue factor pathway’). In the vasculature, it is exposed to the plasma upon vessel injury and complexes with factor (F) VII to form the prothrombinase complex, leading to FX activation and ultimately thrombin formation (Figure 2). Interestingly, TF is expressed not only by endothelial cells or vascular smooth muscle cells but also by neutrophils and monocytes. Currently, monocytes are regarded as the prime source of TF expression and activity in venous thrombosis and atherosclerosis/−thrombosis. Upon stimulation via the ADP/P2X7 axis, macrophages produce inflammasome‐dependent endosomal ROS, form filipodia, transport TF to the extracellular surface and shed TF‐rich, procoagulant microparticles (Rothmeier et al., 2015). Deletion of TF specifically in LysM‐positive immune cells prevents the initiation of deep vein thrombosis in mouse models (von Bruhl et al., 2012; Subramaniam et al., 2017). AngII has been shown to increase TF mRNA and protein expression in vascular smooth muscle cells (Taubman et al., 1993). We have found increased mRNA expression of TF in monocytes isolated from mice infused with AngII (unpublished data). Blocking TF activity with the antibody 21E10 prevented both AngII‐induced vascular dysfunction in mice as well as the rolling and adhesion of leukocytes to the endothelium of the carotid (Kossmann et al., 2017). This indicates a causative role for TF in contributing to arterial hypertension, at least in part through the induction of thrombin (FIIa). Heterotypic thrombin generation within the vessel wall was shown to be increased in SHRs and could be blocked by an ACE inhibitor. Smooth muscle cells isolated from SHRs were more sensitive to thrombin‐induced proliferation than SMCs from control rats (Ait Aissa et al., 2015). Additionally, the expression of thrombin receptors (PARs) has been shown to be up‐regulated in the vessel wall of AngII‐infused rats, which was mediated by AT1 receptors and lead to increased susceptibility to thrombin‐induced vasoconstriction (Capers et al., 1997b). Thrombin itself is a strong inducer of MCP‐1 in vascular cells and may thereby increase monocytes adhesion to the endothelium (Wenzel et al., 1995). As early as 1993, it was shown that in the presence of AngII, thrombin‐induced platelet aggregation was significantly higher in patients with uncontrolled hypertension than in treated hypertensives and control individuals (Touyz and Schiffrin, 1993). Blocking FIIa activity by the direct thrombin inhibitor hirudin prevented endothelial leukocyte rolling and adhesion and improved vascular dysfunction in AngII‐induced hypertension. Similar to antagonizing TF activity, it attenuated vascular ROS formation and markers of vascular inflammation (Kossmann et al., 2017). Interestingly, the TF‐mediated thrombin activity appears to be propagated by an amplification loop via the coagulation factor FXI in hypertension. That pathway was proposed earlier in coagulation research (Gailani and Broze Jr, 1991) and can be exploited therapeutically to perform safe thromboprophylaxis (Buller et al., 2015). Blocking FXI synthesis prevented localized thrombin generation in platelets and lowered pro‐inflammatory platelet‐monocyte complexes, the vascular accumulation of myeloid cells, ROS formation in the vessel wall and ultimately dampened the blood pressure increase in models of hypertension (Kossmann et al., 2017). This unexpected role of FXI in mediating myeloid cell driven inflammation and vascular damage in hypertension needs to be addressed by further research. Besides driving thrombin generation on platelets, the protease function of FXIa could also contribute to non‐canonical signalling pathways that directly affect inflammatory damage. For instance, the pro‐inflammatory role of FXI could also relate to its novel function in the cleavage of prochemerin (Ge et al., 2018), which might be involved in hypertension‐induced vascular inflammation as well.
Figure 2.
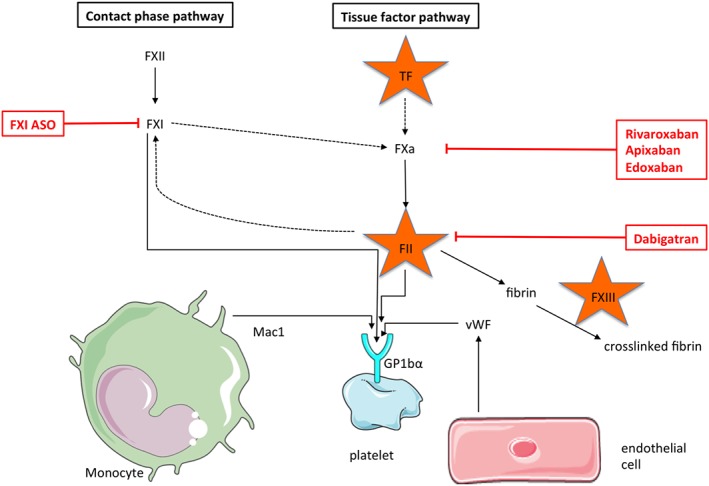
Coagulation factors and their connection to monocyte activation. This figure illustrates the contact phase as well as the tissue factor (TF) pathway of coagulation in a simplified fashion. The framed drug names indicate pharmaceuticals that have already been shown to effectively prevent or treat thrombotic disease in clinical trials. The coagulation factors TF, FII and FXIII are encircled by the star symbol because they are either expressed by monocytes or have a direct effect on monocyte functionality with regard to hypertension. vWF, von Willebrand factor.
