

Abstract
Background and Purpose
The adaptive immune response and IL‐17A contribute to renal damage in several experimental models of renal injury.
Experimental Approach
To evaluate the role of the adaptive immune response, 5/6 nephrectomy was performed in wildtype DBA/1J mice and in recombination‐activating gene‐1 (RAG‐1) deficient mice that lack B and T‐cells. To assess the role of IL‐17A, we carried out 5/6 nephrectomy in IL‐17A deficient mice. Flow cytometric analysis, immunohistochemistry and RT‐PCR were used.
Key Results
Infiltration of CD3+ T‐cells in the remnant kidney was increased after 5/6 nephrectomy in wildtype mice, along with a robust induction of IL‐17A production in CD4+ T and γδ T‐cells. After 5/6 nephrectomy, wildtype mice developed albuminuria in the nephrotic range over 10 weeks. This was accompanied by severe glomerular sclerosis and tubulointerstitial injury, and as well as renal mRNA expression of markers of inflammation and fibrosis (the chemokine CCL2, plasminogen activator inhibitor‐1; PAI‐1 and neutrophil gelatinase‐associated lipocalin; NGAL). Unexpectedly, RAG‐1 deficient mice and IL‐17A deficient mice developed renal injury, similar to that in wildtype mice. No differences were found for albuminuria, glomerular sclerosis, tubulointerstitial injury and mRNA expression of CCL2, PAI‐1 and NGAL. Mortality did not differ between the three groups.
Conclusions and Implications
Numbers of CD3+ T‐cells and IL‐17A+ lymphocytes infiltrating the kidney were increased after 5/6 nephrectomy. In contrast to other experimental models of renal injury, genetic deficiency of the adaptive immune system or of IL‐17A did not attenuate induction or progression of chronic kidney disease after 5/6 nephrectomy.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- CKD
chronic kidney disease
- FoxP3
forkhead box P3
- FSC
forward scatter
- GR1
granulocyte receptor 1
- NGAL
neutrophil gelatinase‐associated lipocalin
- PAI‐1
plasminogen activator inhibitor‐1
- PAS
periodic acid–Schiff
- RAG‐1
recombination‐activating gene‐1
- SSC
side scatter
- Treg
regulatory T‐cells
Introduction
Recent data suggest that chronic kidney disease (CKD) is not only mediated by haemodynamic injury but also by innate and adaptive immune responses (Lehners et al., 2014). Development of CKD is accompanied by T‐cell accumulation in the kidney and immunosuppressive therapies directed against T‐cells restrict disease (Fujihara et al., 1998), but the mechanisms causing inflammation in CKD remain elusive. Moreover, which cell population causes the renal injury is not well‐defined. We recently described a massive infiltration of CD3 + T‐cells in the remaining kidney after 5/6 nephrectomy (Lehners et al., 2014); The technique of 5/6 nephrectomy is an excellent experimental model of human CKD, because it reproduces many features, including loss of renal function, proteinuria, glomerular and tubulointerstitial lesions, which are characteristic for the evolution and progression of human kidney disease. Over the last 50 years, this model has led to the discovery of key pathophysiological pathways as well as to the design of therapeutic strategies to slow the progression of CKD, such as the widely clinically used renin‐angiotensin inhibitors (Anderson et al., 1986).
The recombination‐activating gene‐1 (RAG‐1) is essential to the generation of B and T lymphocytes, the two cell types that are crucial components of the adaptive immune system. Therefore, RAG‐1 knockout mice can be used to evaluate the role of the adaptive immune system in models of disease. In 2005, a novel T helper cell subset that produces the unique cytokine IL‐17, designated TH17 cells, was described. IL‐17A is the most widely expressed member of the IL‐17 family and has been implicated in the pathogenesis of many inflammatory diseases, such as rheumatoid arthritis, psoriasis, multiple sclerosis, asthma and inflammatory bowel disease (Krebs and Panzer, 2018; Krebs et al., 2017; Veldhoen, 2017). In addition, a causative role of IL‐17A has been shown in hypertension (Norlander et al., 2016; Saleh et al., 2016), glomerulonephritis (Paust et al., 2009) and other renal disease models such as cyclosporine and cisplatin induced renal injury (Chan et al., 2014; Chiasson et al., 2017). It is of interest that deficiency or inhibition of IL‐17A was protective in all the models. An increased frequency of TH17 cells has been described in progressive renal fibrosis (Mehrotra et al., 2015). However, the causative role of IL‐17A in CKD induced by 5/6 nephrectomy is unknown.
Most mice strains are resistant to 5/6 nephrectomy (Ma and Fogo, 2003; Viau et al., 2010). We and others have recently described that FVB/N mice develop severe renal injury in response to 5/6 nephrectomy (Benndorf et al., 2009; Fraune et al., 2012; Lehners et al., 2014). In the present study, we used mice of the DBA/1J strain and describe for the first time (i) that this strain also develops severe renal injury after 5/6 nephrectomy, (ii) that renal injury is accompanied by a robust invasion of the remnant kidney by IL‐17A producing γδ T‐cells and CD4 + T‐cells, but (iii) that neither B cells, T‐cells nor IL‐17A are required for the full development of renal damage after 5/6 nephrectomy.
Methods
Mice and experimental groups
All animal care and experimental procedures have been approved by the local animal committee and were in accord with national and institutional animal care guidelines. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). IL‐17A deficient (IL‐17A−/−) mice were provided by Y. Iwakura (Nakae et al., 2002). IL‐17A−/− and RAG‐1−/− were backcrossed for at least 10 generations with DBA/1J mice, as previously described (Hünemörder et al., 2015). Experiments were performed in male mice at an age of 10 to 12 weeks.
5/6 Nephrectomy
In total, 39 wildtype, 42 IL‐17A deficient mice and 28 RAG‐1 deficient mice were used in the study. Following anaesthesia with isoflurane (induction 5%, maintenance 3%), a small flank incision was used to obtain retroperitoneal access in order to expose the left kidney; 2/3 of this kidney was removed as described earlier (Fraune et al., 2012). For analgesia tramadol (10 mg/kg) was given subcutaneously at the time of surgery and three days in the drinking water (1 mg/mL). The removed tissue was weighed with a precision balance (ED153; Sartorius, Germany). Fourteen days later, 5/6 nephrectomy was completed by contralateral uni‐nephrectomy. One week after the second surgery, urinary albumin and creatinine were measured. Only mice with an albumin‐to‐creatinine ratio over 0.5 mg·mg−1 (indicating successful induction of renal disease) were included in the experiment. Age‐matched, untouched wildtype animals were used as the control group (n = 8). At the end of the experiment, 12 weeks after the second nephrectomy, under deep anaesthesia with isoflurane (5%) and tramadol (10 mg/kg, s.c.), heparinized blood (20 IU heparin/mL) was withdrawn by cardiac puncture and the remnant kidneys were removed for examination. In this model, some animals (8 WT; 6 RAG‐1‐/‐ and 9 IL‐17A‐/‐ mice) died within 1‐2 days after the second nephrectomy and the remainder of animals excluded (Table 2) did not develop CKD (Lehners et al., 2014).
Table 2.
