

Abstract
Pulmonary hypertension (PH) is a rare, progressive pulmonary vasculopathy characterized by increased mean pulmonary arterial pressure, pulmonary vascular remodelling and right ventricular failure. Current treatments are not curative, and new therapeutic strategies are urgently required. Clinical and preclinical evidence has established that inflammation plays a key role in PH pathogenesis, and recently, inflammasomes have been suggested to be central to this process. Inflammasomes are important regulators of inflammation, releasing the pro‐inflammatory cytokines IL‐1β and IL‐18 in response to exogenous pathogen‐ and endogenous damage‐associated molecular patterns. These cytokines are elevated in PH patients, but whether this is a consequence of inflammasome activation remains to be determined. This review will briefly summarize current PH therapies and their pitfalls, introduce inflammasomes and the mechanisms by which they promote inflammation and, finally, highlight the preclinical and clinical evidence for the potential involvement of inflammasomes in PH pathobiology and how they may be targeted therapeutically.
Linked Articles
This article is part of a themed section on Immune Targets in Hypertension. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.12/issuetoc
Abbreviations
- 4‐PBA
4‐phenyl butyric acid
- 6MWD
6‐min walk distance
- AIM
absent in melanoma
- ASC
apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain (CARD)
- BALF
bronchoalveolar lavage fluid
- CARD
caspase activation and recruitment domain
- COPD
chronic obstructive pulmonary disease
- DAMP
damage‐associated molecular pattern
- EC
endothelial cell
- ERA
endothelin receptor antagonist
- ET‐1
endothelin‐1
- IFI
interferon (IFN)‐γ inducible protein
- IPF
idiopathic pulmonary fibrosis
- LRR
leucine rich repeat
- mPAP
mean pulmonary arterial pressure
- NACHT/NOD
nucleotide‐binding and oligomerization domain
- NLR
NOD‐like receptor
- NLRC
NOD‐like receptor family, CARD‐containing protein
- NLRP
NOD‐like receptor family, pyrin domain (PYD)‐containing protein
- PAMP
pathogen associated molecular pattern
- PASMC
pulmonary artery smooth muscle cell
- PDTC
pyrrolidine dithiocarbamate
- PH
pulmonary hypertension
- PPHN
persistent pulmonary hypertension of the new‐born
- PRR
pattern recognition receptor
- PSGL
P‐selectin glycoprotein ligand
- PVR
pulmonary vascular resistance
- PYD
pyrin domain
- RV
right ventricular
- RVSP
right ventricular systolic pressure
- TLR
toll‐like receptor
- Treg
regulatory T‐cell
Introduction
Pulmonary hypertension (PH), defined as increased mean pulmonary arterial pressure (mPAP) ≥25 mmHg at rest by right heart catheterization (Galie et al., 2015), is a rare but incurable pulmonary vasculopathy, primarily affecting the distal pulmonary arteries. Associated morbidity and mortality are unacceptably high, with 5‐year survival standing at almost 50% (Humbert et al., 2010; Benza et al., 2012; Ling et al., 2012; Hoeper et al., 2017). Specifically, PH is characterized by muscularization of the pulmonary vascular wall, resulting in a progressive increase in resistance and right heart pressure‐overload, ultimately leading to right heart failure and death (Galie et al., 2015; Ryan et al., 2015). Underpinning the increased mPAP is an interplay between an aberrant vascular endothelial homeostasis and the remodelling that occurs in the vasculature and heart (Lai et al., 2014). Because right ventricular (RV) function is a major determinant of prognosis (Fine et al., 2013), the poor outcomes associated with PH are likely to be due to the fact that currently approved drugs are primarily pulmonary vasodilators with limited ability to address the underlying pathological remodelling, although they almost certainly offer some RV support (Wilkins et al., 2005; Seferian and Simonneau, 2013). Therefore, to advance therapy, new treatments which target remodelling in the pulmonary vasculature and/or RV are urgently required.
An increasing body of evidence suggests that the immune system plays a central role in the pathogenesis of PH, particularly in terms of the pulmonary vascular remodelling (Rabinovitch et al., 2014). Although this phenomenon was recognized more than two decades ago (Tuder et al., 1994; Voelkel et al., 1994; Humbert et al., 1995), advancement in our understanding of the inflammatory pathways involved in PH has been slow. Recently, inflammasomes have emerged as key contributors to disease progression, with supportive evidence from both preclinical and clinical studies (Soon et al., 2010; Ross et al., 2012; Villegas et al., 2013; Groth et al., 2014; Cero et al., 2015; Tang et al., 2015; Morisawa et al., 2016; Parpaleix et al., 2016; Yin et al., 2017). Inflammasomes are key components of macrophage‐mediated immunity and important regulators of inflammation (Martinon et al., 2002), releasing the pro‐inflammatory cytokines IL‐1β and IL‐18 in response to pathogen‐ and damage‐associated molecular patterns (PAMPs and DAMPs) (Krishnan et al., 2014). While this line of research is undoubtedly in its infancy, further elucidation of these inflammatory pathways and strategies aimed at alleviating their pathogenicity may represent a novel approach to treat PH and address the current deficit. This review will briefly summarize current PH therapies and their pitfalls, introduce inflammasomes and the mechanisms by which they promote inflammation and highlight both the preclinical and clinical evidence for the potential involvement of inflammasomes in PH pathobiology and how they may be targeted therapeutically.
Classification and current therapies for pulmonary hypertension
Pulmonary hypertension encompasses a large and aetiologically distinct group of pathologies, outlined in the most recent ‘updated clinical classification of PH’ (Simonneau et al., 2013). In addition to aetiological dissimilarity, four different classes of PH functional severity exist; ranging from mild, class I PH, which does not affect daily activity, to the most severe, class IV PH, where patients are unable to partake in any activity without symptoms and are likely to experience discomfort even at rest (Taichman et al., 2009).
Haemodynamic assessment by right heart catheterization is a prerequisite to confirm PH diagnosis (Rich and Rich, 2014). Other measures such as the 6‐min walk distance (6MWD; shown to be significantly reduced in PH patients compared to healthy controls) and echocardiography are used to assess exercise and functional capacity (Bossone et al., 2013; Demir and Kucukoglu, 2015). As treatments are not curative, clinical management of patients is primarily on a symptomatic basis, aiming to improve quality of life. In this sense, the functional class (I‐IV) is more important in determining treatment options than the WHO category, despite the fact that most drugs are only officially approved for group 1 PH (Trammell et al., 2015). Group 1 PH, or pulmonary arterial hypertension (PAH) in particular, includes a wide range of idiopathic aetiologies, but despite this, these patients share a collection of clinically presenting pathologies, including pulmonary artery smooth muscle cell (PASMC) and pulmonary endothelial cell (EC) proliferation and dysfunction, as well as vasoconstriction and in situ thrombosis (Montani et al., 2014), elements which are present in other PH categories as well. As this diverse group has many similarities in terms of clinical manifestation, the treatment and management of these patients are also similar.
Therapies currently used for the management of PH target one of three pathways and are categorized based on their mechanism of action. The notion that pulmonary vascular remodelling is central to the disease is somewhat of a paradigm‐shift compared to initial understanding. Previously, the dysregulation of endothelial mediators was thought more pertinent in PH than remodelling, a concept which led to the subsequent development of all currently used and approved therapies (Montani et al., 2014). Thus, a reduction in the vasodilator mediators, prostacyclin (PGI2; Tuder et al., 1999) and NO (Giaid and Saleh, 1995), combined with an up‐regulation of vasoconstrictor endothelin‐1 (ET‐1; Giaid et al., 1993), causes an overwhelming pathological shift towards a constricted pulmonary arterial vasculature.
Currently approved PH therapeutics targeting these pathways include PGI2 analogues (e.g. epoprostenol, treprostinil and iloprost) and prostacyclin (IP) receptor agonists (e.g. selexipag) which activate the endogenous Gs‐coupled IP receptor to increase intracellular cAMP and hence induce vasodilatation (Tuder et al., 1999; Mitchell et al., 2014). Secondly, there are two main classes of drugs which exploit the NO pathway; the PDE5 inhibitors (e.g. sildenafil, tadalafil and vardenafil) and soluble GC (GC‐1/GC‐2; Alexander et al., 2017c) stimulators (e.g. riociguat). Under normal physiological conditions, NO activates GC‐1 and/or GC‐2 to increase cGMP in PASMCs. cGMP is subsequently broken down by PDE5, but elevated cGMP levels must be maintained in order for the vessel to remain dilated and patent (Qian and Fulton, 2013). Hence, the main mechanism of action of PDE5 inhibitors is to stop breakdown of cGMP to induce vasodilatation. As such, PDE5 inhibitors rely mainly on endogenous NO to exert their effects, although they are also able to potentiate natriuretic peptide activity to ameliorate PH (Zhao et al., 2003; Preston et al., 2004; Klinger et al., 2006; Baliga et al., 2008). Conversely, GC‐1/GC‐2 stimulators increase cGMP production independently of intrinsic NO bioavailability. In addition to these NO modulating drugs, inhaled NO is utilized specifically in a unique category of PH, persistent pulmonary hypertension of the new‐born (PPHN). Normally, the high pulmonary vascular resistance (PVR) seen in utero falls shortly after birth, but in PPHN infants, there is a sustained increase in PVR with concomitantly normal or low systemic vascular resistance which persists post‐partum (Nair and Lakshminrusimha, 2014). Inhaled NO is the first line therapy in this setting due to its high specificity as a pulmonary vasodilator with minimal effect on systemic resistance (Nair and Lakshminrusimha, 2014; Abman et al., 2015). Finally, endothelin receptor antagonists (ERAs) block the actions of ET‐1 at its two cognate Gq‐coupled receptors, ETA and ETB; both of which are present on PASMCs while only ETB receptors are expressed on pulmonary ECs. Activation of both ETA and ETB receptors on the PASMCs initiates a contractile response, via inositol triphosphate signalling, leading to sarcoplasmic reticulum‐dependent Ca2+ release (Giaid et al., 1993). Conversely, binding of ET‐1 to ETB receptors located on the pulmonary ECs aids in vasodilatation by promoting clearance of ET‐1 as well as increasing production of NO and PGI2 (Seo et al., 1994). Despite the fact that ETB receptor activity can be different depending on the localization of the receptor, in practice, dual antagonists (e.g. bosentan) appear to have similar efficacy in PH to those selective for ETA receptors (e.g. ambrisentan), with both non‐selective and ETA receptor selective ERAs used in PH patients (Hoeper et al., 2016; Lajoie et al., 2017).