Further downstream of the coagulation cascade, Abdalla et al. (2004) showed that the transglutaminase coagulation factor XIII may crosslink the AT1 receptors on monocytes and would, therefore, be required to allow the full activation of monocytes by AngII. This is intriguing, because non‐conventional roles for FXIII have also been demonstrated to be important for collagen crosslinking in the process of wound healing and scar formation post myocardial infarction, the major endpoint of the hypertension sequela (Nahrendorf et al., 2006).
Altogether, a new area of research is opened up with regard to novel extracellular roles of coagulation factors in hypertension that could also serve as drugable immunotargets (Figure 2). Clinical evidence comes from the recent COMPASS trial. Here, the addition of a very low, non‐anticoagluant dose of the FXa inhibitor rivaroxaban to standard therapy with acetylsalicylic acid reduced cardiovascular disease (CVD) events in secondary prevention (Eikelboom et al., 2017). It will be challenging – and potentially rewarding – to explore the possible vasoprotective effects of targeting coagulation factors, in particular with regard to monocytes and hypertension.
NADPH oxidase in monocytes as putative target
NADPH oxidase is a core component of the innate immune response and abundantly expressed in mononuclear phagocytes (neutrophil granulocytes, monocytes and macrophages) but also by other immune cells as well as endothelial cells, smooth muscle cells and advential cells, like fibroblasts, perivascular adipocytes and pericytes. The multiple roles of NADPH oxidase in CVD and hypertension have been extensively reviewed before. For stringency reasons, we also do not expand on the important other sources of ROS in this review, such as an uncoupled NO synthase, the mitochondrial respiratory chain or xanthin oxidases (Li et al., 2014).
The NADPH oxidase in myelomonocytes deserves specific attention (Cathcart, 2004). Like their counterparts in the myeloid lineage, the neutrophil granulocytes, monocytes contain a phagocyte‐type NADPH oxidase with the catalytic subunit gp91phox (nox2) and anchoring unit p22phox, together making the cytochrome b558. To be activated, regulatory subunits need to translocate from the cytosol to the membrane, including the p67phox and the p47phox, together with the small GTPase rac1 (Figure 1). The activation process in neutrophils is slightly different, with the GTPase rac2 substituting the rac1 (Zhao et al., 2003), priming these innate cells for rapid release of superoxide in the so‐called oxidative burst (Leusen et al., 1996), essential for microbial killing and neutrophil extracellular trap formation (Fuchs et al., 2007).
In a seminal paper, Rajagopalan et al. showed that the phagocyte‐type NADPH oxidase (NOX) in the vasculature produces superoxide in response to AngII. Both NOX‐derived ROS formation and endothelial dysfunction could be blocked in vivo by an AT1 receptor blocker in AngII‐infused rats (Rajagopalan et al., 1996). Expression of gp91phox, p22phox as well as NOX1 was increased in response to AngII, and concomitantly, endothelial NOS becomes dysfunctional in a ROS‐sensitive manner, a process called eNOS uncoupling (Mollnau et al., 2002). This disbalance between superoxide and NO is regarded as the main redox‐biochemical mechanism of endothelial dysfunction in hypertension. Pharmacological targeting of the gp91phox by a docking sequence peptide blocking the catalytic activity of the enzyme (gp91ds‐tat) prevented AngII‐induced infiltration of ED1+ myelomonocytes and attenuated medial hypertrophy in large arteries, independently of blood pressure effects (Liu et al., 2003). The same group has shown that AngII‐infused mice treated with the gp91ds‐tat, which attenuated NADPH‐oxidase‐derived superoxide formation, were largely devoid of blood pressure increase (Rey et al., 2001). These data suggest that NADPH oxidase from mononuclear cells might be causally involved in the blood pressure increase in response to AngII. Indeed, when transgenic mice were depleted of monocytes, blunted hypertension could only be re‐established by reconstitution with gp91phox competent monocytes, not with monocytes isolated from gp91phox y/− mice (Wenzel et al., 2011). p47phox−/− mice lacking an important regulatory subunit of the phagocyte‐type NADPH oxidase had blunted the increase in blood pressure in response to AngII. This was accompanied by a loss of NADPH oxidase‐derived superoxide formation (Landmesser et al., 2002). Similar findings were obtained in mice deficient in the gp91phox homologue in vascular smooth muscle cells, NOX1. Nox1y/− had normal blood pressure at baseline but a significantly blunted hypertensive response to AngII. This was paralleled by a lack of superoxide formation and preserved NO‐dependent vasorelaxation, whereas medial hypertrophy was similar in nox1y/− compared to nox1y/+ mice (Matsuno et al., 2005). Interestingly, gp91phox