Number of mice
| Control | Wildtype | RAG‐1−/− | IL‐17A−/− | |
|---|---|---|---|---|
| All mice | 8 | 39 | 28 | 42 |
| Excludeda | – | 22 (56%) | 16 (57%) | 19 (45%) |
| Remaining | 8 | 17 | 12 | 23 |
| End of experiment | 8 | 15 (88%) | 9 (75%) | 17 (74%) |
Albumin‐to‐creatinine ratio <0.5 mg·mg−1 or death after surgery.
Albuminuria, plasma, urine
Mice were placed into metabolic cages for a 6 h urine collection (Krebs et al., 2012, 2014) every second week after the 5/6 nephrectomy. At the end of the experimental period, animals were killed and blood collected, as described above. Plasma urea‐N and urinary creatinine were measured by an autoanalyser (Hitachi 717; Roche, Mannheim, Germany). Albumin in urine was measured by ELISA (Bethyl Laboratories, Montgomery, USA).
Histopathological analysis
The remnant kidney tissues were fixed with 4% neutral buffered formalin, embedded in paraffin and sectioned at 1 μm thickness. Sections were then deparaffinized and stained for light microscopy with periodic acid–Schiff (PAS) (renal tissue). Glomerular injury was evaluated histologically using a semiquantitative scale with 0 indicating no injury, 1 mild injury in less than a third of the glomerular tuft, 2 damage of more than a third of the glomerular tuft and 3 damage of the whole glomerulus. Under 200× magnification, 20 glomeruli per animal were analysed (Fraune et al., 2012; Lange et al., 2013). Glomerular size was measured with Axiovision LE (Carl Zeiss MicroImaging, Jena, Germany). Renal infiltration by CD3+ T‐cells was examined immunohistochemically as described (Krebs et al., 2014); 1.5 μm thick sections were used. Cells were visualized immunohistochemically using anti‐CD3 antibodies (polyclonal antibody, product number A 0452, DakoCytomation, USA), anti‐forkhead box P3 (FoxP3) (eBioscience, Schwerte, Germany), anti‐F4/80 (BMA Biomedicals, Augst, Switzerland) and anti‐granulocyte receptor 1 (GR1) (Ly6G, Hycult, Uden, The Netherlands). For detection, ZytoChem‐Plus AP Polymer kit was used (Zytomed, Berlin, Germany). Infiltration of cells was counted in 20 high power fields per section. Scoring was performed in a blinded fashion.
Real‐time quantitative RT‐PCR
Total RNA from kidney cortex was isolated using the RNeasy kit (Qiagen, USA). Real‐time quantitative PCR was performed using the Applied Biosystems ABI Prism system and SYBR Green JumpStart taq Ready Mix (Sigma, Germany). Mouse‐specific PCR primers were used. The levels of mRNA expression in each sample were normalized to 18S rRNA expression (Krebs et al., 2014).
Flow cytometry
Previously described methods for leukocyte isolation from murine kidneys were used (Weiss et al., 2016). Kidneys were minced and digested with collagenase D and DNase (Roche, Mannheim, Germany) at 37°C for 45 min. To get a single cell suspension, kidneys were then dissociated using the gentleMACS Dissociater (Miltenyi Biotec, Bergisch Gladbach, Germany) and centrifuged at 300× g at 4°C for 8 min. Percoll gradient (37% Percoll; GE Healthcare, Chalfont St. Giles, Great Britain) centrifugation was performed at 500× g at room temperature for 20 min to further purify the cells. Subsequently, erythrocytes were lysed with ammonium chloride. For flow cytometry analysis, fluorochrome‐conjugated antibodies were used (CD45 (30‐F11), CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7), γδTCR (GL3), NK1.1 (PK136)). For intracellular staining, samples were processed using a commercial intranuclear staining kit (FoxP3 kit; eBioscience). Staining of intracellular IL‐17A (TC11‐18H10.1) and IFNγ (XMG1.2) was performed as recently described (Krebs et al., 2014). In brief, cells were activated by incubation for 4 h with phorbol 12‐myristate 13‐acetate (50 ng·mL−1; Sigma) and ionomycin (1 μg·mL−1; Calbiochem‐Merck, Darmstadt, Germany). After 30 min of incubation, Brefeldin A (10 μg·mL−1; Sigma) was added. LIVE/DEAD staining (Invitrogen Molecular Probes, Carlsbad, USA) was used to exclude dead cells during flow cytometry. Absolute numbers of cells in the kidney cell suspensions were determined by comparing the ratio of CD45+ cell events to counting bead events (Countbright; Invitrogen Molecular Probes) by flow cytometry analysis. Number of cells were expressed per gram kidney weight. Samples were acquired on a Becton & Dickinson LSRII System using the Diva software. Data analysis was performed with FlowJo (Tree Star, USA). After gating for single cells by forward scatter (FSC)‐W and FSC‐A, followed by gating for living cells, leukocytes isolated from the kidney were determined via CD45 surface staining.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). For the statistical analysis, GraphPad Prism 6.01 was used. All data are expressed as mean ± SEM. The Kolmogorov–Smirnov test was used to test for normal distribution of the values. In case of normal distribution for multiple comparisons one‐way ANOVA and post hoc analysis by Newman–Keuls Multiple Comparison test was performed. If the data were not normally distributed, Kruskall–Wallis with Dunn's test was used. For comparison of two parameters, unpaired t‐test for normally distributed and Mann–Whitney test for not normally distributed parameters were used. Differences were considered statistically significant at P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Detection of TH17 cells in a model of CKD
As a first step, we measured IL‐17A producing cells in the kidneys of mice 2 weeks after 5/6 nephrectomy. The gating strategy is shown in Figure 1. Cells were first plotted for granularity [side scatter (SSC‐A)] and size (FSC‐A) (Figure 1A). Only single cells were gated by FSC‐W and FSC‐A (Figure 1B). From these, only living cells were examined (Figure 1C). Out of the living cells, lymphocytes were determined by SSC‐A and CD45high expression (Figure 1D). Intracellular cytokine staining was performed. IL‐17A+ cells were gated as shown in Figure 1E and F. Quantification revealed that 0.75% of the renal CD45+ T‐cells in control mice produced IL‐17A, whereas this was increased 4.5‐fold in mice after 5/6 nephrectomy (Figure 1G). Next, we examined which cells expressed IL‐17A and found that mainly CD4+ and γδ T‐cells were producing IL‐17A, whereas CD8+ T‐cells only marginally produced IL‐17A (Figure 1H). The increased percentage of IL‐17A+ cells was significant for each of the three cell types. IL‐17A staining in NK cells and NKT cells was extremely low. The number of IFNγ+ T‐cells was also increased after 5/6 nephrectomy (Figure 1I).
Figure 1.