Therapeutic limitations
The introduction of these drugs has undoubtedly improved treatment over the last 20 years. Indeed, these therapies have proven efficacy to combat the increased mPAP, albeit by only ~10% (Galie et al., 2005), and they do extend survival and diminish morbidity by reducing breathlessness and increasing exercise capacity (Demir and Kucukoglu, 2015). However, despite this, they do not offer a cure, and PH patients must continue to take these drugs for the remainder of their lives. While many agents, including ERAs and PDE5 inhibitors, show benefits on pulmonary vascular remodelling in preclinical studies (Jeffery and Wanstall, 2001), they have negligible impact on remodelling in PH patients (Pogoriler et al., 2012). Therefore, despite years of research, current therapies have reached somewhat of an efficacy plateau. For this reason, it is paramount that new targets and pathways in the setting of PH are identified, to facilitate further improvement in disease outcome.
Inflammation and immunity in pulmonary hypertension: what is known?
Inflammation and immunity have long been considered as major underlying factors in the pathogenesis of PH; indeed, evidence dating back over 20 years hints at this possibility (Table 1). Tuder et al. (1994) revealed that plasma levels of the pro‐inflammatory cytokines IL‐1 and IL‐6 are elevated in PH patients, and Humbert et al. (1995) showed that monocrotaline‐induced PH in rats was ameliorated by the use of an IL‐1 antagonist. The link between PH and altered immunity is also illustrated by the fact that group 1 PH encompasses PAH associated with both autoimmune disorders such as systemic lupus erythematosus and systemic sclerosis (Bazan et al., 2018), as well as human immunodeficiency virus infection (Correale et al., 2015). In addition to this, PAH patients and indeed PH patients more generally (Pugliese et al., 2015; Kuebler et al., 2018) display signs of a chronic inflammatory state even without an associated immune‐related condition, most clearly delineated by elevated circulating cytokine levels and perivascular inflammatory infiltrates (Stacher et al., 2012). While it is well established that the pathogenesis, and poor prognosis, of PH is primarily due to the remodelling that occurs in the pulmonary vasculature along with RV dysfunction, it is now becoming evident that this remodelling is not simply a consequence of imbalanced vasoreactivity but may indeed be driven by aberrant immune responses (Rabinovitch et al., 2014). Therefore, in order to advance therapy, researchers have recently sought to identify alternative pathways that may be involved in the remodelling and inflammatory aspects of disease progression.
Table 1.
Clinical and preclinical evidence for inflammation in pulmonary hypertension
| Target | Preclinical | Clinical |
|---|---|---|
| NF‐κB | Inhibition improves PH (Kumar et al., 2012; Hosokawa et al., 2013; Wang et al., 2013; Farkas et al., 2014; Li et al., 2014) | – |
| IL‐1 | Inhibition improves PH (Voelkel et al., 1994; Parpaleix et al., 2016) | Increased levels in patients (Humbert et al., 1995; Soon et al., 2010) |
| IL‐18 | Inhibition improves PH (Morisawa et al., 2016) | Increased levels in patients (Ross et al., 2012) |
| IL‐6 | Inhibition improves PH (Golembeski et al., 2005; Savale et al., 2009; Steiner et al., 2009; Hashimoto‐Kataoka et al., 2015; Parpaleix et al., 2016) | Increased levels in patients (Humbert et al., 1995; Selimovic et al., 2009; Soon et al., 2010; Gitto et al., 2012; Matura et al., 2015) |
| Other cytokines/chemokines/growth factors | – | Increased levels of MCP‐1 (Itoh et al., 2006); VEGF, PDGF, TGF‐β1 (Selimovic et al., 2009); IL‐2, ‐4, ‐8, ‐10, ‐12p70, TNF‐α (Soon et al., 2010) IL‐8, TNF‐α in PPHN (Gitto et al., 2012); CXCL10 (Ross et al., 2012); TNF‐α (Matura et al., 2015) |
| Inflammatory cells | Depletion of T‐cells worsens PH (Tamosiuniene et al., 2011; Chu et al., 2015); macrophages improves PH (Zaloudikova et al., 2016) | Infiltrates comprised of macrophages, T and B lymphocytes, dendritic and mast cells (Tuder et al., 1994; Savai et al., 2012; Stacher et al., 2012) |
| P2X7/ATP | Inhibition improves PH (Yin et al., 2017) | – |
| Inflammasomes | Inhibition improves PH (Villegas et al., 2013; Cero et al., 2015; Tang et al., 2015) | – |
| Survival | – | Elevated cytokines predict (Soon et al., 2010) |
| Remodelling | – | Inflammatory infiltrates correlate with vessel wall thickness (Stacher et al., 2012) |
A growing body of evidence, in the form of both clinical and preclinical studies, links various aspects of inflammation to human and experimental PH (Table 1). In fact, numerous studies have demonstrated that perivascular inflammatory infiltrates comprised of macrophages, T and B lymphocytes, dendritic and mast cells are present in the occlusive plexiform lesions in the pulmonary vasculature of PH patients (Figure 1; Tuder et al., 1994; Humbert et al., 1995; Soon et al., 2010; Stacher et al., 2012; Rabinovitch et al., 2014). In addition to this, Stacher et al. (2012) examined the degree of inflammatory cell infiltration in explanted lungs from PH patients and used this information to calculate perivascular inflammation scores. They found that in PH patients, perivascular inflammation scores positively correlated with vascular intima plus media and adventitia thickness as well as a trend towards a correlation with increased mPAP. Taken together, these studies demonstrate the importance of inflammation to pulmonary vascular remodelling. Intriguingly, manipulation of some of the aforementioned immune cell types confers protection or worsens progression in animal models of PH. Specifically, a recent study in rats found that depletion of alveolar macrophages, by clodronate‐containing liposomes, attenuated hypoxia‐induced PH (Zaloudikova et al., 2016), but conversely, regulatory T‐cells (Tregs) appear to limit vascular endothelial injury and prevent PH. As such, Tamosiuniene et al. (2011) found that T‐cell deficient athymic nude rats developed worsened PH in response to VEGF inhibition, which could be rescued by adoptive T‐cell transfer. Moreover, Chu et al. (2015) revealed that adoptive transfer of Tregs into hypoxic mice protected them from PH as well as vascular and RV remodelling. In addition, these animals displayed reduced expression of pro‐inflammatory cytokines, IL‐1β and IL‐6, and the chemokine CCL2 in the lungs. Thus, it appears that macrophages are detrimental, whereas certain T‐cell populations may be beneficial in the resolution or prevention of PH. In addition, Wang et al. (2013) revealed that inhibition of the pro‐inflammatory cytokine TNF‐α in rats attenuates monocrotaline‐induced PH, with the involvement of NF‐κB.
Figure 1.
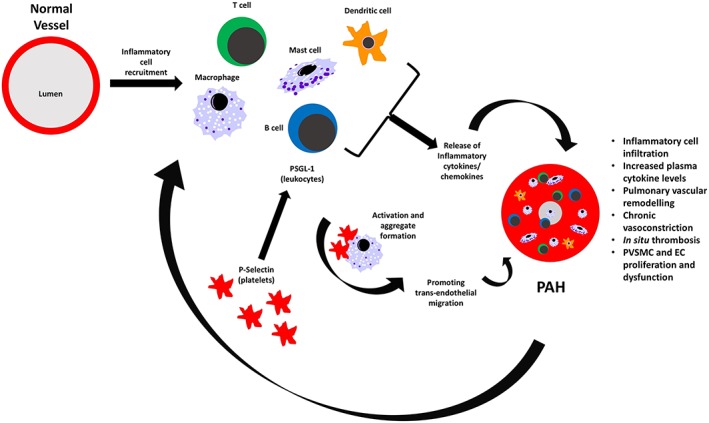
Inflammation in pulmonary hypertension. Proposed mechanisms by which inflammation contributes to PH pathogenesis. Recruitment of inflammatory cells such as macrophages, T and B lymphocytes, dendritic and mast cells leads to the release of pro‐inflammatory cytokines IL‐1β, IL‐2, IL‐6, IL‐8, IL‐12, IL‐18 and TNF‐α, MCP‐1 and CXCL10. Platelet P‐selectin binds PSGL‐1 expressed on leukocytes to induce multicellular aggregation and trans‐endothelial migration. Here, inflammatory cells take up residence to form perivascular inflammatory infiltrates seen in the plexiform lesions of PH patients. This results in further cytokine release, pulmonary vascular remodelling, cell proliferation and thrombosis causing an ongoing exacerbation cycle.
Similar links are evident in PH patients, with circulating levels of many cytokines including, IL‐1β, IL‐2, IL‐4, IL‐6, IL‐8, IL‐10, IL‐12, IL‐18 and TNF‐α, as well as the chemokines CCL2 and CXCL10 (Humbert et al., 1995; Itoh et al., 2006; Soon et al., 2010; Ross et al., 2012), being elevated compared to healthy controls; a strong indicator that altered immunity is at play. Indeed, Soon et al. (2010) found that increased levels of the pro‐inflammatory cytokines IL‐2, IL‐6, IL‐8 and IL‐12, and perhaps surprisingly, the anti‐inflammatory cytokine IL‐10, correlate with worse outcome and patient survival. Interestingly, in this study, these cytokines served as a better predictor of prognosis than traditional measures like 6MWD and haemodynamic parameters (Soon et al., 2010), although this remains to be substantiated.
Another emerging concept is that interactions between platelets, leukocytes and endothelial cells contribute to the inflammatory processes underpinning PH. Indeed, these cells are a recognized source of inflammatory mediators (Thomas and Storey, 2015). NO and PGI2 inhibit platelet activation and aggregation, but both molecules are reduced in PH (Giaid and Saleh, 1995; Tuder et al., 1999), allowing aberrant platelet aggregation and thrombus formation to occur (Thomas and Storey, 2015). In addition to their interactions with NO and PGI2, platelets may also serve as a source of IL‐1β in PH, as they have been shown to process and release IL‐1β in dengue fever patients, as well as increase endothelial permeability in this infection (Hottz et al., 2013). Activated platelets also play an integral role in leukocyte adhesion, rolling and subsequent extravasation in atherosclerosis (Lievens and von Hundelshausen, 2011). The adhesion molecule, P‐selectin, is expressed on activated platelets, and endothelial cells and its ligand P‐selectin glycoprotein ligand 1 (PSGL‐1) is expressed on leukocytes (Dole et al., 2005; von Hundelshausen and Weber, 2007). P‐selectin–PSGL‐1 interactions lead to the formation of platelet‐leukocyte multicellular aggregates and contribute to the recruitment of leukocytes to sites of endothelial injury and inflammation (Dole et al., 2005; von Hundelshausen and Weber, 2007; Thomas and Storey, 2015; Coenen et al., 2017). Although many adhesion molecules are involved in this process, P‐selectin is of particular interest with respect to PH since P‐selectin levels have been shown to be elevated in PH patients (Semenov et al., 2000). The importance of this mechanism in the development of atherosclerosis is demonstrated by the fact that mice genetically deficient in P‐selectin are protected from atherosclerotic lesion development (Dong et al., 2000). As atherosclerotic lesions are known to include inflammatory cell infiltrates (Libby, 2012), and a similar inflammatory milieu is present within the plexiform lesions of PH patients (Tuder et al., 1994; Stacher et al., 2012), it is plausible that platelet and endothelial adhesion molecules play a role in the trans‐endothelial migration of immune cells here too (Figure 1), although the precise mechanism in this setting remains to be elucidated. Encouragingly, in terms of inflammatory processes and potential translation into new therapies, similar findings are made in animal models of PH, as are observed in PAH and PH patients more generally, and will be described below. This indicates that the use of these models will be instrumental in elucidating inflammatory aspects of disease progression and provides a strong rationale for their continued use.