y/− mice had lower blood pressure than their gp91phox y/+ counterparts at baseline and a lower blood pressure in response to AngII resulting in a pulse pressure that was not different between the groups (Wang et al., 2001). Importantly, vascular NADPH oxidase‐mediated superoxide formation, nitrotyrosine staining and medial hypertrophy were absent in AngII‐infused gp91phox y/− mice, similar to the findings obtained with the gp91ds‐tat (Rey et al., 2001; Liu et al., 2003) and by depletion of gp91phox competent myelomonocytic cells (Wenzel et al., 2011). In a recent study by Sag et al. using mice with conditional knockout of gp91phox specifically in myelomonocytic cells, the findings of Wang et al. regarding blood pressure response could be corroborated, underpinning a central role for NOX2 inside vascular monocyte/macrophages for blood pressure regulation and vascular tone (Sag et al., 2017). Targeting intrinsic antioxidative pathways inside monocytes has been proven effective as well, at least in the experimental setting. The induction of HO‐1 in monocytes suppressed AngII‐induced superoxide formation and lowered the expression of CCR2, the receptor for MCP‐1, and attenuated the chemotactic response. Administration of downstream products of HO‐1, bilirubin and carbon monoxide was equally effective, suggesting the antioxidant properties of HO‐1 as a putative immune target on monocytes (Morita et al., 2003).
Immune modulation as therapeutic target in cardiovascular disease
The findings of the COMPASS indirectly implicate an involvement of the immune system in the progression of CVD. However, there is a growing body of evidence indicating that an activated immune system might also be targeted by anti‐inflammatory therapy (Figure 1). Based on the findings that individuals with elevated high sensitive C‐reactive protein levels are at increased risk of CVD events that might benefit from a statin therapy despite normal LDL cholesterol levels (Ridker et al., 1997; Ridker et al., 2008), trials using metothrexate (CIRT trial) or the IL‐1β antagonist canakinumab (CANTOS trial) have been performed to test the effectiveness of direct anti‐inflammatory drugs to lower CVD events and mortality in individuals with a history of coronary artery disease and/or myocardial infarction (Ridker, 2009; Ridker et al., 2017). While results of the CIRT trial are not yet available, the CANTOS trial showed a significant reduction in nonfatal myocardial infarction or stroke and cardiovascular death with canakinumab versus placebo, on top of optimal medical care. Interestingly, the individuals who had the biggest benefit from IL‐1β antagonism were those who experienced the biggest drop in hsCRP levels (Ridker et al., 2018). It has to be tested, however, if essential arterial hypertension also can be treated or prevented by an anti‐inflammatory strategy. Evidence from patients with autoimmune disease at least suggests that this promise could hold true. In a small single centre study, treatment with the immunosuppressant mycophenolate mofetil significantly lowered blood pressure in patients with rheumatoid arthritis or psoriasis who had concomitant arterial hypertension (Herrera et al., 2006). Treating individuals suffering from rheumatoid arthritis with the TNF‐α inhibitor, infliximab, significantly reduced ambulatory blood pressure (Yoshida et al., 2014). These data have encouraged leading scientist in the field to promote interventional clinical trials to test whether arterial hypertension can specifically be treated by an anti‐inflammatory therapy (Leslie, 2018). It remains to be established if myelomonocytic cells qualify as a feasible target for such an approach.
Anti‐inflammatory effects of established antihypertensive drugs: monocytes as targets
While the concept of a specific anti‐inflammatory therapy for hypertension is only developing, there is already a lot of evidence that established antihypertensive drugs also have anti‐inflammatory properties (Figure 1). This includes inhibitory actions on myelomonocytic cells. For example, angiotensin‐converting enzyme (ACE) inhibition has been shown to prevent arterial NF‐κB activation, MCP‐1 expression and macrophage infiltration in a rabbit model of early accelerated atherosclerosis (Hernandez‐Presa et al., 1997). More specifically, Napoleone et al. (2000) demonstrated that ACE inhibitors may down‐regulate TF synthesis in monocytes. Expression of the essential receptor for MCP‐1 on monocytes, CCR2, was blunted by AT1 receptor blockers in hypertensive mice (Ishibashi et al., 2004), and AT1 receptor blockade reduces vascular TF in AngII‐induced cardiac vasculopathy (Muller et al., 2000a). In a groundbreaking paper, Clozel and co‐workers (1991) demonstrated that the extent of subendothelial monocyte and macrophage accumulation paralleled the extent of endothelial dysfunction in SHRs and that both were attenuated by treating the animals with ACE inhibitors.