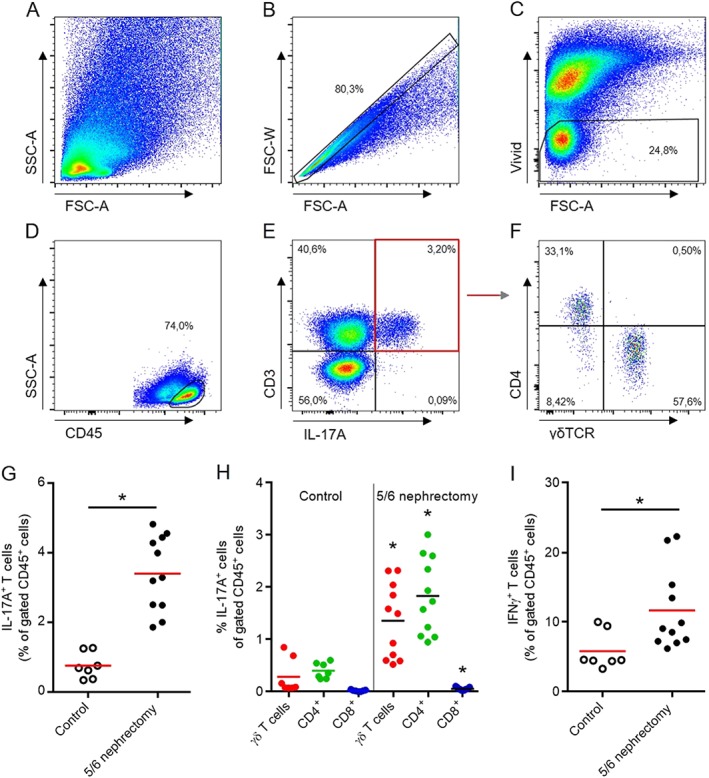
Detection of IL‐17A expressing lymphocytes in the kidney. Flow cytometry gating strategy of leukocytes isolated from the kidney, 2 weeks after 5/6 nephrectomy (A–F). Intracellular cytokine staining revealed that 0.75% of the infiltrating CD45+ T‐cells in control mice produced IL‐17A, whereas this was increased to 3.41% in mice after 5/6 nephrectomy (G). Mainly CD4+ and γδ T‐cells produced IL‐17A, whereas CD8+ cells only marginally produced IL‐17A. Compared to control mice, the increased percentage was significant for each of the three cell types (H). The number of IFNγ+ cells was increased after 5/6 nephrectomy (I). Number of mice examined was controls n = 7, 5/6 nephrectomy n = 11. *P < 0.05, significantly different from control.
Induction of CKD in wildtype, RAG‐1 and IL‐17A deficient mice
CKD was initiated by removal of around 2/3 of the left kidney. The amount of kidney tissue removed was not different between wildtype, RAG‐1 deficient and IL‐17A deficient mice indicating that the surgical procedure to induce renal injury was identical in all groups (Table 1). Albuminuria increased immediately after 5/6 nephrectomy. Some (about 20%) of the mice die within 2 days after 5/6 nephrectomy and some of the mice do not develop CKD, as shown previously by us (Lehners et al., 2014). To avoid studying mice without renal injury, mice with an albumin‐to‐creatinine ratio below 0.5 mg·mg−1 one week after the second nephrectomy were excluded from the experiment. As shown in Table 2, the number of excluded or dead mice was not significantly different between the three groups. In addition, the number of mice that died during the 12 weeks of observation did not differ between the three groups.
Table 1.
Body and kidney weight
| Wildtype | RAG‐1−/− | IL‐17A−/− | |
|---|---|---|---|
| Body weight at surgery (g) | 21.50 ± 0.50 | 22.16 ± 0.50 | 20.55 ± 0.63 |
| Rel. removed kidney weight at surgery (mg·g−1) | 3.24 ± 0.11 | 3.48 ± 0.09 | 3.20 ± 0.12 |
All data are expressed as mean ± SEM.
Albuminuria
Albuminuria is shown in Figure 2 with logarithmic scaling. Throughout the experiment, albuminuria was increased but no differences between the three groups were found, at any time point (Figure 2). IL‐17A−/− mice on the DBA/1J background have no basal renal phenotype (Hünemörder et al., 2015). In addition, to exclude the possibility that that RAG‐1 deficient mice on the DBA/1J background have a renal phenotype, the albumin/creatinine ratio was evaluated in RAG‐1 deficient mice before renal ablation and no difference was detected compared to wildtype controls (wildtype 0.51 ± 0.13 mg·mg−1, n = 12, RAG‐1−/− 0.41 ± 0.03 mg·mg−1, n = 24, P> 0.05).
Figure 2.
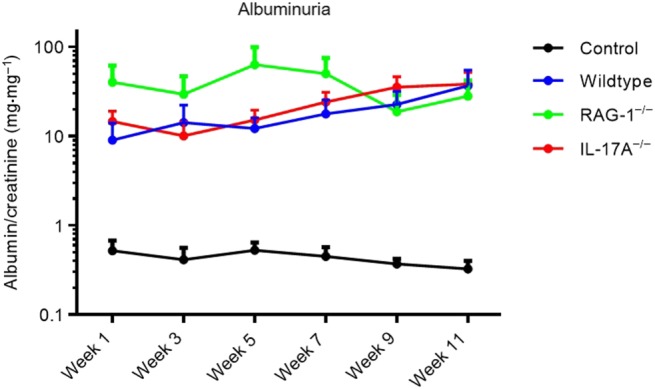
Albuminuria. Albumin/creatinine ratio 1, 3, 5, 7, 9 and 11 weeks after 5/6 nephrectomy is shown with logarithmic scaling. Throughout the experiment, albuminuria was increased, but no differences were found between the three groups, at any time point.