A new approach to inflammation in pulmonary hypertension
In addition to the clinical data (Soon et al., 2010; Ross et al., 2012; Groth et al., 2014), many preclinical studies, involving depletion or alteration of immune cells, cytokines or their signalling pathways have been shown to modify PH pathobiology (Voelkel et al., 1994; Kumar et al., 2012; Li et al., 2014; Cero et al., 2015; Tang et al., 2015; Morisawa et al., 2016; Parpaleix et al., 2016; Zaloudikova et al., 2016), highlighting the important role of altered immunity in PH (as described above; Table 1). The inflammasome, a key component of the innate immune system, is emerging as a novel target in the treatment of PH, because preclinical studies modulating cells or pathways associated with the inflammasome show particular promise (Gasse et al., 2009; Villegas et al., 2013; Tang et al., 2015; Parpaleix et al., 2016; Yin et al., 2017). Inflammasome components are most highly expressed in macrophages (Heng and Painter, 2008) but have also been described in endothelial cells (Xiang et al., 2011; Xia et al., 2014), neutrophils, dendritic cells (Guarda et al., 2011), platelets (Hottz et al., 2013) and lung epithelial cells (De Nardo et al., 2014). The observation that depletion of macrophages attenuates PH (Zaloudikova et al., 2016) and inflammasome components are most highly expressed in these cells (Heng and Painter, 2008) suggests macrophages may play a critical role in PH development, potentially mediated via the inflammasome.
Inflammasome activation and targets in disease
Inflammasomes are a family of multi‐protein complexes, first identified by Martinon et al. (2002), which revolutionized the understanding of immune function. They are key players in macrophage‐related immunity and important regulators of inflammation, responding to danger signals and releasing the pro‐inflammatory cytokines IL‐1β and IL‐18 (Mariathasan et al., 2006; Martinon et al., 2006; Halle et al., 2008). Inflammasomes comprise of three subunits and, depending upon the particular subunit composition, display selectivity towards a unique array of activation signals. Each inflammasome contains a distinct pattern recognition receptor (PRR) and this defines the inflammasome type and selectivity (Man and Kanneganti, 2015). To date, inflammasome‐forming PRRs identified include NLRP1 and NLRP3 [nucleotide‐binding oligomerization domain (NOD)‐like receptor (NLR) family, leucine rich repeat (LRR) and pyrin domain (PYD)‐containing proteins; Krishnan et al., 2016]. These NLRP‐type inflammasomes are comprised of an NLR structure [a central nucleotide‐binding and oligomerization domain (NACHT) flanked by leucine‐rich repeats at the C‐terminus and at the N‐terminus a PYD]. In addition to these most widely known NLRP‐type inflammasomes, another PRR known as NLRC4 [NOD‐like receptor family CARD (caspase activation and recruitment domain)‐containing protein 4] also forms an inflammasome. Furthermore, there are non‐NLR type inflammasome‐forming PRRs such as absent in melanoma (AIM) 2 and IFN‐γ inducible protein 16 (IFI16; Figure 2; Coll et al., 2015). Following detection of pathogen‐ and damage‐ associated molecular patterns (PAMPs and DAMPs), the PRR is auto‐activated which allows it to recruit the heterodimeric adaptor molecule, ASC (apoptosis‐associated speck‐like protein containing a CARD), which is common to a number of inflammasome varieties (Figure 2; Lu et al., 2014). Cytoplasmic ASC monomers are then reorganized into a single large ‘speck’, which is considered a hallmark of inflammasome assembly (Man and Kanneganti, 2015). Finally, the ASC speck recruits the cysteine protease pro‐caspase‐1, which, by means of proximity‐induced auto‐cleavage yields active caspase‐1 (Malik and Kanneganti, 2017).
Figure 2.
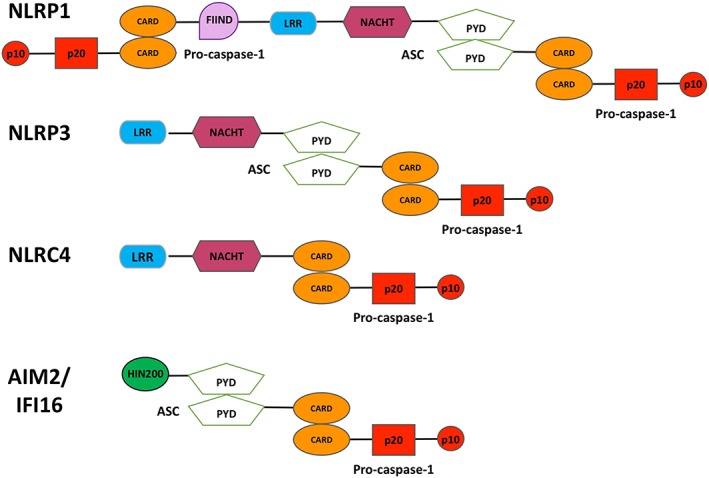
Inflammasome isoforms and subunit compositions. The main inflammasome isoforms described to date include NLRP1, NLRP3, NLRC4, AIM2 and IFI16; the subunit composition differs depending on the variant. NLRP3, AIM2 and IFI16 inflammasomes require the adaptor molecule ASC for homotypic oligomerization. Conversely, NLRC4 contains its own CARD domain and can recruit pro‐caspase‐1 independently of ASC. NLRP1 contains both a CARD and PYD and can therefore either recruit caspase‐1 independently of ASC using its own CARD, or recruit caspase‐1 in an ASC‐dependent manner, similarly to the NLRP3, AIM2 and IFI16 inflammasomes. FIIND, function to find domain; HIN200, dsDNA binding domain.
Inflammasome assembly and activation
By far the most widely studied and best characterized inflammasome isoform to date, and the focus here, is the NLRP3 inflammasome (also known as NALP3). In order for the NLRP3 inflammasome to initiate its activity, a two‐step process is required, beginning with signal I (priming) and then subsequently signal II (activation; Figure 3; Bauernfeind et al., 2009). Priming is induced by engagement of toll‐like receptors (TLRs) or inflammatory cytokine receptors on the cell surface by exogenous PAMPs [e.g. microbial components (Ishii et al., 2008), pore‐forming toxins (Malik and Kanneganti, 2017), silica crystals or asbestos fibres (Dostert et al., 2008; Franchi et al., 2009)] and/or endogenous DAMPs [e.g. uric acid crystals (Martinon et al., 2006), pro‐inflammatory cytokines like TNF‐α (Chow et al., 2014)]. Following this initial detection, NF‐κB signal transduction up‐regulates expression of the inflammasome components NLRP3, ASC and pro‐caspase‐1, and pro‐inflammatory cytokine precursors pro‐IL‐1β and pro‐IL‐18, completing the priming (signal I) step (Figure 3; Bauernfeind et al., 2009). The NLRP3 inflammasome is then equipped for activation, an event which involves inflammasome oligomerization. A number of pathways have been postulated to be involved in triggering inflammasome activation including lysosomal rupture and release of enclosed microcrystals (Martinon et al., 2006; Gasse et al., 2009), ROS generation (Zhou et al., 2010) and high extracellular ATP concentrations (resulting from cellular damage) acting at P2X7 purinoceptors (Mariathasan et al., 2006; Franchi et al., 2009; Riteau et al., 2010; Krishnan et al., 2014; Shao et al., 2015). Once these subsequent danger signals have been recognized, activation of the NLRP3 inflammasome is initiated by oligomerization of NLRP3 monomers via homotypic PYD–PYD reactions, which then interact with the PYD of the adaptor molecule ASC (Sborgi et al., 2015). Once activated, the inflammasome recruits the zymogen, pro‐caspase‐1 using the CARD region in ASC, and several pro‐caspase‐1 subunits cluster around the inflammasome, resulting in auto‐cleavage to active caspase‐1 heterodimers which each comprise a p20 (20 kDa) and p10 (10 kDa) subunit (Malik and Kanneganti, 2017). Active caspase‐1 then cleaves inactive pro‐IL‐1β (31 kDa) and pro‐IL‐18 (24 kDa) to their active forms IL‐1β (17.5 kDa) and IL‐18 (18 kDa) respectively (Figure 3; Dinarello, 2002). Additionally, inflammasome generated caspase‐1 promotes a highly inflammatory type of cell death known as pyroptosis (Maltez et al., 2015). The IL‐1 family of cytokines are ‘early response’ mediators of immunity and promote the release of a number of secondary cytokines which lie further downstream in the inflammasome activation cascade (Labow et al., 1997; Dinarello, 2002; Cahill and Rogers, 2008; Mills et al., 2013). These include IL‐6, which is released from macrophages and T‐cells in response to IL‐1β stimulation (Cahill and Rogers, 2008), as well as IL‐2 and IL‐12 which are produced downstream of IL‐18 (Dinarello, 2002).
Figure 3.