The finding that monocytes could represent a treatable target in arterial hypertension in humans was made 20 years ago. Monocytes isolated from hypertensive patients secreted significantly higher levels of IL‐1β in response to AngII than monocytes isolated from controls. IL‐1β levels correlated significantly with blood pressure level. Likewise, IL‐1β as well as IL‐6 and TNF‐α levels secreted by monocytes were higher in those exposed to LPS, revealing their inflammatory phenotype in hypertension (Dorffel et al., 1999). AngII induces the migration of isolated human monocytes, a process that involves pro‐inflammatory ERK1/2, Src and p38 MAPK‐dependent pathways and that can be abolished by AT1 receptor blockade (Kintscher et al., 2001). High dose treatment of patients afflicted by metabolic syndrome with the AT1 receptor blocker telmisartan induced PPAR‐γ target genes in monocytes, such as CD36 (Bahr et al., 2011). Interestingly, the AT1 receptor blocker losartan given to high risk patients with terminal renal failure significantly reduced the number of CD14+CD16+ inflammatory monocytes compared to controls (Merino et al., 2012). These findings suggest that guideline recommended antihypertensive drugs directly affect monocyte phenotype and behaviour. This implies that pharmacological targeting of myelomonocytic cells could contribute to improve treatment of humans with arterial hypertension that may even be additive to currently used medication.
Conclusion
Monocytes play a key role in the inflammatory continuum of arterial hypertension. Their molecular and cellular (inter)actions are multifaceted and can potentially be exploited for pharmacotherapy of high blood pressure. Promising targets are (i) cytokines such as IL‐1β, IL‐6, IL‐12 or IFN‐γ that either are released by or act upon myelomonocytic cells; (ii) chemokine signalling such as the enigmatic MCP‐1/CCR2 axis to block monocyte recruitment and down the line also adhesion and infiltration into the vasculature and (iii) phagocyte type NADPH oxidase or antioxidant enzymes to interfere with the ROS formation of myelomonocytic cells. Excitingly, pleiotropic effects of thrombin inhibition or other novel anti‐thrombotic regimens may also contribute to the anti‐inflammatory therapy of hypertension. Altogether, lowering the inflammatory burden imposed on the organism by inflammatory myelomonocytic cells may help to lower the global burden of disease imposed on mankind by arterial hypertension.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018) and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Acknowledgements
This work was supported by the Bundesministerium für Bildung und Forschung (BMBF 01EO1503), the German Research Foundation (DFG WE 4361/7‐1), the Center for Translational Vascular Biology at the University Medical Center Mainz and the Boehringer Ingelheim Foundation. Graphical icons depicting monocytes, endothelial cells and platelets in Figures 1 and 2 were taken from Servier Medical Art (https://smart.servier.com/terms‐of‐use/), published under Creative Commons Attribution 3.0 Unported License.
Conflict of interest
The author declares no conflicts of interest.
Wenzel P. (2019) Monocytes as immune targets in arterial hypertension, British Journal of Pharmacology, 176, 1966–1977, doi: 10.1111/bph.14389.