Renal function, cholesterol and leukocytes
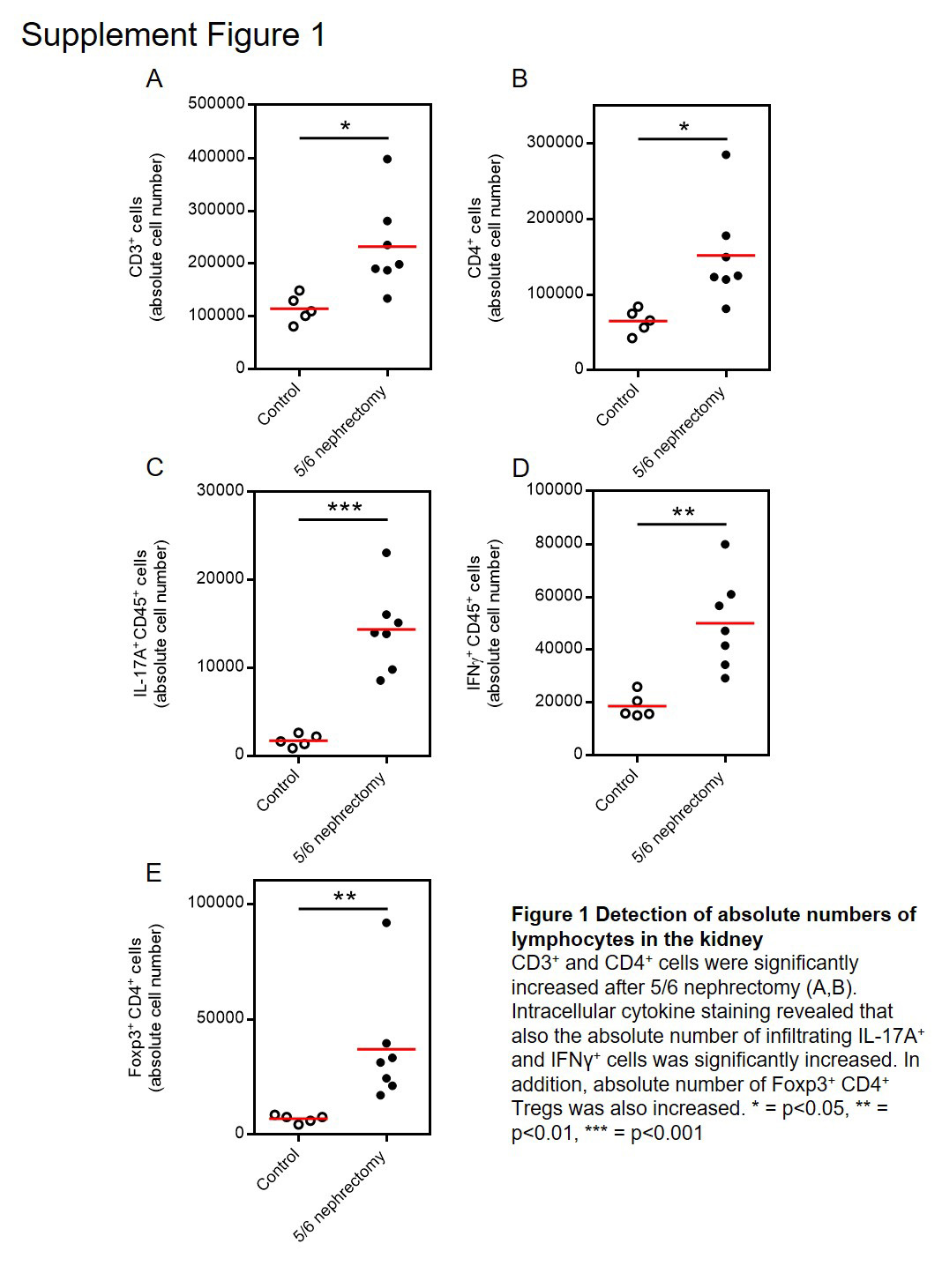
At the end of the experiment, 12 weeks after 5/6 nephrectomy, plasma urea‐N was increased, indicating reduced renal function, but no difference was detected between the three groups (Figure 3A). Plasma cholesterol levels were increased, indicating that albuminuria was in the nephrotic range, but again, no significant difference was found between the three groups (Figure 3B). Flow cytometry analysis of leukocytes isolated from the kidney revealed a significantly increased number of CD3+ T‐cells in wildtype mice after 5/6 nephrectomy compared to control mice, whereas no CD3+ T‐cells were detected in RAG‐1 deficient mice (Figure C and D). Immunohistochemistry showed an increased frequency of CD3+ T‐cells in wildtype mice and no cells were found in RAG‐1 knockout mice. Interestingly, a smaller number of CD3+ T‐cells was found in the kidneys of IL‐17A deficient mice compared to wildtype mice after renal ablation. As shown in Figure 3F, no CD3+ IL‐17A+ lymphocytes were detected in the kidneys of IL‐17A deficient mice, confirming the knockout of the gene as shown in the upper right square of representative blots of a wildtype and an IL‐17A−/− mouse. To demonstrate that the relative increase of CD3+ T‐cells, of IL‐17A+ and IFNγ+ cells reflected an increase in absolute cell numbers, we added count beads during the isolation of cells from the kidney. Absolute numbers calculated on the basis of count beads showed similar results for relative and absolute cell numbers (Supporting Information Figure S1A–D). In addition, we also examined FoxP3+ CD4+ regulatory T‐cells (Tregs) and found these cells to be significantly up‐regulated in the kidney after 5/6 nephrectomy as shown in Supporting Information Figure S1E. NK and NKT cells were evaluated in the kidney by flow cytometry as shown in Figure 3G. CD3−NKp46+ NK cells are shown in the circle and CD3+NKp46+ NKT cells in the square. The numbers of renal NK cells did not change after 5/6 nephrectomy in wildtype and IL‐17A−/− mice and were increased in RAG‐1 deficient mice (Figure 3H). NKT cells behaved like T‐cells and were increased in wildtype and IL‐17A deficient and absent in RAG‐1 knockout mice (Figure 3I). The number of Treg cells was evaluated by immunohistochemistry. An increased number of FoxP3+ cells was found in wildtype and IL‐17A deficient mice, and no cells were detected in RAG‐1 deficient mice (Figure 3J).
Figure 3.
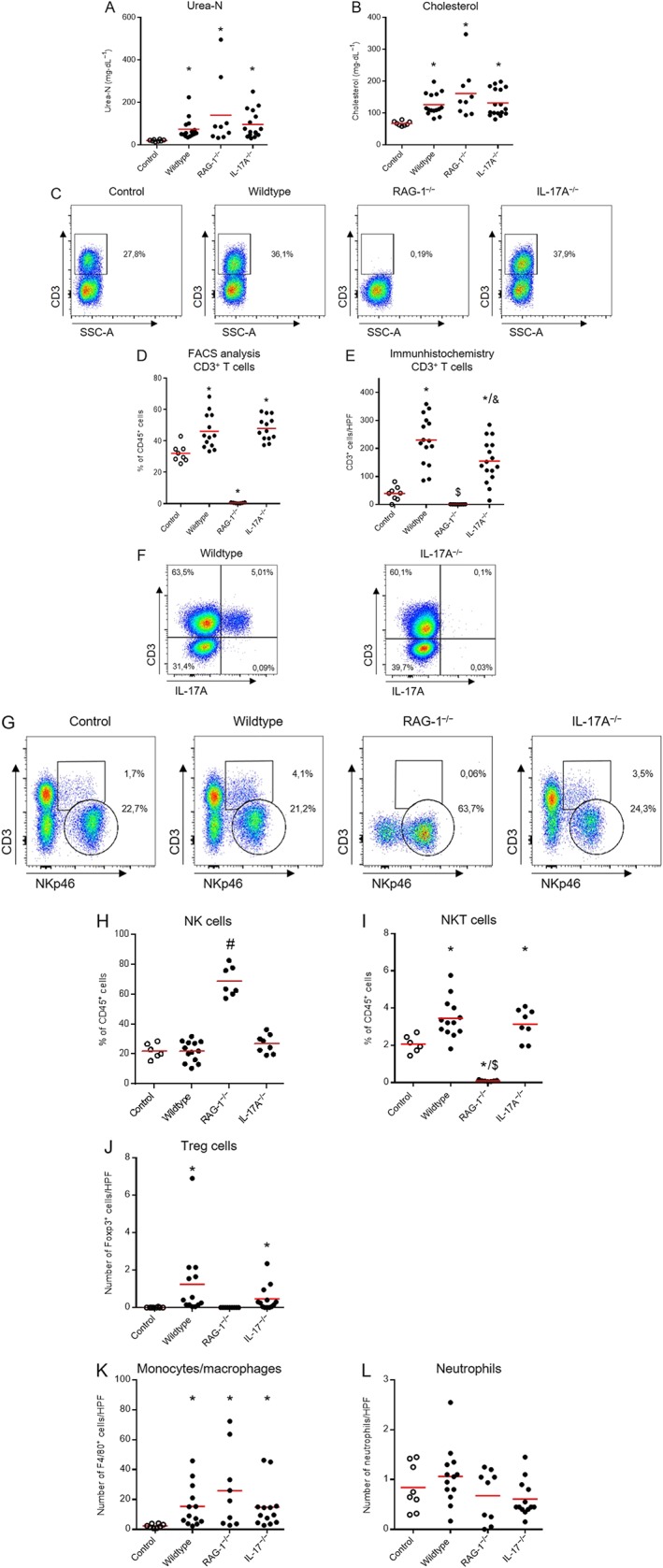
Renal function, plasma cholesterol and inflammatory cells. At the end of the experiment, an increased plasma urea‐N was found indicating reduced renal function (A). Elevated plasma cholesterol levels indicated the nephrotic range of albuminuria (B). In a subset of animals, leukocytes were isolated from the kidney for analysis of T‐cells. Figure 3C shows representative blots of CD3+ T‐cells and Figure 3D quantification. A significantly increased frequency of infiltrating CD3+ T‐cells was found in the kidney after 5/6 nephrectomy in wildtype and IL‐17A−/− mice, whereas no CD3+ T‐cells were detectable in RAG‐1 deficient mice. CD3 immunohistochemistry data are shown in Figure 3E. An increased number of CD3+ cells was found in wildtype mice, and no cells were found in RAG‐1 knockout mice. The number of infiltrating T‐cells was slightly reduced in IL‐17A deficient mice. IL‐17A knockout was confirmed by the absence of CD3+ IL‐17A+ cells compared to wildtype mice as shown in the right upper squares of Figure 3F. Flow cytometry data for NK (circle) and NKT (square) cells isolated from the kidney are shown in Figure 3G. Number of renal NK cells did not change after 5/6 nephrectomy in wildtype and IL‐17A−/− mice and was increased in RAG‐1 deficient mice. NKT cells increased after 5/6 nephrectomy and were absent in RAG‐1 deficient mice (I). The number of Treg cells in kidney samples, evaluated by immunohistochemistry, was increased in wildtype