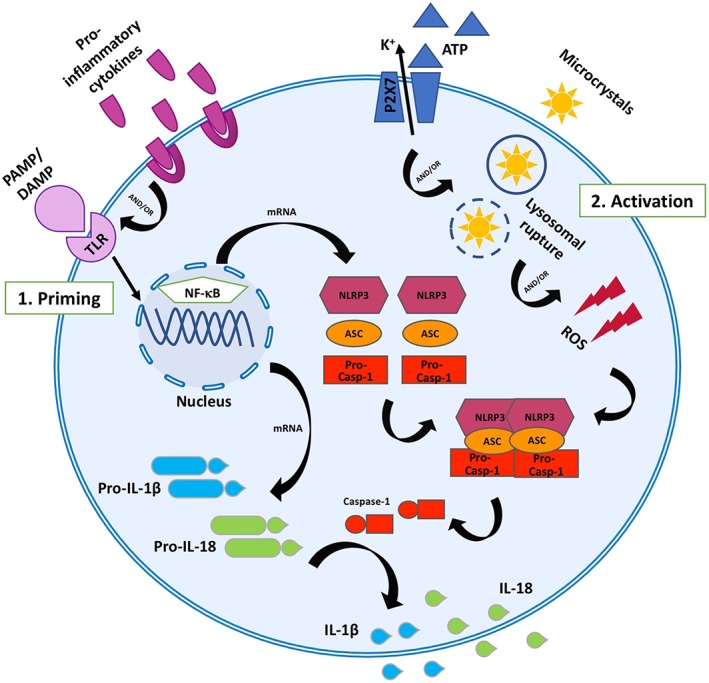
NLRP3 inflammasome activation. Schematic representation of activators and effectors of the NLRP3 inflammasome. The NLRP3 inflammasome comprises the pattern recognition receptor, NLRP3, along with the adaptor molecule, ASC, and pro‐caspase‐1. NLRP3 inflammasome activation requires two steps. (1) Priming is induced by engagement of TLRs and pro‐inflammatory cytokine receptors on the cell surface by exogenous PAMPs (e.g. microbial components, silica crystals, asbestos fibres or pore‐forming toxins), endogenous DAMPs (e.g. ATP, microcrystals or ROS) or pro‐inflammatory cytokines such as TNF‐α; this results in NF‐κB‐mediated up‐regulation of NLRP3, ASC, pro‐caspase‐1, pro‐IL‐1β and pro‐IL‐18 gene expression. (2) Activation occurs when further DAMPs are detected by NLRP3. This leads to oligomerization of NLRP3 subunits and recruitment of ASC and pro‐caspase‐1. Pro‐caspase‐1 then undergoes auto‐cleavage into p10 and p20 subunits, which heterodimerize to form active caspase‐1. Caspase‐1 then processes pro‐IL‐1β and pro‐IL‐18 into their active, pro‐inflammatory cytokine forms IL‐1β and IL‐18.
NLRP3 inflammasome pathophysiology and pharmacology
The NLRP3 inflammasome has been shown to play a role in a large number of disease states affecting various organ systems, including the kidneys (Anders and Muruve, 2011), liver (Szabo and Csak, 2012) and as a consequence of ageing (Youm et al., 2013). More interestingly, of relevance to PH, increased NLRP3 inflammasome activity has been associated with cardiovascular and metabolic diseases such as gout (Martinon et al., 2006), atherosclerosis (Duewell et al., 2010), arteritis (Chen et al., 2015) obesity and type 2 diabetes (Wen et al., 2012; Liu et al., 2015), as well as lung disorders including chronic obstructive pulmonary disease (COPD; Yang et al., 2015), asthma (Simpson et al., 2014; Kim et al., 2017) and cystic fibrosis (Iannitti et al., 2016); and fibrotic disorders like idiopathic pulmonary fibrosis (IPF) and substance‐induced pulmonary fibrosis (e.g. asbestos and silica crystals; Dostert et al., 2008). The level of understanding in each condition varies, but many examples have implications for PH.
As alluded to earlier, endothelial cells play an important role in the inflammatory cell infiltration seen in atherosclerosis, a phenomenon that is likely to parallel what occurs in PH. The actions of the NLRP3 inflammasome may also contribute to the endothelial dysfunction seen in PH patients. In fact, the NLRP3 inflammasome has been associated with endothelial dysfunction in a number of cardiovascular pathologies. Liu et al. (2015) found that activation of the NLRP3 inflammasome induces endothelial dysfunction in obese rats, and moreover, Chen et al. (2015) revealed that lysosomal destabilization in coronary arteritis induces NLRP3 inflammasome activation which subsequently results in endothelial dysfunction. Finally, cardiovascular insult in the form of homocysteine or inflammatory LPS leads to NLRP3‐dependent pyroptosis in endothelial cells, ultimately contributing to endothelial dysfunction (Xi et al., 2016). Given that endothelial dysfunction is a major contributor to the pathogenesis of PH and the NLRP3 inflammasome contributes to this in other diseases, it is possible that the NLRP3 inflammasome may contribute to PH pathogenesis in this manner.
Gout is characterized by deposition of uric acid crystals in joints and periarticular tissues (Martinon et al., 2006) and, as previously mentioned, uric acid crystals are known activator signals for NLRP3 activity. Interestingly, a similar pathology is seen in a murine model of IPF, where bleomycin instillation produces uric acid crystals in the lung which subsequently induce NLRP3 inflammasome activation (Gasse et al., 2009). Similarly, inhalation of other particulate substances (e.g. asbestos or silica) induces pulmonary fibrosis and leads to NLRP3 inflammasome activation and IL‐1β release in the lung, by means of lysosomal rupture and ROS production (Dostert et al., 2008). Furthermore, IPF patients exhibit elevated extracellular ATP (released from injured cells) in their bronchoalveolar lavage fluid (BALF) compared to healthy controls, which acts as a major NLRP3 danger signal (Riteau et al., 2010), an observation that can be reproduced by bleomycin administration in mice. Interestingly, dos Santos et al. (2015) found that the cytoskeletal intermediate filament, vimentin, is an essential element for bleomycin and asbestos‐induced lung injury and NLRP3 inflammasome activation. The link between pulmonary fibrotic disorders and the NLRP3 inflammasome is strong evidence that this inflammasome also contributes to PH. As PH often occurs as a result of pulmonary fibrosis, it is possible the NLRP3 inflammasome plays a similar and synergistic role in the two conditions (Riteau et al., 2010). Perhaps the lung disorder most strongly tied to NLRP3 inflammasome activity, and closely linked to the pathology seen in PH, is COPD. Notably, in mice, exposure to cigarette smoke leads to increased caspase‐1 activity as well as IL‐1β and IL‐18 release in the lungs, and genetic disruption or pharmacological inhibition of the P2X7 receptor, a known NLRP3‐related activation stimulus, ameliorates this effect (Eltom et al., 2011). Moreover, similar findings are noted in patients, with increased caspase‐1 detected in the lungs of emphysema patients and smokers compared to non‐smokers (Eltom et al., 2011). Additionally, the known inflammasome activator, uric acid, is increased in BALF from COPD patients (Wanderer, 2008). Interestingly, mice overexpressing IL‐1β exhibit a COPD‐like phenotype including lung inflammation and fibrosis, and mice genetically deficient in IL‐1β are protected from cigarette smoke induced pathology (Lappalainen et al., 2005).
A lack of selective inflammasome inhibitors is one reason these pathways have been so difficult to study, not only in PH, but also other disorders. Compounds which inhibit inflammasome activity, albeit non‐selectively include various micro‐RNAs, ellagic acid, the SOD mimetic (MnTE‐2‐PyP), 4‐phenyl butyric acid (4‐PBA) and type I interferons, which have been used in multiple sclerosis for many years (Villegas et al., 2013; Shao et al., 2015; Tang et al., 2015; Zeng et al., 2017). There are a number of other approaches to modulate inflammasome activity at different levels within its signalling cascade. Firstly, targeting inflammasome priming by inhibiting NF‐κB‐mediated transcription of inflammasome subunits (NLRP3, ASC and caspase‐1) and pro‐inflammatory cytokine precursors (pro‐IL‐1β and IL‐18), with compounds such as pyrrolidine dithiocarbamate (PDTC), and the selective, synthetic NF‐κB inhibitor, IMD‐0354 (Hosokawa et al., 2013; Farkas et al., 2014). Secondly, inflammasome activation signals can be targeted to mitigate oligomerization. Depending on the condition, and particular stimuli, such approaches include P2X7 receptor inhibition with A‐740003 (Yin et al., 2017) or limiting production of ROS with a SOD mimetic (Villegas et al., 2013). Another potential inflammasome‐directed target is caspase‐1. Indeed, there are a number of low MW caspase‐1 inhibitors that have been trialled in humans for inflammatory conditions such as psoriasis and rheumatoid arthritis. Unfortunately, these trials were terminated early due to the risk of liver toxicity or generally poor efficacy (MacKenzie et al., 2010). As such, the newer caspase‐1 inhibitor VX‐765 was developed to improve safety and efficacy (MacKenzie et al., 2010). Another well‐known inflammasome target is IL‐1, for which two specific drugs are available to block its activity, anakinra and canakinumab, have prior approved uses in humans. Indeed, the former is currently used to treat rheumatoid arthritis and the latter is licensed for a number of auto inflammatory syndromes. Canakinumab has additionally undergone recent evaluation in patients with inflammatory vascular (i.e. coronary artery) disease (Van Tassell et al., 2017; Ridker et al., 2017b). Therefore, both of these drugs may represent a novel opportunity to repurpose for use in PH. Finally, IL‐6 is produced downstream of IL‐1β (Cahill and Rogers, 2008), and the human monoclonal antibody targeting the IL‐6 receptor (IL‐6R), tocilizumab, is currently undergoing early phase testing in human PAH (Hernandez‐Sanchez et al., 2018). Although all these compounds do interrupt inflammasome activity to some extent or at some level, a truly selective inflammasome inhibitor did not emerge until 2015 when Coll et al. (2015) described a highly selective low MW inhibitor of the NLRP3 inflammasome, MCC950. They demonstrated that MCC950 selectively inhibits NLRP3 inflammasome activation both in vitro and in vivo. Although MCC950 has not been trialled in humans to date, it does show promise as a therapeutic agent in terms of safety and efficacy in several preclinical models of disease, including cardiovascular and metabolic disorders and lung‐associated conditions (Krishnan et al., 2016; Primiano et al., 2016; Kim et al., 2017; Pavillard et al., 2017; van der Heijden et al., 2017; van Hout et al., 2017), which will be described in more detail below.
Inflammasomes in pulmonary hypertension
The concept of inflammasome involvement in PH is particularly new, with studies examining this unique aspect of inflammation only emerging over the last 5 years or so. There is currently a lack of evidence for involvement of other inflammasome isoforms, apart from NLRP3, in PH and this review will therefore focus on this isoform. As outlined below, preclinical and clinical evidence for NLRP3 inflammasome activity and downstream signalling in the pathobiology of PH is now strong. Pharmacological manipulation of the priming, assembly, activation and subsequent downstream signalling of the NLRP3 inflammasome in the context of PH will be addressed, beginning at the upstream end and working downward.
Potential sites of inflammasome modulation in pulmonary hypertension
Furthest upstream are the signals that initiate the priming stage of inflammasome activity. These include a variety of exogenous and endogenous signals in the form of PAMPs and DAMPs; the distinct combination of which can initiate activity in different inflammasome isoforms through activation of TLRs (Kahlenberg et al., 2005; Becker and O'Neill, 2007). To date, there is no evidence of specifically modulating TLR activity with respect to inflammasomes in PH, but since this pathway is so far upstream, it is probably not the most desirable target. Following the initiation of priming through TLR‐mediated detection of PAMPs and DAMPs, an important pathway to complete this step is NF‐κB (Figure 3). This transcription factor drives the expression of the inflammasome subunits NLRP3, ASC and pro‐caspase‐1 as well as the inflammatory cytokine precursors, pro‐IL‐1β and pro‐IL‐18 (Bauernfeind et al., 2009; Krishnan et al., 2016). Interestingly, Kumar et al. (2012) found that cardiac‐specific genetic deletion of NF‐κB in mice is able to prevent RV hypertrophy and increased right ventricular systolic pressure (RVSP; a surrogate for mPAP used in rodents) induced by monocrotaline, and moreover, Li et al. (2014) showed that genetic NF‐κB deletion in the lung prevented monocrotaline‐induced lung fibrosis and attenuated increased RVSP in mice. In addition, pharmacological inhibition of NF‐κB with the selective, synthetic inhibitor IMD‐0354 or with PDTC decreased RVSP and reversed pulmonary vascular remodelling and RV hypertrophy in monocrotaline (Hosokawa et al., 2013) and sugen‐hypoxic rats (Farkas et al., 2014; Figure 4).