References
- AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U (2004). Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 119: 343–354. [DOI] [PubMed] [Google Scholar]
- Abu Nabah YN, Losada M, Estelles R, Mateo T, Company C, Piqueras L et al (2007). CXCR2 blockade impairs angiotensin II‐induced CC chemokine synthesis and mononuclear leukocyte infiltration. Arterioscler Thromb Vasc Biol 27: 2370–2376. [DOI] [PubMed] [Google Scholar]
- Ait Aissa K, Lagrange J, Mohamadi A, Louis H, Houppert B, Challande P et al (2015). Vascular smooth muscle cells are responsible for a prothrombotic phenotype of spontaneously hypertensive rat arteries. Arterioscler Thromb Vasc Biol 35: 930–937. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr IN, Tretter P, Kruger J, Stark RG, Schimkus J, Unger T et al (2011). High‐dose treatment with telmisartan induces monocytic peroxisome proliferator‐activated receptor‐γ target genes in patients with the metabolic syndrome. Hypertension 58: 725–732. [DOI] [PubMed] [Google Scholar]
- Bautista LE, Vera LM, Arenas IA, Gamarra G (2005). Independent association between inflammatory markers (C‐reactive protein, interleukin‐6, and TNF‐α) and essential hypertension. J Hum Hypertens 19: 149–154. [DOI] [PubMed] [Google Scholar]
- Buller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE et al (2015). Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372: 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B et al (2017). MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest 127: 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capers Q, Alexander RW, Lou P, De Leon H, Wilcox JN, Ishizaka N et al (1997a). Monocyte chemoattractant protein‐1 expression in aortic tissues of hypertensive rats. Hypertension 30: 1397–1402. [DOI] [PubMed] [Google Scholar]
- Capers Q, Laursen JB, Fukui T, Rajagopalan S, Mori I, Lou P et al (1997b). Vascular thrombin receptor regulation in hypertensive rats. Circ Res 80: 838–844. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Mascio G, Ajmone‐Cat MA, D'Andrea I, Cifelli G, Madonna M et al (2012a). Role of neuroinflammation in hypertension‐induced brain amyloid pathology. Neurobiol Aging 33: 205.e219–e229. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Mascio G, D'Andrea I, Fardella V, Bell RD, Branchi I et al (2012b). Hypertension induces brain beta‐amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 60: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathcart MK (2004). Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis. Arterioscler Thromb Vasc Biol 24: 23–28. [DOI] [PubMed] [Google Scholar]
- Chamarthi B, Williams GH, Ricchiuti V, Srikumar N, Hopkins PN, Luther JM et al (2011). Inflammation and hypertension: the interplay of interleukin‐6, dietary sodium, and the renin‐angiotensin system in humans. Am J Hypertens 24: 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I (1999). Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277. [DOI] [PubMed] [Google Scholar]
- Clozel M, Kuhn H, Hefti F, Baumgartner HR (1991). Endothelial dysfunction and subendothelial monocyte macrophages in hypertension. Effect of angiotensin converting enzyme inhibition. Hypertension 18: 132–141. [DOI] [PubMed] [Google Scholar]
- Cybulsky MI, Gimbrone MA Jr (1991). Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science 251: 788–791. [DOI] [PubMed] [Google Scholar]
- D'Andrea A, Rengaraju M, Valiante NM, Chehimi J, Kubin M, Aste M et al (1992). Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J Exp Med 176: 1387–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL (2005). Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II‐infused macrophage colony‐stimulating factor‐deficient mice: evidence for a role in inflammation in angiotensin‐induced vascular injury. Arterioscler Thromb Vasc Biol 25: 2106–2113. [DOI] [PubMed] [Google Scholar]
- de Kloet AD, Liu M, Rodriguez V, Krause EG, Sumners C (2015). Role of neurons and glia in the CNS actions of the renin‐angiotensin system in cardiovascular control. Am J Physiol Regul Integr Comp Physiol 309: R444–R458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S et al (2017). MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ Res 121: 930–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia‐Aguilar J, Hibbs ML et al (1990). ICAM‐1 (CD54): a counter‐receptor for Mac‐1 (CD11b/CD18). J Cell Biol 111: 3129–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorffel Y, Latsch C, Stuhlmuller B, Schreiber S, Scholze S, Burmester GR et al (1999). Preactivated peripheral blood monocytes in patients with essential hypertension. Hypertension 34: 113–117. [DOI] [PubMed] [Google Scholar]
- Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O et al (2017). Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med 377: 1319–1330. [DOI] [PubMed] [Google Scholar]
- Fan X, Sibalic V, Niederer E, Wuthrich RP (1996). The proinflammatory cytokine interleukin‐12 occurs as a cell membrane‐bound form on macrophages. Biochem Biophys Res Commun 225: 1063–1067. [DOI] [PubMed] [Google Scholar]
- Ferguson AV (2009). Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinology 89: 370–376. [DOI] [PubMed] [Google Scholar]
- Folkow B, Hallback M, Lundgren Y, Sivertsson R, Weiss L (1973). Importance of adaptive changes in vascular design for establishment of primary hypertension, studied in man and in spontaneously hypertensive rats. Circ Res 32 (Suppl 1): 2–16. [DOI] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al (2007). Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176: 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B et al (2017). Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med 23: 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailani D, Broze GJ Jr (1991). Factor XI activation in a revised model of blood coagulation. Science 253: 909–912. [DOI] [PubMed] [Google Scholar]