and IL‐17A knockout, and no cells were detected in RAG‐1 knockout mice (J). F4/80+ monocytes/macrophages increased in all groups after 5/6 nephrectomy. No significant differences were found for GR1+ neutrophils (L). Number of mice examined was controls n = 8, wildtype n = 15, RAG‐1−/− n = 9, IL‐17A−/− n = 16–17. In the flow cytometry analysis, the number was smaller (controls n = 6–8, wildtype n = 13, RAG‐1−/− n = 7, IL‐17A−/− n = 8–13) as flow cytometry analysis was not carried out for all mice. Number of mice examined in immunohistochemistry was controls n = 8, wildtype n = 13–15, RAG‐1−/− n = 9–13, IL‐17A−/− n = 9–16). *P < 0.05, significantly different from versus control, $ P < 0.05, significantly different from wildtype and IL‐17A−/−, & P < 0.05, significantly different from wildtype, # P < 0.05, significantly different from control and wildtype.
In a subset of mice, we have examined by flow cytometry whether there is a compensatory increase of renal IFNγ+ CD4+ or CD8+ cells in IL‐17A−/− mice. The number of IFNγ+ CD4+ and CD8+ cells was not increased in IL‐17A knockout mice, and no cells were detected in RAG‐1−/− mice as shown in Supporting Information Table S1. This makes it unlikely that IFNγ+ T‐cells could take over the function of IL‐17A in the IL‐17A deficient mice.
To further evaluate innate immune cells, monocyte/macrophages and neutrophils were evaluated by immunohistochemistry using F4/80 and GR1 as markers. The results are shown in Figure 3K and L. An increased number of F4/80+ monocyte/macrophages was found after 5/6 nephrectomy without difference between the three genotypes. The number of GR1+ neutrophils was not different to controls after 5/6 nephrectomy.
As shown in Table 3, spleen weight was significantly increased in wildtype mice after 5/6 nephrectomy compared to control mice. Due to the lack of T‐ and B cells, RAG‐1 deficient mice have a small spleen. This was also found in the present study. RAG‐1 deficient mice had a significantly decreased spleen weight compared to control as well as wildtype and IL‐17A deficient mice after 5/6 nephrectomy (Table 3).
Table 3.
Body and spleen weight
| Control | Wildtype | RAG‐1−/− | IL‐17A−/− | |
|---|---|---|---|---|
| Body weight at kill (g) | 24.64 ± 0.61 | 22.80 ± 0.44 | 22.63 ± 1.35 | 21.81 ± 0.73 |
| Spleen weight rel. to body weight (mg·g‐1) | 3.56 ± 0.14 | 5.93 ± 0.30* | 1.34 ± 0.22* , # | 4.82 ± 0.42 |
All data are expressed as mean ± SEM.
P < 0.05, significantly different from control.
P < 0.05, significantly different from wildtype and IL‐17A−/−.
Renal injury
Whereas control animals presented a normal renal histology, PAS‐stained kidney sections after 5/6 nephrectomy revealed severe histopathological abnormalities as shown in Figure 4A. Glomeruli with mild injury are shown in the upper images, whereas severely damaged glomeruli are shown in the lower images. Enlarged glomeruli and capillary obsolescence was observed. The typical morphological changes are highlighted in the lower panel of the wildtype mice. The dark circle shows sclerosis. The glomerular and capillary architecture is completely lost. Plasma insudation is marked with an asterisk. The dashed circle shows matrix expansion and fibrosis. The capillary structure is still identifiable. Semiquantitative analysis of these changes showed no significant difference between the three groups (Figure 4B). In addition, also, no difference was found for glomerular size (Figure 4C). Interestingly, glomerular size was only numerically increased in the RAG‐1 deficient mice. The reason is unclear. An increased number of intratubular proteinaceous casts, the morphological correlate of proteinuria, were observed in mice after 5/6 nephrectomy. Quantification by counting revealed an increased number of casts after 5/6 nephrectomy, but no difference between the three genotypes (Figure 4D). In addition, no difference was found for tubulointerstitial injury assessed by scoring (Figure 4E).
Figure 4.