Figure 4.
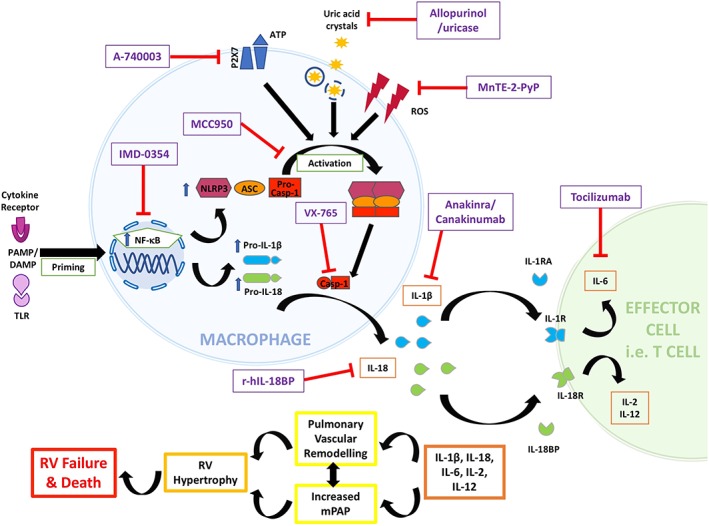
Involvement of the NLRP3 inflammasome and potential sites of modulation in pulmonary hypertension. Numerous drugs are able to modulate NLRP3 inflammasome activity at various levels in PH; from activation to downstream cytokines. Priming is followed by assembly and activation of the NLRP3 inflammasome, resulting in subsequent pro‐inflammatory cytokine generation (Figure 3). The actions of these cytokines are thought to contribute to pulmonary vascular remodelling (Figure 1), which occurs alongside increased mPAP. These effects lead to RV compensatory hypertrophy and eventually RV decompensation, failure and death. Inhibitory drugs are in purple squares. IMD‐0354, selective NF‐κB inhibitor; A‐740003, P2X7 receptor inhibitor; Allopurinol/uricase, uric acid inhibitors; MnTE‐2‐PyP, SOD mimetic; MCC950, selective, low MW NLRP3 inflammasome activation inhibitor; VX‐765, caspase‐1 inhibitor; Anakinra, recombinant IL‐1 decoy receptor, IL1‐RA; Canakinumab, human monoclonal antibody targeting IL‐1β; r‐hIL‐18BP, recombinant IL‐18 decoy receptor, IL‐18BP; Tocilizumab, human monoclonal antibody targeting IL‐6 receptor (IL‐6R).
Following priming, a number of other pathways promote NLRP3 activation; one such signal is high extracellular ATP levels, acting at the P2X7 purinoceptor (Mariathasan et al., 2006; Franchi et al., 2009; Riteau et al., 2010). Indeed, the P2X7 receptor inhibitor A‐740003 has been utilized in a rat model of PH with positive effects, reducing RVSP, reversing RV hypertrophy and pulmonary vascular remodelling as well as attenuating NLRP3 inflammasome up‐regulation (Yin et al., 2017; Figure 4). Another NLRP3 activating stimulus is ROS, and interestingly, reducing superoxide through the use of the SOD mimetic, MnTE‐2‐PyP also attenuates PH and NLPR3 expression in hypoxic mice (Villegas et al., 2013; Figure 4). It is important to note that, as both the P2X7 receptor and SOD are widespread physiologically and diverse in their actions, these approaches may be simultaneously exerting beneficial effects through mechanisms distinct from NLRP3. An additional NLRP3 inflammasome activation signal is microcrystal lysosomal rupture (Martinon et al., 2006). Indeed, Gasse et al. (2009) illustrated that minimizing uric acid crystal levels in mice with the inhibitors allopurinol or uricase attenuates bleomycin‐induced pulmonary fibrosis, inflammation and increased IL‐1β levels (Figure 4). Conversely, they revealed that exogenous addition of uric acid microcrystals reproduces the lung injury induced by bleomycin.
Another potential inflammasome target is caspase‐1 but, so far, there is no human evidence available for the use of low MW caspase‐1 inhibitors in any cardiovascular or lung disorders. However, there is some evidence that the newer caspase‐1 inhibitor VX‐765 is beneficial in a rat model of myocardial infarction (Yang et al., 2017). Excitingly, this orally bioavailable compound has also recently been trialled for the treatment of epilepsy in humans (MacKenzie et al., 2010) and it could, therefore, be potentially repurposed for therapeutic utility in PH (Figure 4).
Further support for a role of the NLRP3 inflammasome in PH comes in the form of up‐regulated NLRP3 expression and activation in the lungs of mice exposed to a chronic hypoxia model of PH. Such effects that are attenuated by a SOD mimetic, presumably via reduced ROS formation, an established activation signal for the NLRP3 inflammasome (Villegas et al., 2013). Additionally, inhibition of endoplasmic reticulum stress with 4‐PBA prevents monocrotaline‐induced PH via a reduction in NLRP3 inflammasome activity in rats (Zeng et al., 2017). Moreover, NLRP3 inflammasome inhibition by ellagic acid ameliorates monocrotaline‐induced PH in rats, as well as vascular remodelling, RV hypertrophy and expression of NLRP3, caspase‐1 and IL‐1β (Tang et al., 2015). It is important to note, however, that while the SOD mimetic, ellagic acid and 4‐PBA did inhibit NLRP3 inflammasome activity in these studies, they may have had other non‐specific effects, targeting other inflammasomes or inflammatory pathways. Furthermore, NLRP3−/− and Caspase‐1−/− mice are protected from acute bleomycin‐induced lung fibrosis and inflammation (Gasse et al., 2009). Taken together, these findings suggest that selectively inhibiting NLRP3 inflammasome activity may be of therapeutic benefit in PH.
In addition to promising results from modulating the inflammasome itself, other studies have looked at altering the downstream cytokines generated as a result of inflammasome activity (Figures 3 and 4). A pathway of particular recent interest in cardiovascular disease has been the use of IL‐1 receptor activity blockers such as anakinra (a recombinant version of the endogenous IL‐1 receptor antagonist, IL1‐RA; Granowitz et al., 1992) and canakinumab (a human monoclonal antibody targeted at IL‐1β; Alten et al., 2008; Figure 4). In contrast to the low MW caspase‐1 inhibitors, the structure of these agents necessitates subcutaneous administration (Granowitz et al., 1992; Alten et al., 2008) and implicitly blocks only the IL‐1β half of the inflammasome‐driven IL‐1β/IL‐18 cytokine pathway. Nonetheless, the recently published results of the CANTOS (Ridker et al., 2017b) trial support the idea that targeting IL‐1β produces an anti‐inflammatory effect in patients, at least in the context of secondary prevention following myocardial infarction (despite no reduction in overall mortality). A secondary analysis also revealed that patients receiving canakinumab had a significantly lower incidence of lung cancer compared to placebo, and the associated mortality was also reduced. Interestingly, patients that developed lung cancer exhibited increased IL‐6 levels, and these were lowered following treatment with canakinumab (Ridker et al., 2017a). These observations parallel the pathobiology of PH which is characterized by marked increases in circulating IL‐6 and is increasingly thought of as a hyper‐proliferative, anti‐apoptotic, metabolically disturbed disorder (Boucherat et al., 2017). Notwithstanding, there was a higher incidence of fatal infection and sepsis in those patients receiving canakinumab compared to placebo (Ridker et al., 2017b). Given that blocking IL‐1β will not only suppress the aberrant inflammation known to exist in cardiovascular disease (Libby, 2012) but also compromise the infection‐fighting capacity of the immune system in general, this is not entirely surprising. Nonetheless, these findings are very promising as they provide a ‘proof of concept’ for the use of drugs that modulate inflammasome‐related cytokines in human vascular disease and endorse further investigation into this pathway. Indeed, a pilot study, run by Virginia Commonwealth University, USA, is currently underway to investigate the efficacy and safety of another IL‐1 modulator, anakinra, in PH patients, with results expected by April 2019 (clinical trials US identifier: NCT03057028). Intriguingly, long before inflammasomes were discovered, Voelkel et al. (1994) found that treatment with an IL‐1 receptor antagonist improved monocrotaline‐induced, but not hypoxia‐induced PH in rats; more recently, anakinra was found to attenuate both monocrotaline‐induced PH in rats and hypoxia‐induced PH in mice (Parpaleix et al., 2016; Figure 4). Given the differences between experimental models and species seen in animal studies, it will be interesting to see if anakinra produces positive effects in PH patients.
The alternate cytokine pathway triggered by the inflammasome involves IL‐18. To date, little information exists on the modulation of IL‐18 activity in the setting of PH and that available is somewhat conflicting. On one hand, Morisawa et al. (2016) showed that genetic IL‐18 disruption suppresses hypoxia‐induced PH in mice, but conversely, Bruns et al. (2016) concluded that genetic IL‐18 deletion alone is not sufficient to prevent RV hypertrophy and increased RVSP in response to hypoxia, suggesting that therapeutic approaches targeting both inflammasome cytokines, IL‐18 and IL‐1β would be more efficacious. A human recombinant version of the endogenous IL‐18 binding protein (IL‐18BP), a decoy receptor (r‐hIL‐18BP), was developed and underwent safety and efficacy evaluation in healthy volunteers and rheumatoid arthritis patients (Tak et al., 2006). This compound has shown promise in a rat model of experimental autoimmune myocarditis (Chang et al., 2014), but the conflicting results following blockade of IL‐18 alone in PH models raises the question as to whether this is a therapeutic avenue worth exploring further. Regardless, such studies do imply the involvement of both of the primary cytokines produced by the NLRP3 inflammasome, IL‐1β and IL‐18, in the pathogenesis of PH.