- Ge X, Yamaguchi Y, Zhao L, Bury L, Gresele P, Berube C et al (2018). Prochemerin cleavage by factor XIa links coagulation and inflammation. Blood 131: 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR (2003). Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19: 71–82. [DOI] [PubMed] [Google Scholar]
- Goldblatt H (1947). The renal origin of hypertension. Physiol Rev 27: 120–165. [DOI] [PubMed] [Google Scholar]
- Gordon S, Todd J, Cohn ZA (1974). In vitro synthesis and secretion of lysozyme by mononuclear phagocytes. J Exp Med 139: 1228–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe M, Auch‐Schwelk W, Zakrzewicz A, Regitz‐Zagrosek V, Bartsch P, Graf K et al (1997). Angiotensin II‐induced leukocyte adhesion on human coronary endothelial cells is mediated by E‐selectin. Circ Res 81: 804–811. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwani SC (2018). Macrophages under pressure: the role of macrophage polarization in hypertension. Transl Res 191: 45–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashmat S, Rudemiller N, Lund H, Abais‐Battad JM, Van Why S, Mattson DL (2016). Interleukin‐6 inhibition attenuates hypertension and associated renal damage in Dahl salt‐sensitive rats. Am J Physiol Renal Physiol 311: F555–F561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Presa M, Bustos C, Ortego M, Tunon J, Renedo G, Ruiz‐Ortega M et al (1997). Angiotensin‐converting enzyme inhibition prevents arterial nuclear factor‐κB activation, monocyte chemoattractant protein‐1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 95: 1532–1541. [DOI] [PubMed] [Google Scholar]
- Herrera J, Ferrebuz A, MacGregor EG, Rodriguez‐Iturbe B (2006). Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17: S218–S225. [DOI] [PubMed] [Google Scholar]
- Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R et al (2018). Myeloid CD11c(+) antigen‐presenting cells ablation prevents hypertension in response to angiotensin II plus high‐salt diet. Hypertension 71: 709–718. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S et al (2004). Critical role of monocyte chemoattractant protein‐1 receptor CCR2 on monocytes in hypertension‐induced vascular inflammation and remodeling. Circ Res 94: 1203–1210. [DOI] [PubMed] [Google Scholar]
- Karbach SH, Schonfelder T, Brandao I, Wilms E, Hormann N, Jackel S et al (2016). Gut microbiota promote angiotensin II‐induced arterial hypertension and vascular dysfunction. J Am Heart Assoc 5: pii: e003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Berliner JA, Nadler JL (1996). Angiotensin II increases monocyte binding to endothelial cells. Biochem Biophys Res Commun 226: 862–868. [DOI] [PubMed] [Google Scholar]
- Kintscher U, Wakino S, Kim S, Fleck E, Hsueh WA, Law RE (2001). Angiotensin II induces migration and Pyk2/paxillin phosphorylation of human monocytes. Hypertension 37: 587–593. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Lagrange J, Jackel S, Jurk K, Ehlken M, Schonfelder T et al (2017). Platelet‐localized FXI promotes a vascular coagulation‐inflammatory circuit in arterial hypertension. Sci Transl Med 9: pii: eaah4923. [DOI] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M et al (2013). Angiotensin II‐induced vascular dysfunction depends on interferon‐γ‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 33: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Kurihara T, Shimizu‐Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ et al (2012). Neutrophil‐derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation 126: 3070–3080. [DOI] [PubMed] [Google Scholar]
- Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I et al (2009). Regulatory T cells ameliorate angiotensin II‐induced cardiac damage. Circulation 119: 2904–2912. [DOI] [PubMed] [Google Scholar]
- Lagrange J, Kossmann S, Kiouptsi K, Wenzel P (2018). Visualizing leukocyte rolling and adhesion in angiotensin II‐infused mice: techniques and pitfalls. J Vis Exp. 10.3791/56948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H et al (2002). Role of p47 (phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DL, Sturgis LC, Labazi H, Osborne JB Jr, Fleming C, Pollock JS et al (2006). Angiotensin II hypertension is attenuated in interleukin‐6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940. [DOI] [PubMed] [Google Scholar]
- Leslie M (2018). Restraining immunity could lower high blood pressure. Science 359: 966–967. [DOI] [PubMed] [Google Scholar]
- Leusen JH, Verhoeven AJ, Roos D (1996). Interactions between the components of the human NADPH oxidase: intrigues in the phox family. J Lab Clin Med 128: 461–476. [DOI] [PubMed] [Google Scholar]
- Li H, Horke S, Forstermann U (2014). Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237: 208–219. [DOI] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair‐Rohani H et al (2012). A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380: 2224–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP (2004). Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol 24: 1359–1366. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ (2003). NAD (P) H oxidase mediates angiotensin II‐induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol 23: 776–782. [DOI] [PubMed] [Google Scholar]