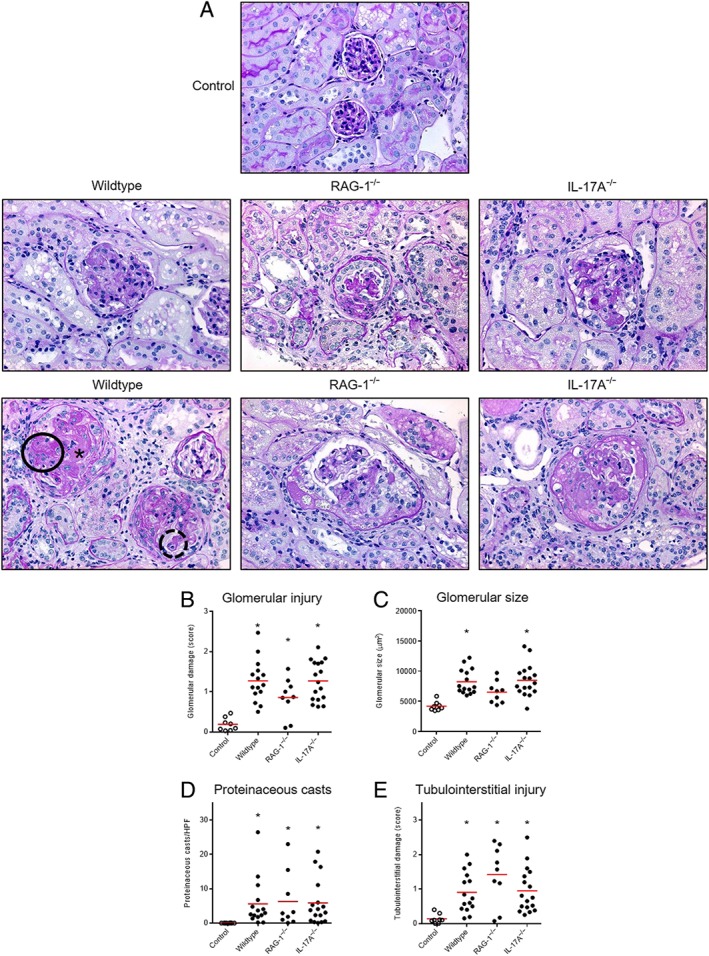
Renal injury. Representative micrographs of periodic acid–Schiff‐stained sections (magnification 400x) 12 weeks after 5/6 nephrectomy are shown (A). Compared with controls, enlarged glomeruli with mesangial expansion and sclerosis were found in all three groups. Glomeruli with mild injury are shown in the upper images, whereas severely damaged glomeruli are shown in the lower images. Scoring of glomerular injury (B), measurement of glomerular size (C), counting of intratubular proteineous casts (D) and scoring of tubulointerstitial injury (E) revealed no difference between the three groups. Number of mice examined was controls n = 8, wildtype n = 15, RAG‐1−/− n = 9, IL‐17A−/− n = 17. *P < 0.05, significantly different from control.
Markers of renal injury
Reverse transcriptase (RT)‐PCR analysis of mRNA derived from the whole kidney cortex was performed in a subset of experiments and showed increased expression of markers of renal injury such as neutrophil gelatinase‐associated lipocalin (Lcn2, NGAL), plasminogen activator inhibitor‐1 (Serpine1, PAI‐1) and the chemokine Ccl2 in all experimental groups, subjected to 5/6 nephrectomy (Figure 5 first row). In addition, the defining cytokines of TH17 and TH1 responses were evaluated. Il17a was 50‐fold up‐regulated in wildtype mice confirming the flow cytometry data. RAG‐1 deficient mice (that lack TH17 cells) showed significantly less Il17a expression than wildtype mice and no expression was detected in IL‐17A knockout mice. The presence of Il17a in RAG‐1 knockout mice points to an innate source of IL‐17A in these mice. Compared to Il17a, Ifnγ expression was only mildly up‐regulated after renal ablation compared to controls and the increase was only significant in wildtype mice (3.9‐fold compared to control mice). One may speculate that the mild increase of Ifnγ expression seen in the RAG‐1−/− mice could be derived from the NK cells that are up‐regulated in these mice as shown above in Figure 3H. FoxP3 is the master regulator of Treg cells. It was significantly increased in wildtype and IL‐17A−/− mice, and no expression was found in RAG‐1 deficient mice that lack Treg cells. Il6 and chemokine Cxcl1 were significantly increased in all groups after renal ablation.
Figure 5.
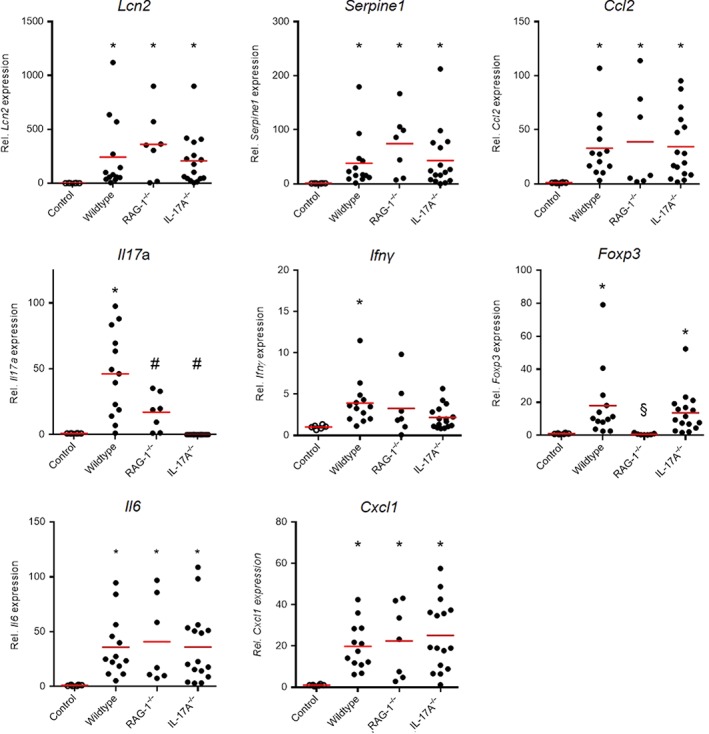
Expression of markers of renal injury. Renal mRNA expression data of inflammatory markers at the end of the experiment are shown in Figure 5. Serpine1, Lcn2 and Ccl2 were significantly increased after renal ablation compared to controls, and no difference was found between the three groups. The cytokines defining TH17 and TH1 sub‐types, Il17a and Ifnγ, were significantly up‐regulated in wildtype mice. An increased expression for the master regulator of Treg cells, Foxp3 was found in wildtype and IL‐17A−/− mice. In addition, Il6 and Cxcl1 were significantly up‐regulated in all three genotypes after renal ablation. Number of mice examined was controls n = 6, wildtype n = 13, RAG‐1−/− n = 7, IL‐17A−/− n = 16. *P < 0.05, significantly different from control, # P < 0.05, significantly different from wildtype, § P < 0.05, significantly different from wildtype and IL‐17A−/−.
Discussion
For many years, it was believed that the glomerular injury in CKD is merely the result of an increased intraglomerular pressure. Recent experimental and clinical data have shown that inflammatory mechanisms also importantly affect the progression of CKD (Wenzel et al., 2016); 5/6 nephrectomy, a model frequently used in experimental kidney research, was used in the present study for CKD induction. This model exhibits all the clinical hallmarks of CKD, including substantial albuminuria, decreased renal function, tubulointerstitial injury and glomerulosclerosis. Moreover, the 5/6 nephrectomy performed by removal of kidney tissue allows quantification of the induction of renal injury. The amount of removed kidney tissue was nearly identical in wildtype, RAG‐1 and IL‐17A knockout mice, indicating that the primary experimental stimulus to induce CKD did not differ between wildtype and both genotypes.
The present study aimed to assess the potential benefit of lack of T‐ and B cells or IL‐17A deficiency in a mouse model of CKD. Immune‐mediated glomerular and tubulointerstitial diseases encompass a heterogeneous group of disorders that cause inflammation within the glomerulus and other compartments of the kidney. The kidneys are frequently targeted by pathogenic immune responses against renal autoantigens or by local manifestations of systemic autoimmunity (Kurts et al., 2013). RAG‐1 knockout mice are a useful tool to examine the role of the adaptive immune system on development and course of various forms of renal disease. For example, cisplatin‐induced renal injury was markedly decreased in RAG‐1 deficient mice (Nozaki et al., 2011) showing that the adaptive immune system aggravates nephrotoxicity induced by cisplatin. IL‐17A producing cells initiate, perpetuate and sustain inflammatory processes by inducing proinflammatory cytokines and chemokines and recruiting other inflammatory cells and orchestrating tissue inflammation (Thorenz et al., 2017). Strikingly, neither deficiency of the adaptive immune system nor of IL‐17A affected the induction and course of progressive renal injury. This was unexpected, because there was a marked infiltration of CD3+ T‐cells in the kidney after 5/6 nephrectomy (Lehners et al., 2014).