The early response IL‐1 family of cytokines released by inflammasomes promote the release of secondary cytokines lying further downstream (Labow et al., 1997; Dinarello, 2002; Cahill and Rogers, 2008; Mills et al., 2013); akin to the primary cytokines, most of these have also been implicated in PH. Firstly, IL‐1β stimulation promotes IL‐6 release from macrophages and T‐cells (Cahill and Rogers, 2008), and secondly, IL‐2 and IL‐12 are produced downstream of IL‐18 (Dinarello, 2002). Interestingly, Parpaleix et al. (2016) found that expression of IL‐1β, together with IL‐6, is up‐regulated in hypoxic mice, and IL‐1 receptor signalling is necessary for the development and progression of PH, implicating not only IL‐1β but also the IL‐1β/IL‐6 axis in this disease. Moreover, pharmacological IL‐6 blockade (Hashimoto‐Kataoka et al., 2015) or genetic deletion (Savale et al., 2009) protects mice from hypoxia‐induced PH, and conversely, lung‐specific IL‐6 overexpression spontaneously produces PH in mice, an effect worsened by chronic hypoxia (Steiner et al., 2009). Finally, subcutaneous injection of recombinant IL‐6 also induces PH in rats (Miyata et al., 2001) and mice (Golembeski et al., 2005). Importantly, these experimental studies are supported by clinical findings (as alluded to above), in which plasma concentrations of the inflammasome‐associated cytokines, IL‐1β, IL‐2, IL‐6, IL‐12 and IL‐18 were significantly increased in PH patients compared to healthy controls. Of these cytokines, increased IL‐6 and IL‐12 levels correlate with worse outcome and survival (Humbert et al., 1995; Soon et al., 2010; Ross et al., 2012; Groth et al., 2014). This body of evidence surrounding IL‐6 in PH led to the current UK‐wide clinical trial (TRANSFORM‐UK), examining the efficacy and safety of a 6‐month course of tocilizumab, a human monoclonal antibody targeting the IL‐6R in a small number of group 1 PH patients (Hernandez‐Sanchez et al., 2018). If this study proves successful, a large‐scale phase II trial will take place to fully elucidate the efficacy of this approach. Although in the very early stages, this is an exciting development in PH research, and it will be interesting to see if the results from this trial support the strong body of preclinical evidence for IL‐6 blockade, as a treatment strategy for PH.
As the efficacy of IL‐18 blockade alone in PH is unclear, and the CANTOS trial demonstrated that pan IL‐1β blockade is associated with an increased incidence of fatal infection (Ridker et al., 2017b), it is reasonable to hypothesize that inhibition of a specific inflammasome subtype is likely to be a safer therapeutic strategy. This is one of the reasons that the recently identified selective NLRP3 inflammasome inhibitor, MCC950 (Coll et al., 2015), might be ideal as a therapeutic agent. The ability to selectively inhibit activation of NLRP3 whilst sparing the activity of other inflammasomes (and their ability to produce IL‐1β and IL‐18) may offer therapeutic benefit, but still allow appropriate response to infection if necessary. Moreover, since MCC950 has a relatively short half‐life and is orally bioavailable, in the setting of infection, it could be withdrawn to allow immune function to restore quickly. While the precise mechanism of action of MCC950 remains to be elucidated, Coll et al. (2015) successfully demonstrated that MCC950 selectively inhibits activation (but not priming) of the NLRP3 inflammasome both in vitro and in vivo. Since then, it has shown promising results as a therapeutic agent, being utilized in numerous preclinical models of disease. Those of particular interest in the context of PH include cardiovascular and metabolic disorders such as hypertension (Krishnan et al., 2016), myocardial infarction (van Hout et al., 2017), atherosclerosis (van der Heijden et al., 2017) and obesity (Pavillard et al., 2017) along with lung‐associated conditions like asthma (Kim et al., 2017) and pulmonary inflammation (Primiano et al., 2016). The fact that the NLRP3 inflammasome is implicated in other lung and cardiovascular diseases and that MCC950 treatment shows promise in these conditions, taken together with the knowledge that inflammasome related cytokines are elevated in PH patients (Soon et al., 2010; Ross et al., 2012) and that NLRP3 activation occurs in animal models of PH (Villegas et al., 2013; Cero et al., 2015; Tang et al., 2015; Zeng et al., 2017), lends heavily to the possibility that specific targeting of NLRP3, may be beneficial in PH.
These studies support a role, in particular, for the NLRP3 inflammasome in PH. By contrast, Cero et al. (2015) found that ASC−/−, but not NLRP3−/− mice, were protected from hypoxia‐induced PH. Additionally, there is some indirect evidence that the NLRP1 inflammasome could contribute to PH pathogenicity. Kovarova et al. (2012) found that administration of a lethal toxin from anthrax in mice led to pyroptosis induced by NLRP1 activation, causing acute lung injury and death, but mice genetically deficient in NLRP1 were protected from these effects. Intriguingly, gain of function polymorphisms in the NLRP1 gene appear to be associated with asthma in a cohort of Brazilian children (Leal et al., 2018). Taken together, these data suggest that the NLRP1 inflammasome plays a role in lung disease, but whether this holds true in PH remains to be seen. The observations of Cero et al. (2015), along with the indirect evidence for a role of the NLRP1 inflammasome in PH, are interesting as it raises the possibility that even though there is abundantly more information currently available on the NLRP3 inflammasome, it is not the sole contributor to the pathogenesis of PH. Since the ASC component is common to a number of inflammasome complexes aside from NLRP3, namely, NLRP1, AIM2 and IFI16 (Lee et al., 2016), these other inflammasomes, or indeed a combination of inflammasomes, may contribute to PH. Unfortunately, due to the lack of selective inhibitors available for other inflammasome varieties, teasing out the contribution of each could be difficult. Mice deficient in NLRP1 have been developed and utilized in other disease models (Kovarova et al., 2012), so further studies could use these mice to help substantiate the above findings and uncover the possible contribution of these inflammasomes.
Conclusions
Inflammasomes, particularly NLRP3, have emerged as promising drug targets in the setting of PH. Both clinical and preclinical evidence support the thesis that modulating the inflammasome, at multiple levels, confers beneficial effects in the disease. Such modulation may encompass disrupting upstream signals such as the P2X7 receptors and NF‐κB signal transduction, through to depletion of macrophages and interruption of inflammasome‐related cytokines (e.g. IL‐1β, IL‐6 and IL‐18), as well as inflammasome components (i.e. NLRP3, ASC and caspase‐1). In reality, not all of these targets are therapeutically viable, as targeting too far upstream, such as at the level of priming, is likely to have unpredictable side effects. The effectiveness of anakinra and canakinumab in auto‐inflammatory disorders, and proof‐of‐concept results from the CANTOS trial, coupled with evidence for the pathogenicity of IL‐1β in PH, suggests that drugs modifying inflammasome‐generated cytokines might be an efficacious strategy to treat the disease. This is an exciting new avenue in the PH field, and as such, future studies should aim to elucidate the precise nature, and function, of inflammasome(s) involved in the pathogenesis of PH to develop novel and more effective therapies.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c, 2017d).
Conflict of interest
A.H. has been a consultant/advisory board member for Bayer AG, Serodus ASA and Palatin Technologies Inc.
Acknowledgements
This work is supported by the Commonwealth Scholarship Commission, UK and the Australian Government Research Training Program (RTP).
Scott T. E., Kemp‐Harper B. K., and Hobbs A. J. (2019) Inflammasomes: a novel therapeutic target in pulmonary hypertension?, British Journal of Pharmacology, 176, 1880–1896, 10.1111/bph.14375.
References
- Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK et al (2015). Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation 132: 2037–2099. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: 17–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: 225–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: 272–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: 130–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alten R, Gram H, Joosten LA, van den Berg WB, Sieper J, Wassenberg S et al (2008). The human anti‐IL‐1 beta monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof‐of‐concept study in patients with rheumatoid arthritis. Arthritis Res Ther 10: R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders HJ, Muruve DA (2011). The inflammasomes in kidney disease. J Am Soc Nephrol 22: 1007–1018. [DOI] [PubMed] [Google Scholar]
- Baliga RS, Zhao L, Madhani M, Lopez‐Torondel B, Visintin C, Selwood D et al (2008). Synergy between natriuretic peptides and phosphodiesterase 5 inhibitors ameliorates pulmonary arterial hypertension. Am J Respir Crit Care Med 178: 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al (2009). Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan IS, Mensah KA, Rudkovskaia AA, Adonteng‐Boateng PK, Herzog EL, Buckley L et al (2018). Pulmonary arterial hypertension in the setting of scleroderma is different than in the setting of lupus: a review. Respir Med 134: 42–46. [DOI] [PubMed] [Google Scholar]
- Becker CE, O'Neill LA (2007). Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol 29: 239–248. [DOI] [PubMed] [Google Scholar]
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD (2012). An evaluation of long‐term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142: 448–456. [DOI] [PubMed] [Google Scholar]
- Bossone E, D'Andrea A, D'Alto M, Citro R, Argiento P, Ferrara F et al (2013). Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr 26: 1–14. [DOI] [PubMed] [Google Scholar]
- Boucherat O, Vitry G, Trinh I, Paulin R, Provencher S, Bonnet S (2017). The cancer theory of pulmonary arterial hypertension. Pulm Circ 7: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns DR, Buttrick PM, Walker LA (2016). Genetic ablation of interleukin‐18 does not attenuate hypobaric hypoxia‐induced right ventricular hypertrophy. Am J Physiol Lung Cell Mol Physiol 310: L542–L550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Rogers JT (2008). Interleukin (IL) 1β induction of IL‐6 is mediated by a novel phosphatidylinositol 3‐kinase‐dependent AKT/IκB kinase ɑ pathway targeting activator protein‐1. J Biol Chem 283: 25900–25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cero FT, Hillestad V, Sjaastad I, Yndestad A, Aukrust P, Ranheim T et al (2015). Absence of the inflammasome adaptor ASC reduces hypoxia‐induced pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol 309: L378–L387. [DOI] [PubMed] [Google Scholar]
- Chang H, Wang Y, Li G, Zhang L, Zhang GW, Liao YC et al (2014). Effect of hydrodynamics‐based delivery of IL‐18BP fusion gene on rat experimental autoimmune myocarditis. Clin Exp Med 14: 397–408. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y et al (2015). Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta 1853: 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow MT, Duret H, Andrews DM, Faveeuw C, Moller A, Smyth MJ et al (2014). Type I NKT‐cell‐mediated TNF‐ɑ is a positive regulator of NLRP3 inflammasome priming. Eur J Immunol 44: 2111–2120. [DOI] [PubMed] [Google Scholar]
- Chu Y, Xiangli X, Xiao W (2015). Regulatory T cells protect against hypoxia‐induced pulmonary arterial hypertension in mice. Mol Med Rep 11: 3181–3187. [DOI] [PubMed] [Google Scholar]
- Coenen DM, Mastenbroek TG, Cosemans J (2017). Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood 130: 2819–2828. [DOI] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz‐Planillo R, Inserra MC et al (2015). A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale M, Palmiotti GA, Lo Storto MM, Montrone D, Foschino Barbaro MP, Di Biase M et al (2015). HIV‐associated pulmonary arterial hypertension: from bedside to the future. Eur J Clin Invest 45: 515–528. [DOI] [PubMed] [Google Scholar]