- Liu Z, Morgan S, Ren J, Wang Q, Annis DS, Mosher DF et al (2015). Thrombospondin‐1 (TSP1) contributes to the development of vascular inflammation by regulating monocytic cell motility in mouse models of abdominal aortic aneurysm. Circ Res 117: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al (2012). Interferon‐γ signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Mateo T, Abu Nabah YN, Abu Taha M, Mata M, Cerda‐Nicolas M, Proudfoot AE et al (2006). Angiotensin II‐induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol 176: 5577–5586. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M et al (2005). Nox1 is involved in angiotensin II‐mediated hypertension: a study in Nox1‐deficient mice. Circulation 112: 2677–2685. [DOI] [PubMed] [Google Scholar]
- McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ (1998). Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo. Circulation 98: 2765–2773. [DOI] [PubMed] [Google Scholar]
- McMaster WG, Kirabo A, Madhur MS, Harrison DG (2015). Inflammation, immunity, and hypertensive end‐organ damage. Circ Res 116: 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino A, Alvarez‐Lara MA, Ramirez R, Carracedo J, Martin‐Malo A, Aljama P (2012). Losartan prevents the development of the pro‐inflammatory monocytes CD14+CD16+ in haemodialysis patients. Nephrol Dial Transplant 27: 2907–2912. [DOI] [PubMed] [Google Scholar]
- Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M et al (2002). Effects of angiotensin II infusion on the expression and function of NAD (P) H oxidase and components of nitric oxide/cGMP signaling. Circ Res 90: E58–E65. [DOI] [PubMed] [Google Scholar]
- Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT et al (2015). M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol 309: H906–H917. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Tabas I (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, Imai T, Yamaguchi T, Sugiyama T, Katayama S, Yoshino G (2003). Induction of heme oxygenase‐1 in monocytes suppresses angiotensin II‐elicited chemotactic activity through inhibition of CCR2: role of bilirubin and carbon monoxide generated by the enzyme. Antioxid Redox Signal 5: 439–447. [DOI] [PubMed] [Google Scholar]
- Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A et al (2000b). NF‐κB inhibition ameliorates angiotensin II‐induced inflammatory damage in rats. Hypertension 35: 193–201. [DOI] [PubMed] [Google Scholar]
- Muller DN, Mervaala EM, Dechend R, Fiebeler A, Park JK, Schmidt F et al (2000a). Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II‐induced cardiac vasculopathy. Am J Pathol 157: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahrendorf M, Hu K, Frantz S, Jaffer FA, Tung CH, Hiller KH et al (2006). Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation 113: 1196–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoleone E, Di Santo A, Camera M, Tremoli E, Lorenzet R (2000). Angiotensin‐converting enzyme inhibitors downregulate tissue factor synthesis in monocytes. Circ Res 86: 139–143. [DOI] [PubMed] [Google Scholar]
- Ndisang JF, Mishra M (2013). The heme oxygenase system selectively suppresses the proinflammatory macrophage m1 phenotype and potentiates insulin signaling in spontaneously hypertensive rats. Am J Hypertens 26: 1123–1131. [DOI] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018). The immunology of hypertension. J Exp Med 215: 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen F (1971). Correlation between infiltration of mononuclear cells and production of connective tissue in acute hypertensive vascular disease. Acta Pathol Microbiol Scand C 79: 15–21. [DOI] [PubMed] [Google Scholar]
- Olsen F (1980). Transfer of arterial hypertension by splenic cells from DOCA‐salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C Immunol 88: 1–5. [DOI] [PubMed] [Google Scholar]
- Ozawa Y, Kobori H, Suzaki Y, Navar LG (2007). Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292: F330–F339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV et al (2000). Angiotensin II induces leukocyte‐endothelial cell interactions in vivo via AT(1) and AT(2) receptor‐mediated P‐selectin upregulation. Circulation 102: 2118–2123. [DOI] [PubMed] [Google Scholar]
- Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB (2000). Angiotensin II stimulates endothelial vascular cell adhesion molecule‐1 via nuclear factor‐κB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol 20: 645–651. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK et al (1996). Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ (2001). Novel competitive inhibitor of NAD (P) H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res 89: 408–414. [DOI] [PubMed] [Google Scholar]
- Ridker PM (2009). Testing the inflammatory hypothesis of atherothrombosis: scientific rationale for the cardiovascular inflammation reduction trial (CIRT). J Thromb Haemost JTH 7 (Suppl 1): 332–339. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH (1997). Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 336: 973–979. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ et al (2008). Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med 359: 2195–2207. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C et al (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ et al (2018). Relationship of C‐reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391: 319–328. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND (2004). Evolution of renal interstitial inflammation and NF‐κB activation in spontaneously hypertensive rats. Am J Nephrol 24: 587–594. [DOI] [PubMed] [Google Scholar]