We used a number of quantitative and semiquantitative measures to gauge the extent of renal injury induced by 5/6 nephrectomy. Renal injury, as assessed by scoring of glomerular and tubulointerstitial changes in PAS‐stained renal sections, was confirmed by up‐regulation of markers of renal injury and fibrosis such as CCL2, PAI‐1 and NGAL. The increased infiltration of CD3+ T‐cells was confirmed in the present study by flow cytometry analysis and immunohistochemistry.
Furthermore, in the present study, we found also an increased frequency of IL‐17A producing T‐cells in the remnant kidney and that CD4+ and γδ T‐cells were the main producers of IL‐17A. In glomerulonephritis and hypertension, the γδ and CD4+ T‐cells are major IL‐17 producing cells in the kidney (Turner et al., 2012; Riedel et al., 2016; Saleh et al., 2016). Interestingly, albuminuria was identical in all three groups 1 week after 5/6 nephrectomy indicating that absence of T and B cells, as well as a lack of IL‐17A, had no effect on the acute responses to renal mass reduction and induction of CKD. The profibrotic effects of IL‐17A have been studied in the lung (Wilson et al., 2010), heart (Zhou et al., 2014), liver (Tan et al., 2013) and in hypertensive end‐organ damage (Wu et al., 2014).. An antifibrotic effect of IL‐17A blockade was found in angiotensin II‐infused mice (Saleh et al., 2016). Despite these antifibrotic effects in other organs, we did not see antifibrotic effects of IL‐17A deficiency in the kidney after 5/6 nephrectomy. Cell and organ‐specific differences might induce different cytokine‐dependent pathophysiological changes and thus could explain the difference in the effects of IL‐17A deficiency. As the gut microbiome strongly influences TH17 responses (Tremaroli and Bäckhed, 2012), variations in the composition of the gut microbiome in these studies may have also contributed to the different effects of IL‐17A deficiency on fibrosis. Our finding that IL‐17A deficiency did not reduce renal fibrosis in CKD is, however, compatible with recent data from Thorenz et al. who reported that IL‐17A deficient mice were not protected against renal fibrosis induced by ischaemia/reperfusion injury (Thorenz et al., 2017).
Mycophenolic acid is an immunosuppressant drug used to prevent rejection in organ transplantation. It leads to a relatively selective inhibition of DNA replication in T‐cells and B cells by inhibition of inosine‐5′‐monophosphate dehydrogenase. Mycophenolic acid is known to ameliorate progression of CKD after 5/6 nephrectomy (Fujihara et al., 1998; Romero et al., 1999), leading to the conclusion that therapy directed against B and T‐cells would be a valid pharmacological approach in CKD. However, our data raise doubts about this conclusion, suggesting that the protective effects of mycophenolic acid in these earlier studies were caused by other drug effects than inhibition of B and T cells. It is conceivable that the adaptive immune system has proinflammatory and anti‐inflammatory effects in the progression of renal disease in the 5/6 nephrectomy model. CD4+ T‐cells can polarize into proinflammatory TH17 and TH1 cells but also into anti‐inflammatory regulatory T‐cells. As RAG‐1 deficient mice lack all subsets of T cells, the results found in mice lacking B and T cells may be caused by a balanced deficiency of proinflammatory and anti‐inflammatory cell types. However, such a mechanism does not explain why selective deficiency of IL‐17A failed to improve renal outcome in the present study. RAG‐1 deficient mice lack TH1 cells and as no protection was found in RAG‐1 deficient mice after 5/6 nephrectomy, it is unlikely that IFNγ deficiency would be protective. The relative number of NK cells is up‐regulated in RAG‐1 deficient mice, most likely due to the lack of T‐cells in these mice. It is an interesting question whether the NK cells can take over T‐cell function in RAG‐1 deficient mice and cause renal injury in the present study. Use of a double knockout mouse for RAG‐1 and the cytokine receptor common γ chain would allow an assessment of the effects of a deficiency of NK cells. However, this mouse is currently not available on the DBA/1J background and this mouse also lacks innate lymphoid cells that may play a role in renal disease as shown recently by us (Riedel et al., 2017). Depletion of NK cells in RAG‐1 deficient mice could be another approach to examine whether indeed NK cells participate in or even cause renal injury. Administration of depleting antibodies over a period of 12 weeks is challenging but should be performed in future experiments, if it is feasible.
However, 5/6 nephrectomy induced albuminuria in the nephrotic range and severe glomerular and tubulointerstitial injury, as shown in Figures 2 and 4. This is most likely due to the fact that the haemodynamic stress is so severe in this model (in which only 1/6 of the kidneys are filtering all the blood) that manipulating the adaptive immune system is unable to provide adequate protection. Alternatively, the adaptive immune system did modify renal injury but its effects were overcome by the haemodynamic forces, inducing severe albuminuria and glomerular sclerosis. The present study examined the role of the adaptive immune system in the CKD model of 5/6 nephrectomy. Alongside, we also found an activated innate immune system as shown by an increased infiltration of monocyte/macrophages in the remnant kidney and increased expression of two chemokines, CCL2 and CXCL1. Further experiments are necessary to pinpoint the contribution of these factors to the progression of CKD. We recently described a role for myeloperoxidase in the progression of CKD in mice (Lehners et al., 2014).
In summary, 5/6 nephrectomy induced CKD in mice with a marked influx of CD3+ T‐cells and IL‐17A producing CD4+ and γδ T‐cells. However, genetic absence of T and B cells or of IL‐17A did not provide protection or modify renal injury. These data indicate that T and B cells, as well as IL‐17A, do not contribute to the induction and progression of renal damage in the model of 5/6 nephrectomy.
Author contributions
A.R., R.K., A.C., M.B., S.K. and U.O.W. performed the experiments and the analysis of data. A.R., R.K., A.C., M.B., S.K., H.E., H.‐W.M. and U.O.W. wrote the manuscript. H.E., H.‐W.M. and U.O.W. planned and interpreted the experiments. U.O.W. and H.E. obtained funding for the project.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Detection of absolute numbers of lymphocytes in the kidney.
{kind=link}
Table S1 Percentage of IFNγ+ T‐cells in the kidney.
Acknowledgements
We thank S. Gatzemeier for excellent technical assistance and Thomas Kamradt, Jena, Germany for RAG‐1 mice on DBA/1J background.
The study was supported by German Research Foundation grants We 1688/17‐1 to U.W. and the SFB 1192 to U.W., H.E. and H.‐W.M.
Rosendahl A., Kabiri R., Bode M., Cai A., Klinge S., Ehmke H., Mittrücker H.‐W., and Wenzel U. O. (2019) Adaptive immunity and IL‐17A are not involved in the progression of chronic kidney disease after 5/6 nephrectomy in mice, British Journal of Pharmacology, 176, 2002–2014, 10.1111/bph.14509.