- De Nardo D, De Nardo CM, Latz E (2014). New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol 184: 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir R, Kucukoglu MS (2015). Six‐minute walk test in pulmonary arterial hypertension. Anatolian J Cardiol 15: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA (2002). The IL‐1 family and inflammatory diseases. Clin Exp Rheumatol 20: S1–S13. [PubMed] [Google Scholar]
- Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD (2005). Activated platelets induce Weibel‐Palade‐body secretion and leukocyte rolling in vivo: role of P‐selectin. Blood 106: 2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong ZM, Brown AA, Wagner DD (2000). Prominent role of P‐selectin in the development of advanced atherosclerosis in ApoE‐deficient mice. Circulation 101: 2290–2295. [DOI] [PubMed] [Google Scholar]
- dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales‐Nebreda L et al (2015). Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 6: 6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464: 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S et al (2011). P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One 6: e24097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas D, Alhussaini AA, Kraskauskas D, Kraskauskiene V, Cool CD, Nicolls MR et al (2014). Nuclear factor κB inhibition reduces lung vascular lumen obliteration in severe pulmonary hypertension in rats. Am J Respir Cell Mol Biol 51: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine NM, Chen L, Bastiansen PM, Frantz RP, Pellikka PA, Oh JK et al (2013). Outcome prediction by quantitative right ventricular function assessment in 575 subjects evaluated for pulmonary hypertension. Circ Cardiovasc Imaging 6: 711–721. [DOI] [PubMed] [Google Scholar]
- Franchi L, Eigenbrod T, Nunez G (2009). Cutting edge: TNF‐ɑ mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: 792–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D et al (2005). Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353: 2148–2157. [DOI] [PubMed] [Google Scholar]
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al (2015). 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46: 903–975. [DOI] [PubMed] [Google Scholar]
- Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V et al (2009). Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179: 903–913. [DOI] [PubMed] [Google Scholar]
- Giaid A, Saleh D (1995). Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 333: 214–221. [DOI] [PubMed] [Google Scholar]
- Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H et al (1993). Expression of endothelin‐1 in the lungs of patients with pulmonary hypertension. N Engl J Med 328: 1732–1739. [DOI] [PubMed] [Google Scholar]
- Gitto E, Pellegrino S, Aversa S, Romeo C, Trimarchi G, Barberi I et al (2012). Oxidative stress and persistent pulmonary hypertension of the newborn treated with inhaled nitric oxide and different oxygen concentrations. J Matern Fetal Neonatal Med 25: 1723–1726. [DOI] [PubMed] [Google Scholar]
- Golembeski SM, West J, Tada Y, Fagan KA (2005). Interleukin‐6 causes mild pulmonary hypertension and augments hypoxia‐induced pulmonary hypertension in mice. Chest 128: 572s–573s. [DOI] [PubMed] [Google Scholar]
- Granowitz EV, Porat R, Mier JW, Pribble JP, Stiles DM, Bloedow DC et al (1992). Pharmacokinetics, safety and immunomodulatory effects of human recombinant interleukin‐1 receptor antagonist in healthy humans. Cytokine 4: 353–360. [DOI] [PubMed] [Google Scholar]
- Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC (2014). Inflammatory cytokines in pulmonary hypertension. Respir Res 15: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P et al (2011). Differential expression of NLRP3 among hematopoietic cells. J Immunol 186: 2529–2534. [DOI] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid‐β. Nat Immunol 9: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto‐Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T et al (2015). Interleukin‐6/interleukin‐21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A 112: E2677–E2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TS, Painter MW (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9: 1091–1094. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Sanchez J, Harlow L, Church C, Gaine S, Knightbridge E, Bunclark K et al (2018). Clinical trial protocol for TRANSFORM‐UK: a therapeutic open‐label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm Circ 8 2045893217735820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N et al (2017). Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J 50. PII: 1700740. [DOI] [PubMed] [Google Scholar]
- Hoeper MM, McLaughlin VV, Dalaan AM, Satoh T, Galie N (2016). Treatment of pulmonary hypertension. Lancet Respir Med 4: 323–336. [DOI] [PubMed] [Google Scholar]
- Hosokawa S, Haraguchi G, Sasaki A, Arai H, Muto S, Itai A et al (2013). Pathophysiological roles of nuclear factor κB (NF‐kB) in pulmonary arterial hypertension: effects of synthetic selective NF‐κB inhibitor IMD‐0354. Cardiovasc Res 99: 35–43. [DOI] [PubMed] [Google Scholar]
- Hottz ED, Lopes JF, Freitas C, Valls‐de‐Souza R, Oliveira MF, Bozza MT et al (2013). Platelets mediate increased endothelium permeability in dengue through NLRP3‐inflammasome activation. Blood 122: 3405–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot‐Keros L et al (1995). Increased interleukin‐1 and interleukin‐6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med 151: 1628–1631. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V et al (2010). Survival in patients with idiopathic, familial, and anorexigen‐associated pulmonary arterial hypertension in the modern management era. Circulation 122: 156–163. [DOI] [PubMed] [Google Scholar]
- Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M et al (2016). IL‐1 receptor antagonist ameliorates inflammasome‐dependent inflammation in murine and human cystic fibrosis. Nat Commun 7: 10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S (2008). Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3: 352–363. [DOI] [PubMed] [Google Scholar]
- Itoh T, Nagaya N, Ishibashi‐Ueda H, Kyotani S, Oya H, Sakamaki F et al (2006). Increased plasma monocyte chemoattractant protein‐1 level in idiopathic pulmonary arterial hypertension. Respirology (Carlton, Vic) 11: 158–163. [DOI] [PubMed] [Google Scholar]
- Jeffery TK, Wanstall JC (2001). Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther 92: 1–20. [DOI] [PubMed] [Google Scholar]
- Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR (2005). Potentiation of caspase‐1 activation by the P2X7 receptor is dependent on TLR signals and requires NF‐κB‐driven protein synthesis. J Immunol 175: 7611–7622. [DOI] [PubMed] [Google Scholar]
- Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC et al (2017). Role for NLRP3 inflammasome‐mediated, IL‐1β‐dependent responses in severe, steroid‐resistant asthma. Am J Respir Crit Care Med 196: 283–297. [DOI] [PubMed] [Google Scholar]
- Klinger JR, Thaker S, Houtchens J, Preston IR, Hill NS, Farber HW (2006). Pulmonary hemodynamic responses to brain natriuretic peptide and sildenafil in patients with pulmonary arterial hypertension. Chest 129: 417–425. [DOI] [PubMed] [Google Scholar]
- Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z et al (2012). NLRP1‐dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol 189: 2006–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D et al (2016). Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt‐induced hypertension in mice. Br J Pharmacol 173: 752–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR (2014). IL‐1β and IL‐18: inflammatory markers or mediators of hypertension? Br J Pharmacol 171: 5589–5602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler WM, Bonnet S, Tabuchi A (2018). Inflammation and autoimmunity in pulmonary hypertension: is there a role for endothelial adhesion molecules? (2017 Grover Conference Series). Pulm Circ 8 2045893218757596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R et al (2012). Cardiac‐specific genetic inhibition of nuclear factor‐κB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol 302: H1655–H1666. [DOI] [PubMed] [Google Scholar]
- Labow M, Shuster D, Zetterstrom M, Nunes P, Terry R, Cullinan EB et al (1997). Absence of IL‐1 signaling and reduced inflammatory response in IL‐1 type I receptor‐deficient mice. J Immunol 159: 2452–2461. [PubMed] [Google Scholar]
- Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT (2014). Pulmonary arterial hypertension: the clinical syndrome. Circ Res 115: 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie AC, Bonnet S, Provencher S (2017). Combination therapy in pulmonary arterial hypertension: recent accomplishments and future challenges. Pulm Circ 7: 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K (2005). Interleukin‐1β causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol 32: 311–318. [DOI] [PubMed] [Google Scholar]
- Leal VNC, Genov IR, Mallozi MC, Sole D, Pontillo A (2018). Polymorphisms in inflammasome genes and risk of asthma in Brazilian children. Mol Immunol 93: 64–67. [DOI] [PubMed] [Google Scholar]
- Lee S, Suh GY, Ryter SW, Choi AM (2016). Regulation and function of the nucleotide binding domain leucine‐rich repeat‐containing receptor, pyrin domain‐containing‐3 inflammasome in lung disease. Am J Respir Cell Mol Biol 54: 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wei C, Kim IK, Janssen‐Heininger Y, Gupta S (2014). Inhibition of nuclear factor‐κB in the lungs prevents monocrotaline‐induced pulmonary hypertension in mice. Hypertension 63: 1260–1269. [DOI] [PubMed] [Google Scholar]
- Libby P (2012). Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 32: 2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lievens D, von Hundelshausen P (2011). Platelets in atherosclerosis. Thromb Haemost 106: 827–838. [DOI] [PubMed] [Google Scholar]
- Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS et al (2012). Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 186: 790–796. [DOI] [PubMed] [Google Scholar]
- Liu P, Xie Q, Wei T, Chen Y, Chen H, Shen W (2015). Activation of the NLRP3 inflammasome induces vascular dysfunction in obese OLETF rats. Biochem Biophys Res Commun 468: 319–325. [DOI] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR et al (2014). Unified polymerization mechanism for the assembly of ASC‐dependent inflammasomes. Cell 156: 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie SH, Schipper JL, Clark AC (2010). The potential for caspases in drug discovery. Curr Opin Drug Discov Devel 13: 568–576. [PMC free article] [PubMed] [Google Scholar]
- Malik A, Kanneganti TD (2017). Inflammasome activation and assembly at a glance. J Cell Sci 130: 3955–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM et al (2015). Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity 43: 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Kanneganti TD (2015). Regulation of inflammasome activation. Immunol Rev 265: 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M et al (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐β. Mol Cell 10: 417–426. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006). Gout‐associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241. [DOI] [PubMed] [Google Scholar]
- Matura LA, Ventetuolo CE, Palevsky HI, Lederer DJ, Horn EM, Mathai SC et al (2015). Interleukin‐6 and tumor necrosis factor‐ɑ are associated with quality of life‐related symptoms in pulmonary arterial hypertension. Ann Am Thorac Soc 12: 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KH, Dungan LS, Jones SA, Harris J (2013). The role of inflammasome‐derived IL‐1 in driving IL‐17 responses. J Leukoc Biol 93: 489–497. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Ahmetaj‐Shala B, Kirkby NS, Wright WR, Mackenzie LS, Reed DM et al (2014). Role of prostacyclin in pulmonary hypertension. Global Cardiol Sci Pract 2014: 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Ito M, Sasajima T, Ohira H, Kasukawa R (2001). Effect of a serotonin receptor antagonist on interleukin‐6‐induced pulmonary hypertension in rats. Chest 119: 554–561. [DOI] [PubMed] [Google Scholar]