- Rothmeier AS, Marchese P, Petrich BG, Furlan‐Freguia C, Ginsberg MH, Ruggeri ZM et al (2015). Caspase‐1‐mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Invest 125: 1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag CM, Schnelle M, Zhang J, Murdoch CE, Kossmann S, Protti A et al (2017). Distinct regulatory effects of myeloid cell and endothelial cell NAPDH oxidase 2 on blood pressure. Circulation 135: 2163–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieffer B, Schieffer E, Hilfiker‐Kleiner D, Hilfiker A, Kovanen PT, Kaartinen M et al (2000). Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation 101: 1372–1378. [DOI] [PubMed] [Google Scholar]
- Schulz C, Gomez Perdiguero E, Chorro L, Szabo‐Rogers H, Cagnard N, Kierdorf K et al (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90. [DOI] [PubMed] [Google Scholar]
- Shen XZ, Li Y, Li L, Shah KH, Bernstein KE, Lyden P et al (2015). Microglia participate in neurogenic regulation of hypertension. Hypertension 66: 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV et al (2000). Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac‐1 (CD11b/CD18). J Exp Med 192: 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquest K, Powell N, Luci C, van Rooijen N, Hidalgo A, Geissmann F et al (2011). Monocytes control natural killer cell differentiation to effector phenotypes. Blood 117: 4511–4518. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K et al (2017). Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood 129: 2291–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida T, Naito AT, Nomura S, Nakagawa A, Higo T, Hashimoto A et al (2015). Complement C1q‐induced activation of β‐catenin signalling causes hypertensive arterial remodelling. Nat Commun 6: 6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori K, Ito H, Suzuki T (2000). Effects of the AT1 receptor antagonist on adhesion molecule expression in leukocytes and brain microvessels of stroke‐prone spontaneously hypertensive rats. Am J Hypertens 13: 1233–1241. [DOI] [PubMed] [Google Scholar]
- Taubman MB, Marmur JD, Rosenfield CL, Guha A, Nichtberger S, Nemerson Y (1993). Agonist‐mediated tissue factor expression in cultured vascular smooth muscle cells. Role of Ca2+ mobilization and protein kinase C activation. J Clin Invest 91: 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu BC, Lee C, Sun H, Lejeune W, Recinos A 3rd, Ju X et al (2009). An adventitial IL‐6/MCP1 amplification loop accelerates macrophage‐mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest 119: 3637–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL (1993). Effects of angiotensin II and endothelin‐1 on platelet aggregation and cytosolic pH and free Ca2+ concentrations in essential hypertension. Hypertension 22: 853–862. [DOI] [PubMed] [Google Scholar]
- Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW et al (1999). Angiotensin II induces vascular cell adhesion molecule‐1 expression in rat vasculature: a potential link between the renin‐angiotensin system and atherosclerosis. Circulation 100: 1223–1229. [DOI] [PubMed] [Google Scholar]
- van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G et al (2016). Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue‐resident macrophages. Immunity 44: 755–768. [DOI] [PubMed] [Google Scholar]
- von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M et al (2012). Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209: 819–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ et al (2001). Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res 88: 947–953. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K et al (2005). Leukocyte engagement of platelet glycoprotein Ibα via the integrin Mac‐1 is critical for the biological response to vascular injury. Circulation 112: 2993–3000. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Rossmann H, Muller C, Kossmann S, Oelze M, Schulz A et al (2015). Heme oxygenase‐1 suppresses a pro‐inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J 36: 3437–3446. [DOI] [PubMed] [Google Scholar]
- Wenzel UO, Fouqueray B, Grandaliano G, Kim YS, Karamitsos C, Valente AJ et al (1995). Thrombin regulates expression of monocyte chemoattractant protein‐1 in vascular smooth muscle cells. Circ Res 77: 503–509. [DOI] [PubMed] [Google Scholar]
- Wilson C (1963). Management of renal hypertension. Biochem Clin 2: 275–283. [PubMed] [Google Scholar]
- Wu KL, Chan SH, Chan JY (2012). Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation 9: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Takeuchi T, Kotani T, Yamamoto N, Hata K, Nagai K et al (2014). Infliximab, a TNF‐α inhibitor, reduces 24‐h ambulatory blood pressure in rheumatoid arthritis patients. J Hum Hypertens 28: 165–169. [DOI] [PubMed] [Google Scholar]
- Zeller T, Schurmann C, Schramm K, Muller C, Kwon S, Wild PS et al (2017). Transcriptome‐wide analysis identifies novel associations with blood pressure. Hypertension 70: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA et al (2016). Interleukin‐1 receptor activation potentiates salt reabsorption in angiotensin II‐induced hypertension via the NKCC2 co‐transporter in the nephron. Cell Metab 23: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Carnevale KA, Cathcart MK (2003). Human monocytes use Rac1, not Rac2, in the NADPH oxidase complex. J Biol Chem 278: 40788–40792. [DOI] [PubMed] [Google Scholar]