References
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017). The concise guide to pharmacology 2017/18: Other proteins. Br J Pharmacol 174 (Suppl. 1): S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Rennke HG, Brenner BM (1986). Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 77: 1993–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benndorf RA, Krebs C, Hirsch‐Hoffmann B, Schwedhelm E, Cieslar G, Schmidt‐Haupt R et al (2009). Angiotensin II type 2 receptor deficiency aggravates renal injury and reduces survival in chronic kidney disease in mice. Kidney Int 75: 1039–1049. [DOI] [PubMed] [Google Scholar]
- Chan AJ, Alikhan MA, Odobasic D, Gan PY, Khouri MB, Steinmetz OM et al (2014). Innate IL‐17A‐producing leukocytes promote acute kidney injury via inflammasome and Toll‐like receptor activation. Am J Pathol 184: 1411–1418. [DOI] [PubMed] [Google Scholar]
- Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM (2017). Regulatory T‐cell augmentation or interleukin‐17 inhibition prevents calcineurin inhibitor‐induced hypertension in mice. Hypertension 70: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraune C, Lange S, Krebs C, Hölzel A, Baucke J, Divac N et al (2012). AT1 antagonism and renin inhibition in mice: pivotal role of targeting angiotensin II in chronic kidney disease. Am J Physiol Renal Physiol 303: F1037–F1048. [DOI] [PubMed] [Google Scholar]
- Fujihara CK, Malheiros DM, Zatz R, Noronha IL (1998). Mycophenolate mofetil attenuates renal injury in the rat remnant kidney. Kidney Int 54: 1510–1519. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hünemörder S, Treder J, Ahrens S, Schumacher V, Paust HJ, Menter T et al (2015). TH1 and TH17 cells promote crescent formation in experimental autoimmune glomerulonephritis. J Pathol 237: 62–71. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C, Fraune C, Schmidt‐Haupt R, Turner JE, Panzer U, Quang MN et al (2012). CCR5 deficiency does not reduce hypertensive end‐organ damage in mice. Am J Hypertens 25: 479–486. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer‐Schwesinger C et al (2014). Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii‐induced hypertension. Hypertension 63: 565–571. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Panzer U (2018). Plasticity and heterogeneity of Th17 in immune‐mediated kidney diseases. J Autoimmun 87: 61–68. [DOI] [PubMed] [Google Scholar]
- Krebs CF, Schmidt T, Riedel JH, Panzer U (2017). T helper type 17 cells in immune‐mediated glomerular disease. Nat Rev Nephrol 13: 647–659. [DOI] [PubMed] [Google Scholar]
- Kurts C, Panzer U, Anders HJ, Rees AJ (2013). The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol 13: 738–753. [DOI] [PubMed] [Google Scholar]
- Lange S, Fraune C, Alenina N, Bader M, Danser AH, Frenay AR et al (2013). Aliskiren accumulation in the kidney: no major role for binding to renin or prorenin. J Hypertens 31: 713–719. [DOI] [PubMed] [Google Scholar]
- Lehners A, Lange S, Niemann G, Rosendahl A, Meyer‐Schwesinger C, Oh J et al (2014). Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am J Physiol Renal Physiol 307: F407–F417. [DOI] [PubMed] [Google Scholar]
- Ma LJ, Fogo AB (2003). Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 64: 350–355. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP (2015). Th‐17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT‐1R antagonism. Kidney Int 88: 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I et al (2002). Antigen‐specific T cell sensitization is impaired in IL‐17‐deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17: 375–387. [DOI] [PubMed] [Google Scholar]
- Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L et al (2016). Interleukin‐17A regulates renal sodium transporters and renal injury in angiotensin II‐induced hypertension. Hypertension 68: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki Y, Nikolic‐Paterson DJ, Yagita H, Akiba H, Holdsworth SR, Kitching AR (2011). Tim‐1 promotes cisplatin nephrotoxicity. Am J Physiol Renal Physiol 301: F1098–F1104. [DOI] [PubMed] [Google Scholar]
- Paust HJ, Turner JE, Steinmetz OM, Peters A, Heymann F, Holscher C et al (2009). The IL‐23/Th17 axis contributes to renal injury in experimental glomerulonephritis. J Am Soc Nephrol 20: 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel JH, Becker M, Kopp K, Duster M, Brix SR, Meyer‐Schwesinger C et al (2017). IL‐33‐mediated expansion of type 2 innate lymphoid cells protects from progressive glomerulosclerosis. J Am Soc Nephrol 28: 2068–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel JH, Paust HJ, Krohn S, Turner JE, Kluger MA, Steinmetz OM et al (2016). IL‐17F promotes tissue injury in autoimmune kidney diseases. J Am Soc Nephrol 27: 3666–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F, Rodriguez‐Iturbe B, Parra G, Gonzalez L, Herrera‐Acosta J, Tapia E (1999). Mycophenolate mofetil prevents the progressive renal failure induced by 5/6 renal ablation in rats. Kidney Int 55: 945–955. [DOI] [PubMed] [Google Scholar]
- Saleh MA, Norlander AE, Madhur MS (2016). Inhibition of interleukin 17‐A but not interleukin‐17F signaling lowers blood pressure and reduces end‐organ inflammation in angiotensin II‐induced hypertension. JACC Basic Transl Sci 1: 606–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z, Qian X, Jiang R, Liu Q, Wang Y, Chen C et al (2013). IL‐17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol 191: 1835–1844. [DOI] [PubMed] [Google Scholar]
- Thorenz A, Völker N, Bräsen JH, Chen R, Jang MS, Rong S et al (2017). IL‐17A blockade or deficiency does not affect progressive renal fibrosis following renal ischaemia reperfusion injury in mice. J Pharm Pharmacol 69: 1125–1135. [DOI] [PubMed] [Google Scholar]
- Tremaroli V, Bäckhed F (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489: 242–249. [DOI] [PubMed] [Google Scholar]
- Turner JE, Krebs C, Tittel AP, Paust HJ, Meyer‐Schwesinger C, Bennstein SB et al (2012). IL‐17A production by renal gammadelta T cells promotes kidney injury in crescentic GN. J Am Soc Nephrol 23: 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M (2017). Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 18: 612–621. [DOI] [PubMed] [Google Scholar]
- Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, Mori K et al (2010). Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest 120: 4065–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Rosendahl A, Czesla D, Meyer‐Schwesinger C, Stahl RA, Ehmke H et al (2016). The complement receptor C5aR1 contributes to renal damage but protects the heart in angiotensin II‐induced hypertension. Am J Physiol Renal Physiol 310: F1356–F1365. [DOI] [PubMed] [Google Scholar]
- Wenzel U, Turner JE, Krebs C, Kurts C, Harrison DG, Ehmke H (2016). Immune mechanisms in arterial hypertension. J Am Soc Nephrol 27: 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW et al (2010). Bleomycin and IL‐1β‐mediated pulmonary fibrosis is IL‐17A dependent. J Exp Med 207: 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L et al (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen‐activated protein kinase. Circ Res 114: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SF, Yuan J, Liao MY, Xia N, Tang TT, Li JJ et al (2014). IL‐17A promotes ventricular remodeling after myocardial infarction. J Mol Med 92: 1105–1116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Detection of absolute numbers of lymphocytes in the kidney.
Table S1 Percentage of IFNγ+ T‐cells in the kidney.