- Montani D, Chaumais MC, Guignabert C, Gunther S, Girerd B, Jais X et al (2014). Targeted therapies in pulmonary arterial hypertension. Pharmacol Ther 141: 172–191. [DOI] [PubMed] [Google Scholar]
- Morisawa D, Hirotani S, Oboshi M, Nishimura K, Sawada H, Eguchi A et al (2016). Interleukin‐18 disruption suppresses hypoxia‐induced pulmonary artery hypertension in mice. Int J Cardiol 202: 522–524. [DOI] [PubMed] [Google Scholar]
- Nair J, Lakshminrusimha S (2014). Update on PPHN: mechanisms and treatment. Semin Perinatol 38: 78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpaleix A, Amsellem V, Houssaini A, Abid S, Breau M, Marcos E et al (2016). Role of interleukin‐1 receptor 1/MyD88 signalling in the development and progression of pulmonary hypertension. Eur Respir J 48: 470–483. [DOI] [PubMed] [Google Scholar]
- Pavillard LE, Canadas‐Lozano D, Alcocer‐Gomez E, Marin‐Aguilar F, Pereira S, Robertson AAB et al (2017). NLRP3‐inflammasome inhibition prevents high fat and high sugar diets‐induced heart damage through autophagy induction. Oncotarget 8: 99740–99756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoriler JE, Rich S, Archer SL, Husain AN (2012). Persistence of complex vascular lesions despite prolonged prostacyclin therapy of pulmonary arterial hypertension. Histopathology 61: 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston IR, Hill NS, Gambardella LS, Warburton RR, Klinger JR (2004). Synergistic effects of ANP and sildenafil on cGMP levels and amelioration of acute hypoxic pulmonary hypertension. Exp Biol Med (Maywood) 229: 920–925. [DOI] [PubMed] [Google Scholar]
- Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD et al (2016). Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP‐456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol (Baltimore, Md: 1950) 197: 2421–2433. [DOI] [PubMed] [Google Scholar]
- Pugliese SC, Poth JM, Fini MA, Olschewski A, El Kasmi KC, Stenmark KR (2015). The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am J Physiol Lung Cell Mol Physiol 308: L229–L252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Fulton D (2013). Post‐translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front Physiol 4: 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M, Guignabert C, Humbert M, Nicolls MR (2014). Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 115: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich JD, Rich S (2014). Clinical diagnosis of pulmonary hypertension. Circulation 130: 1820–1830. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C et al (2017a). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ (2017b). Effect of interleukin‐1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double‐blind, placebo‐controlled trial. Lancet 390: 1833–1842. [DOI] [PubMed] [Google Scholar]
- Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L et al (2010). Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med 182: 774–783. [DOI] [PubMed] [Google Scholar]
- Ross DJ, Strieter RM, Fishbein MC, Ardehali A, Belperio JA (2012). Type I immune response cytokine‐chemokine cascade is associated with pulmonary arterial hypertension. J Heart Lung Transplant 31: 865–873. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, Tian L et al (2015). Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol 31: 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L et al (2012). Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 897–908. [DOI] [PubMed] [Google Scholar]
- Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S et al (2009). Impact of interleukin‐6 on hypoxia‐induced pulmonary hypertension and lung inflammation in mice. Respir Res 10: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S et al (2015). Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo‐electron microscopy. Proc Natl Acad Sci U S A 112: 13237–13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seferian A, Simonneau G (2013). Therapies for pulmonary arterial hypertension: where are we today, where do we go tomorrow? Eur Respir Rev 22: 217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimovic N, Bergh CH, Andersson B, Sakiniene E, Carlsten H, Rundqvist B (2009). Growth factors and interleukin‐6 across the lung circulation in pulmonary hypertension. Eur Respir J 34: 662–668. [DOI] [PubMed] [Google Scholar]
- Semenov AV, Kogan‐Ponomarev M, Ruda M, Komarov AL, Panchenko EP, Chazova IE et al (2000). Soluble P‐selectin – a marker of platelet activation and vessel wall injury: increase of soluble P‐selectin in plasma of patients with myocardial infarction, massive atherosclerosis and primary pulmonary hypertension. Ter Arkh 72: 15–20. [PubMed] [Google Scholar]
- Seo B, Oemar BS, Siebenmann R, von Segesser L, Luscher TF (1994). Both ETA and ETB receptors mediate contraction to endothelin‐1 in human blood vessels. Circulation 89: 1203–1208. [DOI] [PubMed] [Google Scholar]
- Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C (2015). NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol 6: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A et al (2013). Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34–D41. [DOI] [PubMed] [Google Scholar]
- Simpson JL, Phipps S, Baines KJ, Oreo KM, Gunawardhana L, Gibson PG (2014). Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 43: 1067–1076. [DOI] [PubMed] [Google Scholar]
- Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD et al (2010). Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122: 920–927. [DOI] [PubMed] [Google Scholar]
- Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV et al (2012). Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB (2009). Interleukin‐6 overexpression induces pulmonary hypertension. Circ Res 104: 236–244 228p following 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Csak T (2012). Inflammasomes in liver diseases. J Hepatol 57: 642–654. [DOI] [PubMed] [Google Scholar]
- Taichman DB, McGoon MD, Harhay MO, Archer‐Chicko C, Sager JS, Murugappan M et al (2009). Wide variation in clinicians' assessment of New York Heart Association/World Health Organization functional class in patients with pulmonary arterial hypertension. Mayo Clin Proc 84: 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Bacchi M, Bertolino M (2006). Pharmacokinetics of IL‐18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur J Drug Metab Pharmacokinet 31: 109–116. [DOI] [PubMed] [Google Scholar]
- Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L et al (2011). Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Chen GX, Liang MY, Yao JP, Wu ZK (2015). Ellagic acid prevents monocrotaline‐induced pulmonary artery hypertension via inhibiting NLRP3 inflammasome activation in rats. Int J Cardiol 180: 134–141. [DOI] [PubMed] [Google Scholar]
- Thomas MR, Storey RF (2015). The role of platelets in inflammation. Thromb Haemost 114: 449–458. [DOI] [PubMed] [Google Scholar]
- Trammell AW, Pugh ME, Newman JH, Hemnes AR, Robbins IM (2015). Use of pulmonary arterial hypertension‐approved therapy in the treatment of non‐group 1 pulmonary hypertension at US referral centers. Pulm Circ 5: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L et al (1999). Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 159: 1925–1932. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Groves B, Badesch DB, Voelkel NF (1994). Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 144: 275–285. [PMC free article] [PubMed] [Google Scholar]
- van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B et al (2017). NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E‐deficient mice – brief report. Arterioscler Thromb Vasc Biol 37: 1457–1461. [DOI] [PubMed] [Google Scholar]
- van Hout GP, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA et al (2017). The selective NLRP3‐inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J 38: 828–836. [DOI] [PubMed] [Google Scholar]
- Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C et al (2017). Interleukin‐1 blockade in recently decompensated systolic heart failure: results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 10. PII: e004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas LR, Kluck D, Field C, Oberley‐Deegan RE, Woods C, Yeager ME et al (2013). Superoxide dismutase mimetic, MnTE‐2‐PyP, attenuates chronic hypoxia‐induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid Redox Signal 18: 1753–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Tuder RM, Bridges J, Arend WP (1994). Interleukin‐1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am J Respir Cell Mol Biol 11: 664–675. [DOI] [PubMed] [Google Scholar]
- von Hundelshausen P, Weber C (2007). Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 100: 27–40. [DOI] [PubMed] [Google Scholar]
- Wanderer AA (2008). Interleukin‐1β targeted therapy in severe persistent asthma (SPA) and chronic obstructive pulmonary disease (COPD): proposed similarities between biphasic pathobiology of SPA/COPD and ischemia‐reperfusion injury. Isr Med Assoc J: IMAJ 10: 837–842. [PubMed] [Google Scholar]
- Wang Q, Zuo XR, Wang YY, Xie WP, Wang H, Zhang M (2013). Monocrotaline‐induced pulmonary arterial hypertension is attenuated by TNF‐ɑ antagonists via the suppression of TNF‐ɑ expression and NF‐κB pathway in rats. Vascul Pharmacol 58: 71–77. [DOI] [PubMed] [Google Scholar]
- Wen H, Ting JP, O'Neill LA (2012). A role for the NLRP3 inflammasome in metabolic diseases – did Warburg miss inflammation? Nat Immunol 13: 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MR, Paul GA, Strange JW, Tunariu N, Gin‐Sing W, Banya WA et al (2005). Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med 171: 1292–1297. [DOI] [PubMed] [Google Scholar]
- Yang W, Ni H, Wang H, Gu H (2015). NLRP3 inflammasome is essential for the development of chronic obstructive pulmonary disease. Int J Clin Exp Pathol 8: 13209–13216. [PMC free article] [PubMed] [Google Scholar]
- Yang XM, Downey JM, Cohen MV, Housley NA, Alvarez DF, Audia JP (2017). The highly selective caspase‐1 inhibitor VX‐765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J Cardiovasc Pharmacol Ther 22: 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, You S, Liu H, Chen L, Zhang C, Hu H et al (2017). Role of P2X7R in the development and progression of pulmonary hypertension. Respir Res 18: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A et al (2013). Canonical Nlrp3 inflammasome links systemic low‐grade inflammation to functional decline in aging. Cell Metab 18: 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X et al (2016). Caspase‐1 inflammasome activation mediates homocysteine‐induced pyrop‐apoptosis in endothelial cells. Circ Res 118: 1525–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL (2014). Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol 184: 1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G et al (2011). Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J Immunol 187: 4809–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaloudikova M, Vytasek R, Vajnerova O, Hnilickova O, Vizek M, Hampl V et al (2016). Depletion of alveolar macrophages attenuates hypoxic pulmonary hypertension but not hypoxia‐induced increase in serum concentration of MCP‐1. Physiol Res 65: 763–768. [DOI] [PubMed] [Google Scholar]
- Zeng M, Sang W, Chen S, Liu Y, Zhang H, Kong X (2017). Inhibition of ER stress by 4‐PBA protects MCT‐induced pulmonary arterial hypertension via reducing NLRP3 inflammasome activation. Int J Clin Exp Pathol 10: 4162–4172. [Google Scholar]
- Zhao L, Mason NA, Strange JW, Walker H, Wilkins MR (2003). Beneficial effects of phosphodiesterase 5 inhibition in pulmonary hypertension are influenced by natriuretic peptide activity. Circulation 107: 234–237. [DOI] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010). Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11: 136–140. [DOI] [PubMed] [Google Scholar]